

Abstract
Two natural product-like inhibitors of TNF-α have been identified using structure-based virtual screening. These compounds represent only the third and fourth examples of direct target of TNF-α by a small molecule and display comparable potency to the strongest TNF-α inhibitor reported to date.
Keywords: inhibitors, tumor necrosis factor, virtual screening, natural products, drug discovery
Tumor necrosis factor α (TNF-α) is a multifunctional cytokine that acts as a central biological mediator for critical immune functions, including inflammation, infection, and antitumour responses.[1] Dysregulation of TNF-α has been implicated in cases of tumorigenesis, diabetes, and especially in autoinflammatory diseases such as rheumatoid arthritis, psoriatic arthritis and Crohn’s disease.[2] The synthetic antibodies etanercept, infliximab, and adalimumab approved for the treatment of inflammatory diseases bind to TNF-α directly, preventing its association with the tumor necrosis factor receptor (TNFR).[3] However, their potential to cause serious side effects such as eliciting an autoimmune anti-antibody response or the weakening of the body’s immune defenses to opportunistic infections, has stimulated the development of alternative small molecule-based therapies to TNF-α inhibition.[4] Most such small molecule inhibitors reported in the literature target TNF-α indirectly.[5–8]
To our knowledge, the only small molecules capable of antagonizing TNF-α directly are the polysulfonated naphthylurea suramin and its analogues,[9] and the indole-linked chromone designated SPD304 (Figure 1).[10] Unfortunately, the low potency and poor selectivity of suramin coupled with its tendency to cause adverse side effects renders it unsuitable for anti-TNF-α therapies.[11] Furthermore, SPD304 containing the toxic 3-alkylindole moiety was found to be metabolized by cytochrome P450 enzymes via a similar dehydrogenation pathway as the potent pneumotoxin 3-methylindole, producing reactive electrophilic iminium species capable of conjugating protein and/or DNA targets.[12] Therefore, the development of relatively less toxic small molecule inhibitors of TNF-α for therapeutic applications remains a highly desirable goal.
Figure 1.

Chemical structures of small molecule TNF-α inhibitors quinuclidine 1, indoloquinolizidine 2 and SPD304.
Natural products (NPs) have been refined over evolutionary time scales for optimal interactions with biomolecules. Not surprisingly, NPs have represented a cornerstone of pharmaceutical research, as they offer a diverse range of chemical scaffolds, bioactive substructures, and potentially lower toxicity profiles.[13] Historically, many approved drugs have been NPs, while numerous others were derived from or inspired by a NP template.[14] Encouraged by these ideas, and by the relative dearth of potent and non-toxic small molecule inhibitors directly targeting TNF-α, we sought to apply high-throughput, ligand docking-based virtual screening methods to identify TNF-α inhibitors from a natural product chemical libraries. We used the X-ray co-crystal structure of TNF-α dimer with SPD304 (PDB code: 2AZ5)[10] as the molecular model for our investigation.
Like most protein-protein interfaces, the binding pocket of the TNF-α dimer is relatively large and featureless, and lacks clearly-defined binding crevices or mechanism-based contacts.[15] The binding site is mostly hydrophobic, consisting primarily of glycine, leucine and tyrosine residues. Not unexpectedly, the binding interaction of small molecule SPD304 to TNF-α has been described to be predominantly hydrophobic and shape-driven.[10] Small-molecule inhibitors of TNF-α should thus be relatively hydrophobic and large enough to contact both subunits of the TNF-α dimer simultaneously, in order to prevent the binding of the third subunit forming the biologically active trimer complex.
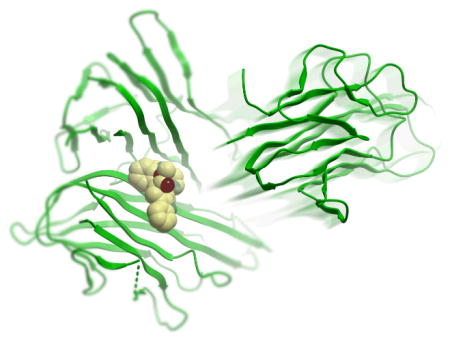
Over 20,000 compounds from a chemical library of natural product/natural product-like structures[16] were screened in silico. The continuously flexible ligands were docked to a grid representation of the receptor and assigned a score reflecting the quality of the complex according to the internal coordinate mechanics (ICM) method [ICM-Pro 3.6-1d molecular docking software (Molsoft)].[18] The highest-scoring 16 compounds from the virtual screening results were tested in a preliminary ELISA to assess their ability to inhibit the binding of TNF-α to TNFR-1. Two chemically distinct structures, the pyrazole-linked quinuclidine 1 and the indolo[2,3-a]quinolizidine 2, emerged as the top candidates (Figure 1). The binding poses of these two compounds overlap well with the crystallographic pose of SPD304 to TNF-α (Figure 2). Like SPD304, compounds 1 and 2 are large enough to contact residues from both subunits of the TNF-α dimer, thus occupying and blocking the binding site for the third TNF-α subunit.
Figure 2.

Low-energy binding conformations of a) 1, b) 2 and c) SPD304 bound to TNF-α dimer generated by virtual ligand docking. The two subunits of the TNF-α dimer are depicted in ribbon form and are colored in contrasted purple (subunit A) and red (subunit B). The small molecules are depicted as a ball-and-stick model showing carbon (yellow), hydrogen (grey), oxygen (red), nitrogen (blue), and fluoride (green) atoms. Hydrogen bonds are depicted as dotted lines. The binding pocket of the TNF-α dimer is represented as a translucent green surface.
In the top-scoring binding mode of 1 to the TNF-α dimer, the pyrazole-linked quinuclidine substructure occupies the hydrophobic binding pocket, and the dioxolane oxygen atom of 1 forms a hydrogen bond with the backbone amino group of Gly121 of TNF-a subunit B (Figure 2a). Compound 2 is not predicted to occupy the region of space close to Gly121 of subunit B, but instead forms a hydrogen bond with the side-chain hydroxyl group of Tyr151 of subunit B through its imidazole functionality (Figure 2b). Common features of the predicted binding modes of 1, 2 and SPD304 are the extended hydrophobic ring systems that are in contact with the β-strand (Leu120–Gly121–Gly122) of TNF-α subunit A, and the presence of polar functional groups orientated away from the binding pocket and exposed to the aqueous environment. Interestingly, whereas the indole substructures of 2 and SPD304 (Figure 2c) are located in a similar region of space, their orientations with respect to the β-strand of subunit A are different. The lack of salt bridges or hydrogen bonding networks in our models of 1 and 2 with TNF-α is consistent with previous findings that the interaction between the small molecule SPD304 and TNF-α is primarily hydrophobic and shape-driven.10 The calculated binding scores of −34.7 and −36.4 for 1 and 2 respectively reflect a strong interaction between the compounds and the dimer complex. As a reference, we calculated the binding score of SPD304 to be −32.9. The predicted binding coordinates of SPD304 in the binding pocket are within 1.0 Å root-mean-square deviation of the reported values based on the protein X-ray crystal structure.[10]
The quinuclidine core of 1 is present in a variety of natural products, such as the antimalarial cinchona alkaloids.[18] Natural products containing the indolo[2,3-a]quinolizidine scaffold of 2 include the alkaloids geissoschizine, deplancheine, corynantheidine, and yohimbane.[19] Waldmann and co-workers employed a biology-orientated synthetic approach to generate indolo[2,3-a]quinolizidine inhibitors of mycobacterial protein tyrosine phosphatase B.[20] To the best of our knowledge, no TNF-α-binding activity nor any other biological activity of 1 or 2 has been reported in the literature.
To validate the results of our molecular modeling, we performed dose-response experiments with compounds 1 and 2 to determine their half-maximal inhibitory concentration (IC50) values against the TNF-α–TNFR-1 interaction using an ELISA (Figure 3). Encouragingly, indoloquinolizidine 2 (IC50 = ca. 10 μM) was found to be more active than SPD304, the most potent small molecule TNF-α inhibitor reported to date (IC50 = 22 μM by a comparable ELISA).[10,21] Quinuclidine 1 was moderately active against TNF-α with an IC50 value of approximately 50 μM.
Figure 3.
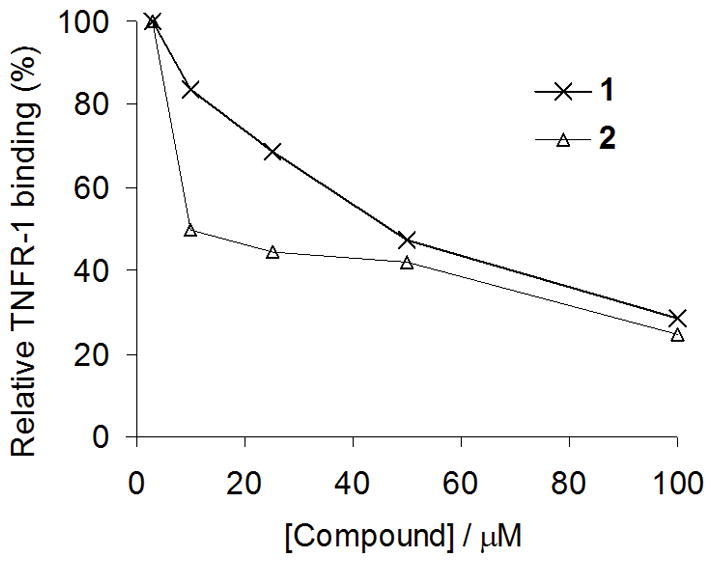
Compound inhibition of TNFR-1 binding to immobilized TNF-α (ELISA). Microtitre plates coated with TNF-α were incubated with TNFR-1 together with 1 or 2 at the indicated concentrations. TNFR-1 binding was detected using anti-TNFR antibody and horseradish peroxidase-conjugated secondary antibody. Approximate IC50 values; 1: 10 μM, 2: 50 μM.
We next investigated the ability of compounds 1 and 2 to inhibit TNF-α signaling in human cells. TNF-α solutions pre-incubated with the test compound were added to HepG2 cells, which were stably transfected with the NF-κB–luciferase gene. The inhibition of TNF-α-induced NF-κB signaling by the test compound was detected by monitoring the reduction in the luciferase activity of the cell lysates (Figure 4). Surprisingly, indoloquinolizidine 2 (IC50 > 30 μM) was found to be less active than quinuclidine 1 (IC50 = ca. 5 μM) in the cellular luciferase assay, despite showing greater potency in the cell-free ELISA. Notably, 2 exhibited a similar IC50 value similar to that of to SPD304 (IC50 = ca. 3 μM) as measured by our system, although it was less potent than SPD304 at higher concentrations. We hypothesize that the markedly reduced activity of 2 in cell culture could be because of its low bioavailability resulting from either poor cellular uptake or metabolic degradation of 2.[22]
Figure 4.
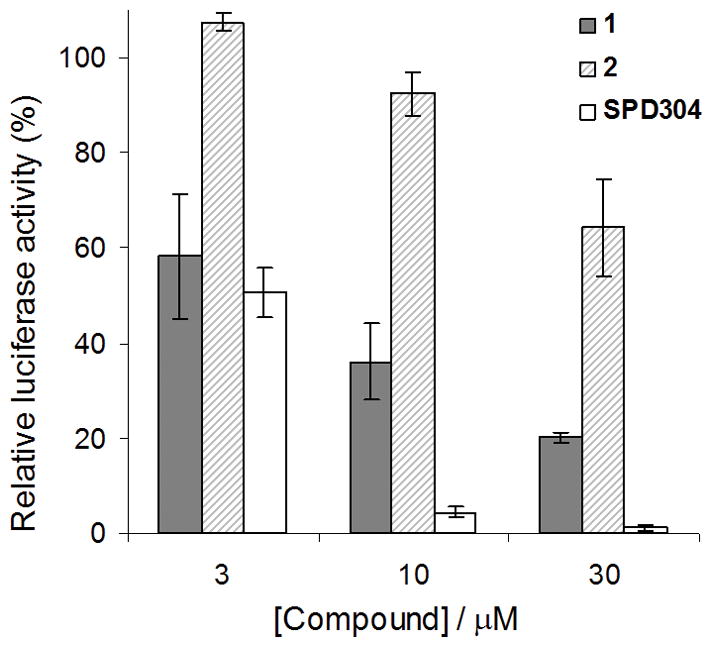
Compound inhibition of cellular TNF-α-induced NF-κB activity. HepG2 cells stably transfected with the NF-κB–luciferase gene were stimulated with TNF-α pre-incubated with the indicated concentrations of 1, 2, or SPD304. Cell lysates were analyzed for luciferase activity to determine the extent of NF-κB inhibition. Approximate IC50 values; 1: 5 μM, 2: >30 μM, SPD304: 3 μM.
In conclusion, we have discovered two small molecules TNF-α inhibitors from a natural-product and natural-product-like chemical library using structure-based design. The identification of quinuclidine 1 and quinolizine 2 represents, to the best of our knowledge, only the third and fourth examples of the direct targeting of TNF-α by a small molecule. Importantly, indoloquinolizidine 2 (IC50 = ca. 10 μM) was found to be more potent against TNF-α in the ELISA compared to SPD304, the strongest small molecule TNF-α inhibitor reported to date. Quinuclidine 1 (IC50 = ca. 5 μM) displayed a comparable activity to SPD304 (IC50 = ca. 3 μM) against cellular TNF-α induced NF-κB signaling. We are currently conducting computer-based hit-to-lead optimization to generate further analogues for in vitro testing.
Acknowledgments
This work was supported by the Area of Excellence Scheme established under the University Grants Committee of the Hong Kong Special Administrative Region, China (AoE/P-10/01), the University of Hong Kong (University Development Fund), the University of Hong Kong Seed Funding Programme for Applied Research, and the University of Hong Kong Seed Funding Programme for Basic Research.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Daniel Shiu-Hin Chan, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176.
Dr. Ho-Man Lee, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176.
Fang Yang, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176.
Prof. Dr. Chi-Ming Che, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176
Dr. Catherine C. L. Wong, Department of Chemical Physiology, The Scripps Research Institute, La Jolla, California, USA
Prof. Ruben Abagyan, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, California, USA
Dr. Chung-Hang Leung, Email: duncanl@hku.hk, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176
Dr. Dik-Lung Ma, Email: edmondma@hku.hk, Department of Chemistry and Open Laboratory of Chemical, Biology of the Institute of Molecular Technology for Drug, Discovery and Synthesis, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, Fax: (+852) 2915 5176
References
- 1.Wajant H, Pfizenmaier K, Scheurich P. Cell Death Diff. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 3.Chatzantoni K, Mouzaki A. Curr Top Med Chem. 2006;6:1707–1714. doi: 10.2174/156802606778194217. [DOI] [PubMed] [Google Scholar]
- 4.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Nat Rev Drug Discov. 2003;2:736–746. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 5.Haraguchi S, Day NK, Kamchaisatian W, Beigier-Pompadre M, Stenger S, Tangsinmankong N, Sleasman JW, Pizzo SV, Cianciolo GJ. AIDS Res Ther. 2006;3:8. doi: 10.1186/1742-6405-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen H, McCann PP. Pharmacol Ther. 1997;75:69–75. doi: 10.1016/s0163-7258(97)00023-5. [DOI] [PubMed] [Google Scholar]
- 7.Lee MR, Dominguez C. Curr Med Chem. 2005;12:2979–2994. doi: 10.2174/092986705774462914. [DOI] [PubMed] [Google Scholar]
- 8.a) Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi F. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]; b) Leung CH, Grill SP, Lam W, Gao W, Sun HD, Cheng YC. Mol Pharmacol. 2006;70:1946–1955. doi: 10.1124/mol.106.028480. [DOI] [PubMed] [Google Scholar]
- 9.a) Alzani R, Cortin A, Grazioli L, Cozzi E, Ghezzi P, Marcucci F. J Biol Chem. 1993;268:12526–12529. [PubMed] [Google Scholar]; b) Mancini F, Toro CM, Mabilia M, Giannangeli M, Pinza M, Milanese C. Biochem Pharmacol. 1999;58:851–859. doi: 10.1016/s0006-2952(99)00150-1. [DOI] [PubMed] [Google Scholar]
- 10.He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, Fung AD, Farrington G, Eldredge JK, Day ES, Cruz LA, Cachero TG, Miller SK, Friedman JE, Choong IC, Cunningham BC. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- 11.McGeary RP, Bennett AJ, Tran QB, Cosgrove KL, Ross BP. Mini Rev Med Chem. 2008;8:1384–1394. doi: 10.2174/138955708786369573. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Yost GS. Chem Res Toxicol. 2008;21:374–385. doi: 10.1021/tx700294g. [DOI] [PubMed] [Google Scholar]
- 13.a) Ertl P, Roggo S, Schuffenhauer A. J Chem Inf Model. 2008;48:68–74. doi: 10.1021/ci700286x. [DOI] [PubMed] [Google Scholar]; b) Breinbauer R, Vetter IR, Waldmann H. Angew Chem Int Ed. 2002;41:2878–2890. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 14.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 15.Toogood PL. J Med Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s. [DOI] [PubMed] [Google Scholar]
- 16.Obtained from AnalytiCon Discovery. See the Supporting Information for details.
- 17.Totrov M, Abagyan R. Proteins Suppl. 1997;29:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman TS, Rúveda EA. Angew Chem Int Ed. 2005;44:854–885. doi: 10.1002/anie.200400663. [DOI] [PubMed] [Google Scholar]
- 19.Saxton JE. Nat Prod Rep. 1997;14:559–590. [Google Scholar]
- 20.Corrêa IR, Jr, Nören-Müller A, Ambrosi H-D, Jakupovic S, Saxena K, Schwalbe H, Kaiser M, Waldmann H. Chem – Asian J. 2007;2:1109–1126. doi: 10.1002/asia.200700125. [DOI] [PubMed] [Google Scholar]
- 21.In a side-by-side experiment, we measured the inhibition of TNFR-1 binding by SPD304 (10 μM) to be 34%.
- 22.Using Molsoft’s molecular property prediction tool, we calculated 2 to be less “drug-like” than 1. See the Supporting Information for details.