

Abstract
Ferredoxin (Fd) interacts with ferredoxin-NADP+ reductase (FNR) to transfer two electrons to the latter, one by one, which will finally be used to reduce NADP+ to NADPH. The formation of a transient complex between Fd and FNR is required for the electron transfer, and extensive mutational and crystallographic studies have been reported to characterize such protein-protein interaction. However, some aspects of the association mechanism still remain unclear. Moreover, in spite of their structural differences, flavodoxin (Fld) can replace Fd in its function and interact with FNR to transfer electrons with only slightly lower efficiency. Although crystallographic structures for the FNR:Fd association have been reported, experimental structural data for the FNR:Fld interaction are highly elusive. We have modeled here the interactions between FNR and both of its protein partners, Fd and Fld, using surface energy analysis, computational rigid-body docking simulations, and interface side-chain refinement. The results, consistent with previous experimental data, suggest the existence of alternative binding modes in these electron transfer proteins.
Keywords: Protein-protein association, electron transfer, binding energy landscapes, computational docking
INTRODUCTION
Many key processes in biological systems rely on electron transfer (ET) reactions between proteins, requiring formation of a complex that allows the optimal orientation between the redox centers. Once this complex is formed, the ET reaction takes place rapidly, thus avoiding side reactions in which electrons could be released out of the system producing unwanted by-products. The molecular requirements for these processes are not fully understood yet, but the main guidelines can be envisaged as: complementary of protein interaction surfaces, weak interactions among the proteins so that the complex remains stable only the necessary time for ET, and finally, close distance and adequate geometry between the redox cofactors that exchange electrons. Such type of processes are taken place in the photosynthetic ET chain that addresses two electrons from Photosystem I (PSI), via two independent Ferredoxin (Fd) molecules to the flavin-dependent Ferredoxin-NADP+ reductase (FNR), which finally catalyzes NADP+ reduction.1,2 In some algae and cyanobacteria (as Anabaena PCC 7119) an FMN-dependent protein, Flavodoxin (Fld), replaces Fd 3 in the ET from PSI to FNR under iron deficient conditions.2 The FMN cofactor is exposed to the solvent and is susceptible to undergo two one-electron redox reactions.4
Crystallographic FNR three-dimensional (3D) structures have been reported from several sources.5–7 The structure shows two domains: the FAD-binding domain in which the cofactor is accommodated (residues 1–138, Anabaena numbering), and the NADP+-binding domain, which is responsible for the binding of the nucleotide substrate (residues 139 to 303).6 Anabaena Fd is an 11 kDa acidic protein that contains a S2Fe2 centre.8,9 Anabaena Fld consists of a 169 amino acid chain that has a non-covalently bound FMN as redox centre.10 Crystal structures have also been reported for the FNR:Fd complexes from Anabaena11 and maize,12 however, no 3D model for the FNR:Fld interaction is yet available.
Extensive mutational studies have been carried out in the FNR region around the FAD, confirming that it is involved in binding to Fd and Fld.2,13–16 Thus, for efficient interaction of Anabaena FNR with Fd and Fld, positions R16, K72, and specially K75, have been clearly shown to be needed as positively charged side-chains, while positions L76 and L78 are strongly required to be hydrophobic side-chains.13,15,17,18 Altered complex stability is the major determinant for the observed decreased reactivity of mutants at these positions. Similarly, a number of residues of the Anabaena Fd surface have been shown to play a moderate effect on the complex stability and ET in its interaction with the reductase, being crucial for the interaction the negatively charged residue E94, the hydrophobic one F65, and S47, which is directly coordinating the S2Fe2 redox center. 3,19–21 Finally, site-directed mutagenesis studies on the Fld surface have suggested several residues that might cooperatively contribute to the orientation and optimization of the FNR:Fld interaction, although the data suggest that this interaction might be less specific than the FNR:Fd one.2,13,15,17,22–26
These mutational studies are, to some extent, consistent with the x-ray structural data of the complex between FNR and Fd,11,12 and with a homology-based model for the FNR:Fld complex.27 However, the x-ray structure does not fully explain the structural role of protein-protein complex formation on the ET mechanism. For instance, the dependence of the ET rate on the ionic strength has been proposed to be due to a reorganization of the complex,28 but no structural evidence is yet available. Moreover, the two available structures of the FNR:Fd complex, Anabaena and maize, have slightly different mutual orientation, being the redox centers located in different relative spatial locations. If these structures truly represent optimal conformations for efficient ET, it is difficult to explain why the redox centers are in such different locations. Recently, it has been proposed that perhaps complexes involved in ET reactions do not require as much specific requirements as other types of protein-protein interactions, suggesting that the bound state could be formed by dynamic ensembles instead of single conformations.29
As a complement to structural data, a number of computational docking methods have been reported to study the phenomenon of protein-protein association.30 The goal of these docking algorithms is to predict the geometry of a protein-protein complex starting from its unbound components. In the recent Critical Assessment of PRedicted Interactions (CAPRI; http://capri.ebi.ac.uk), many of these methods have been blindly evaluated, and some of them were able to correctly predict the structure of ~ 90 % of the proposed targets.31–33 In this direction, rigid-body docking simulations were reportedly applied to a known ET system, the complex formed by cytochrome c and cytochrome c oxidase.34 There, the docking results were consistent with experimental data (including evidence of alternative binding sites) and confirmed the transient nature of the interaction. Actually, the rigid-body docking distributions showed the importance of electrostatics in bringing the redox groups into close proximity, subsequently optimizing the ET reaction. But rather than providing a specific “lock-and-key” fit, those docking results were more consistent with the idea of a “pseudospecific docking surface” for ET proteins. In order to understand better this type of transient interactions, it is necessary to explore other different ET systems, and to apply extended docking approaches that include more refined energy functions. With this goal, we have used here a combination of computational tools such as rigid-body docking, binding energy rescoring and interface refinement, together with kinetic, spectroscopic, and mutational data, in order to characterize the structural and energetic determinants of the interaction between FNR and its redox partners Fd and Fld.
METHODS
Generation of rigid-body docking conformations
Rigid-body docking was performed using ICM software (www.molsoft.com), with the protocols described elsewhere.32,35,36 Translation/orientation of the all-atom ligand was sampled in a completely unrestricted way, by Monte Carlo movements and local minimization using interaction potentials pre-calculated on a 3-D grid from the receptor. Definition of receptor and ligand was arbitrary: for practical reasons, we usually considered the larger molecule as the receptor (FNR), and the smaller as the ligand (Fd or Fld), but this definition did not affect to the final results. Cofactors were included in the calculations as part of the rigid-body molecules. ‘Soft’ rigid-body docking simulations were run, in which van der Waals energy was truncated to a maximum of +1.0 in order to overcome the numerous local steric penalties of the interacting side-chains arisen from the rigid-body assumption. Typically, rigid-body docking simulations ran for 3 hours in ~30 CPUs. Rigid-body docking resulting conformations were sorted by total binding energy, which included van der Waals, electrostatics and hydrogen-bonding potentials plus ASA-based desolvation, with ASPs derived from octanol/water transfer experiments and further optimized for protein docking.36 Lowest-energy 400 rigid-body docking poses were clustered (to remove similar solutions with ligand interface Cα atoms RMSD < 4 Å), and the resulting conformations (typically 250–300) were further refined to optimized their interfaces.
Location of protein interaction sites
We used the resulting rigid-body docking solutions (sorted by total binding energy including desolvation) to predict the location of putative protein-protein interaction sites. Normalized Interface Propensity (NIP) values for every residue were computed analyzing their frequency of appearance at the protein-protein interface in the 100 lowest-energy rigid-body docking orientations.36 As established in previous benchmarks, residues with NIP > 0.4 are potentially involved in protein interactions in 80 % of the cases.
Refinement of the rigid-body docking interfaces
Refinement of interface side-chains of the lowest-energy rigid-body docking poses were performed by Biased-Probability Monte Carlo sampling of the ligand interface side-chains together with restrained translation/rotation minimization of the ligand in order to accommodate the new side-chain conformations.32,35 The cutoff value for the van der Waals binding energy was increased to +1.5 (as compared to +1.0 in the previous rigid-body docking step), since now ligand interface side-chains could move away from heavily clashing conformations. Refinement took around 10 minutes per rigid-body docking conformation. The final refined structures were sorted using the same energy function as in the revaluation of rigid-body docking solutions. As during the phase of rigid-body docking, we have not used here any knowledge-based restraint such as distance between cofactors or experimental mutational data.
RESULTS AND DISCUSSION
Modeling the FNR:Fd interaction: rigid-body docking and interface refinement
We performed completely unrestricted rigid-body docking simulations using the crystallographic coordinates of FNR (PDB 1que) and Fd (PDB 1fxa) from Anabaena, including the coordinates of the cofactors (FAD and S2Fe2, respectively). In the resulting docking poses, the Pearson’s correlation coefficient r between the total energy and each of the two major energy components, electrostatics and desolvation, is r = 0.71 and r = 0.31, respectively, which indicates that most of the variability in the docking energy can be explained by the electrostatics term. Similar results have been found in other ET systems, such as the complex between cytochrome c and cytochrome c oxidase.34 On the structural side, the residues with significant NIP values, as derived from the rigid-body docking, define potential binding sites. Thus, FNR residues with high NIP values are located in the same area than the FAD cofactor, mainly around its isoalloxazine ring (Figure 1A). This area has been proposed by numerous biochemical studies as the most likely binding interface to Fd.2 Indeed, a comparison with the two available x-ray structures of FNR in complex with Fd (PDB codes 1ewy and 1gaq, for Anabaena and maize complexes, respectively) indicates that the predicted binding site is located in the crystallographic interfaces (Figure 1A). Similarly, Fd residues with high NIP values are located in the same area than the S2Fe2 cofactor (Figure 1B). This predicted binding site in Fd is compared to the crystallographic interfaces in Anabaena and maize FNR:Fd complexes. Although the interfaces are slightly different in these two complexes, the predicted site is certainly overlapping the real interfaces.
Figure 1.

Computational analysis of FNR:Fd binding. (A) FNR residues with NIP > 0.4 (from FNR:Fd docking) are shown in red, and FAD cofactor in yellow. For comparison, the crystallographic positions of Fd in Anabaena (green) and in maize (blue) FNR:Fd complexes are shown, with their S2Fe2 cofactor in CPK. (B) Fd residues with NIP > 0.4 (from FNR:Fd docking) are shown in red, with S2Fe2 cofactors in CPK (S yellow, Fe cyan). For comparison, the crystallographic positions of FNR in Anabaena (green) and in maize (blue) complexes are shown, along with their FAD cofactors. (C) Lowest-energy FNR:Fd docking solutions after docking and refinement: center of coordinates of S2Fe2 in top 100 solutions is represented as balls (the ten lowest-energy conformations are represented in red). All solutions are superimposed onto the unbound FNR (in white ribbon; FAD in yellow stick). For comparison, it is also shown the crystallographic position of S2Fe2 in Anabaena (green CPK) and maize (blue CPK) complexes. (D) Lowest-energy model for FNR:Fd docking. Position of the residues experimentally known to be critically important for the interaction (FNR in white, with residue labels in blue; Fd in red, with residue labels in black). For comparison, Anabaena and maize complex x-ray structures are shown in green and blue, respectively.
Figure 2A shows the inter-molecular distance between the FAD and S2Fe2 cofactors in all docking poses as a function of the binding energy. A clear funnel-shaped distribution can be observed, where the lowest-energy docking solutions are the ones that have the shortest distance between cofactors. This is remarkable, since the distance between cofactors has not been included as a restraint for docking at all. Similar results are obtained when the coordinates of the cofactors are not included in the simulations (not shown), indicating that the attractive interactions between the two polypeptide chains is sufficient to locate the cofactors at a distance suitable for the ET. However, when comparing these results with the crystallographic structure of the Anabaena FNR:Fd complex (Figure 2B), the lowest-energy docking solutions are not amongst the closest to the x-ray structure. Moreover, the best near-native docking orientation (RMSD 2.2 Å) is ranked 253 (scoring energy = −40.4), as can be seen in Table 1. This could be consequence of the incorrect conformation of the interacting side-chains of the rigid-body molecules, or because of inaccuracy of the energy function (which is not representing the exact Gibbs energy, but an empirical binding energy based on physical terms and previously optimized for protein docking). However, the fact that the lowest-energy conformations obtained from the unrestricted docking simulations had the shortest distances between cofactors (indeed shorter than in the x-ray structure) cannot be just a serendipitous outcome. Instead, this suggests that these low-energy docking solutions may be alternative conformations, different from the ones represented by the crystallographic structures, which could be formed during the rigid-body approach (encounter complex) and perhaps contribute to the ET reaction (as discussed below). It is interesting to analyze the location of the Fd S2Fe2 group in the different docking conformations, as compared to the position found in the crystallographic structures. As can be seen in Supplementary Figure 1A, in the lowest-energy solutions, the S2Fe2 group is around 10 Å RMSD from the one in the Anabaena complex structure, and although other solutions are found where the S2Fe2 group is closer to the crystallographic structure, all of them have significantly higher energies. Interestingly, the location of the S2Fe2 group seems to converge to a location closer to the one in the maize complex (Supplementary Figure 1B). We will explore this later on.
Figure 2.

Resulting conformations for unbound-unbound FNR:Fd rigid-body docking and refinement. (A) Binding energy vs. distance between redox centers (FAD and S2Fe2) of the interacting proteins (dots: rigid-body docking; open circles: interface refinement). The refined docking conformation geometrically closest to the Anabaena complex (RMSD 2.4 Å) is shown as a gray closed circle. (B) Binding energy vs. RMSD of interface ligand atoms with respect to the x-ray structure of the complex, when the receptors are superimposed (dots: rigid-body docking; open circles: interface refinement). (C) Binding energy plus restraint-based pseudo-energy vs. distance between cofactors (a gray closed circle represents the docking orientation nearest to the x-ray structure). (D) Binding energy plus restraint-based pseudo-energy vs. ligand RMSD.
Table 1.
Docking results
| RANK | Energy | RMSD (Å)a | Cofactor distance (Å)b | |
|---|---|---|---|---|
| Rigid-body docking FNR/Fd | 1 | −68.5 | 14.8 (20.7) | 5.6 |
| 46 | −52.8 | 14.6 (4.3) | 6.2 | |
| 253 | −40.4 | 2.2 (15.0) | 6.9 | |
| 406 | −36.4 | 13.7 (4.5) | 6.4 | |
| 553 | −34.0 | 2.4 (13.9) | 4.3 | |
|
| ||||
| Dock/ref FNR/Fd | 1 | −79.1 | 14.8 | 6.4 |
| 2 | −64.9 | 15.0 | 7.4 | |
| 3 | −63.8 | 9.1 | 5.0 | |
| 102 | −49.3 | 2.4 | 7.9 | |
|
| ||||
| Dock/ref FNR K72E/Fd | 1 | −57.0 | 15.1 | 6.8 |
| 49 | −39.6 | 2.3 | 7.5 | |
|
| ||||
| Dock/ref FNR K75E/Fd | 1 | −53.9 | 15.1 | 7.1 |
| 213 | −19.0 | 3.0 | 8.7 | |
|
| ||||
| Dock/ref FNR/Fld | 1 | −52.4 | 5.9c | 4.2 |
| 4 | −47.9 | 4.0c | 4.0 | |
RMSD for ligand Cα atoms with respect to the equivalent ones in Anabaena FNR/Fd X-ray structure (in brackets, RMSD from maize X-ray structure).
Minimal interatomic distance between cofactors
RMSD from CPR X-ray structure.
The 400 lowest-energy rigid-body docking solutions were further refined as described in Methods. As can be seen in Figure 2A, the funnel-shaped distribution of intermolecular FAD-S2Fe2 distance values vs. energy improved after interface refinement. This reinforces the correlation between binding energy and distance between cofactors. However, in spite of the side-chain refinement, the docking solutions near the crystallographic structure of the Anabaena complex (Figure 2B) had still higher energies than many others, being the best near-native solution ranked 102 with a scoring energy of −49.3 (Table 1). This indicates that the apparently higher energies of these near-crystallographic solutions were not merely caused by inaccurate geometries derived from the rigid-body assumption, suggesting that alternative low-energy orientations may exist either forming a native bound ensemble or an encounter complex, where the distance between cofactors is smaller than in the crystallographic complex.
Figure 1C shows the position of the S2Fe2 group in the 100 lowest-energy solutions after refinement. Many of them, especially the ten lowest-energy solutions (in red), clustered around the position of the S2Fe2 group in the maize crystallographic structure. These lowest-energy solutions have varied FNR:Fd orientations, and different from the ones in the crystallographic structures. Actually, the three distinct lowest-energy docking solutions (after removing similar conformations that are < 2 Å RMSD from each other) are rotated approximately 120° with respect to each other along an imaginary axis that goes through the redox centers (Supplementary Figure 2). It is very interesting that in spite of the variation in the orientation of the polypeptide chain, the position of the S2Fe2 group with respect to the FAD is very similar in all these solutions, which indicates that a number of different energetically-favored orientations could bring the cofactors at suitable distance for the ET. Moreover, the fact that the S2Fe2 group is located close to that in the maize structure (Supplementary Figure 1B) suggests that this can be one of the most favorable positions for the ET. Remarkably, the lowest-energy orientation after rigid-body docking is also the rank 1 solution after interface refinement (Figure 1D), with redox centers at 6.4 Å distance. Also interesting is the refined docking solution ranked 3, which shows even shorter distance between redox centers (5.0 Å).
Role of FNR residues K72 and K75 on Fd binding
It is known that some mutations on the FNR surface and in the close environment of the flavin ring affect to the interaction with Fd. Some of these key FNR residues are K72 and K75. Thus, it has been shown that replacement of any of these two residues by a glutamate inhibits the ability of Fd binding, being the effect especially relevant in the case of the K75E mutation.2,13,15 Since the structures of K72E and K75E FNRs were known,27 we studied the docking of these FNR mutants to Fd.
In Figure 3A are shown the results of the rigid-body docking and interface refinement for the K72E FNR mutant (PDB 1g02). As can be seen, the energy values of the lowest-energy solutions were worse than those in the wild-type. Interestingly, the lowest-energy solution after refinement was 1.2 Å RMSD similar to the lowest-energy solution for WT FNR, in spite of their different energies (−57.0 and −79.1, respectively). The closest solution to the Anabaena complex had also higher energy (−39.6) than in the case of WT FNR docking (−49.3). The refined docking landscapes (Figure 3A) were clearly less funnel-shaped than those of the WT (Figure 2A). This indicates that the K72E mutation has an overall negative effect on the FNR:Fd docking energies, in agreement with the experimental results.
Figure 3.
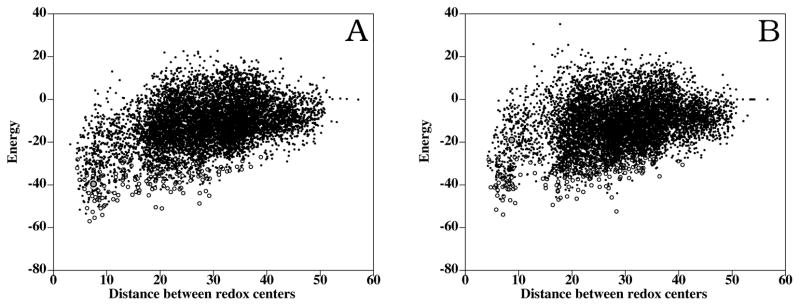
Rigid-body docking (dots) and interface refinement (open circles) of FNR mutants and Fd. (A) FNR K72E docking: distance between FAD and S2Fe2 cofactors vs. energy. The docking conformation closest to the Anabaena complex (RMSD 2.3 Å) is shown as a gray closed circle. (B) FNR K75E docking: distance between FAD and S2Fe2 cofactors vs. energy. The docking conformation closest to the Anabaena complex (RMSD 4.8 Å) is shown as a gray closed circle.
Docking results obtained for the K75E FNR mutant (PDB 1qgy) are shown in Figure 3C. As in the case of the K72E mutant, the energy values after refinement were less funnel-shaped (Figure 3C). The docking conformations found here were significantly different from the ones found when using the WT FNR. Now, the closest solution to the Anabaena complex had a RMSD value of 3.0 Å, and a higher energy (−19.0) than in the WT docking. The lowest-energy solution, which was 1.3 Å RMSD similar to the lowest-energy solution for WT FNR, had also higher energy (−53.9). In addition, some of the low-energy solutions in the case of WT FNR docking were not found amongst the lowest-energy solutions here. This clearly indicates that the K75E mutation is preventing the formation of the favorable orientations found in the WT, which is also consistent with the experimental results. For test purposes, additional docking simulations (not shown) were performed using a model of K75E FNR, built from the WT structure by replacing the K75 residue by a glutamic acid and later minimizing the structure. The main difference between the crystal structure and the model structure is that modeling does not foresee the formation of the salt-bridge between the modeled E75 residue and K72, which was however formed in the crystal structure.27 In the docking performed with the model, the closest solution to the Anabaena complex had a RMSD value of 4.8 Å, and a higher energy (−35.8) than in the WT FNR docking. The lowest-energy solution, which is 2.2 Å RMSD similar to the lowest-energy solution for WT FNR, had also higher energy (−58.8). This clearly indicates that, in addition to the effect of replacing the positive charge of K75 by a negative one, the formation of the salt-bridge between K75E and K72 contributes to further debilitate the FNR:Fd interaction.
FNR:Fd model: comparison to experimental data
Extensive analysis on the Anabaena system indicated that specific electrostatic and hydrophobic interactions between FNR and Fd surface side-chains were required for an efficient interaction.2,13–15,17–21,23 Most of the FNR residues located around the FAD cofactor (i.e. positively charged R16, K72 and K75; apolar Y303 stacking the flavin ring; and to some extent negatively charged E139 and E301) were key for the productive interaction with the protein partners.15,27,37–39 These studies also proved the contribution of V136 and, especially, L76 and L78 to the stabilization of interaction with Fd 17,18, while the FNR residues situated in the FNR NADP+-binding domain, namely K138, R264, K290, K294, V300 and T302, have in general less effect on the processes of recognition and ET than those situated in the FAD-binding domain.13,40
Interestingly, all of the residues on the FNR surface that the experimental analysis has shown to be involved in the interaction with Fd showed positive NIP values in the docking models here presented (Table 2). Moreover, from the nine residues that had significant NIP value (>0.4) and were experimentally tested (i.e. R16, L76, L78, V136, K138, E139, V300, E301, T302), eight of them (Table 2) were shown to be important (either moderately or critically) for the interaction with Fd (89% positive predictive value). Surprisingly, although K75 and K72 residues were revealed as important and even critical from site-directed mutagenesis studies when replaced by Glu, they are not among the residues with highest NIP values. However, we have already seen the importance of these residues by using the structures of the K72E and K75E FNR mutants in the docking to Fd (Figure 3). The data obtained from these calculations clearly show that these mutations have an overall negative effect on the docking energy landscape, in agreement with the experimental results. This validates the reliability of our models and confirms the experimental results.27 It is also noticeable with regard to position of K75 that more conservative mutations showed much less drastic effect in the enzyme ability to interact and transfer electrons to Fd,15 which might explain its position in the NIP values. As a general comment, NIP values predict well the unspecific tendency of a given residue to be at the interface, but they cannot predict residues that are forming highly specific interactions with the partner protein, as it seems to be the case of the K72 and K75 residues.
Table 2.
Residues involved in FNR:Fd interaction as experimentally determined by site-directed mutagenesis. Comparison with the experimental and theoretical structural models.
| Intermolecular pairs a
|
|||
|---|---|---|---|
| NIP | Anabaena x-ray | Docking model b | |
| FNR residuesc | Fd residues a | ||
| R16 (++)d | 0.55 | D67, D69, Q70 | N9, E12, H16, G34, Y35, D36, L37, P38 |
| K72 (++)d | 0.09 | - | E32 |
| K75 (++)e | 0.28 | S47, T48, E94, Y98 | E24, D28, E31, E32 |
| L76 (++)f | 0.40 | A45, C46, F65, Y98 | E31, D36, F39, R42 |
| L78 (++)f | 0.58 | A45, S64, F65, L66 | E31, D36, F39, S40, C41, R42 |
| V136 (+)f | 0.57 | S64, F65, L66, D67, D68, Q70 | D36, L37, P38, F39, S40, T48 |
| K138 (++)d | 0.53 | L66, D68 | S40, S47, T48, H92, K93, E94, E95, Y98 |
| E139 (+)g,h,h | 0.61 | Q60, S61, D62, S64 | S40, A45, C46, S47, T48, F65, E94, Y98, S2Fe2 |
| R264 (+)i | 0.21 | Y25, R42, A43, G44, D62, C79, V80 | A43, D62 |
| K290 (+)d | 0.25 | - | D67, D68 |
| K294 (++)d | 0.28 | - | F65, D67, D69, Q70, L97, Y98 |
| E301 (+)i | 0.48 | S61, D62, Q63, S64 | F39, S40, C41, A45, C46, T48 |
| T302 (+) k | 0.50 | A43, G44, D62, Q63, S64 | S40, C41, R42, A43, A45 |
| Y303 (++)l | 0.12 | C41, A43, G44, A45, D62, Q63 | S40, C41, A43 |
| Fd residuesm | FNR residuesa | ||
| E32 (+)n | 0.28 | - | K72, K75 |
| F39 (+)n | 0.52 | - | N13, S59, I60, G61, L76, R77, L78, T133, V136, G137, E301, FAD |
| S40 (+)o | 0.53 | FAD | G57, S59, L78, V136, G137, K138, E139, M140, V300, E301, T302, Y303, FAD |
| C41 (+)n | 0.61 | Y303, FAD | L78, E267, V300, E301, T302, Y303, FAD |
| R42 (+)p | 0.49 | R264 | L76, L78, T302, FAD |
| C46 (+)n | 0.28 | L76, FAD | E139, V300, E301 |
| S47 (++)q | 0.16 | K75 | K138, E139, V300 |
| T48 (+)o | 0.19 | K75 | V136, G137, K138, E139, M140, |
| D62 (+)q | 0.24 | E139, R264, E267, K293, V300, E301, T302, Y303 | R264 |
| F65 (++)p | 0.19 | S59, L76, R77, L78, V136, FAD | E139, K293, K294, W298, H299, V300 |
| D67 (+)r | 0.02 | V12, N13, R16, P135, V136 | K290, D291, K293, K294 |
| D69 (+)r | −0.12 | V12, N13, R16 | K294 |
| Q70 (+)q | −0.06 | N13, R16, V136 | K294 |
| E94 (++)p | 0.09 | K75 | G137, K138, E139, M140, L141 |
Two residues are considered to interact if any of their non-hydrogen atoms are at a distance ≤ 6 Å.
Docking model ranked 1 after rigid-body docking and refinement.
(++) Residues critically important for the interaction, when upon mutation the binding affinity changed at least one order of magnitude with respect to wild type, that is relative Kbind ≤0.1 or ≥10.0 as determined by difference spectroscopy; (+) Residues moderately important for the interaction, when upon mutation the binding affinity changed at least 20% with respect to wild type, that is relative Kbind ≤0.8 or ≥1.2; (−) Residues whose mutation does not affect the interaction (i.e. 0.8 < relative Kbind <1.2).
Data from Martínez-Júlvez et al. (1999).13
Data from Martínez-Júlvez et al. (1998).15
Data from Martínez-Júlvez et al. (2001).18
Data from Hurley et al. (2000).28
Data from Faro et al. (2002).38
Data from Martínez-Júlvez et al. (1998).14
Data from Medina et al. (1998).37
Data from Faro et al. (2002).40
Data from Nogues et al. (2004).39
(++) Residues critically important for the interaction, when upon mutation show relative kobs ≤0.1 or ≥10.0 as determined by laser flash photolysis; (+) Residues moderately important for the interaction, when relative kobs ≤0.8 or ≥1.2.
Data from Hurley et al. (2002).23
Data from Weber-Main et al. (1998).42
Data from Hurley et al. (1993).19
Data from Hurley et al. (1997).3
Data from Hurley et al. (1996).41
Similar experimental analyses have been reported for mutations on side-chains of the Anabaena Fd surface.23 The reported data showed that E32, F39, S40, C41, R42, C46, T48, D62, D67, D69 and Q70 side-chains moderately contribute to complex stability and ET with the reductase, whereas S47, F65 and E94 have been shown to be critical for the overall reaction to take place. 3,19,23,41,42 In our docking simulations, although these critical residues present positive NIP values (Figure 1B), S47, F65 and E94 are not among the residues with the highest NIP values (Table 2). Again, this is also consistent with the experimental results, which indicates that the effect of site-directed mutagenesis of S47, F65 and E94 side-chains on the strength of the binding interaction with FNR23 cannot account for the four orders of magnitude decrease in ET reactivity observed for these mutants. These results indicate that S47, F65 and E94 may rather be responsible for a precise orientation between both proteins for an efficient ET.23
It is noticeable that in the lowest-energy docking and refinement model (Figure 1D), all of the residues experimentally found to be moderately or critically important for the interaction are located in the interface, whereas in the published x-ray structure, only 82% of the residues experimentally important for the interaction are actually in the interface (Table 2). Thus, the docking model (which of course has been generated without using any mutational data at all) seems to satisfy better the experimental data as compared to the x-ray structure. In this sense, we have computed distance restraints based on the experimentally important residues (moderately or critically) using pyDockRST,43 and have used them to rescore the docking solutions. In Supplementary Figure 1C,D it can be seen that many docking poses satisfy the experimental restraints better than the nearest docking orientation to the x-ray structure. Interestingly, the rank 1 solution obtained after combining binding energy and restraint-based scoring (Figure 2C,D) is exactly the same one found by binding energy alone, with the redox centers at very short distance. This proves that our energy-based docking method provides models that correlate optimally with the experimental restraints, even better than the crystallographic structure.
Interestingly, the rigid-body docking pose that best satisfies the experimental restraints has one of the shortest distances between redox centers, and there is almost direct contact between them (this solution has similar conformation to rank 1 after energy-based scoring). Similarly, the refined docking pose that best satisfies the experimental restraints (data not shown) has the shortest distance between redox centers, although it shows different orientation with respect to our energy-based rank 1 solution. This correlation between satisfaction of experimental restraints and distance between cofactors is very interesting. However, neither of these docking orientations has optimal binding energy, so we have not considered them as our best models (as previously reported,43 the best models usually arise from a fine balance between energy and restraints). What it is clear is the existence of different orientations that satisfy the energetic, electron transfer and experimental restraints. This suggests the existence of alternative conformations, which could pivot around the Fd S2Fe2 group (Supplementary Figure 2). Whether this represents a change of orientation caused by the experimental conditions (for instance, ionic strength can affect to the orientation of the two molecules), 28 or it is some kind of on-off mechanism to let the Fd accepting and donating electrons by different proteins, is something that will need further experimental work.
Modeling the FNR:Fld interaction
We performed completely unrestricted rigid-body docking simulations using the coordinates of unbound FNR (PDB 1que) and Fld (PDB 1flv) from Anabaena (the coordinates of the cofactors, FAD and FMN respectively, were also included in the calculations). A general analysis of the docking energy values (the Pearson’s correlation coefficient r between the total energy and each of the two major energy components, electrostatics and desolvation, is r = 0.73 and r = 0.22, respectively) shows that most of the variability in the docking energy can be explained by the electrostatics term, as in the case of FNR:Fd docking. On the structural side, we found potential binding sites in the surfaces of both proteins, based on the NIP values derived from the docking energy landscapes (Figure 4A,B). The FNR residues with higher NIP values were located around the FAD cofactor (Figure 4A). This region in the FNR surface has been proposed as the expected binding interface to Fld, and indeed is very similar to the crystallographic binding site to Fd. Similarly, Fld residues with higher NIP values from the docking simulations were located in the same area than the FMN cofactor (Figure 4B).
Figure 4.

Computational analysis of FNR:Fld binding. (A) FNR residues with NIP > 0.4 (from FNR:Fld docking) are shown in red, with FAD cofactor in yellow; for comparison, we show the crystallographic position of the FMN domain of the CPR (cyan), when FNR and CPR FAD domain are superimposed. (B) Fld residues with NIP > 0.4 (from FNR:Fld docking) are shown in red, with FMN cofactor in yellow; for comparison, we show the position of FAD domain in CPR (cyan) when Fld and CPR FMN domain are superimposed. (C) FNR:Fld docking and refinement results (FNR in white; Fld in red, solution ranked 1; in yellow, solution ranked 4 after refinement - ranked 1 after addition of experimental restraints -). For comparison, it is also shown the FMN-binding domain of CPR (in cyan), after superimposing FNR and CPR FAD domain. (D) Docking solution ranked 4 after refinement (ranked 1 after addition of experimental restraints), showing the residues that are experimentally known to be critically important for the interaction (FNR in white, with residue labels in blue; Fld in yellow, with residues in red, and labels in black).
In Figure 5A is shown the inter-molecular distance between the FAD and FMN cofactors in all docking poses as a function of the binding energy. Although there is certain funnel-shaped distribution (indeed remarkable, as no inter-cofactor distance restraints have been used during the docking at all), there are no docking solutions with as good energies as the ones found in the FNR:Fd interaction. Actually, the lowest-energy solutions appear geometrically more spread. There is currently no crystallographic structure available for the FNR:Fld complex, but we can compare the docking results with the available structure of cytochrome P450 reductase (CPR),27,44 a multi-domain protein that has two domains highly homologous to FNR and Fld, respectively. For each docking solution, we have superimposed the FNR molecule onto the FAD-binding domain of CPR, and then we have calculated the RMSD of the Fld interface Cα atoms with respect to the FMN-binding domain of the CPR. The results are shown in Figure 5B. There is a certain funnel-shaped distribution, where the lowest-energy docking solutions are oriented as the corresponding domains in the CPR structure.
Figure 5.

FNR:Fld rigid-body docking and interface refinement. (A) Binding energy vs. inter-molecular distance between FAD and FMN from FNR and Fld, respectively (dots: rigid-body docking; open circles: interface refinement). (B) Binding energy vs. RMSD of Fld interface Cα atoms with respect to the FMN domain of CPR, when FNR and CPR FAD domain are superimposed (black: rigid-body docking; open circles: interface refinement). (C) Binding energy plus restraint-based pseudo-energy vs. distance between cofactors. (D) Binding energy plus restraint-based pseudo-energy vs. ligand RMSD.
We next proceeded to refine the interfaces of the lowest-energy rigid-body docking solutions. The energy values of the docking orientations with the closest inter-molecular distance between cofactors generally improved (Figure 5A). The lowest-energy docking solution after refinement (scoring energy of −52.4) brings the cofactors FAD and FMN at a distance of 4.2 Å (Figure 4C). In this refined docking solution, the orientation between the FNR and Fld molecules is very similar to that of the FAD and FMN-binding domains in CPR (5.9 Å RMSD between Fld and CPR FMN domain). Interestingly, conformation ranked 4 after refinement (scoring energy of −47.9) is only 4.0 Å RMSD with respect to CPR and has the redox centers at 4.0 Å distance (Figure 4C). Other low-energy docking orientations are very close to the CPR structure and have their cofactors at a distance of < 6 Å (Figure 5A,B). This suggests that different orientations with short distance between cofactors may be populated at the bound state and thus contribute to the ET reaction.
FNR:Fld model: comparison to experimental data
Replacement of the FNR residues above described to be important for the interaction to Fd produced in general more drastic effects in the processes involving ET with Fld, suggesting that each individual residue does not contribute in the same extent to complex formation with both partners.13 All of the residues on the FNR surface that are experimentally known to be involved in the interaction with Fld showed positive NIP values (Table 3). Moreover, all the FNR residues that had significant NIP (red residues in Figure 4A) and were experimentally tested (i.e. R16, K75, L76, L78, V136, K138, E139) resulted to be important for the interaction with Fld (100% positive predictive value; Table 3). Additionally, the different NIP values obtained for the same FNR residues, with either Fd or Fld (Table 2,3), are consistent with the experimental observations that indicated that although FNR uses the same site for interaction with Fd and Fld each individual residue does not participate to the same extent in the interaction with each of the protein partners.13 On the Fld side, the experimental data indicate that E16, E20, I59, E61, D65, E67, I92, Y94 and D96 contribute moderately to the orientation and optimization of the FNR:Fld interaction, whereas T12, W57 and N58 seem to play a more important role.22,24–26 On top of that, FMN is also critical for the interaction with FNR.26 Interestingly, all these important residues, including FMN, have significant NIP values > 0.4 (100% sensitivity; Table 3), and on the other side, all residues with significant NIP that have been experimentally tested are important for the interaction (100% positive predictive value; Table 3). In general, all the residues exhibiting high NIP values are in the direct environment of the FMN (Figure 4B). However, it is somehow unclear whether the residues stacking the FMN flavin ring, W57 and Y94, are truly affecting the protein binding, or if on the contrary, the observed altered kinetic ET properties when they are mutated can be explained in terms of altered flavin accessibility and/or thermodynamic parameters. 24–26,45
Table 3.
Residues involved in FNR:Fld interaction as experimentally determined by site-directed mutagenesis. Comparison with the theoretical docking model.
| Intermolecular pairs a
|
||
|---|---|---|
| NIP | Docking model b | |
| FNR residuesc | Fld residues a | |
| R16 (++)d | 0.88 | V36, S37, Q38, Q63, S64, D65, W66, E67, G68, L69 |
| K72 (++)d | 0.24 | E16 |
| K75 (++)e | 0.40 | T10, Q11, T12, G13, K14, E16, FMN |
| L76 (++)f | 0.57 | T10, Q11, T12 |
| L78 (++)f | 0.56 | Q11, T12, W57, N58 |
| V136 (+)f | 0.67 | Q11, N58, E61, S64, D65, E67 |
| K138 (++)d | 0.53 | N58, E61, E67 |
| E139 (++)g | 0.45 | W57, N58, I59, G60, E61, N97 |
| R264 (+)h | 0.08 | D90, I92, G93, Y94, D146, N147 |
| K290 (+)d | 0.21 | D96, N128, D129 |
| K294 (+)d | 0.27 | D129 |
| E301 (+)i | 0.25 | N58, I59, Y94, FMN |
| Y303 (++)j | 0.03 | Y94, FMN |
| Fld residuesc | FNR residues a | |
| T12 (++)k | 0.62 | K75, L76, R77, L78, FAD |
| E16 (+)k | 0.33 | K72, K75 |
| E20 (+)k | 0.13 | - |
| W57 (++)l,m | 0.51 | L78, E139, FAD |
| N58 (++)k | 0.41 | S59, L78, V136, G137, K138, E139, M140, V300, E301, FAD |
| I59 (+)f,m | 0.24 | E139, M140, K293, W298, H299, V300, E301 |
| E61 (+)k | 0.13 | V136, G137, K138, E139, M140 |
| D65 (+)k | 0.34 | K4, N13, Y15, R16, P17, V136 |
| E67 (+)k | 0.18 | H7, V10, V12, N13, R16, V136, G137, K138 |
| I92 (+)f,g | 0.25 | R264 |
| Y94 (+)l | 0.44 | R264, E267, V300, E301, T302, T303 |
| D96 (+)k | 0.07 | S86, N289, K290, K293 |
| FMN (++)m | 0.60 | K75, V300, E301, T302, Y303, FAD |
Two residues are considered to interact if any of their non-hydrogen atoms are at a distance ≤ 6 Å.
Docking model ranked 4 after rigid-body docking and refinement. Rank 1 after adding experimental restraints.
(++) Residues are critically important for the interaction, when upon mutation show relative Kbind ≤0.1 or ≥10.0 by difference spectroscopy; (+) Residues are moderately important for the interaction, when relative Kbind ≤0.8 or ≥1.2.
Data from Martínez-Júlvez et al. (1999).13
Data from Martínez-Júlvez et al. (1998).15
Data from Nogues et al. (2003).17
Data from Faro et al. (2002).38
Data from Martínez-Júlvez et al. (1998).14
Data from Medina et al. (1998).37
Data from Nogues et al. (2004).4
Data from Nogues et al. (2005).22
Data from Casaus et al. (2002).25
Data from Goñí et al., in press.26
The docking results confirm that a mutual orientation between FNR and Fld similar to the FAD and FMN binding domains in CPR is highly probable.27 Based on docking solution ranked 4 after docking and refinement, we can propose a possible model for the FNR:Fld complex (Figure 4D). This model fits well with the experimental data (indeed remarkable, since we have not used any experimental restraint during the docking at all). As much as 96% of the residues (both from FNR and Fld) that have been experimentally determined to be important (moderately or critically) are located at the protein-protein interface (Table 3). A previous model, created by superimposition of FNR and Fld molecules on the equivalent domains of CPR,27 explains a smaller percentage (85 %) of the experimentally determined important residues (not shown). Interestingly, all Fld residues that are critically important for the interaction to FNR are in contact with the FAD cofactor in our docking model (in the previous CPR-based model, only the Fld FMN was in contact with the FNR FAD).
We have also evaluated the docking solutions with the distance restraints derived from the experimental mutational data, using pyDockRST.43 In Supplementary Figure 3A,B we can see the good correlation between restraints and inter-cofactor distance or ligand RMSD, although is not as clear as in the FNR:Fd docking. Noticeably, the best solution after combining energy and restraints (Figure 5C,D) is exactly our refined model ranked 4 (Figure 4D). This indeed validates our selected model for FNR:Fld interaction. Interestingly, the refined docking solution that better satisfies the experimental restraints (not shown) has the shortest distance between cofactors. However, its orientation is completely different from either our energy-based model or the CPR-based model (15.9 Å RMSD from CPR FMN domain). Moreover, it has worse binding energy, so again, we prefer to favor our refined model ranked 4 that had also the best scoring with energy plus restraints (Figure 5C,D). Having said so, it is clear that many solutions exist that satisfy energetic, electron transfer and experimental restraints.
The models clearly show that the FMN surface in contact with FNR is quite significant and that ET between FAD and FMN might take place to a much shorter distance than in the case of FNR and Fd redox centers. Such observation suggests that probably only the flavin atoms will be directly responsible for ET. Moreover, our docking models show that Fld could orientate in different ways on the FNR surface without significantly altering the distance between the methyl groups of FAD and FMN. Thus, in the different models obtained with similar binding energy the residues with larger surface in contact with FNR are those in the direct environment of the FMN group, such as the FMN group itself and Q11, T12, W57, Y94 or D146 (Table 3). If the main requirement for ET is the proximity of the redox centers in a non-polar environment, this could be achieved very efficiently by many of the low-energy docking conformations found in the computational simulations. The absence of a strong funnel shape in the rigid-body docking energy landscape (as compared to that of FNR:Fd interaction) could be due to intrinsic error in the energy values of the near-native states arisen from the incorrect conformation of the rigid-body side-chains, as we have discussed above. Indeed, the refinement of the ligand interface side-chains improved the energy landscape: the refined lowest-energy conformation was found at 5.9 Å RMSD with respect to the corresponding domains in CPR, and the one ranked 4 was only 4.0 Å RMSD with respect to CPR (Figure 5B). However, the refined docking landscape still has many orientations of similarly low energy (as compared to the case of FNR:Fd), some of which in spite of their different orientation, have the cofactors at a very short distance. Perhaps the existence of orientations where the cofactors can be very close (much closer than in the FNR:Fd case) makes very efficient the global ET even if other non-productive orientations are present either in the encounter complex or in the native state ensembles. This might explain why mutagenesis of the individual residues has not revealed any specific one that is critical for the efficient interaction with FNR and why subtle changes in the Fld surface electrostatic potential and dipole moment still produce complexes that allow ET.22
Rigid-body docking landscape optimized for electron transfer?
The rigid-body docking results for Anabæna FNR:Fd interaction show a good correlation between binding energy and distance between the redox centers (Figure 2A). The lowest-energy docking poses have short distances between the FAD isoaloxazine and the S2Fe2 centre (4.0 to 5.0 Å), which would be optimal for ET between FNR and Fd. Moreover, there is a good funnel-shaped distribution where the overall cofactor-cofactor distance decreases as the binding energy becomes stronger. We found several docking conformations similar to the available crystallographic complex structures, although they are not ranked as the lowest-energy solutions. For instance, conformation ranked 253 is 2.2 Å RMSD from the Anabaena x-ray structure, and the distance between cofactors is 6.9 Å, similar to the one in the x-ray complex (6.7 Å). Interestingly, in conformation ranked 553 (2.4 Å RMSD from the x-ray complex), the distance between cofactors is 4.3 Å (significantly shorter than in the x-ray complex). It is also noticeable that we found a low-energy docking conformation (rank 46) similar to the x-ray structure of the FNR:Fd maize complex (RMSD 4.3 Å), with distance between cofactors of 6.2 Å (similar to the one in the x-ray structure: 5.9 Å). There is also conformation ranked 406 (RMSD from maize x-ray complex 4.5 Å), where distance between cofactors is 4.0 Å. All of these docking conformations found both in the computer simulations and in the x-ray experiments have the redox groups at a distance where ET would be possible, although the mutual orientations of the interacting proteins are quite different. The fact that the orientations that best fit the experimental restraints are also those ones with short distance between cofactors (Supplementary Figure 2C,D) gives additional validity to these orientations. We do not know yet whether the different spatial arrangements of the residues around the redox centers would have some effect on the efficiency of the ET process, nor which are the optimal orientations of the molecules. One explanation is that the low-energy docking solutions may represent encounter complexes during the rigid-body approach. In this sense, the x-ray structure of the Anabaena complex could represent a more stable and perhaps better oriented for ET that is badly ranked in the docking results due to incorrect geometry of the interacting surfaces of the unbound subunits. These rigid-body docking landscapes are likely to represent a “loose” population of conformations during the approach of the molecules rather than that of the final “lock-and-key” fitting. However, we should not dismiss these alternative FNR:Fd orientations with close redox centers that are strongly favored by the docking energy (Figure 2A), and which are unlikely to arise just from an artifact or noise in the energy function. It is also possible that any of these alternative conformations, in which redox centers are at adequate distances (such as the x-ray structures, docking poses, and other possible ones), would contribute similarly to the overall ET process. Perhaps the two different x-ray structures found for Anabaena and maize complexes are two particular conformations, of the many possible ones, which have been specifically stabilized according to the given crystallization conditions. Indeed, the reported experimental conditions (ionic strength, additives, pH, …) of the crystallization were different in the two complexes, and actually, in the case of Anabaena, there is a second FNR molecule in the crystallographic cell that is likely arising from crystal packing and thus involved in artificial interactions with Fd. In any case, our energy description cannot predict the behavior of the molecules in the crystal lattice. As we have discussed earlier, the low-energy/low-distance solutions found in the docking simulations might be part of an encounter complex, but we could also speculate that they represent alternative rearrangements that could be part of a dynamic ensemble. Ubbink and collaborators have detected by NMR a similar behavior in other ET systems, where dynamic ensembles, as opposed to single conformations, contribute to the ET process.29 Indeed, low-population alternative binding modes in equilibrium have been experimentally found in transient interactions,46 which correlates well with rigid-body ensembles formed by electrostatics.47 Perhaps this loose association mode is general for highly transient complexes like the ones involved in ET reactions, where no tight binding is needed for the reaction to occur. This could explain why it is so difficult to obtain crystallographic data on these types of complexes.
A limited number of three-dimensional structures of analogous systems to FNR:Fd are available. These include bovine adredoxin reductase:adredoxin,48 rubredoxin:rubredoxin reductase from P. aeruginosa 49 or the FAD- and Fe2S2 containing subunits of fumarate reductase from E. coli.50 Interestingly, in these complexes completely different orientations between the flavin and the iron-sulfur center are observed, with minimal inter-cofactor distance ranging between 6 and 12 Å. FNR-, Fd-and Fld-like domains play also an important role in cellular metabolism in a large family of enzymes that probably arose due to an ancestral gene fusion event.51 These proteins, including phthalate dioxygenase reductase, NADPH-cytochrome P450 reductase, nitric oxide synthase, sulfite reductase, NADPH-dependent diflavin oxidoreductase 1, sulfite reductase or methionine synthase reductase, are found in both prokaryotes and eukaryotes (including mammals).44,51–54 The x-ray structures reported for some of these multi-domain proteins show different relative orientation between flavins, as in NADPH-cytochrome P450 reductase and nitric oxide synthase. Additionally, the structural analysis of sulfite reductase indicated a disordered orientation of the Fld-like domain that can be interpreted as the result of multiple allowed conformations of this domain with regard to the FNR one.51 All this observations are compatible with the presence of alternative orientations either in a dynamic ensemble or as encounter complexes able to evolve towards productive ET interactions, similar to those proposed here for the interaction between FNR and its protein partners.
CONCLUSION
We have modeled the interaction between FNR and its redox partners Fd and Fld, using rigid-body docking simulations and interface refinement. The best docking models found for the FNR:Fd interaction would allow an efficient ET between the redox centers. The binding interfaces in the interacting proteins are similar to the ones found in two available crystallographic structures for the FNR:Fd complexes of Anabaena and maize. However, the intermolecular orientation is slightly different, in the models, and also in the two crystallographic structures, which opens the possibility of alternative orientations that could all contribute to the ET reaction. As for the FNR:Fld interaction, one of the goals of the present work was to propose a model for it, since the 3D structure of this complex has not been determined yet. The best of our models is structurally similar to the mutual orientation of FAD and FMN binding domains in cytochrome P450 reductase. Interestingly, we found also other conformations of similar energy that bring both flavin centers at ET distance. The models are consistent with previous experimental results, and they will be of essential interest for designing new experimental approaches in order to probe residues of Fld with special role in protein function. This work shows how protein-protein docking simulations can help to explain complex biochemical and biophysical phenomena involving protein-protein interactions.
Supplementary Material
Resulting conformations for unbound-unbound FNR:Fd rigid-body docking and refinement. (A) Binding energy vs. RMSD of S2Fe2 atoms with respect to the corresponding ones in Anabaena complex, when receptors are superimposed. (B) Binding energy vs. RMSD of S2Fe2 atoms with respect to the corresponding ones in maize complex. (C) Percentage of satisfied experimental restraints vs. distance between redox centers (the docking orientation closest to the x-ray structure is shown as a gray closed circle). (D) Percentage of satisfied experimental restraints vs. ligand RMSD.
Computational analysis of FNR:Fd binding. The three distinct (i.e. > 2 Å RMSD) lowest-energy refined solutions are shown in ribbon, with the S2Fe2 in CPK (red: rank 1; yellow: rank 2; magenta: rank 3). FNR is represented in white, with FAD in cyan. The Anabaena crystallographic complex is also shown in green.
FNR:Fld rigid-body docking and refinement. (A) Percentage of satisfied experimental restraints vs. distance between redox centers. (B) Percentage of satisfied experimental restraints vs. ligand RMSD.
Acknowledgments
We are grateful to Molsoft (www.molsoft.com) for the use of the ICM program. This work has been supported by the Spanish Biotechnology Programme project BIO2004-00279 (Universidad de Zaragoza), the Plan Nacional I+D+I Grant BIO2005-06753, and the Ramón y Cajal Program from the Spanish Ministry of Education and Science.
References
- 1.Carrillo N, Ceccarelli EA. Open questions in ferredoxin-NADP+ reductase catalytic mechanism. Eur J Biochem. 2003;270:1900–1915. doi: 10.1046/j.1432-1033.2003.03566.x. [DOI] [PubMed] [Google Scholar]
- 2.Medina M, Gomez-Moreno C. Interaction of Ferredoxin-NADP(+) Reductase with its Substrates: Optimal Interaction for Efficient Electron Transfer. Photosynth Res. 2004;79:113–131. doi: 10.1023/B:PRES.0000015386.67746.2c. [DOI] [PubMed] [Google Scholar]
- 3.Hurley JK, Weber-Main AM, Stankovich MT, Benning MM, Thoden JB, Vanhooke JL, Holden HM, Chae YK, Xia B, Cheng H, Markley JL, Martinez-Julvez M, Gomez-Moreno C, Schmeits JL, Tollin G. Structure-function relationships in Anabaena ferredoxin: correlations between X-ray crystal structures, reduction potentials, and rate constants of electron transfer to ferredoxin:NADP+ reductase for site-specific ferredoxin mutants. Biochemistry. 1997;36:11100–11117. doi: 10.1021/bi9709001. [DOI] [PubMed] [Google Scholar]
- 4.Nogues I, Campos LA, Sancho J, Gomez-Moreno C, Mayhew SG, Medina M. Role of neighboring FMN side chains in the modulation of flavin reduction potentials and in the energetics of the FMN:apoprotein interaction in Anabaena flavodoxin. Biochemistry. 2004;43:15111–15121. doi: 10.1021/bi0483256. [DOI] [PubMed] [Google Scholar]
- 5.Karplus PA, Daniels MJ, Herriott JR. Atomic structure of ferredoxin-NADP+ reductase: prototype for a structurally novel flavoenzyme family. Science. 1991;251:60–66. [PubMed] [Google Scholar]
- 6.Serre L, Vellieux FM, Medina M, Gomez-Moreno C, Fontecilla-Camps JC, Frey M. X-ray structure of the ferredoxin:NADP+ reductase from the cyanobacterium Anabaena PCC 7119 at 1.8 A resolution, and crystallographic studies of NADP+ binding at 2. 25 A resolution. J Mol Biol. 1996;263:20–39. doi: 10.1006/jmbi.1996.0553. [DOI] [PubMed] [Google Scholar]
- 7.Deng Z, Aliverti A, Zanetti G, Arakaki AK, Ottado J, Orellano EG, Calcaterra NB, Ceccarelli EA, Carrillo N, Karplus PA. A productive NADP+ binding mode of ferredoxin-NADP + reductase revealed by protein engineering and crystallographic studies. Nat Struct Biol. 1999;6:847–853. doi: 10.1038/12307. [DOI] [PubMed] [Google Scholar]
- 8.Rypniewski WR, Breiter DR, Benning MM, Wesenberg G, Oh BH, Markley JL, Rayment I, Holden HM. Crystallization and structure determination to 2. 5-A resolution of the oxidized [2Fe-2S] ferredoxin isolated from Anabaena 7120. Biochemistry. 1991;30:4126–4131. doi: 10.1021/bi00231a003. [DOI] [PubMed] [Google Scholar]
- 9.Morales R, Charon MH, Hudry-Clergeon G, Petillot Y, Norager S, Medina M, Frey M. Refined X-ray structures of the oxidized, at 1.3 A, and reduced, at 1. 17 A, [2Fe-2S] ferredoxin from the cyanobacterium Anabaena PCC7119 show redox-linked conformational changes. Biochemistry. 1999;38:15764–15773. doi: 10.1021/bi991578s. [DOI] [PubMed] [Google Scholar]
- 10.Rao ST, Shaffie F, Yu C, Satyshur KA, Stockman BJ, Markley JL, Sundarlingam M. Structure of the oxidized long-chain flavodoxin from Anabaena 7120 at 2 A resolution. Protein Sci. 1992;1:1413–1427. doi: 10.1002/pro.5560011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales R, Charon MH, Kachalova G, Serre L, Medina M, Gomez-Moreno C, Frey M. A redox-dependent interaction between two electron-transfer partners involved in photosynthesis. EMBO Reports. 2000;1:271–276. doi: 10.1093/embo-reports/kvd057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurisu G, Kusunoki M, Katoh E, Yamazaki T, Teshima K, Onda Y, Kimata-Ariga Y, Hase T. Structure of the electron transfer complex between ferredoxin and ferredoxin-NADP(+) reductase. Nat Struct Biol. 2001;8:117–121. doi: 10.1038/84097. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Julvez M, Medina M, Gomez-Moreno C. Ferredoxin-NADP(+) reductase uses the same site for the interaction with ferredoxin and flavodoxin. J Biol Inorg Chem. 1999;4:568–578. [PubMed] [Google Scholar]
- 14.Martinez-Julvez M, Hermoso J, Hurley JK, Mayoral T, Sanz-Aparicio J, Tollin G, Gomez-Moreno C, Medina M. Role of Arg100 and Arg264 from Anabaena PCC 7119 ferredoxin-NADP+ reductase for optimal NADP+ binding and electron transfer. Biochemistry. 1998;37:17680–17691. doi: 10.1021/bi981718i. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Julvez M, Medina M, Hurley JK, Hafezi R, Brodie TB, Tollin G, Gomez-Moreno C. Lys75 of Anabaena ferredoxin-NADP+ reductase is a critical residue for binding ferredoxin and flavodoxin during electron transfer. Biochemistry. 1998;37:13604–13613. doi: 10.1021/bi9807411. [DOI] [PubMed] [Google Scholar]
- 16.Aliverti A, Corrado ME, Zanetti G. Involvement of lysine-88 of spinach ferredoxin-NADP+ reductase in the interaction with ferredoxin. FEBS Lett. 1994;343:247–250. doi: 10.1016/0014-5793(94)80565-2. [DOI] [PubMed] [Google Scholar]
- 17.Nogues I, Martinez-Julvez M, Navarro JA, Hervas M, Armenteros L, de la Rosa MA, Brodie TB, Hurley JK, Tollin G, Gomez-Moreno C, Medina M. Role of hydrophobic interactions in the flavodoxin mediated electron transfer from photosystem I to ferredoxin-NADP+ reductase in Anabaena PCC 7119. Biochemistry. 2003;42:2036–2045. doi: 10.1021/bi0270541. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Julvez M, Nogues I, Faro M, Hurley JK, Brodie TB, Mayoral T, Sanz-Aparicio J, Hermoso JA, Stankovich MT, Medina M, Tollin G, Gomez-Moreno C. Role of a cluster of hydrophobic residues near the FAD cofactor in Anabaena PCC 7119 ferredoxin-NADP+ reductase for optimal complex formation and electron transfer to ferredoxin. J Biol Chem. 2001;276:27498–27510. doi: 10.1074/jbc.M102112200. [DOI] [PubMed] [Google Scholar]
- 19.Hurley JK, Salamon Z, Meyer TE, Fitch JC, Cusanovich MA, Markley JL, Cheng H, Xia B, Chae YK, Medina M, et al. Amino acid residues in Anabaena ferredoxin crucial to interaction with ferredoxin-NADP+ reductase: site-directed mutagenesis and laser flash photolysis. Biochemistry. 1993;32:9346–9354. doi: 10.1021/bi00087a013. [DOI] [PubMed] [Google Scholar]
- 20.Hurley JK, Cheng H, Xia B, Markley JL, Medina M, Gomez-Moreno C, Tollin G. An aromatic amino acid is required at position 65 in Anabaena ferredoxin for rapid electron transfer to ferredoxin-NADP+ reductase. J Am Chem Soc. 1993;115:11698–11701. [Google Scholar]
- 21.Hurley JK, Medina M, Gomez-Moreno C, Tollin G. Further characterization by site-directed mutagenesis of the protein-protein interface in the ferredoxin/ferredoxin:NADP+ reductase system from Anabaena: requirement of a negative charge at position 94 in ferredoxin for rapid electron transfer. Arch Biochem Biophys. 1994;312:480–486. doi: 10.1006/abbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- 22.Nogues I, Hervas M, Peregrina JR, Navarro JA, de la Rosa MA, Gomez-Moreno C, Medina M. Anabaena flavodoxin as an electron carrier from photosystem I to ferredoxin-NADP+ reductase. Role of flavodoxin residues in protein-protein interaction and electron transfer. Biochemistry. 2005;44:97–104. doi: 10.1021/bi048324d. [DOI] [PubMed] [Google Scholar]
- 23.Hurley JK, Morales R, Martinez-Julvez M, Brodie TB, Medina M, Gomez-Moreno C, Tollin G. Structure-function relationships in Anabaena ferredoxin/ferredoxin:NADP(+) reductase electron transfer: insights from site-directed mutagenesis, transient absorption spectroscopy and X-ray crystallography. Biochim Biophys Acta. 2002;1554:5–21. doi: 10.1016/s0005-2728(02)00188-3. [DOI] [PubMed] [Google Scholar]
- 24.Frago S, Goni G, Serrano A, Mayhew SG, Gomez-Moreno C, Medina M. Role of Anabaena Flavodoxin Hydrophobic Residues in Protein-protein Interaction and Electron Transfer to Ferredoxin-NADP+ Reductase. Photosynthesis: Fundamental Aspects to Global Perspectives. 2005;2:544–546. [Google Scholar]
- 25.Casaus JL, Navarro JA, Hervas M, Lostao A, De la Rosa MA, Gomez-Moreno C, Sancho J, Medina M. Anabaena sp. PCC 7119 flavodoxin as electron carrier from photosystem I to ferredoxin-NADP+ reductase. Role of Trp(57) and Tyr(94) J Biol Chem. 2002;277:22338–22344. doi: 10.1074/jbc.M112258200. Epub 22002 Apr 22311. [DOI] [PubMed] [Google Scholar]
- 26.Goñi G, Serrano A, Frago S, Hervas M, Peregrina JR, De la Rosa MA, Gomez-Moreno C, Navarro JA, Medina M. Flavodoxin-mediated Electron Transfer from Photosystem I to Ferredoxin-NADP+ reductase in Anabaena: Role of Flavodoxin Hydrophobic Residues in Protein-Protein Interactions. Biochemistry. 2007 doi: 10.1021/bi7017392. In press. [DOI] [PubMed] [Google Scholar]
- 27.Mayoral T, Martinez-Julvez M, Perez-Dorado I, Sanz-Aparicio J, Gomez-Moreno C, Medina M, Hermoso JA. Structural analysis of interactions for complex formation between Ferredoxin-NADP+ reductase and its protein partners. Proteins. 2005;59:592–602. doi: 10.1002/prot.20450. [DOI] [PubMed] [Google Scholar]
- 28.Hurley JK, Faro M, Brodie TB, Hazzard JT, Medina M, Gomez-Moreno C, Tollin G. Highly nonproductive complexes with Anabaena ferredoxin at low ionic strength are induced by nonconservative amino acid substitutions at Glu139 in Anabaena ferredoxin:NADP+ reductase. Biochemistry. 2000;39:13695–13702. doi: 10.1021/bi001124r. [DOI] [PubMed] [Google Scholar]
- 29.Worrall JA, Reinle W, Bernhardt R, Ubbink M. Transient protein interactions studied by NMR spectroscopy: the case of cytochrome C and adrenodoxin. Biochemistry. 2003;42:7068–7076. doi: 10.1021/bi0342968. [DOI] [PubMed] [Google Scholar]
- 30.Smith GR, Sternberg MJ. Prediction of protein-protein interactions by docking methods. Curr Opin Struct Biol. 2002;12:28–35. doi: 10.1016/s0959-440x(02)00285-3. [DOI] [PubMed] [Google Scholar]
- 31.Mendez R, Leplae R, Lensink MF, Wodak SJ. Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures. Proteins. 2005;60:150–169. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Recio J, Totrov M, Abagyan R. Soft protein-protein docking in internal coordinates. Protein Sci. 2002;11:280–291. doi: 10.1110/ps.19202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Recio J, Abagyan R, Totrov M. Improving CAPRI predictions: optimized desolvation for rigid-body docking. Proteins. 2005;60:308–313. doi: 10.1002/prot.20575. [DOI] [PubMed] [Google Scholar]
- 34.Roberts VA, Pique ME. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J Biol Chem. 1999;274:38051–38060. doi: 10.1074/jbc.274.53.38051. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Recio J, Totrov M, Abagyan R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins. 2003;52:113–117. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Recio J, Totrov M, Abagyan R. Identification of protein-protein interaction sites from docking energy landscapes. J Mol Biol. 2004;335:843–865. doi: 10.1016/j.jmb.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 37.Medina M, Martinez-Julvez M, Hurley JK, Tollin G, Gomez-Moreno C. Involvement of glutamic acid 301 in the catalytic mechanism of ferredoxin-NADP+ reductase from Anabaena PCC 7119. Biochemistry. 1998;37:2715–2728. doi: 10.1021/bi971795y. [DOI] [PubMed] [Google Scholar]
- 38.Faro M, Frago S, Mayoral T, Hermoso JA, Sanz-Aparicio J, Gomez-Moreno C, Medina M. Probing the role of glutamic acid 139 of Anabaena ferredoxin-NADP+ reductase in the interaction with substrates. Eur J Biochem. 2002;269:4938–4947. doi: 10.1046/j.1432-1033.2002.03194.x. [DOI] [PubMed] [Google Scholar]
- 39.Nogues I, Tejero J, Hurley JK, Paladini D, Frago S, Tollin G, Mayhew SG, Gomez-Moreno C, Ceccarelli EA, Carrillo N, Medina M. Role of the C-terminal tyrosine of ferredoxin-nicotinamide adenine dinucleotide phosphate reductase in the electron transfer processes with its protein partners ferredoxin and flavodoxin. Biochemistry. 2004;43:6127–6137. doi: 10.1021/bi049858h. [DOI] [PubMed] [Google Scholar]
- 40.Faro M, Hurley JK, Medina M, Tollin G, Gomez-Moreno C. Flavin photochemistry in the analysis of electron transfer reactions: role of charged and hydrophobic residues at the carboxyl terminus of ferredoxin-NADP(+) reductase in the interaction with its substrates. Bioelectrochemistry. 2002;56:19–21. doi: 10.1016/s1567-5394(02)00039-7. [DOI] [PubMed] [Google Scholar]
- 41.Hurley JK, Schmeits JL, Genzor C, Gomez-Moreno C, Tollin G. Charge reversal mutations in a conserved acidic patch in Anabaena ferredoxin can attenuate or enhance electron transfer to ferredoxin:NADP+ reductase by altering protein/protein orientation within the intermediate complex. Arch Biochem Biophys. 1996;333:243–250. doi: 10.1006/abbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 42.Weber-Main AM, Hurley JK, Cheng H, Xia B, Chae YK, Markley JL, Martinez-Julvez M, Gomez-Moreno C, Stankovich MT, Tollin G. An electrochemical, kinetic, and spectroscopic characterization of [2Fe-2S] vegetative and heterocyst ferredoxins from Anabaena 7120 with mutations in the cluster binding loop. Arch Biochem Biophys. 1998;355:181–188. doi: 10.1006/abbi.1998.0743. [DOI] [PubMed] [Google Scholar]
- 43.Chelliah V, Blundell TL, Fernandez-Recio J. Efficient restraints for protein-protein docking by comparison of observed amino acid substitution patterns with those predicted from local environment. J Mol Biol. 2006;357:1669–1682. doi: 10.1016/j.jmb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frago S, Goñi G, Herguedas B, Peregrina JR, Serrano A, Perez-Dorado I, Molina R, Gomez-Moreno C, Hermoso JA, Martínez-Júlvez M, Mayhew SG, Medina M. Tuning of the FMN binding and oxido-reduction properties by neighboring side chains in Anabaena Flavodoxin. Arch Biochem Biophys. 2007 doi: 10.1016/j.abb.2007.08.024. in press. [DOI] [PubMed] [Google Scholar]
- 46.Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein-protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- 47.Blundell TL, Fernandez-Recio J. Cell biology: brief encounters bolster contacts. Nature. 2006;444:279–280. doi: 10.1038/nature05306. [DOI] [PubMed] [Google Scholar]
- 48.Muller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U. Adrenodoxin reductase-adrenodoxin complex structure suggests electron transfer path in steroid biosynthesis. J Biol Chem. 2001;276:2786–2789. doi: 10.1074/jbc.M008501200. [DOI] [PubMed] [Google Scholar]
- 49.Hagelueken G, Wiehlmann L, Adams TM, Kolmar H, Heinz DW, Tummler B, Schubert WD. Crystal structure of the electron transfer complex rubredoxin rubredoxin reductase of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2007;104:12276–12281. doi: 10.1073/pnas.0702919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iverson TM, Luna-Chavez C, Cecchini G, Rees DC. Structure of the Escherichia coli fumarate reductase respiratory complex. Science. 1999;284:1961–1966. doi: 10.1126/science.284.5422.1961. [DOI] [PubMed] [Google Scholar]
- 51.Gruez A, Pignol D, Zeghouf M, Coves J, Fontecave M, Ferrer JL, Fontecilla-Camps JC. Four crystal structures of the 60 kDa flavoprotein monomer of the sulfite reductase indicate a disordered flavodoxin-like module. J Mol Biol. 2000;299:199–212. doi: 10.1006/jmbi.2000.3748. [DOI] [PubMed] [Google Scholar]
- 52.Correll CC, Batie CJ, Ballou DP, Ludwig ML. Phthalate dioxygenase reductase: a modular structure for electron transfer from pyridine nucleotides to [2Fe-2S] Science. 1992;258:1604–1610. doi: 10.1126/science.1280857. [DOI] [PubMed] [Google Scholar]
- 53.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 54.Wolthers KR, Scrutton NS. Protein interactions in the human methionine synthase-methionine synthase reductase complex and implications for the mechanism of enzyme reactivation. Biochemistry. 2007;46:6696–6709. doi: 10.1021/bi700339v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Resulting conformations for unbound-unbound FNR:Fd rigid-body docking and refinement. (A) Binding energy vs. RMSD of S2Fe2 atoms with respect to the corresponding ones in Anabaena complex, when receptors are superimposed. (B) Binding energy vs. RMSD of S2Fe2 atoms with respect to the corresponding ones in maize complex. (C) Percentage of satisfied experimental restraints vs. distance between redox centers (the docking orientation closest to the x-ray structure is shown as a gray closed circle). (D) Percentage of satisfied experimental restraints vs. ligand RMSD.
Computational analysis of FNR:Fd binding. The three distinct (i.e. > 2 Å RMSD) lowest-energy refined solutions are shown in ribbon, with the S2Fe2 in CPK (red: rank 1; yellow: rank 2; magenta: rank 3). FNR is represented in white, with FAD in cyan. The Anabaena crystallographic complex is also shown in green.
FNR:Fld rigid-body docking and refinement. (A) Percentage of satisfied experimental restraints vs. distance between redox centers. (B) Percentage of satisfied experimental restraints vs. ligand RMSD.