

Abstract
Purpose
BCR-ABL1–like acute lymphoblastic leukemia (ALL) is a recently identified B-cell ALL (B-ALL) subtype with poor outcome that exhibits a gene expression profile similar to BCR-ABL1-positive ALL but lacks the BCR-ABL1 fusion protein. We examined the outcome of children with BCR-ABL1–like ALL treated with risk-directed therapy based on minimal residual disease (MRD) levels during remission induction.
Patients and Methods
Among 422 patients with B-ALL enrolled onto the Total Therapy XV study between 2000 and 2007, 344 had adequate samples for gene expression profiling. Next-generation sequencing and/or analysis of genes known to be altered in B-ALL were performed in patients with BCR-ABL1–like ALL who had available material. Outcome was compared between patients with and those without BCR-ABL1–like ALL.
Results
Forty (11.6%) of the 344 patients had BCR-ABL1–like ALL. They were significantly more likely to be male, have Down syndrome, and have higher MRD levels on day 19 and at the end of induction than did other patients with B-ALL. Among 25 patients comprehensively studied for genetic abnormalities, 11 harbored a genomic rearrangement of CRLF2, six had fusion transcripts responsive to ABL tyrosine kinase inhibitors or JAK inhibitors, and seven had mutations involving the Ras signaling pathway. There were no significant differences in event-free survival (90.0% ± 4.7% [SE] v 88.4% ± 1.9% at 5 years; P = .41) or in overall survival (92.5% ± 4.2% v 95.1% ± 1.3% at 5 years; P = .41) between patients with and without BCR-ABL1–like ALL.
Conclusion
Patients who have BCR-ABL1–like ALL with poor initial treatment response can be salvaged with MRD-based risk-directed therapy and may benefit from identification of kinase-activating lesions for targeted therapies.
INTRODUCTION
By using genome-wide analysis, two groups of investigators identified a subtype of B-cell acute lymphoblastic leukemia (B-ALL) termed “BCR-ABL1–like” or “Ph-like” ALL, which has a gene expression profile similar to that of Philadelphia chromosome (Ph) –positive ALL but lacks BCR-ABL1 fusion protein expressed from t(9;22)(q34.1;q11.2) and accounts for 10% to 15% of childhood B-progenitor ALL.1,2 Similar to Ph-positive ALL, BCR-ABL1–like ALL is characterized by a high frequency of alterations of the IKZF1 gene, which encodes the early lymphoid transcription factor IKAROS, and a high risk of relapse when treated with conventional chemotherapy.1,2
Although Ph-positive ALL is a single genetic entity defined by the presence of the Ph chromosome and the BCR-ABL1 gene fusion, BCR-ABL1–like ALL is a genetically heterogeneous disease. Approximately half the patients with BCR-ABL1–like ALL harbor abnormalities of the cytokine receptor gene CRLF2,3 either as a translocation to the immunoglobulin heavy chain enhancer region (IGH-CRLF2) or as a focal deletion resulting in the expression of a P2RY8-CRLF2 fusion transcript.4,5 Among patients with CRLF2 rearrangement, approximately half have concomitant activating mutations of the Janus kinase genes, JAK1 or JAK2, resulting in activation of JAK-STAT signaling.4–6 Recent transcriptome and whole-genome sequencing genomics of BCR-ABL1–like patients without CRLF2 rearrangements identified a diverse array of genetic alterations that activate cytokine receptor and tyrosine signaling.3,7 Because of the lack of uniform definition and different gene expression classifiers used by various groups of investigators, each study of BCR-ABL1–like ALL comprised a different, albeit overlapping, cohort of patients.
Although the prognostic impact of high CRLF2 expression varied according to the study cohort,8,9 BCR-ABL1–like ALL has been consistently associated with poor treatment outcome in all studies reported to date1,2,9–13 In this study, we examine the clinical heterogeneity and prognostic impact of BCR-ABL1–like ALL treated in the context of an effective risk-directed protocol based on minimal residual disease (MRD) levels measured during remission induction therapy.14
PATIENTS AND METHODS
Patients
From June 2000 to October 2007, 498 evaluable patients (1 to 18 years of age) with newly diagnosed ALL were consecutively enrolled onto the Total Therapy XV study at St. Jude Children's Research Hospital or at Cook Children's Medical Center.14 This study was limited to patients with B-ALL because all BCR-ABL1–like patients have B-lineage immunophenotype. Of the 422 patients enrolled onto the Total Therapy XV study with B-ALL, 344 (81.5%) had adequate samples to screen for the expression profile of BCR-ABL1–like ALL (Fig 1). The protocol was approved by the institutional review boards. Signed informed consent was obtained from the patients who were 18 years old or from the parents or guardians of younger patients, with assent from the patients, as appropriate.
Fig 1.
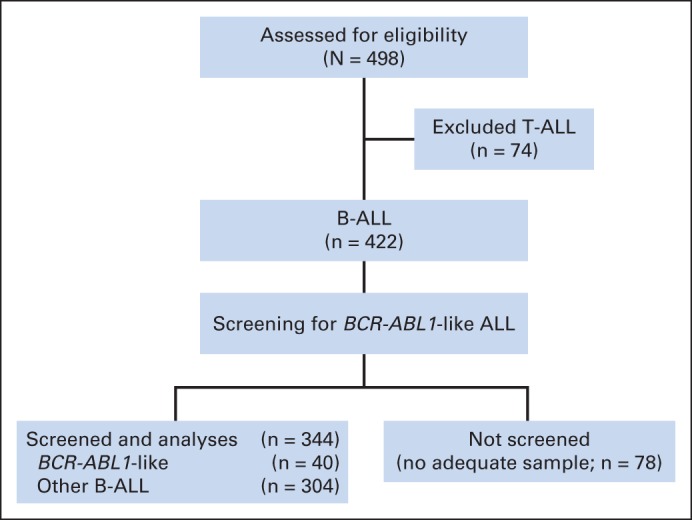
CONSORT diagram. Among the 498 patients enrolled onto the Total Therapy XV trial, 422 had B-cell acute lymphoblastic leukemia (B-ALL), of whom 344 had adequate samples to screen for the expression profile of BCR-ABL1–like ALL. T-ALL, T-cell ALL.
Diagnosis and Risk Classification
The diagnosis of ALL was based on morphologic, immunophenotypic, and genetic features of leukemic blast cells, as described previously.15 MRD was determined by flow cytometry, polymerase chain reaction analysis, or both.16,17 Risk classification was based on presenting age and leukocyte count (National Cancer Institute [NCI] risk group), leukemic cell genotype, and response to remission induction treatment. Patients with B-ALL who were between 1 and 10 years old, had a leukocyte count less than 50 × 109/L (NCI standard risk), and had a leukemic cell DNA index ≥ 1.16 or t(12;21)[ETV6-RUNX1] were provisionally classified as low-risk ALL. Patients with t(9;22)[BCR-ABL1] were considered to have high-risk ALL, and the remaining patients, including those with T-cell ALL and B-ALL with t(1;19)[TCF3-PBX1] were provisionally classified as standard- (intermediate-) risk ALL. The final risk status was determined by the level of MRD during and after remission induction therapy. Any patient with ≥ 1% leukemic cells in the bone marrow on day 19 of remission induction or 0.01% to 0.99% residual leukemia after completion of 6-week induction therapy was considered to have standard-risk ALL. Patients with ≥ 1% residual disease after completion of induction therapy were assigned to the high-risk group.
Treatment
Details of the treatment regimen have been described previously.14 In brief, patients with ≥ 1% residual leukemia in the bone marrow on day 19 of remission induction were given three additional doses of asparaginase. At the end of induction (between days 43 and 46), bone marrow aspiration was performed to assess MRD level, and consolidation therapy with high-dose methotrexate and daily mercaptopurine was given for four courses. Low-risk and standard-risk patients then received risk-directed continuation therapy, and high-risk patients were offered the option of allogeneic hematopoietic stem-cell transplantation. All patients received early triple intrathecal therapy, and none received prophylactic cranial irradiation.
Identification and Genomic Characterization of BCR-ABL1–Like ALL
Single nucleotide polymorphism 500K or single nucleotide polymorphism 6.0 microarrays and U133A gene expression profiling (Affymetrix) were performed in all patients with suitable genomic material available to identify copy number alterations and distinct genetic subgroups by using reference normalization18 and circular binary segmentation,19 as previously described.1,20 Prediction analysis for microarrays was used to identify patients with BCR-ABL1–like ALL with a gene expression profile similar to that of BCR-ABL1 ALL.3,21 Sequence mutation analysis for genes known to be mutated in B-ALL (including IKZF1, PAX5, and JAK2) was performed for all patients with available material, including 16 patients with BCR-ABL1–like ALL, 12 of whom were also studied with next-generation sequencing. Genomic polymerase chain reaction (PCR), Sanger sequencing, and mutation detection were performed as previously described.6,20,22 CRLF2 rearrangements were identified by genomic PCR, real-time PCR, and fluorescent in situ hybridization, as previously described.5 Next-generation sequencing was performed by the St. Jude Children's Research Hospital-Washington University Pediatric Cancer Genome Project, as previously described,23–25 with messenger RNA (mRNA) sequencing in seven patients, whole-genome sequencing in five patients, and both mRNA sequencing and whole-genome sequencing in nine patients (Appendix Table A1, online only). Fusion transcripts were identified from mRNA sequencing data by using CICERO, a novel analysis algorithm (Li et al, manuscript in preparation). Putative fusions were confirmed by real-time PCR and bidirectional Sanger sequencing. Sequence data are deposited at the European Genome Phenome archive, accession EGAS00001000654.
Statistical Analysis
Event-free survival and survival from diagnosis were estimated by the method of Kaplan-Meier, with associated SEs calculated by the method of Peto and Pike. The cumulative incidence functions of relapse were estimated by the method of Kalbfleisch and Prentice,26 and the functions were compared by using Gray's test. Deaths in remission were considered competing events in the estimation of cumulative incidence of relapse. Outcome data updated on August 23, 2013, were used for this analysis. The median follow-up time for patients remaining in continuous remission was 8.2 years (range, 1.3 to 12.9 years). At the time of analysis, 95% of the survivors had had a follow-up visit within 2 years; only 2.0% of the patients lacked a documented contact within the previous 5 years.
RESULTS
Presenting features and treatment outcome of the 344 patients who were evaluated for the gene expression profile of BCR-ABL1–like ALL were not significantly different from those of the 78 patients who were not (data not shown). Of the 344 patients with B-ALL who were evaluated, 40 (11.6%) had BCR-ABL1–like ALL. Table 1 summarizes the clinical and biologic features as well as treatment and outcome of these 40 patients (27 males and 13 females). Their median age was 5.3 years (range, 1.3 to 18.6 years), and median presenting leukocyte count was 7.1 × 109/L (range, 1.7 to 258.3 × 109/L). Compared with other patients with B-ALL, those with BCR-ABL1–like ALL were more likely to be male (P = .04), have Down syndrome (P = .003), and have higher MRD on day 19 (P = .009) and at the end of induction (P = .001; Table 2). There was no significant difference in the distribution of the risk groups between patients with BCR-ABL1–like ALL and those with other B-ALL on the basis of initial risk classification of the protocol (P = .41) or NCI risk classification (P = .61). However, after including response to remission induction based on MRD levels in the risk-classification algorithm, patients with BCR-ABL1–like ALL were more likely to be classified as having standard- or high-risk ALL (P = .02; Table 2).
Table 1.
Clinical and Laboratory Characteristics of the 40 Patients With BCR-ABL1–Like ALL
| Patient | Risk Group | % MRD |
Genomic Lesion | DNA Index ≥ 1.16 | Age at Diagnosis (years) | Sex | Race/Ethnicity† | WBC × 109/L | Current Status | |
|---|---|---|---|---|---|---|---|---|---|---|
| Day 19 | Day 46 | |||||||||
| 1 | Low | < 0.01 | < 0.01 | Not tested | Yes | 6.7 | M | White | 1.7 | First remission for 8.3+ years |
| 2 | Low | < 0.01 | < 0.01 | Not tested | No | 1.29 | F | White | 4.5 | First remission for 10.1+ years |
| 3 | Low | < 0.01 | < 0.01 | Not tested | Yes | 1.8 | M | Other | 4 | First remission for 3.9+ years |
| 4 | Low | < 0.01 | < 0.01 | Not tested | Yes | 2.66 | M | White | 5.3 | Second remission for 5.5+ years after hematologic relapse |
| 5 | Low | < 0.01 | < 0.01 | Not tested | Yes | 3.18 | F | White | 15.5 | Second remission for 6.4+ years after combined hematologic and CNS relapse |
| 6 | Low | < 0.01 | < 0.01 | FLT3 p.Tyr597insGly and Ala680Val | Yes | 4.08 | M | White | 16.8 | First remission for 6.2+ years |
| 7 | Low | < 0.01 | < 0.01 | IL7R p.Val253Gly and JAK1 p.Ala428Pro | No | 2.11 | M | White | 45.1 | First remission for 11.2+ years |
| 8 | Low | < 0.01 | < 0.01 | P2RY8-CRLF2 and JAK2 p.Arg683Ser | No | 3.99 | M | White | 23.3 | First remission for 12.6+ years |
| 9 | Low | 0.01 | < 0.01 | P2RY8-CRLF2 and JAK2 p.Arg683Gly | No | 5.26 | M | White | 4.4 | First remission for 10.0+ years |
| 10 | Low | 0.024 | < 0.01 | P2RY8-CRLF2 | No | 3.94 | M | White | 5 | First remission for 8.4+ years |
| 11 | Low | 0.031 | < 0.01 | NRAS p.Gln61Lys | Yes | 3.26 | F | Hispanic | 7.5 | First remission for 7.6+ years |
| 12 | Low | 0.032 | < 0.01 | SSBP2-PDGFRB | No | 1.9 | F | Black | 33 | First remission for 10.5+ years |
| 13 | Low | 0.042 | < 0.01 | FLT3 p.Ile836del | No | 3.47 | M | White | 10.3 | First remission for 6.1+ years |
| 14 | Low | 0.047 | < 0.01 | P2RY8-CRLF2 | No | 2.88 | F | White | 5.2 | First remission for 8.7+ years |
| 15 | Low | 0.63 | < 0.01 | Not tested | No | 8.11 | F | White | 1.8 | First remission for 7.8+ years |
| 16 | Low | < 0.01 | < 0.01 | No lesion by mRNA sequencing or WGS | Yes | 18.45 | M | White | 5.4 | First remission for 8.6+ years |
| 17 | Standard | < 0.01 | < 0.01 | Not tested | No | 5.95 | F | Black | 4.8 | First remission for 7.8+ years |
| 18 | Standard | 0.034 | < 0.01 | NF1 deletion | No | 3.35 | M | White | 5.9 | First remission for 8.1+ years |
| 19 | Standard | 0.7 | 0.429 | Not tested | Yes | 6.19 | M | White | 4.3 | First remission for 9.1+ years |
| 20 | Standard | 3.01 | 0.1 | Not tested | No | 3.73 | M | White | 2.9 | First remission for 11.2+ years |
| 21 | Standard | 3.8 | < 0.01 | Not tested | Yes | 1.98 | M | Hispanic | 8.3 | First remission for 8.3+ years |
| 22 | Standard | 10 | 0.081 | P2RY8-CRLF2 and JAK2 p.Arg683Gly | No | 3.04 | M | White | 33.6 | First remission for 9.3+ years |
| 23 | Standard | 37.5 | 0.096 | NF1 deletion | No | 5.43 | M | White | 4.1 | Died of hematologic relapse 6.0 years after first remission |
| 24 | Standard | < 0.01 | < 0.01 | Not tested | No | 11.86 | M | White | 2.2 | First remission for 9.9+ years |
| 25 | Standard | < 0.01 | < 0.01 | IL7R p.Ile241_Ile245delinsCysLeuCys | No | 15.76 | F | White | 2.3 | First remission for 11.1+ years |
| 26 | Standard | 0.015 | < 0.01 | P2RY8-CRLF2 and JAK2 p.Arg683Gly | No | 5.35 | M | White | 245.4 | First remission for 7.3+ years |
| 27 | Standard | 0.024 | < 0.01 | Not tested | No | 14.19 | M | Black | 10.4 | Died of combined hematologic and CNS relapse 1.8 years after first remission |
| 28 | Standard | 0.057 | 0.01 | P2RY8-CRLF2 and NRAS p.Gln61Lys | No | 1.87 | F | White | 258.3 | First remission for 9.7+ years |
| 29 | Standard | 0.218 | 0.044 | No lesion by RNA sequencing or WGS | Yes | 16.4 | F | Hispanic | 6 | First remission for 11.4+ years |
| 30 | Standard | 0.927 | < 0.01 | P2RY8-CRLF2,IL7R p.Ser185Cys, NF1 p.Arg1241* and deletion | No | 13.74 | F | White | 2.7 | First remission for 5.8+ years |
| 31 | Standard | 1.83 | 0.01 | Unknown CRLF2 rearrangement | No | 18.55 | M | White | 1.7 | First remission for 6.4+ years |
| 32 | Standard | 2.78 | 0.059 | IGH-CRLF2 and JAK2 p.Arg683Gly | No | 7.66 | M | White | 78.1 | First remission for 12.1+ years |
| 33 | Standard | 8.68 | 0.07 | SSBP2-CSF1R | No | 12.94 | M | White | 103.2 | First remission for 8.5+ years |
| 34 | Standard | 10.5 | 0.133 | Not tested | No | 16.23 | M | Hispanic | 9.8 | Died as a result of an accident 3.1 years after first remission |
| 35 | High | 4.8 | 0.039 | Not tested | No | 1.7 | F | White | 8.1 | First remission for 6.1+ years |
| 36 | High | 16.9 | 3.06 | P2RY8-CRLF2 and JAK1 p.Val658Phe | No | 7.03 | F | Hispanic | 23.5 | Died of respiratory failure after transplantation in first remission for 0.7 years |
| 37 | High | 18.4 | 0.82 | Not tested | No | 7.16 | M | Other | 11.7 | First remission for 5.0+ years |
| 38 | High | 61 | 8.69 | EBF1-PDGFRB | No | 3.97 | M | White | 41.8 | First remission for 9.7+ years |
| 39 | High | 72.1 | 36 | No lesion by mRNA sequencing or WGS | No | 18 | M | White | 6.6 | First remission for 9.5+ years after initial induction failure |
| 40 | High | 73.1 | 6.74 | NUP214-ABL1 | No | 16.32 | M | White | 135.6 | First remission for 7.7+ years |
NOTE. Patients 17, 18, and 21 were classified as having standard-risk leukemia because of CNS3 status, near-haploidy, and high level of minimal residual disease (MRD) at day 19 of remission induction, respectively. DNA index of > 1.16 indicates cases with high hyperdiploidy.
Abbreviations: ALL, acute lymphoblastic leukemia; WGS, whole-genome sequencing.
Genetically determined race/ethnicity using the single nucleotide polymorphism–based ancestry information.
Table 2.
Comparison of Clinical and Biologic Variables Between Patients With and Without BCR-ABL1–Like ALL
| Variable |
BCR-ABL1–Like ALL |
P | |||
|---|---|---|---|---|---|
| Yes |
No |
||||
| No. | % | No. | % | ||
| Age at diagnosis, years | |||||
| 1-10 | 29 | 10.51 | 247 | 89.49 | .21 |
| > 10 | 11 | 16.18 | 57 | 83.82 | |
| Leukocyte count × 109/L | |||||
| < 50 | 35 | 12.92 | 236 | 87.08 | .22 |
| ≥ 50 | 5 | 6.85 | 68 | 93.15 | |
| DNA index | |||||
| ≥ 1.16 | 10 | 11.49 | 77 | 88.51 | 1.00 |
| < 1.16 | 30 | 11.67 | 227 | 88.33 | |
| Sex | |||||
| Male | 27 | 15.08 | 152 | 84.92 | .04 |
| Female | 13 | 7.88 | 152 | 92.12 | |
| Down syndrome | |||||
| No | 35 | 10.48 | 299 | 89.52 | .003 |
| Yes | 5 | 50.00 | 5 | 50.00 | |
| Race/ethnicity* | |||||
| White | 30 | 13.64 | 190 | 86.36 | .557 |
| Black | 3 | 5.66 | 50 | 94.34 | |
| Asian | 0 | 0.00 | 4 | 100.00 | |
| Hispanic | 5 | 10.00 | 45 | 90.00 | |
| Other | 2 | 11.76 | 15 | 88.24 | |
| CNS status | |||||
| CNS1 combined with traumatic tap without blasts | 26 | 10.36 | 225 | 89.64 | .24 |
| CNS3 ≥ 5 WBC/μL with blasts | 2 | 28.57 | 5 | 71.43 | |
| CNS2 < 5 WBC/μL with blasts | 9 | 13.04 | 60 | 86.96 | |
| Traumatic tap with blasts | 3 | 17.65 | 14 | 82.35 | |
| NCI risk group | |||||
| Standard | 26 | 12.50 | 182 | 87.50 | .61 |
| High | 14 | 10.29 | 122 | 89.71 | |
| t(9;22)(BCR-ABL1) | |||||
| Absent | 40 | 11.90 | 296 | 88.10 | .60 |
| Present | 0 | 0.00 | 8 | 100.00 | |
| t(1;19)(TCF3-PBX1) | |||||
| Absent | 40 | 12.54 | 279 | 87.46 | .10 |
| Present | 0 | 0.00 | 25 | 100.00 | |
| t(4;11)(MLL-AFF1) | |||||
| Absent | 40 | 11.73 | 301 | 88.27 | 1.00 |
| Present | 0 | 0.00 | 3 | 100.00 | |
| t(12;21)(ETV6-RUNX1) | |||||
| Absent | 40 | 15.38 | 220 | 84.62 | < .001 |
| Present | 0 | 0.00 | 84 | 100.00 | |
| Ploidy | |||||
| Hyperdiploidy > 50 | 11 | 10.78 | 91 | 89.22 | .72 |
| Others | 29 | 12.78 | 198 | 87.22 | |
| Initial risk classification | |||||
| Low | 24 | 10.48 | 205 | 89.52 | .41 |
| Standard | 16 | 14.81 | 92 | 85.19 | |
| High | 0 | 0.00 | 7 | 100.00 | |
| Final risk classification† | |||||
| Low | 16 | 8.21 | 179 | 91.79 | .02 |
| Standard | 18 | 14.17 | 109 | 85.83 | |
| High | 6 | 27.27 | 16 | 72.73 | |
| MRD on day 19 | |||||
| < 5% | 30 | 9.84 | 275 | 90.16 | .009 |
| ≥ 5% | 9 | 26.47 | 25 | 73.53 | |
| MRD at the end of induction | |||||
| < 0.01% | 24 | 8.60 | 255 | 91.40 | .001 |
| ≥ 0.01% | 16 | 25.81 | 46 | 74.19 | |
Abbreviations: ALL, acute lymphoblastic leukemia; MRD, minimal residual disease; NCI, National Cancer Institute.
Genetically determined race/ethnicity using the single nucleotide polymorphism–based ancestry information.
Based on MRD levels measured on days 19 and 46 of remission induction.
All six patients with high-risk BCR-ABL1–like ALL received hematopoietic stem-cell transplantation. The proportion of patients with BCR-ABL1–like ALL who received a transplantation (15%) was significantly higher than 4.3% for the other patients with B-ALL treated in the same protocol (P = .015). Adverse events among patients with BCR-ABL1–like ALL included hematologic with or without CNS relapse in four patients (No. 4, 5, 23, and 27), two deaths in remission (No. 34 and 36), and one induction failure (No. 39).
None of the 40 patients had t(1;19)(TCF3-PBX1), t(4;11)(MLL-AFF1), or t(12;21)(ETV6-RUNX1); 11 had hyperdiploidy with gain of at least five chromosomes. Of the 40 patients with BCR-ABL1–like ALL identified by gene expression profiling, 11 (27.5%) harbored a genomic rearrangement of CRLF2, including nine patients with P2RY8-CRLF2 and one with IGH-CRLF2. An additional patient had marked CRLF2 overexpression suggestive of rearrangement but was negative for these alterations. Six of these patients with deregulated CRLF2 had concomitant JAK mutations, including JAK2 p.Arg683Gly (four patients), 683Ser (one patient), and JAK1 p.Val658Phe (one patient).
Twenty-five patients with BCR-ABL1–like ALL had suitable material for detailed genomic interrogation and sequencing, including 14 patients who lacked CRLF2 rearrangement. Four patients had fusion transcripts responsive to ABL tyrosine kinase inhibitors, including SSBP2-PDGFRB (patient 12), SSBP2-CSF1R (patient 33), EBF1-PDGFRB (patient 38), and NUP214-ABL1 (patient 40). Two patients had genetic lesions predicted to be responsive to JAK inhibition: IL7R p.Val253Gly and JAK1 p.Ala428Pro (patient 7) and IL7R p.Ile241_Ile245delinsCysLeuCys (patient 25). Seven patients harbored mutations activating Ras signaling (patients 6, 11, 13, 18, 23, 28, and 30). Three patients lacked a kinase activating lesion on mRNA sequencing or whole-genome sequencing (patients 16, 29, and 39). Of the four patients with BCR-ABL1–like ALL with high hyperdiploidy, two had alterations in the Ras pathway with a complex FLT3 mutation (patient 6) and NRAS p.Gln61Lys substitution (patient 11). Collectively, of the 25 patients subjected to detailed genomic profiling and sequencing, 22 (88%) had cytokine receptor, kinase, or Ras alterations.
Seven (27%) of 26 patients with BCR-ABL1–like ALL studied had deletion of IKZF1, a frequency markedly lower than that of patients with BCR-ABL1–like ALL in previous pediatric cohorts.1,7 Seven patients had other lesions involving the B-lineage transcription factor genes PAX5 and EBF1 and, collectively, 11 (42%) had one or more lesions in this pathway.
Despite inferior response to remission induction therapy as shown by overall higher levels of MRD (Table 2), patients with BCR-ABL1–like ALL did not have a significantly inferior event-free survival or overall survival compared with patients with other B-ALL subtypes (Figs 2A and 2B). Even when treatment outcome was analyzed separately within the three risk groups, no significant differences in event-free survival or overall survival between the two groups were noted (Table 3).
Fig 2.

Kaplan-Meier estimates of (A) event-free survival and (B) overall survival for patients with BCR-ABL1–like acute lymphoblastic leukemia (ALL) and other B-cell ALL (B-ALL) subtypes. Five- and 10-year rates are reported as means ± SE.
Table 3.
Comparison of EFS and OS Between Patients With BCR-ABL1–Like ALL and Other Patients With B-ALL, According to Final Risk Group
| Risk Group* | No. of Patients | % EFS (± SE) |
% OS (± SE) |
||||
|---|---|---|---|---|---|---|---|
| Year 5 | Year 10 | P | Year 5 | Year 10 | P | ||
| Low | |||||||
| BCR-ABL1–like | 16 | 100 ± 0.0 | 85.6 ± 14.5 | .36 | 100 ± 0.0 | 100 ± 0.0 | .49 |
| Other B-ALL | 179 | 95.0 ± 1.6 | 92.5 ± 4.2 | 98.3 ± 1.0 | 96.2 ± 3.0 | ||
| Standard | |||||||
| BCR-ABL1–like | 18 | 88.9 ± 7.2 | 83.3 ± 15.2 | .97 | 88.9 ± 7.2 | 83.0 ± 15.3 | .32 |
| Other B-ALL | 109 | 84.3 ± 3.6 | 83.3 ± 6.9 | 92.7 ± 2.6 | 91.6 ± 5.0 | ||
| High | |||||||
| BCR-ABL1–like | 6 | 66.7 ± 17.2 | 66.7 ± 38.5 | .49 | 83.3 ± 13.9 | 83.3 ± 34.0 | .58 |
| Other B-ALL | 16 | 41.3 ± 12.0 | 41.3 ± 22.4 | 74.0 ± 10.9 | 64.8 ± 22.2 | ||
Abbreviations: ALL, acute lymphoblastic leukemia; B-ALL, B-cell ALL; EFS, event-free survival; OS, overall survival.
Based on minimal residual disease levels measured on days 19 and 46 of remission induction.
Consistent with the above results, the cumulative risk of isolated hematologic relapse or any relapse did not differ significantly between patients with BCR-ABL1–like ALL and those with other B-ALL subtypes, regardless of whether these parameters were analyzed including all patients (Figs 3A and 3B) or separately within the three risk groups (Table 4). Similar results for the comparison of event-free survival, overall survival, and cumulative risk of relapse were obtained after exclusion of the 10 patients with Down syndrome or those with specific translocations including t(1;19)(TCF3-PBX1), t(4;11)(MLL-AFF1), t(12;21)(ETV6-RUNX1), and t(9;22)(BCR-ABL1) (data not shown).
Fig 3.

Cumulative risk of (A) isolated hematologic relapse or (B) any relapse for patients with BCR-ABL1–like acute lymphoblastic leukemia (ALL) and other B-cell ALL (B-ALL) subtypes. Five- and 10-year rates are reported as means ± SE.
Table 4.
Comparison of Cumulative Risk of Isolated Hematologic Relapse or Any Relapse Between Patients With BCR-ABL1–Like ALL or Other B-ALL, According to Final Risk Group
| Risk Group* | No. of Patients | % Cumulative Risk of Isolated Hematologic Relapse (± SE) |
% Cumulative Risk of Any Relapse (± SE) |
||||
|---|---|---|---|---|---|---|---|
| Year 5 | Year 10 | P | Year 5 | Year 10 | P | ||
| Low | |||||||
| BCR-ABL1–like | 16 | 0 | 6.7 ± 6.7 | .46 | 0 | 14.4 ± 9.9 | .23 |
| Other B-ALL | 178 | 1.7 ± 1.0 | 4.2 ± 2.2 | 4.0 ± 1.5 | 6.4 ± 2.4 | ||
| Standard | |||||||
| BCR-ABL1–like | 18 | 5.6 ± 5.6 | 5.6 ± 5.6 | .58 | 11.1 ± 7.6 | 11.1 ± 7.6 | .74 |
| Other B-ALL | 109 | 9.3 ± 2.8 | 9.3 ± 2.8 | 13.9 ± 3.4 | 13.9 ± 3.4 | ||
| High | |||||||
| BCR-ABL1–like | 5 | 0 | 0 | .28 | 0 | 0 | .11 |
| Other B-ALL | 15 | 21.3 ± 11.6 | 21.3 ± 11.6 | 42.0 ± 13.9 | 42.0 ± 13.9 | ||
Abbreviations: ALL, acute lymphoblastic leukemia; B-ALL, B-cell ALL.
Based on minimal residual disease levels measured on days 19 and 46 of remission induction.
In multivariable analyses, including known prognostic factors in the Total Therapy XV study (the presence of BCR-ABL1, MRD > 5% on day 19 of induction, MRD > 0.01% on day 46 of induction, IKZF1 alteration, and BCR-ABL1–like ALL), only MRD more than 0.01% on day 46 of induction was independently associated with poor survival (hazard ratio, 5.18; 95% CI, 1.59 to 16.9; P = .006; Appendix Table A2, online only).
Among patients with BCR-ABL1–like ALL, the only prognostic factor is the presence of MRD more than 5% on day 19 of remission induction. The nine patients with MRD more than 5% on day 19 of induction had inferior event-free survival (66.7% ± 14.5% v 96.7% ± 3.3% at 5 years; P = .006) and inferior overall survival (77.8% ± 13.0% v 96.7% ± 3.3%; P = .008) compared with the other 30 patients with lower MRD levels. Event-free survival was not significantly different between the 11 patients with and the 29 patients without rearrangement of CRLF2 (90.9% ± 8.3% v 89.7% ± 5.9% at 5 years and 90.0% ± 15.8% v 76.8% ± 14.0% at 10 years; P = .40). The seven patients with IKZF1 alterations tended to have a poorer event-free survival than the 19 patients without IKZF1 alterations (71.4% ± 15.6% v 100% ± 0.0% at 5 years and 71.4% ± 27.0% v 94.7% ± 8.9% at 10 years; P = .08).
DISCUSSION
This study demonstrates that the adverse prognosis of pediatric BCR-ABL1–like ALL can be improved by effective risk-directed therapy based primarily on MRD levels during and at the end of remission induction therapy. It should be noted that our patient population and their risk assignment may be different from those of previously reported studies. In contrast to prior studies that selectively examined high-risk B-ALL and did not apply MRD measurement for risk-directed therapy,1,2,10,27 this study included all patients with newly diagnosed ALL and used MRD level to direct intensity of therapy. In this regard, patients with BCR-ABL1–like ALL in this study do not have a significantly higher frequency of NCI high-risk ALL at diagnosis compared with patients with other B-ALL subtypes. However, a high proportion of patients with BCR-ABL1–like ALL in this study were subsequently classified as having higher-risk leukemia (standard or high risk according to Total Therapy XV criteria) based on MRD levels during and after completion of remission induction therapy.
Patients with BCR-ABL1–like ALL in Total Therapy XV had the same proportion of high hyperdiploidy as did other patients with B-ALL but lacked other genetic abnormalities with prognostic or therapeutic implications such as t(1;19)(TCF3-PBX1), t(4;11)(MLL-AFF1), and t(12;21)(ETV6-RUNX1). A notable finding was a lower frequency of CRLF2 (27.5%) and IKZF1 (27%) alterations in BCR-ABL1–like ALL than in prior studies (approximately 50% and 70%, respectively). This may, in part, reflect differences in study cohorts, because prior studies selectively examined high-risk ALL, included patients older than 18 years, and were enriched for patients of Hispanic ethnicity (22% to 29%) treated in clinical trials by the Children's Oncology Group.1,3,10 In this regard, in the Children's Oncology Group studies, Hispanic patients with high Native American genetic ancestry have a high frequency of rearrangements of CRLF228 and inherited GATA3 rs3824662 risk allele,29 which were associated with BCR-ABL1–like ALL and inferior treatment outcome. In contrast, the Total Therapy XV study enrolled consecutive patients 1 to 18 years old from all risk groups and had a relatively low frequency (14.5%) of patients with Hispanic ethnicity.
In a risk classification schema based solely on the presenting age, initial leukocyte count, and conventional cytogenetic and molecular genetic features, approximately 60% of our patients would have been treated as having low-risk ALL. Thus, many of the patients with BCR-ABL1–like ALL in the other series with resistant leukemia might not have received intensive treatment because they were not recognized as a result of a lack of response evaluation. By contrast, the Total Therapy XV study used MRD measurements for risk-directed treatment. With this additional information, only 40% of the patients received treatment for low-risk ALL, and the remaining patients were treated with more intensive therapy for standard-risk (45%) or high-risk (15%) ALL.
With improved risk assessment based on MRD measurements, comparable results between BCR-ABL1–like ALL and other B-ALL subtypes were achieved in the Total Therapy XV study; these results extended to all three risk groups (Tables 3 and 4). Therefore, for centers that lack the capability to identify BCR-ABL1–like ALL, excellent overall treatment results can still be achieved, provided that reliable methods for monitoring MRD are available. However, we argue that it is still clinically important to identify BCR-ABL1–like ALL because many of these patients harbor genetic lesions that are sensitive to tyrosine kinase inhibitors (eg, ABL1 and PDGFRB rearrangements) or JAK inhibitors (eg, EPOR, IL7R, JAK2, and SH2B3 that activate JAK-STAT signaling) in preclinical models using cell lines and human leukemic cells.3,7,30,31 The incorporation of these agents in the treatment of patients with targetable lesions could potentially help reduce the intensity of chemotherapy. Although our series had only a limited number of refractory or relapsed patients and was not large enough to capture the full spectrum of targetable lesions, anecdotal reports of refractory BCR-ABL1–like ALL that responded well to tyrosine kinase inhibitors are already emerging.32–34 Because of their high level of MRD at the end of remission induction, six (15%) of the 40 patients with BCR-ABL1–like ALL in this series received hematopoietic stem-cell transplantation, a proportion much higher than 4.3% for the other patients with B-ALL treated in the same protocol.14 Conceivably, identification of patients with BCR-ABL1–like ALL with targetable genetic lesions followed by treatment with tyrosine kinase inhibitors or JAK inhibitors might avoid transplantation in some of these patients, similar to the use of tyrosine kinase inhibitors in BCR-ABL1-positive childhood ALL, which have improved outcome and reduced the number of patients requiring transplantation.35,36 Of note, among four of the six patients with BCR-ABL1–like ALL who received transplantations and were retrospectively tested for genomic lesions in this series, two had fusion transcripts that would respond to ABL tyrosine kinase inhibitors.
Only two of 16 low-risk patients with BCR-ABL1–like ALL treated with an antimetabolite-based regimen in this series (and one of six females 1 to 3 years old with the ZMIZ1-ABL1 fusion treated with conventional therapy in another report)37 had a late relapse after completion of therapy. Both relapsed patients who were initially treated with low-risk therapy in this study are long-term survivors after retrieval chemotherapy. Thus, in the setting of limited resources, screening for BCR-ABL1–like ALL may not be necessary for low-risk patients with negative MRD after remission induction and may be reserved for those with refractory or high-risk ALL, especially patients with high levels of MRD, and for patients with relapsed B-ALL. If our finding of the lack of t(1;19)(TCF3-PBX1), t(4;11)(MLL-AFF1), or t(12;21)(ETV6-RUNX1) in BCR-ABL1–like ALL is confirmed by additional studies, patients with these genetic abnormalities could also be excluded from screening. However, detailed genomic studies were not performed in both relapsed patients with BCR-ABL1–like ALL in the low-risk group in this study. Conceivably, they had genetic lesions responsive to tyrosine kinase inhibitors. Thus, additional studies are still needed to characterize the low-risk patients.
In summary, the advances in cure rates brought about by contemporary MRD-based treatment of childhood ALL have extended to patients with BCR-ABL1–like ALL despite their initial inferior response to treatment. Identification of this ALL subtype and administration of targeted therapy may further improve overall cure rates beyond 90% achieved in some of the contemporary clinical trials38 and improve their quality of life.
Supplementary Material
Glossary Terms
- cumulative incidence of relapse:
the use of competing risk analyses indicated in the presence of competing events (such as death and relapse); the Gray's test is a recommended method to estimate cumulative incidence of relapse.
- genomics:
the scientific discipline in which multiple genes, gene products, or regions of the genome are analyzed via large-scale, high-throughput molecular approaches directed to DNA and RNA. This definition is a deviation from that of the original term, which meant an analysis of the whole genome.
- JAK/STAT pathway:
the pathway usually (not always) activated by cytokine receptors, where binding of a ligand to the cytokine receptor leads to recruitment and subsequent autophosphorylation of JAK proteins (activated state) at the cellular membrane level. Activated JAKs phosphorylate the receptor, creating docking sites for specific signaling proteins, including STAT proteins. When coupled to the activated receptor, STAT proteins are phosphorylated (activated) by JAK proteins. In contrast to cytokine receptor signaling, receptors with intrinsic tyrosine kinase activity (eg, epidermal growth factor receptor, platelet-derived growth factor) may bypass JAK activation and directly phosphorylate STAT proteins. See JAK (Janus kinase) and STAT.
Appendix
Table A1.
Genomic Analysis of BCR-ABL1–Like ALL
| Sample No. | Sample ID | Group | Analysis Type |
Genomic Lesion | |||||
|---|---|---|---|---|---|---|---|---|---|
| Genomic | Sanger | RT-PCR | NGS | RNA Sequencing | WGS | ||||
| 1 | SJHYPER089 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 2 | SJBALL070 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 3 | SJHYPER088 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 4 | SJHYPER201 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 5 | SJBALL159 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 6 | SJHYPER150 | BCR-ABL1–like_non-CRLF2 | Yes | No | No | Yes | No | Yes | FLT3 p.Tyr597insGly and Ala680Val |
| 7 | SJHYPO109 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | Yes | Yes | IL7R p.Val253Gly and JAK1 p.Ala428Pro |
| 8 | SJBALL191 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | Yes | Yes | No | P2RY8-CRLF2 and JAK2 p.Arg683Ser |
| 9 | SJHYPER013 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | Yes | Yes | Yes | P2RY8-CRLF2 and JAK2 p.Arg683Gly |
| 10 | SJBALL206 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | No | No | No | P2RY8-CRLF2 |
| 11 | SJHYPER146 | BCR-ABL1–like_non-CRLF2 | Yes | No | No | Yes | Yes | Yes | NRAS p.Gln61Lys |
| 12 | SJBALL153 | BCR-ABL1–like_non-CRLF2 | Yes | No | Yes | Yes | No | Yes | SSBP2-PDGFRB |
| 13 | SJHYPER120 | BCR-ABL1–like_non-CRLF2 | Yes | No | No | Yes | Yes | Yes | FLT3 p.Ile836del |
| 14 | SJBALL208 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | No | No | No | P2RY8-CRLF2 |
| 15 | SJBALL084 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 16 | SJHYPER021 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | Yes | Yes | No lesion by RNA sequencing or WGS |
| 17 | SJDOWN009 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 18 | SJHYPO123 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | Yes | Yes | NF1 deletion |
| 19 | SJHYPER015 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 20 | SJHYPER053 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 21 | SJHYPER135 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 22 | SJBALL203 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | No | No | No | P2RY8-CRLF2 and JAK2 p.Arg683Gly |
| 23 | SJHYPO146 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | No | Yes | NF1 deletion |
| 24 | SJHYPO137 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 25 | SJBALL063 | BCR-ABL1–like_non-CRLF2 | Yes | No | No | Yes | No | Yes | IL7R p.Ile241_Ile245delinsCysLeuCys |
| 26 | SJBALL083 | BCR-ABL1–like_CRLF2 | Yes | No | Yes | Yes | Yes | No | P2RY8-CRLF2 and JAK2 p.Arg683Gly |
| 27 | SJHYPO140 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 28 | SJHYPO110 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | Yes | Yes | Yes | P2RY8-CRLF2 and NRAS p.Gln61Lys |
| 29 | SJHYPER003 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | Yes | Yes | No lesion by RNA sequencing or WGS |
| 30 | SJBALL101 | BCR-ABL1–like_CRLF2 | Yes | No | Yes | Yes | Yes | No | P2RY8-CRLF2, IL7R p.Ser185Cys, NF1 p.Arg1241* and deletion |
| 31 | SJBALL096 | BCR-ABL1–like_CRLF2 | Yes | Yes | No | No | No | No | Unknown CRLF2 rearrangement |
| 32 | SJBALL195 | BCR-ABL1–like_CRLF2 | Yes | Yes | Yes | Yes | Yes | No | IGH-CRLF2 and JAK2 p.Arg683Gly |
| 33 | SJBALL204 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | Yes | Yes | Yes | No | SSBP2-CSF1R |
| 34 | SJBALL105 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 35 | SJBALL100 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 36 | SJDOWN013 | BCR-ABL1–like_CRLF2 | Yes | No | Yes | Yes | Yes | No | P2RY8-CRLF2 and JAK1 p.Val658Phe |
| 37 | SJBALL071 | BCR-ABL1–like_non-CRLF2 | No | No | No | No | No | No | Not tested |
| 38 | SJBALL012 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | Yes | Yes | No | Yes | EBF1-PDGFRB |
| 39 | SJBALL011 | BCR-ABL1–like_non-CRLF2 | Yes | Yes | No | Yes | Yes | Yes | No lesion by RNA sequencing or WGS |
| 40 | SJBALL085 | BCR-ABL1–like_non-CRLF2 | Yes | No | Yes | Yes | Yes | No | NUP214-ABL1 |
Abbreviations: ALL, acute lymphoblastic leukemia; ID, identification; NGS, next-generation sequencing; RT-PCR, real-time polymerase chain reaction; WGS, whole-genome sequencing.
Table A2.
Multivariable Analyses of Factors Associated With EFS or OS
| Factors | EFS |
OS |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Presence of BCR-ABL1 | 2.28 | 0.69 to 7.54 | .18 | 3.23 | 0.59 to 17.8 | .18 |
| Presence of IZKF1 alteration | 2.08 | 0.81 to 5.29 | .13 | 0.85 | 0.19 to 3.73 | .83 |
| MRD > 5% at day 19 of induction | 2.43 | 0.94 to 6.31 | .07 | 1.31 | 0.37 to 4.62 | .67 |
| MRD > 0.01% at day 46 of induction | 1.93 | 0.79 to 4.70 | .15 | 5.18 | 1.59 to 16.9 | .006 |
| Presence of BCR-ABL1–like subtype | 1.89 | 0.53 to 6.77 | .33 | 1.36 | 0.28 to 6.65 | .70 |
Abbreviations: EFS, event-free survival; HR, hazard ratio; MRD, minimal residual disease; OS, overall survival.
Footnotes
Listen to the podcast by Dr Hunger at www.jco.org/podcasts
Supported in part by Grants No. CA21765, CA36401, and U01 GM92666 from the National Institutes of Health, by the American Lebanese and Syrian Associated Charities, by a grant from the National Health and Medical Research Council (Australia) C.J. Martin Postdoctoral Fellowship (K.G.R.), by a Leukemia and Lymphoma Society Special Fellowship (K.G.R.), by an Alex's Lemonade Stand Foundation Young Investigator Award (K.G.R.), and by Stand Up to Cancer Innovative Research Grant No. SU2C-AACR-IRG0711 (C.G.M.). C.G.M. is a St Baldrick's Scholar and Pew Scholar in the Biomedical Sciences, and C.-H.P. is an American Cancer Society professor.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00137111.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: None Stock Ownership: None Honoraria: None Research Funding: None Expert Testimony: None Patents, Royalties, and Licenses: William E. Evans, 7,741,032 Gamma glutamylhydrolase polymorphisms as predictors of methotrexate response, 5,856,095 Identification of two novel mutant alleles of human TPMT, and diagnostic uses thereof Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Kathryn G. Roberts, Charles G. Mullighan, Ching-Hon Pui
Financial support: Mary V. Relling, William E. Evans, Charles G. Mullighan, Ching-Hon Pui
Administrative support: William E. Evans, James R. Downing, Charles G. Mullighan, Ching-Hon Pui
Provision of study materials or patients: John T. Sandlund, Sima Jeha, Ching-Hon Pui
Collection and assembly of data: Kathryn G. Roberts, Debbie Payne-Turner, Yongjin Li, John T. Sandlund, Sima Jeha, John Easton, Jinghui Zhang, Charles G. Mullighan, Ching-Hon Pui
Data analysis and interpretation: Kathryn G. Roberts, Deqing Pei, Dario Campana, Yongjin Li, Cheng Cheng, John Easton, Jared Becksfort, Jinghui Zhang, Elaine Coustan-Smith, Susana C. Raimondi, Wing H. Leung, Mary V. Relling, William E. Evans, James R. Downing, Charles G. Mullighan, Ching-Hon Pui
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 5.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106:9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts KG, Li Y, Payne-Turner D, et al. Genomic characterization and experimental modeling of BCR-ABL1-like acute lymphoblastic leukemia. American Society of Hematology Annual Meeting 122; 2013. (abstr 232) [Google Scholar]
- 8.Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: A Children's Oncology Group study. Blood. 2012;119:3512–3522. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122:2622–2629. doi: 10.1182/blood-2012-10-462358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: A report from the Children's Oncology Group TARGET Project. Blood. 2013;121:485–488. doi: 10.1182/blood-2012-04-422691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiyokawa N, Iijima K, Yoshihara H, et al. An analysis of Ph-like ALL in Japanese patients. American Society of Hematology Annual Meeting 122; 2013. (abstr 352) [Google Scholar]
- 12.Te Kronnie G, Silvestri D, Vendramini E, et al. Philadelphia-like signature in childhood acute lymphoblastic leukemia: The AEIOP experience. American Society of Hematology Annual Meeting 122; 2013. (abstr 353) [Google Scholar]
- 13.Roberts KG, Payne-Turner D, Pei D, et al. Integrated genomic and mutational profiling of adolescent and young adult all identifies a high frequency of BCR-ABL1-like ALL cases with very poor outcome. American Society of Hematology Annual Meeting 122; 2013. (abstr 825) [Google Scholar]
- 14.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339:605–615. doi: 10.1056/NEJM199808273390907. [DOI] [PubMed] [Google Scholar]
- 16.Coustan-Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96:2691–2696. [PubMed] [Google Scholar]
- 17.Neale GA, Coustan-Smith E, Stow P, et al. Comparative analysis of flow cytometry and polymerase chain reaction for the detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia. 2004;18:934–938. doi: 10.1038/sj.leu.2403348. [DOI] [PubMed] [Google Scholar]
- 18.Pounds S, Cheng C, Mullighan C, et al. Reference alignment of SNP microarray signals for copy number analysis of tumors. Bioinformatics. 2009;25:315–321. doi: 10.1093/bioinformatics/btn624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 20.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 21.Tibshirani R, Hastie T, Narasimhan B, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children's Oncology Group. Blood. 2011;118:3080–3087. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45:1226–1231. doi: 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. New York, NY: Wiley; 2002. [Google Scholar]
- 27.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: Correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45:1494–1498. doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasian SK, Doral MY, Borowitz MJ, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120:833–842. doi: 10.1182/blood-2011-12-389932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120:3510–3518. doi: 10.1182/blood-2012-03-415448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol. 2013;31:e413–e416. doi: 10.1200/JCO.2012.47.6770. [DOI] [PubMed] [Google Scholar]
- 33.Lengline E, Beldjord K, Dombret H, et al. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013;98:e146–e148. doi: 10.3324/haematol.2013.095372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inokuchi K, Wakita S, Hirakawa T, et al. RCSD1-ABL1-positive B lymphoblastic leukemia is sensitive to dexamethasone and tyrosine kinase inhibitors and rapidly evolves clonally by chromosomal translocations. Int J Hematol. 2011;94:255–260. doi: 10.1007/s12185-011-0910-z. [DOI] [PubMed] [Google Scholar]
- 35.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: A Children's Oncology Group study. J Clin Oncol. 2009;27:5175–5181. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): A randomised, open-label, intergroup study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26:123–135. doi: 10.1016/j.blre.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 38.Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50:185–196. doi: 10.1053/j.seminhematol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.