

Abstract
The facultative anaerobe, Bacillus cereus, causes diarrheal diseases in humans. Its ability to deal with oxygen availability is recognized to be critical for pathogenesis. The B. cereus genome comprises a gene encoding a protein with high similarities to the redox regulator, Rex, which is a central regulator of anaerobic metabolism in Bacillus subtilis and other Gram-positive bacteria. Here, we showed that B. cereus rex is monocistronic and down-regulated in the absence of oxygen. The protein encoded by rex is an authentic Rex transcriptional factor since its DNA binding activity depends on the NADH/NAD+ ratio. Rex deletion compromised the ability of B. cereus to cope with external oxidative stress under anaerobiosis while increasing B. cereus resistance against such stress under aerobiosis. The deletion of rex affects anaerobic fermentative and aerobic respiratory metabolism of B. cereus by decreasing and increasing, respectively, the carbon flux through the NADH-recycling lactate pathway. We compared both the cellular proteome and exoproteome of the wild-type and Δrex cells using a high throughput shotgun label-free quantitation approach and identified proteins that are under control of Rex-mediated regulation. Proteomics data have been deposited to the ProteomeXchange with identifier PXD000886. The data suggest that Rex regulates both the cross-talk between metabolic pathways that produce NADH and NADPH and toxinogenesis, especially in oxic conditions.
Introduction
Bacillus cereus is a Gram-positive, facultative-anaerobe, rod-shaped endospore-forming human pathogen. Most of the reported illnesses involving B. cereus are food-borne intoxications, classified as emetic and diarrheal syndromes [1], [2]. Diarrheal disease is due to vegetative outgrowth and secretion of various extracellular factors, including enterotoxins [3]. The most extensively studied diarrheal enterotoxins are hemolysin BL (Hbl), nonhemolytic enterotoxin (Nhe), and cytotoxin K (CytK) [4], [5]. These enterotoxins are secreted via the Sec translocation pathway [6]. Although Hbl, Nhe and CytK are currently considered as the etiologic agents of diarrheal syndrome, other toxins, such as EntA, EntB and EntC, may also contribute to the pathogenicity of B. cereus [7], [8]. To grow and produce virulence factors in the human intestine, B. cereus must adapt its metabolism, and regulates its proteome [9] in response to changes in oxygen availability. Indeed, B. cereus encounters oxic conditions in zones adjacent to the mucosal surface [10] and anoxic condition in the intestinal lumen [11]. Changes in oxygen availability can influence the relative levels of the dinucleotide, NAD+ and NADH, in the cell, and such changes are sensed by the transcriptional regulator, Rex, in B. subtilis [12] as in other Gram-positive bacteria [13], [14], [15], [16], [17], [18], [19]. Depending on the cellular NAD+/NADH ratio, Rex regulators modulate the expression of genes involved in fermentative metabolism, biofilm formation, and oxidative stress [18], [20], [21]. Structural studies of Rex proteins have identified dinucleotide-binding pockets in the C-terminal domain of the protein. NADH binding in this region leads to a conformational change in the Rex homodimer, triggering a displacement of Rex from its recognition sites on DNA, and thus leading to de-repression of the downstream genes [15], [19]. A Rex homologue has been detected in the cellular proteome of B. cereus [9]. Furthermore, we found a canonical Rex binding motif [15] overlapping ResD and Fnr binding motifs [22], [23], [24] in the ldhA promoter region [25]. In B. cereus, ResD, Fnr and LdhA regulate both catabolism and enterotoxin production under both aerobic and anaerobic growth conditions, probably through a regulatory complex [26]. Rex could be, thus, a regulator of both catabolism and toxinogenesis in B. cereus. A role of Rex in toxinogenesis has not yet been reported in B. cereus or in any other organism.
In this study, we show that Rex from B. cereus, like its orthologues, is a transcriptional factor capable of interacting with DNA in an NADH/NAD+-responsive manner. We demonstrate that B. cereus Rex is a key regulator of anaerobic fermentation, aerobic respiration, resistance against external reactive oxygen species, and toxinogenesis, by modulating the cellular and extracellular proteome in an oxygen-dependent manner. This study provides the most comprehensive experimental information on proteins whose synthesis was changed in the presence of Rex. All together, our results offer new information about the metabolic events that maximize B. cereus growth in environments with varying oxygen conditions.
Materials and Methods
Bacterial strains, media, and growth conditions
Escherichia coli TOP 10 (Invitrogen) was used as the host for cloning experiments, and E. coli SCS110 (Stratagene, La Jolla, CA) was used to prepare DNA for B. cereus transformation. B. cereus ATCC 14579 [27] was used as the parent strain for the construction of the rex deletion mutant. E. coli strains were grown at 37°C, with agitation, in Luria broth (LB). B. cereus strains were cultured in batches (three independent cultivations per strain) at two oxygen availabilities, i.e. pO2 = 0% and pO2 = 100% [8], [9]. Each batch culture was inoculated with a subculture grown overnight at an initial optical density at 560 nm (OD560) equal to 0.02. For B. cereus cultivation, the minimal MOD medium was supplemented with 30 mM glucose as the carbon source [28]. Anaerobic and aerobic batch cultures were performed at 37°C in a 2 L bioreactor (BioFlo/CelliGen 115, New Brunswick), and the working volume was maintained at 1.8 L. The pH was kept at a controlled value of 7.2 by automatic addition of 5 M KOH. B. cereus growth was monitored spectrophotometrically at 560 nm and calibrated with cell dry-weight measurements as previously described [25]. B. cereus cells were harvested by centrifugation when they reached their maximal growth rate (µ = µmax) and immediately frozen until proteomic analysis. Supernatants were kept at −20°C for glucose and glucose-by-product assays and exoproteomic analysis.
Construction of the B. cereus Δrex mutant strain
The deletion mutant, ATCC 14579 Δrex, was constructed as follows. Two DNA fragments encompassing the 5′untranslated region (UTR) and 3′UTR of rex (BC 0291) were generated by PCR using primer pairs, rex1F (5′-GCCATGTTAATGTTTCGATGTCT-3′) and rex2R (5′-CCCGGGATCTTTTAGCAGTGGCTTGTGG-3′, SmaI restriction site is underlined), and rex3F (5′-CCCGGGGTTTACTTTTTGAAAAACTATCCACAA-3′, SmaI restriction site is underlined) and rex4R (5′-TGCATTATGTATCGTGCTTTGG-3′), respectively. The resulting 840 bp 3′SmaI and 827 bp 5′SmaI DNA fragments were cloned into the TA cloning vector, pCR4-TOPO (Invitrogen, La Jolla, CA), generating plasmids, pCR4mutrex1 and pCR4mutrex2, respectively. The 840 bp DNA fragment encompassing the 5′UTR region of rex was isolated from pCR4mutrex1 with PstI and SmaI, and subcloned into pCR4mutrex2 to generate pCR4mutrex3. A 1.5 kbp SmaI fragment containing the entire spectinomycin gene, spc [29], was purified from pDIA [25]. This purified fragment was ligated into SmaI-digested pCR4mutrex3. The resulting plasmid, pCR4mutrex4, was digested with EcoRI and the resulting 3067 bp 5′rexUTR-spc-3′rexUTR was subsequently inserted into the EcoRI site of pMAD [30]. The resulting plasmid was introduced into B. cereus cells by electroporation. The rex ORF was deleted and replaced with spc via a double-crossover event [30]. Chromosomal allele exchanges were confirmed by PCR with oligonucleotide primers located upstream and downstream of the DNA regions used for allelic exchange. To complement the rex gene in trans, a DNA fragment of 932 bp encompassing the rex ORF (630 bp) and its promoter region (219 bp) was first PCR amplified using the primer pairs, rexcompF (5′-GGATCCCGTTCGAAAGCGCGTTTACTTG-3′; the BamHI restriction site is underlined) and rexcompR (5′-GAGCTCGATTTTAATTTGGCACTTCGCC-3′; the SacI restriction site is underlined), and then cloned into the pCRXL-TOPO plasmid (Invitrogen). The PCR fragment was then cut with BamHI and SacI and ligated into pHT304 [31], digested with the same restriction enzymes. The integrity of the recombinant vector (pHT304rex) insert was verified by sequencing.
Phenotypic characterization of B. cereus Δrex using the API-50CHB testsystem
The carbohydrate metabolism of the wild-type strain (WT) transformed or not with pHT304, Δrex mutant transformed or not with pHT304 and Δrex mutant transformed with pHT304rex was examined using API 50 CHB strips (BioMérieux SA, France). The results showed that WT and WT(pHT304) did not ferment turanose. In contrast, Δrex and Δrex (pHT304) showed a typical positive reaction in turanose test fermentation. Transformation of Δrex mutant with pHT304rex inhibited the capacity of Δrex to ferment turanose.
Measurement of glucose and by-product concentrations
Enzymatic test kits from Diffchamb (Lyon, France), R-Biopharm (Saint-Didier au Mont-d’Or, France), and Roche (Meylan, France) were used to analyze the glucose, lactate, ethanol, formate, acetate, and succinate concentrations in the supernatants of 4 mL cell cultures obtained after centrifugation at 10,000×g for 5 min (4°C). The specific glucose consumption rate, defined as the differential change in glucose concentration with time, was calculated from the equation, q glucose = µ/Yx, where µ is the specific growth rate (h−1) and Yx is the biomass yield (g.mol carbon substrate−1).
Gene expression analysis by RT-PCR
RT-PCR was performed using SYBR Green technology on a Lightcycler instrument (Roche applied Science) as described previously [32]. The primers used in this study have been described previously [8].
Purification of Rex
The rex ORF was amplified by PCR from B. cereus ATCC 14579 using the oligonucleotides, pET101rexF (5′-ACCATGGATCAGCAAAAGATTCCA-3′) and pET101rexR (5′-TTGTGGATAGTTTTTCAAAAAGTAAAC-3′). The amplicon was introduced as a blunt-end PCR product into pET101/D-TOPO (Invitrogen). The integrity of the inserted sequence was confirmed by DNA sequencing. The resulting construct was transformed into E. coli BL21-CodonPlus(DE3)-RIL strain (Stratagene) for protein production. BL21-CodonPlus(DE3)-RIL cells carrying the pET101-rex expression plasmid were grown in 1 L LB medium containing 100 µg.mL−1 ampicillin, at 30°C with agitation (200 rpm) until the cell density reached an OD600 of about 0.5. After this, 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the culture, and growth was continued for four hours. The cells were harvested by centrifugation (6500 rpm for 10 min at 4°C), washed, and resuspended in 20 mL ice-cold extraction buffer consisting of 50 mM TRIS buffered at pH 8.0, 0.3 M NaCl, 10% glycerol, and a protease inhibitor cocktail (one tablet of Complete Mini, EDTA free, Roche). Cells were then incubated with 0.5 mg/mL lysozyme for 45 min under gentle agitation at 4°C and disrupted by sonication for 5 min at 4°C using a Vibra Cell ultrasonicator (Fisher Bioblock Scientific). The lysates were clarified by centrifugation at 9000 rpm for 30 min at 4°C and loaded onto a 2 mL Co2+-immobilized TALON metal affinity chromatography column (Clontech) equilibrated with the extraction buffer. The column was washed with 30 mL of extraction buffer containing 20 mM imidazole and Rex was eluted with 3 mL extraction buffer containing 200 mM imidazole. The eluted fraction was desalted by dialysis, concentrated using Nanosep 30 kDa-molecular-mass cutoff devices (Omega disc membrane; Pall filtron), and stored at −80°C until analysis. The purity of the protein was estimated to be above 90% by Coomassie blue-stained SDS-PAGE. The protein concentration was determined using a Bradford assay (Interchim) with bovine serum albumin as the reference.
Electrophoretic mobility shift assays (EMSAs)
Nucleic acid fragments containing the promoter regions of ldhA and rex were PCR-amplified using biotinylated forward primers, LdhAF (5′-ACCTGCTAATCCGATGATTG-3′) and RexF (5′-CAAGAATCGTTTCTGCACCG-3′), and nonbiotinylated reverse primers, LdhAR (5′-GGATCCAACTAATCCAGTAC-3′) and RexR (5′-GCTTACCAGAAAGAGATAAG-3′). The DNA used as negative control was a fragment of the ssuRNA BC0007 sequence (NC_004722), which was amplified with the biotinylated ssubioF (5′-GGTAGTCCACGCCGTAAACG-3′) and ssuR (5-GACAACCATGCACCACCTG-3′) primer pair. The 5′-labeled amplicons were purified using the High Pure PCR Product Purification Kit (Roche). Binding reactions were performed for 30 min at 37°C by incubating biotin-labeled DNA fragments (2 nM per reaction) with different amounts of Rex in 10 mM Tris-HCl buffered at pH 7.5, and containing 50 mM KCl, 2.5% glycerol, 5 mM MgCl2 and 5 mg/L poly(dI−dC). The samples were resolved by electrophoresis on a 6% nondenaturing polyacrylamide gel and electrotransferred onto Hybond N+ Nylon membranes (Amersham). Biotin-labeled DNA was detected using the LightShift Chemiluminescent EMSA Kit (Pierce).
Proteomic sample preparation and nanoLC-MS/MS analysis of tryptic peptides
Three independent biological replicates were harvested for each of the two conditions (aerobiosis and anaerobiosis) and two strains (Δrex mutant and its parent strain, ATCC 14579). The extracellular proteins of the 12 cultures were extracted by trichloroacetic acid precipitation [8]. The cellular proteins from the 12 samples were obtained as previously described [9]. The 24 resulting samples were subjected to SDS-PAGE, and then identified after trypsin proteolysis by nanoLC-MS/MS tandem mass spectrometry with an LTQ-Orbitrap XL mass spectrometer as previously described [8], [9]. A total of 131 nanoLC-MS/MS runs were carried out to acquire the whole dataset. The MS/MS spectra were assigned to tryptic peptide sequences with the Mascot Daemon software (version 2.3.2; Matrix Science) with mass tolerances of 5 ppm on the parent ion and 0.5 Da on the MS/MS, fixed modification for carbamidomethylated cysteine, and variable modification for methionine oxidation. Mascot results were parsed with a p-value threshold below 0.05 for peptide identification and proteins were validated when at least two peptides were detected. The number of MS/MS spectra per protein recorded by nanoLC-MS/MS was extracted for each sample. In each condition, proteins were further considered for comparison only if peptides were seen in at least two of the three replicates. The resulting datasets were normalized taking into account the total protein concentration of the corresponding pellet and supernatant. Protein concentrations in B. cereus were determined using the Reducing agent Compatible Detergent Compatible (RCDC) protein assay (Bio-Rad) following the supplier’s instructions. MS/MS spectral counts were compared with the TFold method using the PatternLab software program 2.0.0.13 [33] using a p-value cut-off set at 0.05. Log2 (fold-change) were then calculated for comparisons and only the proteins for which the p-value was lower than the 0.05 cut-off were considered. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org) via the PRIDE partner repository [34] with the dataset identifier PXD000856 and DOI 10.6019/PXD000856.
Exposure of bacteria to H2O2 and viability assays
Oxidative stress resistance of the B. cereus Δrex mutant and the parent strain, ATCC 14579, was assessed by exposing aerobically grown (OD∼0.4) and anaerobically grown (OD∼0.2) cells to 20 and 5 mM H2O2, respectively. Samples were taken prior to oxidative stress (time zero) and after 20 min. Aliquots (100 µl) of the samples were diluted in H2O, appropriate dilutions of the culture were plated onto LB agar, and after overnight incubation at 37°C the colony forming units (CFUs) were counted. All the experiments were performed at least in triplicate, and at least two technical replicates from each dilution step were carried out to determine the number of CFUs.
Results
Expression analysis of rex in B. cereus ATCC 14579 at two different pO2
We identified BC_0291 in the B. cereus ATCC 14579 genome [27] as the homologue to the Bacillus subtilis transcriptional repressor, Rex [12], [35], [36], with 90% sequence identities. We established by 5′RACE PCR (Fig. S1) that a transcriptional start site (G) was located 41 bp from the translational start site (ATG) of the BC_0291 open reading frame. Upstream of this transcriptional start site, we identified a potential housekeeping σA-type promoter, TATACAN(17)TAAACT. The stop codon (TAA) overlapped an inverted repeat (AAAACGCAGAGG(N6)CCTCTGCGTTTT; ΔG = −23.2 kcal/mol) that may be a transcriptional terminator, suggesting that rex was monocistronically transcribed. Using real-time RT-PCR, we investigated the expression of B. cereus rex under aerobiosis and anaerobiosis in early-growing ATCC 14579 cells. The results indicated that rex expression was significantly higher under aerobiosis than under anaerobiosis (log2 fold-change = 1.1, p-value<0.05).
Rex regulates glucose catabolism in B. cereus
Given that Rex orthologues function as NAD+/NADH-responsive regulators of metabolism in several Gram-positive bacteria (such as B. subtilis, S. aureus and S. coelicolor [12], [13], [14], [15], [36], [37]), we sought to determine if B. cereus rex contributed to anaerobic fermentative and aerobic respiratory growth. As shown in Table 1, rex deletion slightly increased the B. cereus growth rate under both anaerobiosis and aerobiosis without a significant change in final biomass. Under anaerobiosis, the increase of growth rate was not related to glucose consumption, suggesting perturbation of the fermentative pathways. In accordance with this hypothesis, the spectra of fermentation end products were significantly modified by the rex deletion (Table 1). We observed that formate, acetate and ethanol production were promoted at the expense of lactate production. In addition, the ethanol-to-acetate ratio increased in Δrex mutant, while NADH recovery levels remained unchanged and ATP yields tended to increase. Under aerobiosis, respiratory Δrex cells secreted ∼8-fold higher amounts of lactate compared with the wild-type cells, while producing lower levels of acetate. We conclude that rex deletion affects the carbon flux through the NADH recycling lactate pathway under both anaerobiosis and aerobiosis (Fig. 1). Complementation for growth of the non-polar rex mutant in B. cereus ATCC 14579 was not possible using a pHT304-based plasmid. This is probably due to the presence of multiple copies of the rex promoter region and/or overexpression of rex [38]. Therefore, to validate the role of Rex in aerobic respiration and anaerobic fermentation in B. cereus, we deleted the rex gene of B cereus F4430/73 [39] and cultured the mutant strain under both aerobiosis and anaerobiosis. The results showed that the growth phenotype of Δrex was the same in strains ATCC 14579 and F4430/73 (Table S1).
Table 1. Results from controlled batch cultures of Δrex mutant and its parent strain, B. cereus ATCC 14579a.
| Anaerobic fermentative growth | Aerobic respiratory growth | |||
| WTb | Δrex | WTb | Δrex | |
| Maximal specific growth rate (µmax) (h−1) | 0.84±0.04 | 0.90±0.02* | 1.22±0.11 | 1.41±0.04* |
| Final biomass (g.liter−1) | 0.78±0.01 | 0.83±0.06 | 2.38±0.12 | 2.31±0.12 |
| Yglucose (g of cells. mol of glucose−1) | 26±1 | 28±1 | 79±1 | 77±1 |
| Maximal specific glucose consumption(mmol.g−1.h−1) | 32±2 | 32±2 | 15±1 | 18±2 |
| Yields of end products (mol.mol glucose−1)c | ||||
| Lactate (Y lactate) | 1.33±0.01 | 1.25±0.02* | 0.05±0.02 | 0.40±0.01* |
| Acetate (Yacetate) | 0.32±0.01 | 0.36±0.01* | 1.05±0.01 | 0.69±0.03* |
| Formate (Yformate) | 0.23±0.01 | 0.27±0.01* | 0.01±0.01 | 0.01±0.01 |
| Ethanol (Yethanol) | 0.13±0.01 | 0.18±0.01* | NZd | NZ |
| Succinate (Ysuccinate) | 0.01±0.01 | 0.01±0.01 | 0.05±0.01 | 0.04±0.01 |
| Ethanol versus Acetate | 0.40 | 0.50* | ||
| ATP yielde | 2.10 | 2.15 | NDf | ND |
| NADH recoveredg | 1.2 | 1.2 | ND | ND |
Cells were grown under anaerobiosis (pO2 = 0%) and full aerobiosis (pO2 = 100%). Data are the means of triplicate measures obtained from three independent cultures.
WT, wild-type parent strain B. cereus ATCC 14579.
Yields of end products were calculated at the stationary phase.
NZ, yield was below 0.01 mol.mol glucose−1.
ATP yield was calculated as moles of ATP produced per mole of consumed glucose, and was equal to Y lactate+Yethanol+2*Yacetate.
ND, not determined.
NADH recovery was calculated as the ratio of pathways producing NADH versus those consuming NADH (producing NAD+), and was equal to (lactate+2×acetate+2×ethanol−formate)/(lactate+2×ethanol).
*p<0.05 versus WT in Student’s t-test.
Figure 1. An overview of the main anaerobic and aerobic glucose catabolic pathways utilized by B. cereus ATCC 14579.

The proteins detected in this study are indicated by their BC number. Protein names and functions are listed in Table S6. The form filling indicates fold-change values that satisfied the Student’s t-test statistical criteria (p-value<0.05) in anaerobiosis (blue) and aerobiosis (yellow). Red and green BC numbers indicated significant increase and decrease, respectively, of abundance level of the proteins in Δrex mutant compared with wild-type. The nicotinamide nucleotides are indicated in red (NADP+/NADPH) or blue (NAD+/NADH).
Rex regulates hydrogen peroxide resistance of B. cereus cells
To evaluate the impact of Rex deficiency on B. cereus resistance to oxidative stress, aerobically and anaerobically grown ATCC 14579 cells were exposed to hydrogen peroxide (20 and 5 mM H2O2, respectively). Figure 2 shows that aerobically grown Δrex cells were less susceptible to H2O2 harmful effects than WT cells, while the anaerobically grown Δrex cells were more susceptible. These data indicate that Rex restricts the resistance to oxidative stress of aerobically grown B. cereus cells, while sustaining a high resistance to oxidative stress of anaerobically growing cells. To validate the role of Rex in the B. cereus resistance to external H2O2, we repeated the experiments using the F4430/73 Δrex strain (Fig. S2). The results showed that Rex deficiency similarly impacts F4430/73 and ATCC 14579 cells while the survival of F4430/73 anaerobically grown cells was strongly higher than the survival of ATCC 14579 cells.
Figure 2. Survival of B. cereus cells towards external hydrogen peroxide under aerobiosis and anaerobiosis.
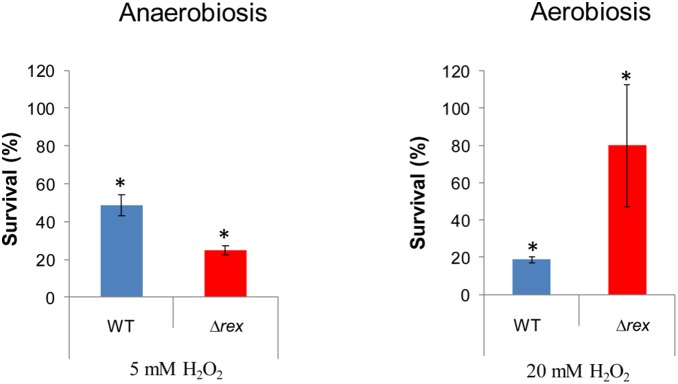
Cells were grown in liquid cultures to the mid-exponential growth phase either aerobically or anaerobically, and subjected to 20 or 5 mM H2O2, respectively. Samples were taken at 0 and 20 min after H2O2 addition. Colony forming units per mL were counted and expressed as (N/No)×100. Error bars represented the standard deviation from three independent measures. Significant differences (p-value<0.05) between mutant and wild-type strains are indicated with asterisks.
DNA binding activity of B. cereus Rex
Previous studies have shown that proteins belonging to the Rex family function as dimers that bind to promoter regions containing DNA motifs with the 5′-TTGTGAAnnnnTTCACAA-3′ consensus sequence [21]. This typical binding motif is missing in the rex regulatory region of B. cereus, as reported for S. aureus and B. subtilis [12], [15]. However, a putative binding motif with two mismatches compared with the known consensus motif was found upstream of the transcription start site of ldhA [25] in B. cereus, as in S. aureus and B. subtilis. To test whether B. cereus Rex binds to the ldhA promoter region, we overexpressed B. cereus rex in E. coli, purified the dimeric His6-tagged recombinant protein (Fig. S3), and performed electrophoretic mobility shift assays (EMSA). As a negative control, we assessed the binding of the Rex protein to the ssu DNA fragment (Fig. 3A). The results showed that a concentration of 0.6 µM Rex led to a complete shift of the ldhA fragment (2 nM). As expected, no shift was observed with the rex DNA fragment. The specificity of the binding was demonstrated by the absence of DNA shift with the negative control. The second EMSA experiment (Fig. 3B) showed that 10 mM NADH impaired Rex binding to the ldhA promoter region while 100 mM NAD+ did not interfere with Rex-DNA complex formation. An NAD+/NADH ratio of five led to an incomplete shift of the ldhA fragment. We conclude that in B. cereus, as in B. subtilis and S. aureus [15], Rex binding activity depends on the NADH/NAD+ ratio.
Figure 3. In vitro binding of Rex to ldhA promoter region determined by EMSA.

Panel A. Biotin-labeled PCR products (2 nM) corresponding to ldhA and rex promoter regions and a negative control (a fragment of ssuRNA BC_0007 sequence) were mixed with increasing concentrations of purified Rex. Panel B. PCR product corresponding to the ldhA promoter region was incubated with 0.6 µM purified Rex and different concentrations of NAD+ and NADH, as indicated.
Global comparative proteomics of B. cereus Δrex and wild-type cells
To investigate the regulatory role of Rex in aerobic respiratory and anaerobic fermentative growth of B. cereus and decipher its role in toxinogenesis, we carried out a comparative proteome analysis from cells harvested in exponential growth phase (µ = µmax). We compared the cellular and extracellular proteomes of aerobically and anaerobically grown Δrex with those of its parental strain, with biological triplicates for each condition. A total of 1,620 proteins were identified in the whole study (Tables S2, S3, S4 and S5), with 1,450 proteins specifically detected in the whole-cell shotgun analysis and 170 further proteins observed in the exoproteome study. These proteins were quantified by spectral count as previously described [8], [9]. The Rex-dependent changes in protein abundance, expressed as log2 of the ratio of a protein’s abundance at a given condition (aerobiosis or anaerobiosis) relative to the wild-type, are presented in Tables S6 and S7, where the proteins with significant abundance changes based on the PatternLab Tfold student t-test (p-value below 0.05) are highlighted while proteins with non-significant changes are in grey color.
(i) Anaerobiosis
Among the whole cellular proteome (Table S6), 145 proteins showed significant abundance level changes in Δrex cells relative to WT (p-values<0.05): 89 proteins were significantly up-regulated and 56 proteins were significantly down-regulated in Δrex cells. In the exoproteome (Table S7), 41 proteins showed significant abundance level changes in Δrex cells relative to WT: 17 proteins were significantly up-regulated and 24 proteins were significantly down-regulated in Δrex cells.
(ii) Aerobiosis
Table S6 indicates that 132 proteins showed significant abundance level changes in Δrex cellular proteome (p-values<0.05): 82 proteins were significantly up-regulated and 50 proteins were significantly down-regulated in Δrex cells. Table S7 indicates that 70 proteins showed significant abundance level changes in Δrex cellular exoproteome (p-values<0.05): 35 were significantly up-regulated and 35 were significantly down-regulated in Δrex cells.
(iii) Global analysis (Fig S4)
Only 8 cellular proteins showed similar behavior under both aerobiosis and anaerobiosis (4 were up-regulated and 4 were down-regulated in Δrex cells). Only 3 extracellular proteins were down-regulated under both anaerobiosis and aerobiosis. However, all the extracellular proteins that were up-regulated under anaerobiosis were also up-regulated under aerobiosis. Taken together, the data indicate that Rex (i) differently modulates the cellular proteome of B. cereus anaerobically and aerobically grown cells in terms of the identity of proteins that show significant abundance level changes, (ii) acts mainly as a repressor at the cellular proteome level under both aerobiosis and anaerobiosis and, has a stronger impact on the exoproteome of aerobically grown than anaerobically grown cells.
Insights into the cellular proteome of the Δrex mutant
The list of cellular proteins showing significant abundance changes (p-values<0.05) due to the absence of Rex includes proteins related to glucose metabolism, amino acid, nucleotide and lipid metabolism, protein folding, response to stress, virulence, cell wall/membrane assembly, transport, and a number of proteins of unknown function under both aerobiosis and anaerobiosis (see Table S6 for details on protein characteristics, the fold-changes and p-values in each comparison).
(i) Impact of Rex on glucose metabolism-related proteins under anaerobic fermentative conditions
Figure 1 shows an overview of the glucose catabolic pathways of B. cereus. As illustrated in Fig. 1, the abundance level of key fermentative enzymes, such L-lactate dehydrogenases (Ldh; BC_1924, BC_4870, BC_4996), pyruvate formate lyase (PflB; BC_0491), alcohol dehydrogenase (BC_4365, BC_2220, BC_0802) and acetate kinase (BC_4634) were not significantly different in the mutant compared with wild-type cells. However, the lack of Rex resulted in a decrease of the abundance of the IIA component of the glucose-specific phosphotransferase (PTS) system (BC_5320, log2 = −0.4; p-value = 0.021). A significant decrease in abundance was also observed for the pyruvate-formate-lyase-activating enzyme, PflA (BC_0492; log2 = −1; p-value = 0.048). PflA is the sole enzyme able to activate the oxygen-sensitive PflB (BC_0491), which converts pyruvate to acetyl-CoA and formate (Fig. 1) [40]. In contrast, anaerobically grown Δrex cells sustain a higher level of phosphoglucomutase (Pgm; BC_4919) than the wild-type cells (log2 = 0.6; p-value = 0.036) and thus, could sustain a higher carbon flux through gluconeogenesis than glycolysis (Fig. 1). As for Pgm, the malate dehydrogenase (Mdh; BC_4592) abundance level was increased in the absence of Rex under anaerobiosis (log2 = 1.7; p-value = 0.017). Mdh is one of the enzymes involved in the NADH-dependent reduction of oxaloacetate to malate, which is the first reaction in the succinate pathway (Fig. 1). The increase of Mdh abundance in the Δrex mutant compared with wild-type was associated with the increase of NADPH-producing malic enzyme (ME; BC_4604; log2 = 0.5; p-value = 0.033), which converts malate into pyruvate. Taken together, our data suggest that Rex may modulate the abundance level of key metabolic enzymes to control carbon flow through fermentative pathways, possibly at the expense of the pathway that couples NADH-recycling Mdh with NADPH-producing ME.
(ii) Impact of Rex on glucose metabolism-related proteins under aerobic respiratory conditions
Figure 1 shows that ME was the sole enzyme showing an abundance level change in both aerobically and anaerobically grown mutant cells (log2 = 0.4; p-value = 0.01 under aerobiosis). 6-Phosphogluconolactonase (Pgl; BC_3368) showed a higher increase than ME (log2 = 1.3; p-value = 0.047) in Δrex aerobically grown cells. The increase of the Pgl abundance level may have an impact on the activity of the NADPH-generating pentose phosphate pathway (PPP) in the Δrex mutant (Fig. 1). Two glycolytic proteins showed significant abundance decreases in aerobically grown mutant cells: the NADPH-producing glyceraldehyde-3-phosphate-dehydrogenase (NADPH-GAPDH; BC_0868; (log2 = −0.5; p-value = 0.012) and pyruvate kinase (PK; BC_4599; (log2 = −0.4; p-value = 0.032), which catalyzes the last steps of glycolysis. The decrease of the NADPH-GAPDH level could increase the carbon flow through the NADH-producing GAPDH (BC_4583, BC_5333) and thus stimulate flux through the upstream glycolysis pathways, especially through the NADH-recycling lactate pathway (Fig. 1). Finally, Rex may modulate glycolytic flux by controlling the abundance level of key enzymes of glycolysis and pentose phosphate pathway when cells are grown under aerobiosis in glucose-containing medium.
(iii) Impact of Rex on other relevant cellular pathways
Cell-wall and cell surface related-proteins. Among the 20 proteins classified in the cell-wall/surface functional group, 8 showed significant abundance level changes (p<0.05) in anaerobically grown Δrex mutant cells while 4 proteins showed significant abundance level changes in aerobically grown cells (Table 2). A protein annotated as a putative prolipoprotein diacylglyceryl transferase (Lgt; BC_5163), which may be required for lipid modification of the cysteine residue present within the lipobox of prolipoproteins [41], showed a significant change of abundance level under both conditions: it was more highly detected in anaerobically grown cells and less detected in aerobically grown cells when Rex was absent.
Table 2. Cellular abundance level changes of cell membrane, transport, stress response, transcriptional regulation, virulence and phage-related proteins in Δrex mutant relative to wild-type under anaerobiosis and aerobiosis.
| Abundance level change and significance | |||||||
| Anaerobiosis | Aerobiosis | ||||||
| Accession no (NP) | Proteinname | Gene number | Functionnal annotation | log2(fold-change)a | p-value | log2(fold-change) | p-value |
| Cell wall, membrane and cell surface proteins | |||||||
| NP_833630.1 | - | BC_3910 | N-acetylglucosaminyl transferase | 1,561 | 0,018 | −1,515 | 0,197 |
| NP_835080.1 | TagA | BC_5419 | N-acetyl-beta-D-mannosaminyltransferase | 1,263 | 0,042 | −0,377 | 0,382 |
| NP_832677.1 | - | BC_2929 | Peptidoglycan N-acetylglucosamine deacetylase | 0,832 | 0,019 | 0,799 | 0,183 |
| NP_835088.1 | - | BC_5427 | Cell wall biosynthesis glycosyltransferase | −1,120 | 0,046 | −0,889 | 0,070 |
| NP_834932.1 | AmsK | BC_5269 | Amylovoran biosynthesis AmsK | −0,713 | 0,112 | 1,595 | 0,001 |
| NP_834404.1 | - | BC_4698 | Choline kinase | 0,949 | 0,032 | 0,214 | 0,199 |
| NP_834827.1 | Lgt | BC_5163 | Prolipoprotein diacylglyceryl transferase | 1,007 | 0,012 | −0,811 | 0,004 |
| NP_834936.1 | - | BC_5273 | UDP-bacillosamine synthetase | 1,322 | 0,033 | 0,632 | 0,110 |
| NP_835093.1 | - | BC_5432 | Bactoprenol glucosyl transferase | −1,218 | 0,032 | 0,536 | 0,306 |
| NP_835019.1 | - | BC_5358 | Collagen adhesion protein | 0,918 | 0,088 | 1,170 | 0,045 |
| NP_830682.1 | - | BC_0896 | S-layer protein | −0,234 | 0,180 | −0,286 | 0,017 |
| Transport-related proteins | |||||||
| NP_829996.1 | SecE | BC_0115 | Preprotein translocase subunit | −0,971 | 0,044 | 0,411 | 0,252 |
| NP_834447.1 | - | BC_4743 | ABC transporter ATP-binding protein | 1,561 | 0,018 | −0,029 | 0,485 |
| NP_834539.1 | - | BC_4839 | ABC transporter ATP-binding protein | 1,585 | 0,031 | 0,856 | 0,115 |
| NP_830141.1 | Uup | BC_0290 | ABC transporter ATP-binding protein | 0,151 | 0,382 | 0,705 | 0,045 |
| NP_830187.1 | - | BC_0348 | Methionine ABC transporter | −1,690 | 0,004 | 2,160 | 0,064 |
| NP_830967.1 | OppD | BC_1182 | Oligopeptide transport ATP-binding protein | 0,651 | 0,002 | −0,044 | 0,421 |
| NP_830660.1 | - | BC_0874 | Arginine ABC transporter ATP-binding protein | −0,396 | 0,349 | −1,218 | 0,024 |
| NP_832817.1 | CutC | BC_3071 | Copper tansport | 0,903 | 0,007 | −0,358 | 0,341 |
| NP_830432.1 | - | BC_0615 | Di-/tripeptide transporter | −0,837 | 0,047 | 0,422 | 0,232 |
| NP_830429.1 | - | BC_0612 | L-lactate permease | 0,748 | 0,026 | −0,644 | 0,261 |
| Stress response-related proteins | |||||||
| NP_831381.1 | - | BC_1603 | Cold shock protein | −1,120 | 0,037 | −0,474 | 0,277 |
| NP_830286.1 | TelA | BC_0447 | Tellurite resistance protein | 0,084 | 0,432 | 0,840 | 0,008 |
| NP_833675.1 | - | BC_3956 | GTP-binding protein TypA/BipA | 0,310 | 0,049 | 0,043 | 0,419 |
| NP_833972.1 | - | BC_4258 | Hydroxyacylglutathione hydrolase | 0,824 | 0,028 | −0,578 | 0,213 |
| NP_833576.1 | - | BC_3855 | Alkaline-shock protein | 0,014 | 0,488 | 0,740 | 0,026 |
| NP_834187.1 | OhrA | BC_4475 | Organic hydroxyperoxide protein A | 0,189 | 0,340 | 2,029 | 0,015 |
| NP_829959.1 | Hsp15 | BC_0062 | Heat shock protein 15 | 0,799 | 0,082 | 1,667 | 0,007 |
| NP_830216.1 | AhpC | BC_0377 | Alkyl hydroperoxide reductase C22 | −0,136 | 0,426 | −1,029 | 0,007 |
| NP_830941.1 | KatE | BC_1155 | Catalase | 1,111 | 0,014 | −0,074 | 0,466 |
| NP_834345.1 | - | BC_4639 | Thiol peroxidase | −0,044 | 0,463 | −0,737 | 0,034 |
| Transcriptional Regulation | |||||||
| NP_833888.1 | ArgR2 | BC_4174 | Arginine biosynthesis repressor | 1,511 | 0,030 | 1,269 | 0,079 |
| NP_833771.1 | - | BC_4053 | Transcriptional regulator, GntR family | 0,895 | 0,019 | 0,227 | 0,399 |
| NP_833799.1 | - | BC_4081 | MarR family transcriptional regulator | 0,029 | 0,464 | 1,903 | 0,001 |
| NP_834861.1 | - | BC_5197 | MarR family transcriptional regulator | 0,345 | 0,029 | 0,604 | 0,123 |
| NP_832113.1 | - | BC_2351 | MerR family transcriptional regulator | 0,485 | 0,276 | 1,170 | 0,045 |
| NP_830532.1 | TenA | BC_0742 | Transcriptional activator | 0,791 | 0,046 | −0,621 | 0,193 |
| NP_834137.1 | - | BC_4425 | Transcriptional regulator | 0,632 | 0,044 | −1,029 | 0,051 |
| NP_831310.1 | - | BC_1531 | Transcriptional regulatory protein | −0,029 | 0,471 | 1,480 | 0,004 |
| NP_831256.1 | ResD | BC_1477 | Transcriptional regulatory protein | 0,356 | 0,008 | −0,599 | 0,119 |
| NP_830596.1 | - | BC_0806 | BigG family transcription antiterminator | 0,485 | 0,276 | 1,922 | 0,000 |
| Virulence-related proteins | |||||||
| NP_834769.1 | HlyI | BC_5101 | Hemolysin I | −0,152 | 0,438 | 2,611 | 0,001 |
| NP_832488.1 | Npr2 | BC_2735 | Bacillolysin | −1,089 | 0,193 | −2,120 | 0,013 |
| NP_833332.1 | HhoA | BC_3600 | Protease | 0,485 | 0,098 | −1,690 | 0,026 |
| NP_831147.1 | - | BC_1366 | SSEB protein | 1,163 | 0,036 | 1,269 | 0,055 |
| Phage-related proteins | |||||||
| NP_831667.1 | - | BC_1894 | Phage protein | 3,023 | 0,014 | −1,358 | 0,286 |
| NP_831632.1 | - | BC_1859 | Phage protein | 2,526 | 0,039 | −0,644 | 0,261 |
| NP_831635.1 | - | BC_1862 | Phage protein | 1,714 | 0,027 | −0,761 | 0,302 |
| NP_831673.1 | - | BC_1901 | Phage protein | 1,727 | 0,012 | −1,889 | 0,205 |
| NP_831676.1 | - | BC_1904 | Phage protein | 1,975 | 0,038 | −0,515 | 0,367 |
Only changes satisfying statistical criteria (p-value<0.05) at least in one growth condition are shown.
The relative amount of each protein was determined using PatternLab software. Numbers in bold indicate that satisfied statistical criteria. See Table S6 for details on all the proteins showing significant abundance level changes in Δrex cells.
Transport-related proteins. The largest change in transport-related protein abundance was observed in anaerobically grown Δrex cells: 8 proteins showed significant abundance level changes (p<0.05). This suggests that the Rex-dependent control of the abundance pattern of transport related-proteins was stronger in anaerobic fermentative conditions than in aerobic respiratory conditions.
Transcriptional regulation-related proteins. Δrex up-regulated the synthesis of 6 and 4 transcriptional regulators under anaerobiosis and aerobiosis, respectively. Among these, we found regulators of the Mer and Mar families, which control many cellular processes including oxidative stress response and virulence [43], [44]. Interestingly, we noted that ResD was significantly up-regulated in anaerobically grown Δrex cells but remained unchanged in aerobically grown Δrex cells. ResD is involved in both metabolism and toxinogenesis in B. cereus [22], [32].
Stress-response related proteins. Among the stress response-related proteins, the antioxidant protein, OhrA [7], [9], was significantly up-regulated in aerobically grown Δrex cells and its abundance remained constant under anaerobiosis (Table 2). Like OhrA, the ribosome-associated heat-shock protein, Hsp15 (BC_0062), showed, under aerobiosis, a significant change in abundance level. This protein could be involved in the recycling of free 50 S ribosomal subunits [45]. Interestingly, we noted significant changes in the abundance patterns of 50 S ribosomal subunits in aerobically grown Δrex cells (Table S6). Unlike aerobically grown cells, anaerobically grown Δrex cells exhibited higher levels of catalase, KatE (BC_1155), lower levels of the BC1603 protein, which is functionally annotated as a cold-shock protein and no significant level change of alkyl hydroperoxide reductase (AhpC, BC_0377). Clearly, Rex controls the abundance pattern of stress-related proteins in an oxygen-dependent manner.
Phage-related proteins. Five phage-related proteins were strongly up-regulated in anaerobically grown Δrex cells (log2>1.5, p-value<0.05) and their abundance remained constant in aerobically grown Δrex cells. The functions of these proteins are unknown but they are considered to be potentially beneficial to B. cereus in facing adverse environmental conditions [42].
Virulence-related proteins. Table 2 shows that rex deletion has a significant impact on the abundance levels of hemolysin I (Hly I), neutral protease Npr2 (BC_2735), protease HhoA (BC_3600), and SSEB (BC_1366) proteins under aerobiosis, all of which are predicted to be secreted virulence factors [4], [46].
Insights into the extracellular proteome of the Δrex mutant
(i) Focus on toxin-related proteins
Although the intracellular abundance level of HlyI was higher in the aerobically grown Δrex mutant strain, the extracellular level of this protein was not significantly different in the mutant compared with wild-type cells (Table S7). This was also the case for EntB, a putative enterotoxin [9]. However, 12 other toxin-related proteins showed a significantly higher abundance level in the exoproteome of aerobically grown Δrex cells than wild-type cells (Table 3). Some of these proteins also showed a higher abundance level under anaerobiosis, but to a lesser extent, i.e. the three components of Nhe (NheA, NheB and NheC), EntA, EntC and the L1 component of Hbl. From these data, we conclude that Rex regulates the toxinogenic profile of the B. cereus exoproteome in response to oxygen availability.
Table 3. Changes in mRNA and protein abundance of toxins and putative toxins induced by Δrex mutation under anaerobic and aerobic growth conditions#.
| Protein name | Log2(fold-change) in B. cereus Δrex vs wild-typea | ||||||
| Gene | Anaerobiosis | Aerobiosis | |||||
| mRNA | Protein | mRNA | Protein | ||||
| cellular | extracellular | s | cellular | extracellular | |||
| CytK | BC_1110 | NS | NSa | NS | NS | NS | 1,72 ( p = 0.001) |
| EntA | BC_5239 | NS | NS | 0,29 ( p = 0.035) | NS | NS | 0,53 ( p = 0.001) |
| EntB | BC_2952 | NS | NS | NS | NS | NS | NS |
| EntC | BC_0813 | NS | NS | 0,37 ( p = 0.042) | −1,29 (p<0.05) | NS | 0,75 ( p = 0.002) |
| EntFM | BC_1953 | NS | NS | NS | −1,49 (p<0.05) | NS | 0,68 ( p = 0.000) |
| HblB | BC_3102 | NS | NS | NS | NS | NS | 1,38 ( p = 0.001) |
| HblB' | BC_3101 | NS | NS | NS | NS | NS | 1,28 ( p = 0.002) |
| HblL1 | BC_3103 | NS | NS | 0,32 ( p = 0.039) | NS | NS | 1,09 ( p = 0.006) |
| HblL2 | BC_3104 | NS | NS | NS | NS | NS | 1,29 ( p = 0.000) |
| HlyI | BC_5101 | NS | NS | NS | NS | 2,61( p = 0.001) | NS |
| HlyII | BC_3523 | NS | NS | NS | NS | NS | 2,45 ( p = 0.000) |
| NheA | BC_1809 | NS | NS | 0,47 ( p = 0.003) | NS | NS | 1,35 ( p = 0.002) |
| NheB | BC_1810 | NS | NS | 0,42 ( p = 0.008) | NS | NS | 1,34 ( p = 0.001) |
| NheC | BC_1811 | NS | NS | 0,83 ( p = 0.001) | NS | NS | 1,70 ( p = 0.001) |
Concerning mRNA levels, each log2(fold-change) represents the mean level of mRNA in the Δrex mutant samples relative to the mean level in the wild-type sample. The mean values were obtained from three measurements done on triplicate independent cultures. Only significant log2 ratios are indicated in bold. Concerning protein levels, the relative amount of each protein in Δrex mutant compared to its parental strain was determined using PatternLab software. Numbers in bold indicate data satisfied statistical criteria (p-value<0.05). The p-values were indicated in brackets. Plus and minus indicate increased and decreased abundance levels, respectively. NS: no significant change was observed. For details, see Tables S6 and S7.
(ii) Other predicted extracellular proteins
Like the toxin-related proteins, many degradative enzymes, flagella components and proteins that are released from the cell wall, are more abundant in the exoproteome of the Δrex mutant strain than wild-type cells, especially under aerobiosis (Table 4). All of these proteins are predicted to be extracellular proteins and, except for flagella components, all of them encompass signal peptides in their N-terminal primary structure and/or transmembrane helices. One metabolism-related protein, the subunit α of F0F1 ATP synthase (AtpA), and three proteins of unknown function (BC_1894, BC_5360 and BC_4062) also showed a higher abundance level in the exoproteome of aerobically grown Δrex cells. Except for BC_1894, which was annotated as a prophage protein and thus could be secreted via holin(-like) pathway [47], these proteins contain a signal peptide, and thus are predicted to be secreted by classical export pathways.
Table 4. Changes in protein abundances in Δrex exoproteome compared to wild-type exoproteome under anaerobiosis and aerobiosis.
| Log2 fold-change and significancea | ||||||||||
| Proteinname | Gene | Accession no. (NP) | Functional annotation | TM domain | Export Signal peptide | Anaerobiosis | Aerobiosis | |||
| log2-fold-change | p-value | log2-fold-change | p-value | |||||||
| Degradative enzymes and adhesins | ||||||||||
| ColC | BC_0556 | NP_830373 | Collagenase | N | Y | 0,4 | 0,03 | 1,9 | 0,00 | |
| NprB | BC_5351 | NP_835012 | Bacillolysin | N | Y | 0,67 | 0,00 | 1,97 | 0,00 | |
| NprP2 | BC_2735 | NP_832488 | Bacillolysin | N | Y | 1,58 | 0,00 | 2,6 | 0,01 | |
| HhoA | BC_3600 | NP_833332 | Protease | N | Y | 0,176 | 0,241 | 0,6 | 0,03 | |
| Tgc | BC_1991 | NP_831760 | Putative murein endopeptidase | N | Y | 0,45 | 0,01 | 1,71 | 0,00 | |
| PlcB | BC_0670 | NP_830483 | phospholipase C | N | Y | 0,6 | 0,00 | 2,25 | 0,00 | |
| PlcA | BC_3761 | NP_833485 | Phosphatidylinositol phosphodiesterase | N | Y | 0,48 | 0,02 | 2,55 | 0,00 | |
| InhA2 | BC_0666 | NP_830479 | Immune inhibitor A precursor | N | Y | 1,59 | 0,01 | 3,43 | 0,00 | |
| PgdA | BC_2929 | NP_832677 | Peptidoglycan deacetylase | N | Y | 0,585 | 0,055 | 0,87 | 0,04 | |
| Sph | BC_0671 | NP_830484 | Sphingomyelin phosphodiesterase | N | Y | 0,82 | 0,03 | 2,82 | 0,00 | |
| Flagella components | ||||||||||
| FlaA | BC_1659 | NP_831436 | Flagellin | N | N | 0,516 | 0,117 | 0,6 | 0,02 | |
| FlgK | BC_1636 | NP_831414 | Flagellar hook-associated protein | N | N | 0,96 | 0,01 | 1,04 | 0,05 | |
| FlgL | BC_1637 | NP_831415 | Flagellar hook-associated protein | N | N | 0,78 | 0,04 | 1,28 | 0,00 | |
| FliD | BC_1638 | NP_831416 | Flagellar capping protein | N | N | 0,526 | 0,063 | 1,22 | 0,00 | |
| FlgB | BC_1641 | NP_831419 | Flagellar basal body rod protein | N | N | 0,111 | 0,411 | 0,9 | 0,01 | |
| Cell wall and membrane-related proteins | ||||||||||
| FtsI | BC_4270 | NP_833984 | Penicillin-binding protein | Y | N | 0,189 | 0,145 | 1,73 | 0,02 | |
| YvgJ1 | BC_2932 | NP_832680 | Phosphoglycerol transferase | Y | N | −0,03 | 0,462 | 1,25 | 0,03 | |
| YvgJ2 | BC_5232 | NP_834895 | phosphoglycerol transferase | Y | N | −0,03 | 0,463 | 0,71 | 0,00 | |
| CwlD | BC_5196 | NP_834860 | N-acetylmuramoyl-L-alanine amidase | N | Y | 0,485 | 0,022 | 0,67 | 0,00 | |
| CwlB | BC_0902 | NP_830688 | N-acetylmuramoyl-L-alanine amidase | N | Y | 1,091 | 0,187 | −1,1 | 0,04 | |
| Metabolism | ||||||||||
| Eno | BC_5135 | NP_834803 | Phosphopyruvate hydratase | N | N | −1,2 | 0,01 | −0,12 | 0,385 | |
| PfkA | BC_4600 | NP_834306 | 6-Phosphofructokinase | N | N | −0,9 | 0,04 | −0,18 | 0,377 | |
| PykA | BC_4599 | NP_834305 | Pyruvate kinase | N | N | −0,15 | 0,429 | −4,6 | 0,01 | |
| Pgi | BC_4898 | NP_834571 | Glucose-6-phosphate isomerase | N | N | −1 | 0,04 | 0,014 | 0,493 | |
| AtpA | BC_3230 | NP_832971 | F0F1 ATP synthase subunit alpha | N | Y | −0,03 | 0,448 | 1,75 | 0,00 | |
| GapA | BC_5140 | NP_834805 | Glyceraldehyde-3-phosphate dehydrogenase | N | N | −0,62 | 0,144 | −2,9 | 0,00 | |
| PtA | BC_5387 | NP_835048 | Phosphotransacetylase | N | N | −1,6 | 0,01 | −2,1 | 0,03 | |
| GapN | BC_0868 | NP_830654 | NADP-dependent glyceraldehyde-3-phosphate dehydrogenase | N | N | −3,6 | 0,00 | −1,32 | 0,054 | |
| FbA | BC_5335 | NP_834997 | Fructose-bisphosphate aldolase | N | N | −0,76 | 0,298 | −3,6 | 0,03 | |
| Hpr | BC_4049 | NP_833767 | Phosphocarrier protein HPr | N | N | −2,4 | 0,03 | −1,18 | 0,079 | |
| Bdh | BC_0868 | NP_830481 | (R,R)-butanediol dehydrogenase | N | N | −3,32 | 0,06 | −3,6 | 0,02 | |
| PtsA | BC_4048 | NP_833766 | Phosphotransferase | N | N | −2,12 | 0,058 | −3,2 | 0,00 | |
| SfcA | BC_4604 | NP_834310 | NAD-dependent malic enzyme | N | N | −0,86 | 0,187 | −1,6 | 0,04 | |
| GlmM | BC_0188 | NP_830056 | Phosphoglucosamine mutase | N | N | −1,32 | 0,058 | −3,8 | 0,00 | |
| GdhA | BC_4162 | NP_833877 | Leucine dehydrogenase | N | N | −1,8 | 0,04 | −2,4 | 0,081 | |
| - | BC_4366 | NP_834078 | Cystathionine beta-lyase | N | N | 0,151 | 0,447 | −2 | 0,03 | |
| AdsS | BC_5468 | NP_835123 | Adenylosuccinate synthetase | N | N | −1,8 | 0,04 | −1,15 | 0,202 | |
| - | BC_3799 | NP_833521 | Aspartate-semialdehyde dehydrogenase | N | N | −2,56 | 0,079 | −3,2 | 0,01 | |
| ThiG | BC_0749 | NP_830539 | Thiazole synthase | N | N | ND | ND | −2,2 | 0,01 | |
| - | BC_0071 | NP_829966 | Phosphoribosyltransferase | N | N | −0,23 | 0,425 | −3,5 | 0,00 | |
| DeoD | BC_1463 | NP_831242 | Purine nucleoside phosphorylase | N | N | −2,6 | 0,02 | −1,06 | 0,185 | |
| - | BC_0015 | NP_829919 | Pyridoxine biosynthesis protein | N | N | −1,8 | 0,01 | −3,6 | 0,00 | |
| - | BC_3981 | NP_833700 | tetrahydrodipicolinate -acetyltransferase | N | N | ND | ND | −3,3 | 0,00 | |
| MoaB | BC_4758 | NP_834462 | Molybdenum cofactor | N | N | −0,94 | 0,187 | −2,3 | 0,02 | |
| FrvB | BC_4571 | NP_834277 | Deblocking aminopeptidase | N | N | −3,2 | 0,00 | −0,92 | 0,215 | |
| PepT | BC_4143 | NP_833858 | Peptidase T | N | N | −1,6 | 0,04 | −1,94 | 0,061 | |
| Stress-related proteins | ||||||||||
| AhpC | BC_0377 | NP_830216 | Alkyl hydroperoxide reductase C22 | N | N | −0,94 | 0,154 | −2,2 | 0,00 | |
| GrpE | BC_4313 | NP_834025 | Chaperon | N | N | −1,4 | 0,04 | −0,38 | 0,335 | |
| GroEL | BC_0295 | NP_830146 | Chaperon | N | N | −0,69 | 0,214 | −2,3 | 0,01 | |
| Dps2 | BC_5044 | NP_834714 | Non-specific DNA-binding protein | N | N | −2,4 | 0,00 | −1 | 0,165 | |
| Dps1 | BC_2011 | NP_831779 | Non-specific DNA-binding protein | N | N | −1 | 0,04 | 0 | 0,499 | |
| Sod | BC_5445 | NP_835106 | Superoxide dismutase [Mn] | N | N | −2,4 | 0,03 | −0,92 | 0,226 | |
| AhpF | BC_0376 | NP_830215 | Alkyl hydroperoxide reductase subunit F | N | N | ND | ND | −2,4 | 0,02 | |
| TerD | BC_0443 | NP_830282 | tellurium resistance protein | N | N | −2,7 | 0,00 | −0,97 | 0,064 | |
| Translation | ||||||||||
| RpsF | BC_5476 | NP_835129 | 30 S ribosomal protein S6 | N | N | −2,3 | 0,01 | −2,32 | 0,073 | |
| RpsG | BC_0126 | NP_830007 | 30 S ribosomal protein S7 | N | N | 0,566 | 0,2 | −1,8 | 0,01 | |
| RpsH | BC_0145 | NP_830025 | 30 S ribosomal protein S8 | N | N | 1,709 | 0,059 | −1,8 | 0,00 | |
| RpsJ | BC_0130 | NP_830010 | 30 S ribosomal protein S10 | N | N | ND | ND | −2,7 | 0,01 | |
| RplJ | BC_0119 | NP_830000 | 50 S ribosomal protein L10 | N | N | −0,58 | 0,16 | −4,3 | 0,00 | |
| RplK | BC_5075 | NP_834743 | 50 S ribosomal protein L11 | N | N | −0,94 | 0,187 | −2,4 | 0,02 | |
| RplL | BC_0120 | NP_830001 | 50 S ribosomal protein L7/L12 | N | N | −1,7 | 0,03 | −1,1 | 0,03 | |
| RpsT | BC_4320 | NP_834032 | 30 S ribosomal protein S20 | N | N | ND | ND | −2 | 0,03 | |
| RplX | BC_0142 | NP_830022 | 50 S ribosomal protein L24 | N | N | −2,2 | 0,03 | −0,79 | 0,154 | |
| RpmD | BC_0149 | NP_830029 | 50 S ribosomal protein L30 | N | N | −1,03 | 0,095 | −2,4 | 0,02 | |
| Frr | BC_3822 | NP_833543 | Ribosome recycling factor | N | N | −1,6 | 0,04 | −0,23 | 0,405 | |
| RaiA | BC_5190 | NP_834854 | SSU ribosomal protein S30P | N | N | −2,3 | 0,04 | −2,6 | 0,02 | |
| EftS | BC_3824 | NP_833545 | Elongation factor Ts | N | N | 1,263 | 0,204 | −3,1 | 0,01 | |
| Transcriptional Regulation | ||||||||||
| AbrB | BC_0042 | NP_829939 | Transcription state regulator | N | N | −1,15 | 0,082 | −3,3 | 0,00 | |
| CodY | BC_3826 | NP_833547 | Transcriptional repressor | N | N | ND | ND | −2,6 | 0,02 | |
| - | BC_3728 | NP_833453 | DNA-binding protein HU | N | N | −0,23 | 0,422 | −2,5 | 0,00 | |
| Other | ||||||||||
| - | BC_1984 | NP_831667 | Phage protein | N | N | 3,113 | 0,134 | 4,77 | 0,01 | |
| - | BC_1012 | NP_830798 | unknown | N | N | −0,81 | 0,291 | −3,5 | 0,00 | |
| - | BC_5360 | NP_835021 | unknown | Y | Y | 0,299 | 0,134 | 2,38 | 0,00 | |
| - | BC_0002 | NP_829890 | unknown | N | N | −4,1 | 0,00 | −3,1 | 0,01 | |
| - | BC_2077 | NP_831845 | unknown | N | N | −4,6 | 0,00 | −0,01 | 0,477 | |
| - | BC_4062 | NP_833780 | unknown | Y | N | 0,31 | 0,363 | 0,9 | 0,01 | |
| - | BC_2705 | NP_832458 | unknown | N | N | −1,89 | 0,084 | −3,6 | 0,00 | |
Only changes satisfying statistical criteria (p-value<0.05) at least in one growth condition are shown.
Each log2 fold-change value represents the mean protein level of the Δrex sample in relation to the wild-type sample. The relative amount of each protein was determined using PatternLab software. Plus and minus symbols indicate up-and down regulation of the protein, respectively. Numbers in bold indicate data that satisfied statistical criteria (p-value<0.05). ND: not detected. For details, see Table S7.
(iii) Predicted cytoplasmic proteins
In contrast to proteins predicted to be classically secreted, many proteins predicted to be cytoplasmic proteins, including metabolic proteins, stress response-related proteins, translation-related proteins, and other proteins were significantly less abundant in the exoproteome of the Δrex mutant than the parental strain (Table 4). We noted that most of the cytoplasmic proteins that showed a significant abundance level decrease under aerobiosis did not show significant abundance level changes under anaerobiosis, and vice-versa (Table 4 and Table S7). Finally, the percentage of predicted cytoplasmic proteins significantly decreased in the Δrex exoproteome compared with the wild-type exoproteome in the presence of oxygen, while the percentage of typically secreted proteins (toxin-related proteins and degradative enzymes and adhesins) was significantly higher (Fig. 4).
Figure 4. Functional distribution of proteins identified in Δrex and wild-type B. cereus exoproteomes under aerobiosis and anaerobiosis.

The diagrams represent the average proportion of each functional protein group based on total spectral counts (Table S5). For readability reasons, standard deviations (below 10%) are not shown.
The absence of Rex does not affect the mRNA level of toxin-related proteins
An explanation for the increased abundance levels of toxin-related proteins in the Δrex mutant exoproteome could be a change in their mRNA levels. We thus tested whether transcription of genes encoding these proteins was altered in the Δrex mutant compared with wild-type. Table 3 indicates that rex deletion significantly down-regulated entC and entFM under aerobiosis, but did not significantly change transcription of all other toxin-related genes. These transcriptomic results are not concordant with the proteomic data. This suggests that toxin-related protein abundance may be mainly modulated at the post-transcriptional level when Rex is lacking.
Discussion
B. cereus Rex is an authentic Rex transcriptional factor because (i) it is able to bind to promoter fragments in vitro, indicating that at least in some cases the effect of Rex is mediated by direct interaction with the promoters, (ii) it impacts the cellular metabolism and oxidative stress tolerance, and (iii) it acts mainly as a repressor at the proteomic level [12], [13], [14], [15], [20], [48]. However, B. cereus Rex also plays an important role in fully aerobic respiratory metabolism and it modulates the toxinogenic profile of exoproteome.
When oxygen was supplied to pH-regulated batch cultures at pO2 = 100%, B. cereus metabolizes glucose to carbon dioxide by oxidation of glycolytic pyruvate in the tricarboxylic acid (TCA) cycle. This reaction produces NADH, which then fuels oxidative phosphorylation to produce ATP, with minimal production of lactate and elimination of carbon excess through excretion of acetate. Rex deficiency facilitates the entry of carbon flow into the NADH-recycling lactate pathway at the expense of pyruvate oxidation into acetyl-CoA (Fig. 1). This promotes lactate production at the expense of acetate without impairment of TCA capacity, since acetate was always produced as a waste [28]. In the absence of oxygen or other external electron acceptors, ATP synthesis occurs at the level of substrate phosphorylation and NADH is reoxidized in terminal step fermentative reactions from pyruvate (Fig. 1). When grown in pH-controlled anaerobic batch cultures (pO2 = 0%), B. cereus produces lactate as the main glucose by-product to satisfy the demand for redox balance. Rex deficiency favored the production of more reduced metabolites, as evidenced by the increase in the ethanol to acetate ratio and the decrease of lactate production. This indicates that Rex regulates the carbon flow distribution at the pyruvate node by favoring carbon flow through the NADH-recycling lactate pathway at the expense of Pfl-dependent fermentative pathways under anaerobiosis. A key question is how Rex controls the carbon flow into the NADH-recycling lactate pathway in B. cereus cells grown in fully aerobic respiratory and anaerobic fermentative conditions? This control process could involve interplay of transcriptional and/or post-transcriptional regulation of key glycolytic enzymes (such as Pgm, BC_4919 under anaerobiosis and PK, BC_4599 under aerobiosis) and post-translational regulation of lactate dehydrogenase. Evidently, further intensive work is required to unravel the complexity of the regulatory network involving Rex. Finally, by controlling the entry of carbon flow into the lactate pathway, Rex controls the availability of glycolytic intermediates for macromolecular synthesis as well as supporting NADPH production under both aerobiosis and anaerobiosis.
In contrast to anaerobic fermentation, aerobic respiration is a major source of reactive oxygen species (ROS) generation [49]. B. cereus, like other facultative anaerobes, uses scavenging systems to control the level of ROS [50]. Sequestration of ROS leads to oxidative deactivation of these scavenging systems, which are then reactivated directly or indirectly depending on NADPH level [51]. By modulating the distribution pattern of antioxidant enzymes (Tables 2 and 4), and possibly NADPH production, Rex may thus modulate the effectiveness of antioxidative defense systems. Specifically, Rex may maximize antioxidant system activity under anaerobiosis while restricting this activity under aerobiosis; this could explain why the resistance of B. cereus cells towards external H2O2, when Rex is absent, is lower under anaerobiosis and higher under aerobiosis.
Except for HlyI, the abundance levels of classical extracellular proteins (such as toxins) were enhanced in B. cereus exoproteome when Rex was absent, especially under aerobiosis. In B. cereus, as in other Gram-positive bacteria, most extracellular proteins are synthesized as precursors, with typical N-terminal signal peptides, and exported from the cytoplasm by the Sec-dependent pathway [52]. This is the case for toxins [6]. However, unlike other toxins, HlyI contains a putative N-terminal twin arginine motif that could target the protein to the Tat secretion pathway instead of the Sec-dependent pathway [53], [54]. Besides classical export routes, B. cereus possesses specialized protein export systems such the flagellar type III system [5], [55], which translocates most flagellar components across the cytoplasmic membrane, and possibly the ESAT-6 secretion system, which mediates the secretion of virulence factors belonging to the WXG100 protein family, such as the protein BC_5360 [56], [57]. A feature of these two specialized secretion systems is the lack of a classical signal peptide to direct the substrate protein for secretion. However, like the Sec-dependent pathway and unlike the TAT-dependent pathway, these protein export systems require chaperones to prevent premature folding, aggregation and degradation by cytoplasmic proteases [58], [59]. Rex may thus influence protein-assisted export by modulating the distribution pattern of the components of export systems as observed in our proteomic data (Table 2, [60]). In addition, it has been reported that chaperones may be targets for intracellular ROS [61]. Keeping the functional integrity of these chaperones, specifically in the context of secretion, may thus depend on Rex-dependent NADPH availability. The role and contribution of NADPH in classical and specialized secretory mechanisms remains an open question that deserves further experiments.
Our results show that many cytoplasmic proteins predicted to be involved in active growth, either directly (such as metabolic enzymes, translational and post-translational-related proteins) or indirectly by preventing inappropriate gene expression (such as AbrB and CodY under aerobiosis [62]), were less abundant in B. cereus exoproteome when Rex was absent. We assume that cell lysis cannot account for changes in the abundance level of these cytoplasmic proteins since: (i) cell viability count did not change (data not shown), (ii) EF-Tu did not accumulate in extracellular medium (Table S7, [63]), and (iii) the abundance pattern of cytoplasmic proteins was dependent on growth conditions and did not always correlate with intracellular abundance changes (Table 2 and 4). Among the cytoplasmic proteins identified in B. cereus exoproteome, several have already been identified in the exoproteome of other bacteria, and many have been described as extracellular moonlighting components playing a role in bacterial virulence [64], [65]. The non classical mechanism of secretion that could explain their release remains to be firmly described [47], [66]. One hypothesis is that signal-less predicted cytoplasmic proteins might be secreted within outer membrane vesicles [63]. Such nanovesicles have been observed in Bacillus anthracis [67]. Interestingly, ATP is the primary metabolic factor involved in secretory vesicle movements. ATP generation is lower in fermenting cells than in non-fermenting cells, according to metabolism. In addition, Rex promotes ATP generation by enhancing NADH-producing pathways at the expense of NADPH pathways. Therefore, if ATP availability is a triggering factor for export of cytoplasmic proteins, this could explain the higher export of cytoplasmic proteins in aerobically grown cells compared to anaerobically grown cells, as well as the substantial decrease of cytoplasmic protein export under aerobiosis when Rex was absent.
In conclusion, Rex in B. cereus plays a pivotal role in controlling the cross-talk between the metabolic networks that control growth, oxidative defense machinery and extracellular accumulation of toxins. In the context of B. cereus natural environment, Rex may thus help the pathogen to (i) maintain its host alive until transmission to the next host can be achieved, by controlling its growth and production of its toxins [68], and (ii) better survive stress conditions under anaerobiosis, albeit to the detriment of maximizing its survival when oxygen is present [10], .
Supporting Information
Genetic organization of rex chromosomal region of B. cereus ATCC 14579. Panel A. Large arrows represent open reading frames identified in strain ATCC 14579, and their orientation shows the transcriptional direction. Panel B. Nucleotide sequence of rex ORF (black capital letters), as well as promoter and terminator regions (blue letters), are shown. Transcriptional start site (+1), putative −35 and −10 motifs and putative terminator are underlined. Start and stop translation codons are in bold.
(TIF)
Survival of B. cereus F4430/73 cells towards external hydrogen peroxide. Cells were grown in liquid cultures to mid-exponential growth phase either aerobically or anaerobically, and subjected to 20 and 10 mM H2O2 stress, respectively. Samples were taken at 0 and 20 min. Colony forming units per mL were counted and expressed as (N/No)×100. Error bars represented the standard deviation from three independent measures. Significant differences (p<0.05) between mutant and wild-type strains are indicated with asterisks.
(TIF)
SDS-PAGE analysis of overproduced and purified B. cereus Rex. Rex purification fractions were analyzed by electrophoresis on an SDS-12% polyacrylamide gel after Coomassie Brillant Blue staining (Panel A) or Imperial Protein Stain (Thermo scientific, Panel B). Position and molecular weights (kDa) of markers (lanes 1) are given on the left. Panel A: Lane 1, standard proteins; Lane 2, soluble whole cell extract from E. coli. Panel B: Lane 1, standard proteins; Lane 2, purified protein after Co2+ IMAC.
(TIF)
Venn diagrams summarizing proteins that showed significant abundance level changes in the cellular and extracellular proteomes of Δ rex compared with wild-type B. cereus cells under anaerobiosis (blue) and aerobiosis (red). Proteins up- and down-regulated by Δrex were distinguished.
(TIF)
Results from controlled batch cultures of Δ rex mutants and its parent strain, B. cereu s F4430/73.
(DOCX)
List of assigned MS/MS spectra from the cellular proteomes.
(XLSX)
List of assigned MS/MS spectra from the exoproteomes.
(XLSX)
List of proteins identified in cellular proteomes and their normalized spectral count.
(XLSX)
List of proteins identified in exoproteomes and their normalized spectral count.
(XLSX)
Fold changes of cellular proteins and their statistical significance.
(XLSX)
Fold changes of exoproteins and their statistical significance.
(XLSX)
Acknowledgments
S.L. held a fellowship from the Institut National de la Recherche Agronomique and the French Region of Provence Alpes Côtes d’Azur and G.C. held a fellowship from the Ministère de la Recherche et de l’Enseignement Supérieur.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. Our MS/MS data are deposited on the PRIDE repository database http://www.ebi.ac.uk/pride/; Username: reviewer73547@ebi.ac.uk; Password: 0EbXtIq9.
Funding Statement
This work was supported by the university of Avignon, the Institut National de la Recherche Agronomique, the Commissariat à l’Energie atomique and the French Region of Provence-Alpes-Côtes d’Azur. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bottone EJ (2010) Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev 23: 382–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stenfors Arnesen LP, Fagerlund A, Granum PE (2008) From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev 32: 579–606. [DOI] [PubMed] [Google Scholar]
- 3. Ceuppens S, Boon N, Uyttendaele M (2013) Diversity of Bacillus cereus group strains is reflected in their broad range of pathogenicity and diverse ecological lifestyles. FEMS Microbiol Ecol. 84: 433–50. [DOI] [PubMed] [Google Scholar]
- 4. Ramarao N, Sanchis V (2013) The pore-forming haemolysins of Bacillus cereus: a review. Toxins (Basel) 5: 1119–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Senesi S, Ghelardi E (2010) Production, secretion and biological activity of Bacillus cereus enterotoxins. Toxins (Basel) 2: 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fagerlund A, Lindback T, Granum PE (2010) Bacillus cereus cytotoxins Hbl, Nhe and CytK are secreted via the Sec translocation pathway. BMC Microbiol 10: 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clair G, Lorphelin A, Armengaud J, Duport C (2013) OhrRA functions as a redox-responsive system controlling toxinogenesis in Bacillus cereus . J Proteomics 94: 527–539. [DOI] [PubMed] [Google Scholar]
- 8. Clair G, Roussi S, Armengaud J, Duport C (2010) Expanding the known repertoire of virulence factors produced by Bacillus cereus through early secretome profiling in three redox conditions. Mol Cell Proteomics 9: 1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clair G, Armengaud J, Duport C (2012) Restricting Fermentative Potential by Proteome Remodeling: An adaptative strategy evidenced In Bacillus cereus. Mol Cell Proteomics 11: M111 013102. [DOI] [PMC free article] [PubMed]
- 10. Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, et al. (2010) Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465: 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moriarty-Craige SE, Jones DP (2004) Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr 24: 481–509. [DOI] [PubMed] [Google Scholar]
- 12. Larsson JT, Rogstam A, von Wachenfeldt C (2005) Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis . Microbiology 151: 3323–3335. [DOI] [PubMed] [Google Scholar]
- 13. Brekasis D, Paget MS (2003) A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J 22: 4856–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T (2006) Regulatory loop between redox sensing of the NADH/NAD(+) ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis . J Bacteriol 188: 7062–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pagels M, Fuchs S, Pane-Farre J, Kohler C, Menschner L, et al. (2010) Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 76: 1142–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sickmier EA, Brekasis D, Paranawithana S, Bonanno JB, Paget MS, et al. (2005) X-ray structure of a Rex-family repressor/NADH complex insights into the mechanism of redox sensing. Structure 13: 43–54. [DOI] [PubMed] [Google Scholar]
- 17. Somerville GA, Proctor RA (2009) At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci . Microbiol Mol Biol Rev 73: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vesic D, Kristich CJ (2013) A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J Bacteriol 195: 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang E, Ikonen TP, Knaapila M, Svergun D, Logan DT, et al. (2011) Small-angle X-ray scattering study of a Rex family repressor: conformational response to NADH and NAD+ binding in solution. J Mol Biol 408: 670–683. [DOI] [PubMed] [Google Scholar]
- 20. Bitoun JP, Nguyen AH, Fan Y, Burne RA, Wen ZT (2011) Transcriptional repressor Rex is involved in regulation of oxidative stress response and biofilm formation by Streptococcus mutans. FEMS Microbiol Lett 320: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, et al. (2012) Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol 194: 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esbelin J, Armengaud J, Zigha A, Duport C (2009) ResDE-dependent regulation of enterotoxin gene expression in Bacillus cereus: evidence for multiple modes of binding for ResD and interaction with Fnr. J Bacteriol 191: 4419–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Esbelin J, Jouanneau Y, Armengaud J, Duport C (2008) ApoFnr binds as a monomer to promoters regulating the expression of enterotoxin genes of Bacillus cereus . J Bacteriol 190: 4242–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zigha A, Rosenfeld E, Schmitt P, Duport C (2007) The redox regulator Fnr is required for fermentative growth and enterotoxin synthesis in Bacillus cereus F4430/73. J Bacteriol 189: 2813–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laouami S, Messaoudi K, Alberto F, Clavel T, Duport C (2011) Lactate dehydrogenase A promotes communication between carbohydrate catabolism and virulence in Bacillus cereus . J Bacteriol 193: 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esbelin J, Jouanneau Y, Duport C (2012) Bacillus cereus Fnr binds a [4Fe-4 S] cluster and forms a ternary complex with ResD and PlcR. BMC Microbiol 12: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, et al. (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis . Nature 423: 87–91. [DOI] [PubMed] [Google Scholar]
- 28. Rosenfeld E, Duport C, Zigha A, Schmitt P (2005) Characterization of aerobic and anaerobic vegetative growth of the food-borne pathogen Bacillus cereus F4430/73 strain. Can J Microbiol 51: 149–158. [DOI] [PubMed] [Google Scholar]
- 29. Murphy E (1985) Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3″) (9). Mol Gen Genet 200: 33–39. [DOI] [PubMed] [Google Scholar]
- 30. Arnaud M, Chastanet A, Debarbouille M (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70: 6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arantes O, Lereclus D (1991) Construction of cloning vectors for Bacillus thuringiensis . Gene 108: 115–119. [DOI] [PubMed] [Google Scholar]
- 32. Duport C, Zigha A, Rosenfeld E, Schmitt P (2006) Control of enterotoxin gene expression in Bacillus cereus F4430/73 involves the redox-sensitive ResDE signal transduction system. J Bacteriol 188: 6640–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carvalho PC, Fischer JS, Chen EI, Yates JR 3rd, Barbosa VC (2008) PatternLab for proteomics: a tool for differential shotgun proteomics. BMC Bioinformatics 9: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, et al. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 41: D1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q, Ou MS, Kim Y, Ingram LO, Shanmugam KT (2010) Metabolic flux control at the pyruvate node in an anaerobic Escherichia coli strain with an active pyruvate dehydrogenase. Appl Environ Microbiol 76: 2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, et al. (2008) Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol Microbiol 69: 466–478. [DOI] [PubMed] [Google Scholar]
- 37. Schau M, Chen Y, Hulett FM (2004) Bacillus subtilis YdiH is a direct negative regulator of the cydABCD operon. J Bacteriol 186: 4585–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sprynski N, Felix C, O'Callaghan D, Vergunst AC (2012) Restoring virulence to mutants lacking subunits of multiprotein machines: functional complementation of a Brucella virB5 mutant. FEBS Open Bio 2: 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spira WM, Goepfert JM (1975) Biological characteristics of an enterotoxin produced by Bacillus cereus . Can J Microbiol 21: 1236–1246. [DOI] [PubMed] [Google Scholar]
- 40. Garrigues C, Goupil-Feuillerat N, Cocaign-Bousquet M, Renault P, Lindley ND, et al. (2001) Glucose metabolism and regulation of glycolysis in Lactococcus lactis strains with decreased lactate dehydrogenase activity. Metab Eng 3: 211–217. [DOI] [PubMed] [Google Scholar]
- 41. Wichgers Schreur PJ, Rebel JM, Smits MA, van Putten JP, Smith HE (2011) Lgt processing is an essential step in Streptococcus suis lipoprotein mediated innate immune activation. PLoS One 6: e22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, et al. (2010) Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holley TA, Stevenson CE, Bibb MJ, Lawson DM (2013) High resolution crystal structure of Sco5413, a widespread actinomycete MarR family transcriptional regulator of unknown function. Proteins 81: 176–182. [DOI] [PubMed] [Google Scholar]
- 44. Newberry KJ, Brennan RG (2004) The structural mechanism for transcription activation by MerR family member multidrug transporter activation, N terminus. J Biol Chem 279: 20356–20362. [DOI] [PubMed] [Google Scholar]
- 45. Jiang L, Schaffitzel C, Bingel-Erlenmeyer R, Ban N, Korber P, et al. (2009) Recycling of aborted ribosomal 50 S subunit-nascent chain-tRNA complexes by the heat shock protein Hsp15. J Mol Biol 386: 1357–1367. [DOI] [PubMed] [Google Scholar]
- 46. Gohar M, Faegri K, Perchat S, Ravnum S, Okstad OA, et al. (2008) The PlcR virulence regulon of Bacillus cereus . PLoS One 3: e2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, et al. (2004) Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev 68: 207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bitoun JP, Liao S, Yao X, Xie GG, Wen ZT (2012) The redox-sensing regulator Rex modulates central carbon metabolism, stress tolerance response and biofilm formation by Streptococcus mutans . PLoS One 7: e44766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seaver LC, Imlay JA (2001) Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli . J Bacteriol 183: 7173–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mols M, Abee T (2011) Primary and secondary oxidative stress in Bacillus. Environ Microbiol. 13: 1387–1394. [DOI] [PubMed] [Google Scholar]
- 51. Mailloux RJ, Lemire J, Appanna VD (2011) Metabolic networks to combat oxidative stress in Pseudomonas fluorescens . Antonie Van Leeuwenhoek 99: 433–442. [DOI] [PubMed] [Google Scholar]
- 52.Schneewind O, Missiakas D (2013) Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim Biophys Acta. [DOI] [PMC free article] [PubMed]
- 53. Yuan J, Zweers JC, van Dijl JM, Dalbey RE (2010) Protein transport across and into cell membranes in bacteria and archaea. Cell Mol Life Sci 67: 179–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goosens VJ, Monteferrante CG, van Dijl JM (2013) The Tat system of Gram-positive bacteria. Biochim Biophys Acta. [DOI] [PubMed]
- 55. Senesi S, Salvetti S, Celandroni F, Ghelardi E (2010) Features of Bacillus cereus swarm cells. Res Microbiol 161: 743–749. [DOI] [PubMed] [Google Scholar]
- 56. Burts ML, Williams WA, DeBord K, Missiakas DM (2005) EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A 102: 1169–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pallen MJ (2002) The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends Microbiol 10: 209–212. [DOI] [PubMed] [Google Scholar]
- 58. Alonzo F 3rd, Xayarath B, Whisstock JC, Freitag NE (2011) Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol Microbiol 80: 1530–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moran-Barrio J, Limansky AS, Viale AM (2009) Secretion of GOB metallo-beta-lactamase in Escherichia coli depends strictly on the cooperation between the cytoplasmic DnaK chaperone system and the Sec machinery: completion of folding and Zn(II) ion acquisition occur in the bacterial periplasm. Antimicrob Agents Chemother 53: 2908–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roberts IN, Lam XT, Miranda H, Kieselbach T, Funk C (2012) Degradation of PsbO by the Deg protease HhoA Is thioredoxin dependent. PLoS One 7: e45713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ezraty B, Aussel L, Barras F (2005) Methionine sulfoxide reductases in prokaryotes. Biochim Biophys Acta 1703: 221–229. [DOI] [PubMed] [Google Scholar]
- 62. Frenzel E, Doll V, Pauthner M, Lucking G, Scherer S, et al. (2012) CodY orchestrates the expression of virulence determinants in emetic Bacillus cereus by impacting key regulatory circuits. Mol Microbiol 85: 67–88. [DOI] [PubMed] [Google Scholar]
- 63. Meneses N, Mendoza-Hernandez G, Encarnacion S (2010) The extracellular proteome of Rhizobium etli CE3 in exponential and stationary growth phase. Proteome Sci 8: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Copley SD (2012) Moonlighting is mainstream: paradigm adjustment required. Bioessays 34: 578–588. [DOI] [PubMed] [Google Scholar]
- 65. Henderson B, Martin A (2013) Bacterial moonlighting proteins and bacterial virulence. Curr Top Microbiol Immunol 358: 155–213. [DOI] [PubMed] [Google Scholar]
- 66. Yang CK, Ewis HE, Zhang X, Lu CD, Hu HJ, et al. (2011) Nonclassical protein secretion by Bacillus subtilis in the stationary phase is not due to cell lysis. J Bacteriol 193: 5607–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rivera J, Cordero RJ, Nakouzi AS, Frases S, Nicola A, et al. (2010) Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc Natl Acad Sci U S A 107: 19002–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. ten Bokum AM, Movahedzadeh F, Frita R, Bancroft GJ, Stoker NG (2008) The case for hypervirulence through gene deletion in Mycobacterium tuberculosis . Trends Microbiol 16: 436–441. [DOI] [PubMed] [Google Scholar]
- 69. Jakesevic M, Aaby K, Borge GI, Jeppsson B, Ahrne S, et al. (2011) Antioxidative protection of dietary bilberry, chokeberry and Lactobacillus plantarum HEAL19 in mice subjected to intestinal oxidative stress by ischemia-reperfusion. BMC Complement Altern Med 11: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marteyn B, Scorza FB, Sansonetti PJ, Tang C (2010) Breathing life into pathogens: the influence of oxygen on bacterial virulence and host responses in the gastrointestinal tract. Cell Microbiol 13: 171–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic organization of rex chromosomal region of B. cereus ATCC 14579. Panel A. Large arrows represent open reading frames identified in strain ATCC 14579, and their orientation shows the transcriptional direction. Panel B. Nucleotide sequence of rex ORF (black capital letters), as well as promoter and terminator regions (blue letters), are shown. Transcriptional start site (+1), putative −35 and −10 motifs and putative terminator are underlined. Start and stop translation codons are in bold.
(TIF)
Survival of B. cereus F4430/73 cells towards external hydrogen peroxide. Cells were grown in liquid cultures to mid-exponential growth phase either aerobically or anaerobically, and subjected to 20 and 10 mM H2O2 stress, respectively. Samples were taken at 0 and 20 min. Colony forming units per mL were counted and expressed as (N/No)×100. Error bars represented the standard deviation from three independent measures. Significant differences (p<0.05) between mutant and wild-type strains are indicated with asterisks.
(TIF)
SDS-PAGE analysis of overproduced and purified B. cereus Rex. Rex purification fractions were analyzed by electrophoresis on an SDS-12% polyacrylamide gel after Coomassie Brillant Blue staining (Panel A) or Imperial Protein Stain (Thermo scientific, Panel B). Position and molecular weights (kDa) of markers (lanes 1) are given on the left. Panel A: Lane 1, standard proteins; Lane 2, soluble whole cell extract from E. coli. Panel B: Lane 1, standard proteins; Lane 2, purified protein after Co2+ IMAC.
(TIF)
Venn diagrams summarizing proteins that showed significant abundance level changes in the cellular and extracellular proteomes of Δ rex compared with wild-type B. cereus cells under anaerobiosis (blue) and aerobiosis (red). Proteins up- and down-regulated by Δrex were distinguished.
(TIF)
Results from controlled batch cultures of Δ rex mutants and its parent strain, B. cereu s F4430/73.
(DOCX)
List of assigned MS/MS spectra from the cellular proteomes.
(XLSX)
List of assigned MS/MS spectra from the exoproteomes.
(XLSX)
List of proteins identified in cellular proteomes and their normalized spectral count.
(XLSX)
List of proteins identified in exoproteomes and their normalized spectral count.
(XLSX)
Fold changes of cellular proteins and their statistical significance.
(XLSX)
Fold changes of exoproteins and their statistical significance.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. Our MS/MS data are deposited on the PRIDE repository database http://www.ebi.ac.uk/pride/; Username: reviewer73547@ebi.ac.uk; Password: 0EbXtIq9.