

Abstract
Recent developments in methods to specifically modify genomic DNA using sequence-specific endonucleases and donor DNA have opened the door to a new therapeutic paradigm for cell and gene therapy of inherited diseases. Sequence-specific endonucleases, in particular transcription activator-like (TAL) effector nucleases (TALENs), have been coupled with polynucleotide small/short DNA fragments (SDFs) to correct the most common mutation in the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) gene, a 3-base-pair deletion at codon 508 (delF508), in induced pluripotent stem (iPS) cells. The studies presented here describe the generation of candidate TALENs and their co-transfection with wild-type (wt) CFTR-SDFs into CF-iPS cells homozygous for the delF508 mutation. Using an allele-specific PCR (AS-PCR)-based cyclic enrichment protocol, clonal populations of corrected CF-iPS cells were isolated and expanded.
Keywords: Sequence-specific endonucleases, TALENs, SFHR, Gene targeting, Cystic fibrosis, iPS cells, Clonal isolation, Nucleofection, Polynucleotides, SDFs
1 Introduction
Sequence-specific modification of genomic DNA in human pluripotent and multipotent stem cells is a strategy that would promote the development of autologous cell and gene therapies for inherited diseases, aging associated diseases, and regenerative medicine. While classical gene-targeting protocols, utilizing cellular DNA repair pathways, have been successfully used for engineering pluripotent mouse embryonic stem cells (mESCs) and immortalized human cell lines, there has been only limited success with human pluripotent stem cell (hPSC) systems for sequence-specific genomic modifications. Classical gene-targeting protocols typically involve the transfection of cells with a plasmid-base gene-targeting vector that is homologous to a genomic target sequence and uses drug selection enrichment strategies to identify and isolate cells that have the desired genomic modification. This approach is, however, highly inefficient where approximately 10−6–10−7 of the surviving cells contain the desired genomic modification. In addition, the classical gene-targeting vectors contain drug-selectable marker genes and other nonnative DNA sequences that are therapeutically undesirable.
Small fragment homologous replacement (SFHR) is one alternative approach that avoids some of the limitations associated with classical gene targeting [1–3]. Polynucleotide small DNA fragments (SDFs) used as donor DNA for the SFHR-mediated, sequence-specific homologous exchange are typically about 200–1,000 base pairs and can be prepared in large quantities by polymerase chain reaction (PCR) amplification. SFHR does not rely on drug selection and thereby avoids the introduction of nonnative DNA sequences. SFHR has been previously shown to successfully modify genomic DNA in mouse ESCs [4] (Gruenert and Emamekhoo, unpublished data), immortalized and primary human epithelial cell line [1, 2, 5–7], hematopoietic stem cells [8, 9], peripheral blood monocytes [10], human lymphoblasts [11], mouse T cells [12], and myoblasts [13–15] of a number of target genes, at efficiencies approaching 1–10 %.
Homologous recombination (HR) in hPSCs, and other mammalian cells, can be dramatically improved by introducing double-strand breaks (DSBs) in the region of the targeted genomic DNA sequences. This was initially demonstrated using a model system based on the yeast I-SceI homing endonuclease to introduce DSBs at ectopic sites in rodent and human cellular genomic DNA [16–18]. However, the I-SceI endonuclease recognition sequences are not naturally found in human genomic DNA and have, therefore, not proven to be a useful therapeutic tool that can be readily implemented to create site-specific double-strand breaks at any desired DNA sequence.
Recent advances in the development of sequence-specific chimeric endonucleases have demonstrated that fusion of the nuclease domain of the FokI endonuclease to DNA sequence-specific zinc-finger or transcription activator-like effector (TALE) binding motifs can generate hybrid zinc-finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs) that introduce DSBs in specific DNA regions [19–24]. The ZF or TALE amino acid motifs are designed as pairs to bind specific target DNA sequences 5′ and 3′ to the proposed FokI cut site. Since the FokI endonuclease is only active as a dimer, the inactive monomeric FokI nuclease domains fused to the 3′ end of each ZF or TALE binding pair will generate DSBs in mammalian cells at specific DNA target sequences where the nuclease domains are in close proximity to dimerize. In this chapter, we describe TALEN enhancement of SDF-potentiated correction of the delF508 mutation in exon 11 of the CFTR gene in human-induced pluripotent stem cells (hiPSCs).
2 Materials
2.1 Tissue Culture
Pluripotent cell lines: CF-induced pluripotent stem (CF-iPS) cell line, CF1-iPS4, homozygous for the delF508 mutation (Suzuki, Sargent, and Gruenert, unpublished data).
Media: TeSR1 (StemCell, Inc, Vancouver, BC, Canada).
Growth substrates: Matrigel (BD Biosciences, San Jose, CA).
Cell dissociation: Accutase (Life Technologies, Grand Island, NY), Dispase (StemCell, Inc).
Culture reagents: Rho-associated kinase (ROCK) inhibitor Y27632 (Sigma, St Louis, MO).
Tissue culture plates: 24-well plate (Corning/Costar, Corning, NY), 60 mm tissue culture dish (Falcon/Becton Dickenson Labware, Franklin Lakes, NJ).
2.2 Preparation of Polynucleotide Small DNA Fragments (SDFs)
Genomic and plasmid DNA: Human genomic DNA or plasmid containing human wild-type (wt) CFTR DNA sequence [6, 7].
PCR oligonucleotide primers: Table 1.
PCR buffer and polymerase: MyTaq HS Mix, 2× (Bioline, Taunton, MA).
Agarose gel electrophoresis: Agarose—Genetic Analysis Grade (Thermo Fisher Scientific, Waltham, MA), ethidium bromide 1 % (10 mg/mL) (Thermo Fisher Scientific).
Table 1.
PCR primers used for generating analytical or preparative products from genomic and genomic DNA-derived PCR templates are indicated
| Primer | Sequence (5′–3′) | Product (bp) |
|---|---|---|
| CF1 (f) CF5 (r) |
GCAGAGTACCTGAAACAGGA CATTCACAGTAGCTTACCCA |
491 (wt)/488 (delF508) |
| CF1B (f) | CCTTCTCTGTGAACCTCTATCA | |
| CF7C (r) | ATAGGAAACACCAAAGATGA | 392 |
| CF8C (r) | ATAGGAAACACCAATGATAT | 389 |
| CF17 (f) | GAGGGATTTGGGGAATTATTTG | |
| CF7C (r) | ATAGGAAACACCAAAGATGA | 330 |
| CF8C (r) | ATAGGAAACACCAATGATAT | 327 |
Primer pair CF1/CF5 was used to generate preparative quantities of SDF by PCR. Analysis of the wtCFTR and delF508-CFTR alleles in genomic DNA was performed by AS-PCR with primer pairs CF1B/CF7C (wt) and CF1B/CF8C (delF508). Primers CF7C and CF8C are allele specific and common to both DNA and RNA analyses; however, when paired with the non-allele-specific primer CF17 in exon10, amplification is specific for RNA-derived cDNA (AS-RT-PCR) crosses intron/exon boundaries. Forward primer = f, Reverse primer = r
2.3 TALEN Preparation
Golden Gate TALEN plasmid kit (Addgene, Cambridge, MA).
Plasmid backbone (MR015, Porteus and Rahdar, unpublished data).
2.4 Electroporation/Nucleofection
Amaxa 4D-Nucleofector X apparatus (Lonza, Allendale, NJ).
P3 Primary Cell 4D-Nucleofector Kit L with pmaxGFP (Lonza).
3 Methods
3.1 Tissue Culture
The CF1-iPS4 cell line and corrected clones are cultured on Matrigel-coated tissue culture plates using TeSR1 medium (StemCell, Inc) with daily medium changes.
Cultures of iPS cells are routinely subcultured for maintenance and expansion, or for clonal purification, by enzymatic dissociation of individual iPS cell colonies with Dispase (StemCell, Inc) or by mechanical isolation. Mechanical isolation is typically used to enrich clonal populations for undifferentiated hiPSC colonies and/or pick candidate corrected clones (see Notes 1 and 2).
Cells are replated onto Matrigel-coated culture plates grown in fresh TeSR1 medium as described above.
The passage number for the CF1-iPS4 hiPS cell line and corrected clones are designated as Pn1 × n2 × n3 - nz, where n1 = the number of passages as a primary culture before reprogramming, n2 = the number of passages the CF1-iPS4 cell line is grown after reprogramming, and n3 = the number of passages clones are grown after transfection/nucleofection with SDF, where the z in nz=the number of manipulations/treatments/modifications that the cells have undergone since their isolation [27] (see Notes 3 and 4).
3.2 Small DNA Fragment (SDF) Preparation
The wtCFTR SDF (491z-SDF), capable of correcting the delF508 mutation, is generated by PCR amplification with primer pair CF1/CF5 (Table 1) using the p491z-plasmid DNA as template [2, 6, 7].
The PCR amplification conditions for generating the wtCFTR 491z-SDF are as follows: A 50 μL reaction solution containing 1.0 μM of each primer, MyTaq HS Mix (Bioline), and 0.02 ng p491z-plasmid DNA is amplified with an initial denaturation for 2 min at 95 °C, followed by denaturation at 95 °C for 30 s; annealing at 55 °C for 30 s; and extension at 72 °C for 1 min for 35 cycles, with a final extension of 3 min at 72 °C (Table 2).
The 491z-SDF is separated from the p491z plasmid template by agarose gel electrophoresis and purified using a silica-based DNA purification protocol [28, 29] (see Note 5).
A second round of PCR amplification is carried out with 2 pg of the 491z-SDF as template. The amplified 491z-SDF is then purified using silica-based purification as indicated above.
Table 2.
PCR amplification conditions for DNA and RNA indicating the denaturation, annealing, and extension temperatures and times as well as amplification cycle number
| Step | Temperature (°C) | Time | Cycles |
|---|---|---|---|
| SDF and non-AS-PCR amplification conditions (CF1/CF5) | |||
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 30 s | 35 |
| Annealing | 55 | 30 s | |
| Extension | 72 | 1 min | |
| Final extension | 72 | 3 min | 1 |
|
| |||
| AS-PCR amplification conditions (CF1B/CF7C or CF8C) | |||
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 30 s | 35 |
| Annealing | 60 | 30 s | |
| Extension | 72 | 1 min | |
| Final extension | 72 | 8 min | 1 |
|
| |||
| AS-RT-PCR amplification conditions (CF17/CF7C or CF8C) | |||
| Reverse transcription | 42 | 1 h | |
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 30 s | 35 |
| Annealing | 52 | 30 s | |
| Extension | 72 | 1 min | |
| Final extension | 72 | 8 min | 1 |
3.3 TALEN Preparation
CFTR exon 11-specific TALENs are designed using the Web-based software, TALE-NT 2.0, https://boglab.plp.iastate.edu/ [23], and the following sequences were selected: TALEN pairs CFTAL-1B, 5′-TCTCAGTTTTCCTGGATTAT; spacer, gcctggcaccattaaagaa; CFTAL-2B, AATATCATTGGTGTTTCCT A-3′ (see Note 6).
Plasmids for expression of the CFTR-B TALENs are constructed using the Golden Gate TALEN assembly method [21] with the Golden Gate TALEN plasmid kit (Addgene) (see Notes 7–10).
A novel plasmid backbone (MR015, Porteus and Rahdar, unpublished data) can be used for optimal expression of the CFTR-B TALENs in mammalian cells.
3.4 Enhancement of SFHR-Mediated CFTR Correction by TALENs
The CF-iPS cells are pretreated with 10 μM of the Rho-associated kinase inhibitor, Y27632 (Sigma), for at least 2 h and harvested as a single cell suspension by treatment with Accutase (Life Technologies).
The wild-type 491z-SDFs alone or in the presence of 1 μg of each CFTAL-1B and CFTAL-2B TALEN expression vector are introduced into CF1-iPS4 cells by Amaxa nucleofection (electroporation) (Lonza) [30] to correct the genomic delF508 mutation.
Two doses of 491z-SDFs, 107 SDFs/cell (4.32 μg) and 2 × 107 SDFs/cell (8.64 μg), are introduced into ~8 × 105 cells with the Amaxa 4D-Nucleofector X apparatus using Solution P3 (solution:supplement = 82:18) and Program CB150 (Lonza). Duplicate nucleofections are carried out for each SDF amount.
Cells from duplicate electroporations are mixed and then plated into two wells of a 24-well plate (Corning/Costar) coated with Matrigel, with TeSR1 medium containing 10 μM Y27632 (see Note 11).
A sample of cells is also nucleofected with a GFP expression plasmid to monitor nucleofection efficiency. The GFP nucleofection control is evaluated by fluorescence microscopy 24 and 48 h post-electroporation to determine the approximate nucleofection efficiencies.
At 3 days post-nucleofection cells in one well of a nucleofection duplicate are dissociated with Accutase to assess the presence of wtCFTR sequences to determine whether the correction is successful. If successful, the other well of the duplicate is dissociated with Dispase and distributed in approximately equal numbers into 12 wells of a 24-well plate coated with Matrigel (see Note 12).
The enrichment process (see below) is initiated by isolating genomic DNA from nucleofecte cells 7–9 days post-nucleofection from the each well of the 12 wells of the 24-well plate containing cells and assaying for the relative amounts of wtCFTR by allele specific (AS)-PCR amplification with primer pairs CF1B/CF7C (wt) or CF1B/CF8C (delF508) [6, 7]. PCR products are assessed by electrophoretic banding on a 2 % agarose gel and visualized after staining with ethidium bromide (see Note 13).
3.5 Isolation of Corrected CF-iPS Cells
The isolation of clonal populations of corrected cells requires initial enrichment by a cyclic assessment protocol that involves redistribution of approximately equal numbers of the cells with the highest wtCFTR signal into multiple wells of a 24-well plate (Fig. 1).
The initial duplicate well of the 24-well plate containing wtCFTR positive CF-iPS cells (see above) is dissociated with Dispase and then distributed in approximately equal numbers into 12 wells of another 24-well plate coated with Matrigel.
After 5–7 days when the cells reach confluence, they are again treated with 10 μM Y27632 for ~2 h before dissociation with Accutase. Approximately, 5/6th of the cells are assayed for wtCFTR by AS-PCR with primer pair CF1B/CF7C. A replicate 24-well plate of the parent plate is created with the remaining 1/6th of the cells. The well that shows the highest concentration of corrected cells by semiquantitative AS-PCR is then harvested by Dispase dissociation at confluence (~3–4 days after replating in the replicate plate) and equally distributed into 12 wells of a new 24-well plate (see Note 14).
The steps outlined in step 3 are repeated until approximately 10 % of the cells are wtCFTR, as determined by semiquantitative AS-PCR (Fig. 2). The well with the highest degree of enrichment is then pretreated with 10 μM Y27632 for ~2 h and harvested with Accutase. The Accutase-treated, single cell suspension is replated on a 60 mm dish coated with Matrigel for the isolation of individual clones of cells that are examined by AS-PCR to identify cells having either a heterozygous or homozygous wtCFTR genotype (Fig. 3) (see Notes 15 and 16).
Fig. 1.

Enrichment protocol to isolate gene-corrected clones. nucleofection: Donor DNA is nucleofected in the presence or absence of TALENs. Enrichment: After determining that there is a population of corrected cells, the cells in a well are dissociated with Dispase and equally distributed as cell clumps into 12 wells of a 24-well plate. Analysis/duplication: Cells are harvested with Accutase as single cells to maintain the same ratio of corrected:uncorrected cells within the population for analysis by AS-PCR and subculture into a duplicate 24-well plate, i.e., an aliquot of cells from each well is placed into the same well of another 24-well plate. Enrichment cycle: The well determined to have the highest proportion of corrected cells is then dissociated with Dispase, and the cell clumps are equally distributed into 12 wells of a new 24-well plate and is followed by another analysis/duplication step that constitutes the beginning of the next cycle in the enrichment cycle (EC) scheme. Isolation: After the nth enrichment cycle (ECn) when the proportion of corrected cells is approximately 10 %, the cells are dispersed with Accutase and plated as single cells to isolate individual colonies
Fig. 2.
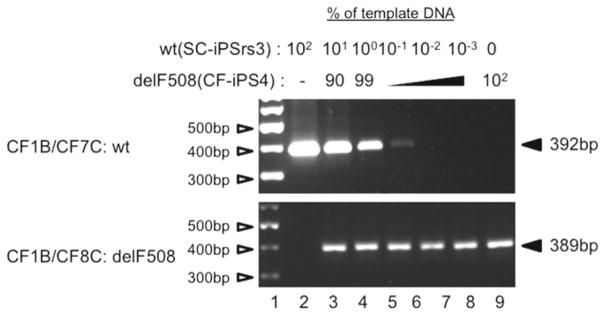
AS-PCR titration analysis for sensitivity of detection. PCR control titration analysis involves the mixing genomic DNA from a nonCF-iPS cell line (SC-iPSrs3) with CF1-iPS4 at varying proportions. The products from the AS-PCR amplification with primer pair CF1B/CF7C (wt) or pair CF1B/CF8C (delF508) are electrophoretically separated on a 2 % agarose gel and visualized by ethidium bromide staining. The intensity of the band represents the proportion of the wtCFTR DNA present within each mixture and reflects the sensitivity of the AS-PCR assay. The marker DNA (Lane 1 ) was the GeneRuler 100 bp plus DNA ladder (Thermo Fisher Scientific, Waltham, MA)
Fig. 3.
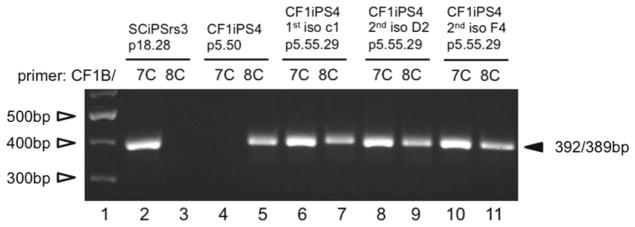
Isolation of corrected clones: AS-PCR analysis performed with primer pairs CF1B/CF7C (wtCFTR) or CF1B/CF8C (delF508 CFTR) on genomic DNA isolated from three independent colonies following limited dilution (Lanes 6–11). The control wtCFTR and homozygote delF508 band patterns were shown using genomic DNA of a nonCF-iPS cell line (SC-iPSrs3) and CF1-iPS4 (delF508) (Lanes 2–5 ). GeneRuler 100 bp plus DNA ladder (Thermo Fisher Scientific) (Lane 1)
Acknowledgments
We would like to thank Dr Michael Yezzi and Carissa Tasto for their expert technical assistance. This study was supported by NIH grants GM075111 AS4 and DK088760; a grant from Cystic Fibrosis Research, Inc; funds from Pennsylvania Cystic Fibrosis, Inc (PACFI); and a grant from the Japan Society for the Promotion of Science (SS).
Footnotes
Subculturing of iPS cells is generally carried out when the cells are subconfluent (30–40 % confluence) and before individual colonies of cells begin to touch each other. Individual iPS cell colonies are isolated by mechanical removal or by enzymatic dissociation with Dispase or Accutase. Manual isolation of iPSC colonies can be very precise, and it is possible to identify and isolate those portions of the cells that appear morphologically identical to the cells that appeared in the initial colony isolate. It is, however, time-consuming and tedious. It is important to note the colony growth rate when choosing which colonies to isolate. Colonies that grow rapidly can be problematic and karyotypically unstable. These colonies should therefore be avoided if possible.
Enzymatic dissociation is generally more time efficient than mechanical isolation but requires attention to detail. If Accutase is used, pre- and posttreatment with Y27632 is required to enhance viability.
The cell passage nomenclature is a bookkeeping tool, where each passage number that is followed by a period represents a specific phenotype, when the cells are exposed to something that will alter the cellular phenotype, e.g., reprogramming factors. At this point the number begins again and increases with each subculture. If the cells are again exposed to a reagent and/or treatment, e.g., mutation correction with an SDF, another period is added, and the numbering is reinitiated and increases with each subculture until the next treatment, etc.
Cells need to be evaluated and screened periodically for their karyotype and their pluripotence features to ensure that they are still pluripotent and genetically stable at a macroscopic level.
To eliminate any residual p491z-plasmid DNA remaining in the amplification solution, the SDF is gel purified.
The TALEN pairs are designed to overlap the mutant target sequences to minimize the potential to cut the donor SDF and the corrected genomic DNA.
The Golden Gate assembly method involves an initial digestion of several different RVD plasmids (<10 RVD plasmids in the Golden Gate Kit) and the backbone plasmid for the first Golden Gate reaction followed by the ligation of different RVD combinations into the backbone plasmid. The RVD plasmids chosen depend on the target nucleotide sequence. Since most target sequences are from 15 to 30 nucleotides, this initial reaction needs to be carried out 2–3 times with different RVD combinations and different backbone vectors.
The next step in this part of the Golden Gate assembly involves harvesting individual white clones and their analysis by colony PCR and sequencing to identify the clone with the correct RVD combination.
A second Golden Gate reaction is carried out with the first reaction products, the RVD plasmid that identifies the final nucleotide (3′ end) in the target sequence, and a backbone vector that can be expressed in human cells. This mixture is digested and ligated to generate the final expression construct. The correct targeting constructs are identified as indicated in Note 8 with an additional diagnostic restriction digestion.
This process needs to be carried for each side of the target FokI endonuclease cut site that is within the spacer sequence (15–30 nucleotides) between the individual TALEN binding sites.
The duplicate electroporations are important for establishing the initial duplicate cultures of nucleofection cells for analysis and for enrichment. Mixing the cells from the individual transfections normalizes the nucleofection efficiency for the individual samples.
While the analytical well of cells is dissociated with Accutase, the dispersed well is dissociated with Dispase. Dissociation with Dispase maintains the cells as clumps and results in higher cell survival overall.
The serial enrichment of populations with wtCFTR is facilitated by choosing the wells with the highest wtCFTR levels as assessed by AS-PCR. The dispersal as cell clumps is likely to favor the distribution of groups of cells that are clonal expansions of individual corrected cells and improve overall viability.
Assessment of the relative amount of wtCFTR DNA in a well can be determined by comparison to a PCR-based standard titration analysis (see Fig. 2). Wild-type and delF508 cells are mixed at different ratios, and the DNA of the mixed populations is used as standards for PCR analysis. The sensitivity of the AS-PCR is limiting, and it is difficult to visualize an ethidium bromide signal below 0.05 %. It is, however, possible to differentiate between individual wells if the proportion of wtCFTR cells is ≥0.05 %.
The number of enrichment cycles is dependent on the efficiency of gene correction in the initial analysis. Enrichment to 5–10 % makes the isolation of an individual corrected clone straightforward when the cells are dissociated and grown as individual cells.
After corrected CF-iPS cell-derived clones are isolated, they need to be further characterized to ensure pluripotence and karyotypic stability and to demonstrate restoration of wtCFTR function.
References
- 1.Gruenert DC, et al. Sequence-specific modification of genomic DNA by small DNA fragments. J Clin Invest. 2003;112(5):637–641. doi: 10.1172/JCI19773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunzelmann K, et al. Gene targeting of CFTR DNA in CF epithelial cells. Gene Ther. 1996;3(10):859–867. [PubMed] [Google Scholar]
- 3.Sargent RG, Kim S, Gruenert DC. Oligo/Polynucleotide-based gene modification: strategies and therapeutic potential. Oligonucleotides. 2011;21(2):55–75. doi: 10.1089/oli.2010.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sangiuolo F, et al. Cftr gene targeting in mouse embryonic stem cells mediated by small fragment homologous replacement (SFHR) Front Biosci. 2008;13:2989–2999. doi: 10.2741/2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goncz KK, et al. Expression of DeltaF508 CFTR in normal mouse lung after site-specific modification of CFTR sequences by SFHR. Gene Ther. 2001;8(12):961–965. doi: 10.1038/sj.gt.3301476. [DOI] [PubMed] [Google Scholar]
- 6.Goncz KK, Gruenert DC. Site-directed alteration of genomic DNA by small-fragment homologous replacement. Methods Mol Biol. 2000;133:85–99. doi: 10.1385/1-59259-215-5:85. [DOI] [PubMed] [Google Scholar]
- 7.Goncz KK, Kunzelmann K, Xu Z, Gruenert DC. Targeted replacement of normal and mutant CFTR sequences in human airway epithelial cells using DNA fragments. Hum Mol Genet. 1998;7(12):1913–1919. doi: 10.1093/hmg/7.12.1913. [DOI] [PubMed] [Google Scholar]
- 8.Goncz KK, et al. Small fragment homologous replacement-mediated modification of genomic beta-globin sequences in human hematopoietic stem/progenitor cells. Oligonucleotides. 2006;16(3):213–224. doi: 10.1089/oli.2006.16.213. [DOI] [PubMed] [Google Scholar]
- 9.Goncz KK, Prokopishyn NL, Chow BL, Davis BR, Gruenert DC. Application of SFHR to gene therapy of monogenic disorders. Gene Ther. 2002;9(11):691–694. doi: 10.1038/sj.gt.3301743. [DOI] [PubMed] [Google Scholar]
- 10.McNab GL, Ahmad A, Mistry D, Stockley RA. Modification of gene expression and increase in alpha1-antitrypsin (alpha1-AT) secretion after homologous recombination in alpha1-AT-deficient monocytes. Hum Gene Ther. 2007;18(11):1171–1177. doi: 10.1089/hum.2007.073. [DOI] [PubMed] [Google Scholar]
- 11.Bedayat B, et al. Sequence-specific correction of genomic hypoxanthine-guanine phosphoribosyl transferase mutations in lymphoblasts by small fragment homologous replacement. Oligonucleotides. 2010;20(1):7–16. doi: 10.1089/oli.2009.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zayed H, McIvor RS, Wiest DL, Blazar BR. In vitro functional correction of the mutation responsible for murine severe combined immune deficiency by small fragment homologous replacement. Hum Gene Ther. 2006;17(2):158–166. doi: 10.1089/hum.2006.17.158. [DOI] [PubMed] [Google Scholar]
- 13.Kapsa R, et al. In vivo and in vitro correction of the mdx dystrophin gene nonsense mutation by short-fragment homologous replacement. Hum Gene Ther. 2001;12(6):629–642. doi: 10.1089/104303401300057324. [DOI] [PubMed] [Google Scholar]
- 14.Kapsa RM, et al. Targeted gene correction in the mdx mouse using short DNA fragments: towards application with bone marrow-derived cells for autologous remodeling of dystrophic muscle. Gene Ther. 2002;9(11):695–699. doi: 10.1038/sj.gt.3301737. [DOI] [PubMed] [Google Scholar]
- 15.Todaro M, et al. Effective detection of corrected dystrophin loci in mdx mouse myogenic precursors. Hum Mutat. 2007;28(8):816–823. doi: 10.1002/humu.20494. [DOI] [PubMed] [Google Scholar]
- 16.Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(4):1968–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91(13):6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sargent RG, Brenneman MA, Wilson JH. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol Cell Biol. 1997;17(1):267–277. doi: 10.1128/mcb.17.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300(5620):764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- 20.Bibikova M, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21(1):289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cermak T, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark KJ, Voytas DF, Ekker SC. A TALE of two nucleases: gene targeting for the masses? Zebrafish. 2011;8(3):147–149. doi: 10.1089/zeb.2011.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle EL, et al. TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40(Web Server issue):W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300(5620):763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z, Gruenert DC. Human CFTR gene sequences in regions flanking exon 10: a simple repeat sequence polymorphism in intron 9. Biochem Biophys Res Commun. 1996;219(1):140–145. doi: 10.1006/bbrc.1996.0195. [DOI] [PubMed] [Google Scholar]
- 26.Zielenski J, et al. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10(1):214–228. doi: 10.1016/0888-7543(91)90503-7. [DOI] [PubMed] [Google Scholar]
- 27.Gruenert DC, et al. Characterization of human tracheal epithelial cells transformed by an origin-defective simian virus 40. Proc Natl Acad Sci U S A. 1988;85(16):5951–5955. doi: 10.1073/pnas.85.16.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boom R, et al. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28(3):495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle JS, Lew AM. An inexpensive alternative to glassmilk for DNA purification. Trends Genet. 1995;11(1):8. doi: 10.1016/s0168-9525(00)88977-5. [DOI] [PubMed] [Google Scholar]
- 30.Maurisse R, et al. Comparative transfection of DNA into primary and transformed mammalian cells from different lineages. BMC Biotechnol. 2010;10:9. doi: 10.1186/1472-6750-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]