

Abstract
Preliminary studies in our laboratory have demonstrated the importance of both the NH2 and COOH terminus scaffolding functions of focal adhesion kinase (FAK). Here, we describe a new small molecule inhibitor, C10 that targets the FAK C-terminus scaffold. C10 showed marked selectivity for cells overexpressing VEGFR3 when tested in isogenic cell lines, MCF7 and MCF7-VEGFR3. C10 preferentially inhibited pancreatic tumor growth in vivo in cells with high FAK-Y925 and VEGFR3 expression. Treatment with C10 led to a significant inhibition in endothelial cell proliferation and tumor endothelial and lymphatic vessel density and decrease in interstitial fluid pressure. These results highlight the underlying importance of targeting the FAK scaffold to treat human cancers.
Keywords: Focal adhesion kinase, vascular endothelial growth factor receptor-3, focal adhesion targeting domain, FAK scaffold inhibition, pancreatic cancer
1. INTRODUCTION
Focal adhesion kinase (FAK) is involved in every aspect of cancer cell progression and metastasis [3, 13, 41]. FAK serves as a scaffold to direct numerous signaling pathways to achieve a wide variety of cellular outcomes such as cell proliferation, motility, invasion, angiogenesis, and survival [22, 42]. The domain architecture of FAK promotes its role as a scaffolding protein where the ability of the N-terminal, FERM and C-terminal, FAT domains of FAK to interact with a variety of binding partners has been clearly demonstrated and these interactions with FAK act in concert to exacerbate tumor aggressiveness [1, 11, 23]. In an attempt to target and inhibit FAK activity, numerous approaches such as RNA interference [7], knockdown [5], antisense oligonucleotides [37], and kinase inhibition [17, 38] have successfully decreased cancer cell survival. FAK kinase inhibitors, PF-562-271, PF-04554878, and GSK2256098 are being currently evaluated in clinical trials [35]. However, destabilizing the FAK scaffold might prove to be an advantageous approach to developing FAK targeted therapeutics than solely focusing on inhibiting the kinase function of FAK [4, 26].
In the pursuit of developing FAK scaffold inhibitors, we previously discovered that VEGFR3 binds to the C-terminal FAT domain of FAK to promote cancer cell survival [14]. We recently developed a novel FAK scaffold inhibitor, C10 (previously compound 29) that disrupted the FAK-VEGFR3 interaction and showed a dramatic increase in potency and binding affinity to the FAT domain of FAK attributed mainly due to its proximity to the tyrosine 925 site in the FAT domain of FAK [16]. FAK-Y925 can be phosphorylated by Src, which in turn creates a docking site for the SH2 domain containing adaptor protein, Grb2 that further activates the Ras/ MAPK pathway [33]. Activation of FAK-Y925 has also been implicated in promoting VEGF induced tumor angiogenesis [27], influencing FAK/ paxillin interactions, and tumor metastasis [6, 19].
FAK is a target in pancreatic adenocarcinoma where a large subset of patient pancreatic tumors display increased levels of FAK where expression significantly correlates with invasive potential, metastatic disease, and poor prognosis [8, 31]. A significant correlation between FAK expression and tumor size in pancreatic cancer patients has also been reported [12]. An increase in copy number of chromosome 8q24 where FAK localizes has been linked to pancreatic cancer disease [10, 24]. Furthermore, increased expression of VEGFR3 and its ligand VEGF-C have been implicated in pancreatic cancer progression [34]. These findings confirm that the FAK pathway is operative in pancreatic cancer and thus disrupting the C-terminal FAK-VEGFR3 scaffold could reduce pancreatic tumor burden. In this study, we tested the efficacy of C10 in a pancreatic cancer model system and found that C10 targets the FAK C-terminal scaffold by inhibiting FAK-Y925 and FAK mediated downstream signaling and induced potent anti-tumor and anti-angiogenic effects in FAK-Y925 and VEGFR3 positive pancreatic cancer cells.
2. MATERIALS AND METHODS
2.1 Chemical synthesis
Compound C10 (previously compound 29) was synthesized and its purity was evaluated as previously described [16].
2.2 Cell culture
MiaPaCa-2-luc and Panc-1-luc human pancreatic cancer cell lines were transfected with retroviral expression vector pMSCV-LucSh (provided by Dr. Andrew M. Davidoff, St. Jude Children's Research Hospital), that contains a luciferase and zeocin resistance fusion gene, was used to create cell lines stably expressing luciferase. The BxPC3 human pancreatic cancer cell line was kindly provided by Dr. Elizabeth Repasky, Roswell Park Cancer Institute, Buffalo, NY. The MCF7 cell line was purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The cell lines were maintained in DMEM supplemented with 10% fetal bovine serum and incubated at 37°C in 5% CO2. The MCF7-VEGFR3 cell line has been previously developed and characterized [21].
2.3 Reagents
The following antibodies were used: FAK mouse monoclonal antibody (clone 4.47). VEGFR3 rabbit polyclonal antibody (Santa Cruz Biotechnology Inc, Santa Cruz, CA). Phosphorylated VEGFR3 rabbit polyclonal antibody (Cell Applications Inc., CA). Phosphorylated Akt (Ser 473), Erk (Thr 202/ Tyr 204), FAK (Tyr 925), paxillin (Tyr 118), Total Akt, Total Erk, Total Grb2, and Total paxillin, rabbit polyclonal antibodies (Cell Signaling Technology Inc., Danvers, MA). Grb2 (Y209) (Abgent, San Diego, CA). FAK (Tyr 861) and GAPDH (Invitrogen, Grand Island, NY).
2.4 Cell proliferation (MTS) assay
Briefly, cell lines were seeded at a density of 5000 cells/well in 96 well plates and allowed to attach overnight. DMSO or C10 were added at the indicated concentrations for 72 h and cell viability was measured as previously described [16].
2.5 Immunoblotting
Cells were seeded at a density such that they were approximately 80% confluent after 24 h. Compound C10 was added at the indicated concentrations and cells were incubated at 37°C for 24 h. Total cell lysates were prepared as previously described [16].
Tumor tissue extracts were prepared from frozen tumor samples according to the TissueLyser LT protocol (Qiagen, Valencia, CA). Homogenized tumor samples were centrifuged at 10,000 rpm for 30 minutes. The supernatant fraction was collected and immunoblotting was performed using the protocol as explained previously [16]. Protein band density measurements were analyzed using the Image J software.
2.6 Wound healing assay
Cells were seeded in 6-well plates and cultured for 24 h to form confluent monolayers. A wound was created in the middle area of the well by dragging a 200 μl pipette tip across the monolayer. Plates were washed to remove cellular debris. Growth medium containing selected concentrations of the drug was added to each well and plates were incubated for 24 h. Wound images were photographed at 0 h and 24 h using an inverted microscope. Percent wound closure was computed using the formula, {(gap width at time 0 – gap width at time 24 h)/ gap width at time 0} * 100.
2.7 Invasion assay
5 * 104 cells were seeded in serum-free medium with or without drugs atop insert membranes coated with matrigel in 24-well plates with 8.0 mm pores (BD Biosciences). 10% FBS was added to the medium in the lower chamber to stimulate cellular invasion for 24 h. Media was aspirated from the inserts and lower chambers. Cotton swabs were used to remove the cells that remained in the inserts and those, which invaded through the matrix, were fixed and stained using 0.1% crystal violet solution containing 50% methanol. Multiple fields of stained cells were counted using light microscopy at 40X magnification.
2.8 In vivo efficacy studies
Animal experiments were approved and monitored by the Institutional Animal Care and Use Committee (IACUC) of RPCI. MiaPaCa-2-luc (2 * 106) or BxPC3 (4 * 106) cells were subcutaneously inoculated into the flanks of 6-8 week old female SCID mice. Once the tumors reached a size of approximately 100 mm3 mice were assigned randomly to different groups before starting vehicle (PBS) (n = 6-10) or compound C10 (n = 6-10) dosing. Tumor volume was calculated using the formula, length * width2 * 0.5. Mice were euthanized at the study endpoint, and tumors were excised, weighed, and analyzed using Western blot for the expression of several proteins.
2.9 Immunohistochemistry
Staining procedures were performed as described previously [20]. A positive and negative control was included in each staining. IHC-stained tissue slides were scanned in an Aperio ScanScope CS and viewed using ImageScope software. Five to eight representative high power fields per slide were evaluated and selected for each stain (Ki67, CD31, and LYVE1). A pathologist (A. W) performed the Aperio Image Analysis algorithms (nuclear algorithm for Ki67 and microvessel algorithm for CD31 and LYVE1) (Aperio Technologies, Inc., Vista, CA). Data were analyzed for statistical significance (p <0.05).
2.10 Tube formation assay
Briefly, 24-well culture plates were coated with Cultrex Basement Membrane Extracts (Trevigen, Gaithersburg, MD) and incubated at 37 °C for 1 h. Next, 5000 HUVEC cells were seeded and incubated with EBM-2 Basal Medium (LONZA) with or without compound C10 for 24 h. Plates were incubated at 37 °C for 6 h and 24 h. At each time point, HUVEC cells were examined for capillary-like network formation and photographed under a light microscope. Images were taken from 7 to 10 different fields in each well. Analysis of tube formation was performed using the Wimasis WimTube Image analysis software (ibidi GmbH, Germany).
2.11 Transwell migration assay
7 * 104 HUVEC cells were seeded per insert with or without compound C10 onto 8 mm pore size polycarbonate filters in a 12-well Boyden chamber (Corning) and incubated for 6 h. The chemotactic migration of cells was induced by 5% FBS or 100 ng/ml FGF2 in the lower chamber. Plates were incubated at 37 °C for 24h. The migrated cells were stained with 0.1% crystal violet staining solution. The stain was extracted with 10% acetic acid solution and absorbance was measured at 590 nm.
2.12 Directed in vivo Angiogenesis Assay (DIVAA™)
Analysis and quantitation of angiogenesis was carried out as per the Cultrex® DIVAA protocol (Trevigen, Gaithersburg, MD). Briefly, 10 mm long surgical-grade silicone tubes (angioreactors) with only one end open were filled with Trevigen's basement membrane extract (BME) mixed with FGF2, either alone or in combination with inhibitors (Avastin or C10) at the indicated concentrations. Once the BME solidified, the angioreactors were surgically implanted subcutaneously in the dorsal flanks of 6-8 week old female SCID mice. After 10 days, the angioreactors were extracted and processed as per the manufacturer's protocol.
2.13 Interstitial fluid pressure (IFP) measurement
MiaPaCa-2-luc cells were subcutaneously inoculated in the flanks of 6-8 week old female SCID mice. Once the tumors reached a size of 100 mm3, compound C10 was administered via intraperitoneal injection once daily for five days a week. IFP was measured after 14 doses of C10 according to the previously described protocol [36].
2.14 Statistical analysis
Comparisons between groups were made using a Students t test. Data were considered significant when p<0.05.
3. RESULTS
3.1 FAK inhibitor C10 preferentially targets FAK-Y925 and VEGFR3 positive cells in vitro
We have previously shown that C10 binds to the FAT domain of FAK (Kd of 18 nM) and disrupts the FAK-VEGFR3 interaction [16]. To further investigate the specificity of C10, we used isogenic cell lines, MCF7 and MCF7-VEGFR3 that differ only in the expression of VEGFR3. As shown in Figure 1A, MCF7-VEGFR3 cells, that overexpress VEGFR3 showed exquisite sensitivity to C10 treatment, whereas a lack of effect on survival was seen in the MCF7 cell line which has very low levels of VEGFR3. We then tested the effects of C10 in pancreatic cancer cells and found that C10 inhibited cell survival in the MiaPaCa-2-luc cell line to a greater extent (average IC50 of 21 μM) as compared to the BxPC3 cell line (average IC50 of 100 μM) (Figure 1B). To investigate the mechanism of C10-induced decrease in cell survival of the aforementioned cell lines, we examined the phosphorylation status of Y861 and Y925 residues located within the C-terminal domain of FAK, downstream signaling partners of FAK, and the phosphorylation of VEGFR3 (Figures 1C and S1). In C10-treated MCF7-VEGFR3 cells, there was a marked decrease of FAK-Y861 phosphorylation with increasing doses of C10 in comparison to the modest decrease seen in the MCF7 cells. Interestingly, basal expression levels of FAK-Y925 phosphorylation was essentially undetectable in MCF7 cells, whereas a stronger expression was observed in MCF7-VEGFR3 cells, which suggested that an increased presence of VEGFR3 could be involved in the activation of FAK-Y925. The change in phosphorylation levels of FAK-Y861, VEGFR3, Akt, and Erk in MCF7 cells was negligible following treatment with C10, whereas a significant decrease in phosphorylation was seen starting at 10 μM of C10 in MCF7-VEGFR3 cells. In the pancreatic cancer cell lines, we found that the expression levels of FAK-Y925 and VEGFR3 were higher in MiaPaCa-2-luc cells than BxPC3 cells. C10 treatment decreased phosphorylation levels of FAK-Y861, FAK-Y925, and VEGFR3 at 1 μM in MiaPaCa-2-luc cells as compared to 60 μM in BxPC3 cells. Phosphorylation of Akt and Erk was also inhibited by C10 in MiaPaCa-2-luc cells than BxPC3 cells. These results demonstrate that the magnitude of response after C10 treatment was greater in cells expressing higher levels of FAK-Y925 and VEGFR3 along with an increased inhibition of FAK C-terminal downstream signaling.
Figure 1. Cell lines with higher FAK-Y925 and VEGFR3 levels are more sensitive to FAK inhibitor C10 in vitro.

Anti-proliferative effects of C10 in (A) isogenic and (B) pancreatic cancer cell lines. Data are represented as mean ± S.D. *p <0.05 (C) MCF7, MCF7-VEGFR3, MiaPaCa-2-luc, and BxPC3 cell lines were exposed to C10 at the indicated concentrations for 24 h. Cell lysates were immunoblotted and probed for the indicated antibodies. GAPDH serves as a loading control.
3.2 FAK inhibitor C10 decreased pancreatic cancer cell motility and invasion
Given FAK's involvement in cell motility and invasion, we first assessed the effect of C10 on cell migration, where MiaPaCa-2-luc cells were treated with increasing doses of C10 (Figure 2A). Analysis of the wound gap at the indicated concentrations, demonstrated that C10 at 5 μM and 10 μM significantly inhibited wound closure as compared to DMSO-treated control cells where the wound gap was closed after 24 h. Next, we determined the effects of C10 on cell invasion where MiaPaCa-2-luc cells were treated with C10 for 24 h at the indicated concentrations (Figure 2B). A 53 % and 24 % decrease in the number of invaded cells at 5μM and 10 μM of C10 respectively was observed in contrast to DMSO-treated control cells. Evidence suggests that the FAT domain of FAK promotes motility and invasion through interactions with paxillin and FAK-Y925 mediated Grb2/Erk/MAPK signaling [15, 27, 28, 30, 32] Because C10 inhibited motility and invasion of MiaPaCa-2-luc cells and decreased FAK-Y925 phosphorylation (Figure 1C), we tested the effects of C10 on paxillin and Grb2 phosphorylation (Figure 2C). C10 caused a dose-dependent decrease in the phosphorylation states of both these proteins. Thus, these results confirm that modulation of the C-terminal FAT domain of FAK by C10 inhibits paxillin and Grb2 phosphorylation, cell motility, and invasion.
Figure 2. FAK inhibitor C10 decreased MiaPaCa-2-luc cell motility and invasion.

(A) Wound healing migration assay of MiaPaCa-2-luc cells treated with DMSO (control) or the indicated concentrations of C10 for 24 h. Lines indicate the gap in wound area. Quantitative representation of the area closed by migrating cells. (n=3). Error bars represent mean ± S.D. *p < 0.05 (B) Representative photomicrographs of invading MiaPaCa-2-luc cells incubated with DMSO (control) or C10 at the indicated concentrations for 24 h. The invaded cells from 7–9 fields for each condition were stained and counted (n=3). Error bars represent mean ± S.D. *p < 0.05 (C) MiaPaCa-2-luc cells were treated with C10 at the indicated concentrations for 24 h. Western blot analysis of paxillin and Grb2 protein expression. GAPDH was used as a loading control.
3.3 Efficacy of FAK inhibitor C10 in pancreatic tumor growth
MiaPaCa-2-luc cells were 60 times more sensitive to C10 than BxPC3 cells in vitro (Figures 1C). To address whether C10 exerts the enhanced selectivity for FAK-Y925 and VEGFR3 in vivo, mice bearing MiaPaCa-2-luc or BxPC3 xenograft tumors were administered C10 and closely monitored for tumor growth (Figures 3A and 3D). C10 exerted a stronger tumor inhibitory effect in MiaPaCa-2-luc tumors (63%) in contrast to the relatively poor response in BxPC3 tumors (37%). Tumor weights of the C10-treated MiaPaCa-2-luc group were substantially less than vehicle-treated controls (Figure 3B). The difference between the tumor weights of the control and C10-treated groups of the BxPC3 model was borderline significant (Figure 3E). Also, no significant difference in body weights was observed between the vehicle and C10-treated arms for both MiaPaCa-2-luc and BxPC3 xenograft models at the dose evaluated (Figures 3C and 3F). From these results, we conclude that C10 sensitized cancer cells with greater levels of FAK-Y925 and VEGFR3 expression.
Figure 3. Enhanced sensitivity of MiaPaCa-2-luc tumors to treatment with FAK inhibitor C10.

Mice (n = 6-10 per group) were inoculated with MiaPaCa-2-luc or BxPC3 pancreatic cancer cells. Vehicle (PBS) or C10 at 20 mg/kg were administered intraperitoneally (IP) once daily for five days a week. (A) MiaPaCa-2-luc or (D) BxPC3 tumor volumes were measured over the course of the study and values represent mean ± S.E.M *p < 0.05. Percent reduction in tumor volume relative to the vehicle-treated group was calculated using the tumor size measurements on the last day of the study. (B) MiaPaCa-2-luc or (E) BxPC3 tumor weights were recorded at the end of the treatment protocol. Error bars represent mean ± S.D. *p <0.05. Body weights of mice bearing (C) MiaPaCa-2-luc or (F) BxPC3 tumors were analyzed to evaluate tolerability to C10-treatment. No statistically significant difference in body weights was observed between the two treatment arms.
3.4 Mechanism of anti-tumor effects mediated by FAK inhibitor C10
To examine the anti-tumor effects exerted by C10 in MiaPaCa-2-luc tumor xenografts, representative tumors from the vehicle and C10-treated arms were harvested and analyzed for proteins associated with the FAK C-terminal pathway by immunoblotting. Consistent with the in vitro findings of the MiaPaCa-2-luc cell line (Figure 1C) Western blot analysis validated a downregulation in FAK-Y861, FAK-Y925, and phosphorylated forms of VEGFR3, Akt, and Erk in the C10-treated group as compared to the vehicle-treated control group (Figure 4A). Immunohistological analysis of MiaPaCa-2-luc tumors revealed a marked decrease in tumor cell proliferation (Ki67 positive cells) and microvessel density of endothelial (CD31) and lymphatic vessels (LYVE1) in C10-treated tumors than the vehicle-treated group (Figure 4B). We also evaluated the mean vascular area and mean vessel perimeter obtained from the CD31 and LYVE1 staining analysis and found that both these parameters were significantly less in the C10-treated group than the vehicle-treated group (Figure S2). These data demonstrate that blocking FAK C-terminal scaffold signaling potentiates anti-tumor efficacy along with a significant effect on tumor endothelial and lymphatic vessels.
Figure 4. C10 treatment decreased FAK and VEGFR3 phosphorylation, FAK downstream signaling, and endothelial and lymphatic microvessel density in MiaPaCa-2-luc tumors.

(A) Protein lysates from MiaPaCa-2-luc tumors were analyzed by Western blot and probed using the indicated antibodies. GAPDH was used as a loading control. Ratio of phosphorylated protein to total protein band density normalized to the control in each case are shown as bar graphs. (B) IHC staining with Ki67, CD31, and LYVE1 antibodies of MiaPaCa-2-luc tumors treated with vehicle (PBS) or C10 (Scale bar = 100 μm). Representative fields from each treatment arm are shown. Quantitative analysis of Ki-67 positive cells and CD31 and LYVE1 microvessel density are presented as bar graphs. Error bars represent mean ± S.D. *p <0.05.
3.5 FAK inhibitor C10 exerted anti-angiogenic effects
Studies have shown that the activation of FAK-Y861[2, 9] and FAK-Y925 [27] lead to increased angiogenesis. Our results so far confirm that C10 not only downregulated FAK-Y861 and FAK-Y925 phosphorylation in vitro (Figure 1C) and in vivo (Figure 4A) but also decreased endothelial and lymphatic vessel density (Figure 4C) in a MiaPaCa-2-luc model system. This prompted us to explore the anti-angiogenic effects of C10. A significant inhibition in the ability of HUVEC's to form a capillary tube-like network was observed as early as 6 h after C10 treatment at 1 μM and at 24 h, a dose of 5 μM and 10 μM of C10 was required to achieve a similar inhibitory effect (Figure 5A). Evaluation of parameters such as total loops (Figure 5A), number of branching points, and total tube length (Figure S3) confirmed a decrease in HUVEC tube formation induced by C10 in a concentration dependent manner. We observed that C10 significantly decreased HUVEC cell motility starting at 1 μM as compared to DMSO or FGF2-treated controls (Figure 5B). We also assessed the impact of C10 on angiogenesis in vivo by using the DIVAA™ assay (Figure 5C). Because Avastin (Bevacizumab) is an anti-angiogenic drug in widespread clinical use, we used it as a negative control in this assay. Although C10 at 10 mg/kg decreased endothelial cell proliferation than DMSO, this difference was not statistically significant. However, C10 at 20 mg/kg dramatically reduced the ability of vascular endothelial cells to migrate and proliferate into the angioreactors as compared to the DMSO control.
Figure 5. Anti-angiogenic activity of FAK inhibitor C10.

(A) HUVEC cells were plated on basement membrane extract coated wells and treated with DMSO (control) or C10 at the indicated concentrations. Capillary tube formation was then assessed at 6 h and 24 h and the number of loops were analyzed from 7–10 fields for each condition. Representative images from three independent experiments are shown. Error bars represent mean ± S.D. *p <0.05. (B) Transwell plates were used to determine HUVEC cell motility following DMSO (control) or C10 treatment at the indicated concentrations for 24 h. FGF2 was included as a positive control. Migrated cells were stained followed by dye extraction and the absorbance (590 nm) was measured for each condition. Data are presented as mean ± S.D. of three experiments. *p <0.05. (C) The effect of C10 on in vivo angiogenesis was evaluated using the directed in vivo angiogenesis assay (DIVAA). Angioreactors (n=4) were prepared and implanted as described in the Materials and methods. Avastin was included as a negative control. The extent of endothelial cell invasion in the angioreactors for each condition was determined by fluorescence quantitation of FITC-Lectin and is reported as relative fluorescent units (R.F.U). Results are shown as mean ± S.D. *p <0.05.
3.6 Treatment with FAK inhibitor C10 decreased interstitial fluid pressure
Finally, because pancreatic cancers are known to have the desmoplastic reaction that creates a barrier to effective drug delivery [25] we sought to determine whether C10 had an effect on interstitial fluid pressure. We measured IFP in MiaPaCa-2-luc tumor bearing mice after 14 doses of C10 at 20 mg/kg (Figure 6). Following C10 treatment, the IFP levels dropped significantly as compared to the vehicle-treated control group. This correlates approximately with the 17 day period by when the mice received 12 doses of C10 after which a clear demarcation in the tumor volume curves was seen between the C10 and vehicle treatment arms (Figure 3A). Together, these findings suggest that by affecting the FAK C-terminal scaffold function, C10 inhibits pancreatic tumor growth by reducing the IFP within the tumor, thereby increasing access to the tumor bed to achieve increased drug accumulation.
Figure 6. FAK inhibitor C10 decreased interstitial fluid pressure.
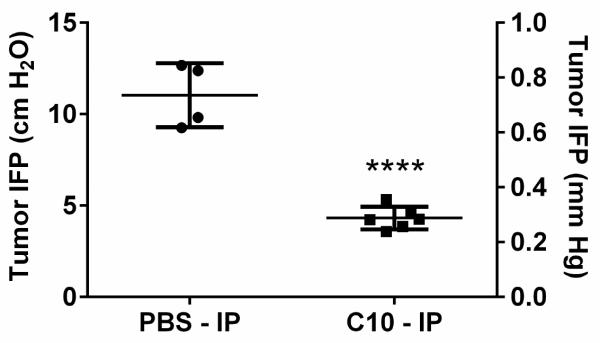
Mice bearing MiaPaCa-2-luc tumors were treated with vehicle (PBS) (n=4) or 14 doses of C10 (n=6), after which the IFP was measured. Each value represents the average of multiple IFP measurements in a single tumor. IFP measurements are represented as both cm H2O and mm Hg. ****p <0.0001 for difference between vehicle (mean = 11.03 cm H2O) and C10-treated (mean = 4.3 cm H2O) groups with one way ANOVA and Dunnett's multiple comparison test.
4. DISCUSSION
The involvement of FAK in cancer has been extensively studied over the past 20 years and various approaches to target FAK have been explored with success in research but limited from a clinical standpoint. In this study, we have broadened the scope of FAK as a molecular target by limiting the activity of FAK's scaffolding function, specifically in this case the C-terminal domain. Using isogenic cell lines and pancreatic cancer cells, we confirmed that FAK inhibitor C10 specifically inhibited survival of cells with higher expression levels of FAK-Y925 and VEGFR3. We then showed that C10 decreased cellular migration and invasion with a decrease in phosphorylated forms of paxillin and Grb2. Our data demonstrated that C10 preferentially decreased tumor burden in cells with higher levels of FAK-Y925 and VEGFR3 in contrast to cells with low levels of these proteins. This suggested that in addition to FAK, the expression of FAK-Y925 and VEGF3 might be important biomarkers to select tumors that are sensitive to FAK C-terminal scaffold inhibition. The mechanism through which C10 achieved anti-tumor effects was by downregulating key tyrosine residues located within the C-terminal domain of FAK and related downstream signaling proteins. Furthermore, we found a significant decrease in tumor cell proliferation and endothelial and lymphatic vessel density following C10 treatment. FAK has been shown to be critical for angiogenesis and reports indicate that FAK promotes tumor angiogenesis [39]. Also, tyrosine residue 925 located within the FAT domain of FAK has been implicated as an angiogenic switch in tumor cells [27]. Hence, we tested the effect of C10 in the context of angiogenesis and found that indeed, the FAK scaffold inhibitor affected endothelial cell function. The results demonstrated that the FAK scaffold inhibitor specifically targeted and inhibited FAK C-terminal signaling and functions to induce anti-tumor and anti-angiogenic effects.
The Achilles' heel of pancreatic cancers is the presence of desmoplastic stroma and sparse vasculature causing a high interstitial fluid pressure within the tumor that in turn hinders drug delivery [18]. Although pancreatic adenocarcinomas are extremely hypovascular, some functional blood vessels do exist as evidenced by the successful delivery of nanoparticle drug formulations such as nab-paclitaxel and PEGPH20 [29, 40]. In fact, recent findings from ongoing studies in our laboratory show that administration of nanoparticles bound to a cyanine dye selectively accumulated in MiaPaCa-2-luc xenograft tumors as compared to the liver and spleen (data not shown). Here, we have shown that the interstitial fluid pressure decreased in C10-treated tumors. Our next steps are to formulate C10-nanoparticles to leverage the accessibility to the pancreatic cancer cell niche.
In conclusion, targeting the FAK scaffold represents a novel approach towards the development of FAK specific molecular therapeutics. Although this approach is in its preliminary stages, it is clear that the role of FAK in cancer is extremely complex and thus, we cannot simply target the ATP-binding site of the FAK enzyme in isolation. Further studies in additional tumor models are required to elucidate the role and extent of FAK scaffold inhibition. In this report, we have shown that a FAK C-terminal scaffold inhibitor successfully abrogated tumor growth and angiogenesis and have also identified potential biomarkers of sensitivity to FAK C-terminal scaffold inhibition. These results aid the design of future FAK C-terminal scaffold inhibitors and highlight the untapped potential of FAK scaffold inhibition.
Supplementary Material
Highlights.
C-terminal FAK scaffold inhibitor C10 selectively targets cancer cells with high FAK-Y925 and VEGFR3 expression levels.
FAK inhibitor C10 inhibited FAK and VEGFR3 signaling and decreased cancer cell motility and invasion.
FAK inhibitor C10 exerts anti-tumor and anti-angiogenic effects.
Treatment with FAK inhibitor C10 decreased interstitial fluid pressure.
Acknowledgements
We thank Dr. Barbara Foster at RPCI for the initial review of the manuscript. We thank Ellen Karasik for technical assistance with immunohistochemistry at Dr. Barbara Foster's laboratory and Dr. Amber Worral and Dr. Wiam Bshara at the Pathology Core at RPCI for their expertise and input regarding the scoring algorithms used for tumor histology analysis.
This work was supported by the National Cancer Institute grant RO1-CA65910 (W.G.C), Photolitec Grant (R.K.P), and Roswell Park Cancer Institute. This work utilized core resources supported by the NCI Cancer Center Support Grant CA016156 (Trump, DL).
Abbreviations
- (FERM)
Four-point-one, ezrin, radixin, moesin
- (FAT)
focal adhesion targeting
- (FGF2)
fibroblast growth factor 2
- (PEGPH20)
PEGylated form of rHuPH20
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest William G. Cance and Elena V. Kurenova are stockholders and founders of CureFAKtor Pharmaceuticals, LLC. University of Florida and Roswell Park Cancer Institute have patents pending and awarded that are based on their work on developing FAK inhibitors.
References
- [1].Abbi S, Guan JL. Focal adhesion kinase: protein interactions and cellular functions. Histol Histopathol. 2002;17:1163–1171. doi: 10.14670/HH-17.1163. [DOI] [PubMed] [Google Scholar]
- [2].Abu-Ghazaleh R, Kabir J, Jia H, Lobo M, Zachary I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem J. 2001;360:255–264. doi: 10.1042/0264-6021:3600255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brunton VG, Frame MC. Src and focal adhesion kinase as therapeutic targets in cancer. Current Opinion in Pharmacology. 2008;8:427–432. doi: 10.1016/j.coph.2008.06.012. [DOI] [PubMed] [Google Scholar]
- [4].Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Science signaling. 2013;6:pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Y-Y, Wang Z-X, Chang P-A, Li J-J, Pan F, Yang L, Bian Z-H, Zou L, He J-M, Liang H-J. Knockdown of focal adhesion kinase reverses colon carcinoma multicellular resistance. Cancer Science. 2009;100:1708–1713. doi: 10.1111/j.1349-7006.2009.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, Takeda K, Ronde P. FAK phosphorylation at Tyr925 regulates crosstalk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell. 2011 doi: 10.1091/mbc.E10-08-0725. mbc.E10-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochem Biophys Res Commun. 2003;311:786–792. doi: 10.1016/j.bbrc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- [8].Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery. 2004;135:555–562. doi: 10.1016/j.surg.2003.10.017. [DOI] [PubMed] [Google Scholar]
- [9].Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA. Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol. 2002;157:149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fiedorek FT, Jr., Kay ES. Mapping of the focal adhesion kinase (Fadk) gene to mouse chromosome 15 and human chromosome 8. Mammalian Genome. 1995;6:123–126. doi: 10.1007/BF00303256. [DOI] [PubMed] [Google Scholar]
- [11].Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11:802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- [12].Furuyama K, Doi R, Mori T, Toyoda E, Ito D, Kami K, Koizumi M, Kida A, Kawaguchi Y, Fujimoto K. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J Surg. 2006;30:219–226. doi: 10.1007/s00268-005-0165-z. [DOI] [PubMed] [Google Scholar]
- [13].Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22:359–374. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- [14].Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66:1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- [15].Giubellino A, Burke TR, Jr., Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets. 2008;12:1021–1033. doi: 10.1517/14728222.12.8.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gogate PN, Ethirajan M, Kurenova EV, Magis AT, Pandey RK, Cance WG. Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction. Eur J Med Chem. 2014;80C:154–166. doi: 10.1016/j.ejmech.2014.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G, Sood AK. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–10983. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- [18].Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- [19].Kaneda T, Sonoda Y, Ando K, Suzuki T, Sasaki Y, Oshio T, Tago M, Kasahara T. Mutation of Y925F in focal adhesion kinase (FAK) suppresses melanoma cell proliferation and metastasis. Cancer Lett. 2008;270:354–361. doi: 10.1016/j.canlet.2008.05.042. [DOI] [PubMed] [Google Scholar]
- [20].Kurenova E, Liao J, He DH, Hunt D, Yemma M, Bshara W, Seshadri M, Cance WG. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget. 2013;4:1632–1646. doi: 10.18632/oncotarget.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kurenova EV, Hunt DL, He D, Fu AD, Massoll NA, Golubovskaya VM, Garces CA, Cance WG. Vascular endothelial growth factor receptor-3 promotes breast cancer cell proliferation, motility and survival in vitro and tumor formation in vivo. Cell Cycle. 2009;8 doi: 10.4161/cc.8.14.9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lechertier T, Hodivala-Dilke K. Focal adhesion kinase and tumour angiogenesis. The Journal of Pathology. 2011;226:404–412. doi: 10.1002/path.3018. [DOI] [PubMed] [Google Scholar]
- [23].Liu G, Guibao CD, Zheng J. Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol Cell Biol. 2002;22:2751–2760. doi: 10.1128/MCB.22.8.2751-2760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mahlamaki EH, Barlund M, Tanner M, Gorunova L, Hoglund M, Karhu R, Kallioniemi A. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosomes Cancer. 2002;35:353–358. doi: 10.1002/gcc.10122. [DOI] [PubMed] [Google Scholar]
- [25].McCarroll J, Teo J, Boyer C, Goldstein D, Kavallaris M, Phillips PA. Potential applications of nanotechnology for the diagnosis and treatment of pancreatic cancer. Frontiers in physiology. 2014;5:2. doi: 10.3389/fphys.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- [27].Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, Cheresh DA, Schlaepfer DD. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006 doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- [28].Petit V, Boyer B, Lentz D, Turner CE, Thiery JP, Valles AM. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J Cell Biol. 2000;148:957–970. doi: 10.1083/jcb.148.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Prutzman KC, Gao G, King ML, Iyer VV, Mueller GA, Schaller MD, Campbell SL. The focal adhesion targeting domain of focal adhesion kinase contains a hinge region that modulates tyrosine 926 phosphorylation. Structure (Camb) 2004;12:881–891. doi: 10.1016/j.str.2004.02.028. [DOI] [PubMed] [Google Scholar]
- [31].Sawai H, Okada Y, Funahashi H, Matsuo Y, Takahashi H, Takeyama H, Manabe T. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Molecular Cancer. 2005;4:37. doi: 10.1186/1476-4598-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scheswohl DM, Harrell JR, Rajfur Z, Gao G, Campbell SL, Schaller MD. Multiple paxillin binding sites regulate FAK function. Journal of molecular signaling. 2008;3:1. doi: 10.1186/1750-2187-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Molecular & Cellular Biology. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schneider M, Buchler P, Giese N, Giese T, Wilting J, Buchler MW, Friess H. Role of lymphangiogenesis and lymphangiogenic factors during pancreatic cancer progression and lymphatic spread. Int J Oncol. 2006;28:883–890. [PubMed] [Google Scholar]
- [35].Schultze A, Fiedler W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents Med Chem. 2011;11:593–599. doi: 10.2174/187152011796817727. [DOI] [PubMed] [Google Scholar]
- [36].Sen A, Capitano ML, Spernyak JA, Schueckler JT, Thomas S, Singh AK, Evans SS, Hylander BL, Repasky EA. Mild elevation of body temperature reduces tumor interstitial fluid pressure and hypoxia and enhances efficacy of radiotherapy in murine tumor models. Cancer Res. 2011;71:3872–3880. doi: 10.1158/0008-5472.CAN-10-4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang X, Cance WG. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005;15:357–362. doi: 10.1097/00008390-200510000-00003. [DOI] [PubMed] [Google Scholar]
- [38].Stokes JB, Adair SJ, Slack-Davis J, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, Stelow EB, Parsons JT, Bauer TW. Inhibition of Focal Adhesion Kinase by PF-562,271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant with Altering the Tumor Microenvironment. Molecular Cancer Therapeutics. 2011 doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tavora B, Batista S, Reynolds LE, Jadeja S, Robinson S, Kostourou V, Hart I, Fruttiger M, Parsons M, Hodivala-Dilke KM. Endothelial FAK is required for tumour angiogenesis. EMBO molecular medicine. 2010;2:516–528. doi: 10.1002/emmm.201000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zachary I, Rozengurt E. Focal adhesion kinase (p125FAK): a point of convergence in the action of neuropeptides, integrins, and oncogenes. Cell. 1992;71:891–894. doi: 10.1016/0092-8674(92)90385-p. [DOI] [PubMed] [Google Scholar]
- [42].Zhao J, Guan J-L. Signal transduction by focal adhesion kinase in cancer. Cancer and Metastasis Reviews. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.