

Abstract
The enhanced cardiac sympathetic afferent reflex (CSAR) contributes to the exaggerated sympatho-excitation in chronic heart failure (CHF). Increased sympatho-excitation is positively related to mortality in CHF patients. However, the potential beneficial effects of chronic CSAR deletion on cardiac and autonomic function in CHF have not been previously explored. Here we determined the effects of chronic CSAR deletion on cardiac remodeling and autonomic dysfunction in CHF. In order to selectively delete the transient receptor potential vanilloid 1 receptor (TRPV1) -expressing CSAR afferents, epicardial application of resiniferatoxin (RTX, 50 μg/ml), an ultrapotent analogue of capsaicin, was performed during myocardium infarction (MI) surgery in rats. This procedure largely abolished the enhanced CSAR, prevented the exaggerated renal and cardiac sympathetic nerve activity and improved baroreflex sensitivity in CHF rats. Most importantly, we found that epicardial application of RTX largely prevented the elevated LVEDP, lung edema and cardiac hypertrophy, partially reduced left ventricular dimensions in the failing heart and increased cardiac contractile reserve in response to β-adrenergic receptor stimulation with isoproterenol in CHF rats. Molecular evidence showed that RTX attenuated cardiac fibrosis and apoptosis and reduced expression of fibrotic markers and TGF β-receptor I in CHF rats. Pressure - volume loop analysis showed that RTX reduced the end diastolic pressure volume relations in CHF rats indicating improved cardiac compliance. In summary, cardiac sympathetic afferent deletion exhibits protective effects against deleterious cardiac remodeling and autonomic dysfunction in CHF. These data suggest a potential new paradigm and therapeutic potential in the management of CHF.
Keywords: cardiac afferents, cardiac sympathetic nerve activity, heart failure, remodeling
INTRODUCTION
Chronic heart failure (CHF) is a serious and debilitating condition with poor survival rates and an increasing level of prevalence. The exaggerated sympatho-excitation that is a hallmark of CHF is a critical factor in the development and progression of the CHF state. Sympatho-excitation targets multiple organs and causes long-term effects that contribute to disease progression. Therapeutic targeting to block excessive sympathetic activation in CHF has become an accepted modality. The mechanism(s) by which sympathetic excitation occurs in the CHF state are not completely understood. Previous studies from this laboratory have shown that the enhanced cardiac sympathetic afferent reflex (CSAR), a sympatho-excitatory reflex originating in the heart, is a critical contributor to the elevated sympathetic tone in CHF1, 2.
It is well known that myocardial ischemia causes the production and release of several metabolites including bradykinin, prostaglandins and protons that stimulate sympathetic afferent nerve endings leading to increases in arterial pressure (AP), heart rate (HR) and sympathetic nerve activity1, 3, 4. In CHF, the CSAR is sensitized and cardiac sympathetic afferents are tonically activated1-3. Recent evidence indicates that transient receptor potential vanilloid 1 (TRPV1)-expressing cardiac afferent fibers are necessary for sensing and triggering the activation of the CSAR and that desensitization of these afferents by systemic administration of resiniferatoxin (RTX), an ultrapotent analogue of capsaicin capable of inducing rapid degeneration of TRPV1-expressing afferent neurons and fibers5-7, can almost completely abolish CSAR activation in adult rats5. Given that a strategy for abolishing the enhanced CSAR-induced sympatho-excitation may be beneficial for preventing the deleterious progression of CHF, we performed epicardial application of RTX at the time of myocardium infarction (MI) surgery in rats in order to 1) investigate potential beneficial effects of selective deletion of TRPV1-expressing CSAR afferents on autonomic dysfunction and 2) determine whether the CSAR control of sympathetic nerve activity (especially cardiac sympathetic nerve activity; CSNA) plays a critical role in deleterious cardiac remodeling in CHF. Furthermore, we investigated the effects of CSAR afferent desensitization on cardiac morphological and hemodynamic function in rats with CHF.
METHODS
Experiments were performed on male Sprague-Dawley rats weighing ~400 to ~500 g. An experimental timeline is shown in Figure S1. In brief, terminal experiments were carried out 9-11 weeks after MI in rats treated with either RTX or vehicle. These experiments were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center and carried out under the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Model of CHF and cardiac sympathetic afferent desensitization
Heart failure was produced by coronary artery ligation as previously described8, 9. Cardiac function in all experimental animals was measured 2 and 6 weeks after MI by echocardiography (VEVO 770, Visual Sonics, Inc.) as previously described8-10. In order to desensitize cardiac sympatheitc afferents, resiniferatoxin (RTX, 1 mg; Sigma Aldrich) was dissolved in 2 ml of ethanol and mixed with 18 ml of Tween in isotonic saline. RTX (50 ug/ml) was painted twice on the entire left and right ventricles with a small brush just prior to coronary ligation. The dose of RTX was determined by our preliminary experiments in which we found nearly complete ablation of TRPV1-expressing cardiac nerve endings on the surface of the rat heart for more than 10 weeks (Figure S2).
General Surgical Preparation for Acute Experiments
At the acute terminal experiments that were performed 9-11 weeks post-MI, rats were anesthetized with urethane (800 mg/kg ip) and α-chloralose (40 mg/kg ip). Renal sympathetic nerve activity (RSNA) and cardiac sympathetic nerve activity (CSNA) were recorded for evaluating basal renal and cardiac sympathetic tones. A detailed surgical description is available in the online-only Data Supplement.
At the end of the experiment, the rat was euthanized with an overdose of pentobarbital sodium. The maximum cardiac or renal nerve activity (Max) occurred 1-2 min after the rat was euthanized as previously described8. The background noise for sympathetic nerve activity was recorded 15-20 min after the rat was euthanized. Using the unit conversion function of Powerlab Chart system, baseline nerve activity was determined as a percent of Max after the noise level was subtracted.
Activation of the CSAR
Epicardial application of capsaicin and bradykinin was used to effectively stimulate the cardiac sympathetic afferents5. The time interval between capsaicin and bradykinin was about 15-20 min to allow the AP, HR and RSNA to return to, and stabilize at their control levels.
Construction of Arterial Baroreflex Curves
Baroreflex curves were generated by measuring the HR and RSNA responses to decreases and increases in AP by intravenous administration of nitroglycerin (25 μg) followed by phenylephrine (10 μg) as previously described2.
Measurement of Left Ventricular Performance
Left ventricular (LV) function was assessed using a Millar PV loop System (Millar Instruments, Houston, Tex, USA). A detailed method description is available in the online-only Data Supplement.
Histopathology
The heart was fixed in 10% phosphate buffered formalin, embedded in paraffin, and cut into 10 μm serial sections from apex to base. Picrosirius red staining was performed to detect myocardial fibrosis. A detailed method description is available in the online-only Data Supplement.
Immunohistochemistry staining of cleaved caspase 3 (CC3) was also performed in paraffin sections from the same heart tissues which were used for picrosirius red staining experiments. A detailed method description is available in the online-only Data Supplement.
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling staining
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining and analysis was performed to evaluate the effect of RTX on cardiac apoptosis. A detailed method description is available in the online-only Data Supplement.
Western Blot Analysis
Protein expressions of alpha smooth muscle actin (α-SMA), fibronectin, Transforming growth factor beta (TGF-β) receptor 1, cleaved caspase 3, caspase 3 and beta 1-adrenergic receptors (β1-AR) in the heart from sham+vehicle, sham+RTX, CHF+vehicle and CHF+RTX rats were assessed by western blot technique. A detailed method description is available in the online-only Data Supplement.
Immunofluorescence labeling of TRPV1 receptors in cardiac nerve endings
To determine the dose-dependent (0.5 μg/ml, 5 μg/ml and 50 μg/ml) and time course (1 week, 4-6 weeks and 9-11 weeks after RTX) effects of RTX on cardiac TRPV1-expressing nerve terminals in the heart, immunofluorescence labeling experiments were conducted in the myocardium obtained from 4 vehicle-treated and 20 RTX-treated rats (each dose or time point includes 4 rats). A detailed method description is available in the online-only Data Supplement.
Statistical Analysis
All values are expressed as mean ± standard error of the mean (SE). Differences between groups were determined by a two-way ANOVA followed by the Tukey post hoc test. P <0.05 was considered statistically significant.
RESULTS
RTX causes loss of TRPV1-expressing cardiac afferents
We examined TRPV1 protein expression in cardiac nerve endings from vehicle and RTX-treated rats using immunofluorescence. In vehicle-treated rats, TRPV1 immunoreactivity was primarily located to the epicardial surface and co-expressed with PGP 9.5 (a nerve terminal marker) (Figure S2A). In RTX-treated rats, TRPV1 immunoreactivity was absent in the epicardium when it was measured 1 week, 4-6 weeks and 9-11 weeks after epicardial application of RTX (50 μg/ml), respectively (Figure S2A), suggesting effective deletion of TRPV1-expressing cardiac afferents during these periods. In another parallel set of experiments, we further confirmed the efficiency of RTX-induced TRPV1-expressing CSAR afferent ablation functionally by epicardial application of capsaicin and bradykinin (10 μg/ml) in sham+RTX rats treated with various doses (0.5 μg/ml, 5 μg/ml and 50 μg/ml) and at various time points (1 week, 4-6 weeks, 9-11 weeks and 6 months after RTX). These data show that both epicardial application of 5 μg/ml and 50 μg/ml RTX largely abolished the cardiovascular responses to acute epicardial application of capsaicin and bradykinin 1 week after RTX in sham rats (Figure S2B). However, in the preliminary experiment, we observed that 40% of sham rats that were treated with epicardial application of RTX at a dose of 5 μg/ml showed partial recovery of the cardiovascular responses to acute epicardial application of capsaicin and bradykinin 10 weeks after RTX whereas all rats treated with 50 μg/ml RTX were still insensitive to capsaicin and bradykinin at this time point. In order to achieve a long-lasting CSAR ablation we chose to use 50 μg/ml RTX in all following experiments. Data from our time course experiments demonstrated that epicardial application of this dose of RTX abolished the cardiovascular responses to acute epicardial application of capsaicin and bradykinin for more than 10 weeks whereas these cardiovascular responses to capsaicin and bradykinin were largely recovered at 6 months after RTX (Figure S2C).
Functional experiments comparing RTX-treated sham and CHF rats 9-11 weeks after RTX were carried out. As shown in Figure 1A-1C, compared to sham+vehicle rats, CHF+vehicle rats exhibit exaggerated MAP, HR and renal sympathetic nerve activity (RSNA) responses to epicardial application of either capsaicin or bradykinin. However, the cardiovascular and sympathetic activation in response to epicardial capsaicin in both sham and CHF rats were almost completely abolished by epicardial pretreatment with RTX. RTX also greatly attenuated the bradykinin-induced CSAR activation in both sham and CHF rats.
Figure 1.
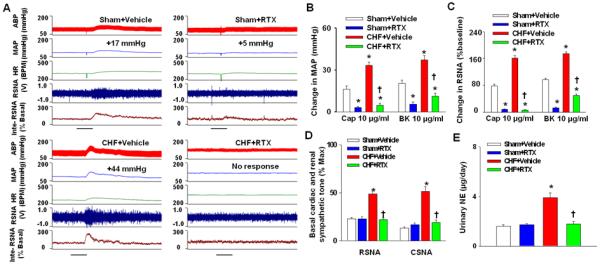
A-C, original tracing (A) and mean data showing the cardiovascular (B) and sympathetic (C) responses to epicardial application of either capsaicin or bradykinin (10μg/ml) in sham+vehicle, sham+RTX, CHF+Vehicle and CHF+RTX rats (n=8-12/each group). Scale bar in A represents 30 seconds. D and E, basal cardiac (n=6-8/each group) and renal sympathetic nerve activity (n=8-12/each group) and urinary NE secretion (E, n=6/each group) in sham+vehicle, sham+RTX, CHF+Vehicle and CHF+RTX rats. Values are mean ± SE. *P<0.05 vs. sham+vehicle; †, P<0.05 vs. CHF+vehicle.
Effect of RTX on body weight, organ weight, and hemodynamics in sham and CHF rats
Morphological and hemodynamic measurements of all groups of rats are summarized in Table 1. The heart weight and lung weight-to-body weight ratios were significantly higher in CHF+vehicle rats compared to sham+vehicle rats, suggesting cardiac hypertrophy and pulmonary congestion in the CHF state. However, after pretreatment with epicardial RTX, there was a significant decrease in both the heart weight and lung weight-to-body weight ratios in CHF rats compared to CHF+vehicle rats, suggesting a protective effect of RTX on cardiac hypertrophy and pulmonary congestion. Importantly, there was no significant difference in infarct size between CHF+vehicle and CHF+RTX rats (Table 1 and Figure S3), which excludes the possibility that the variability in infarct size caused the difference in the heart weight or lung weight between CHF+vehicle and CHF+RTX rats.
Table 1.
Hemodynamic and morphological data in sham and CHF rats following vehicle or RTX treatment
| Parameters | Sham+Vehicle (n=21) |
Sham + RTX (n=20) |
CHF+Vehicle (n=23) |
CHF + RTX (n=25) |
|---|---|---|---|---|
| Body weight, g | 429 ± 7 | 430 ± 7 | 452 ±8 | 440 ± 8 |
| Heart weight, mg | 1438±28 | 1430 ± 30 | 2239± 61* | 1650± 49*† |
| HW/BW, mg/g | 3.4± 0.1 | 3.3 ± 0.1 | 5.0 ± 0.1 * | 3.8 ± 0.1*† |
| WLW/BW, mg/g | 4.4± 0.1 | 4.5 ± 0.1 | 8.7 ± 0.3 * | 5.1 ± 0.2*† |
| SAP, mmHg | 127.9 ± 2.2 | 128.8 ± 2.8 | 115.3± 2.0* | 120.8 ± 2.3* |
| MAP, mmHg | 103.5 ± 2.4 | 105.0 ± 3.1 | 96.7 ± 2.0* | 101.3 ± 2.1 |
| DAP, mmHg | 91.6 ± 2.5 | 92.8 ± 3.2 | 87.3 ± 2.2 | 90.8 ± 2.2 |
| LVEDP mmHg | 5.0± 0.4 | 4.8± 0.4 | 21.3± 1.0* | 8.3± 0.7*† |
| HR, bpm | 357.3 ± 6.1 | 362.0 ±6.8 | 368.9 ± 5.1 | 348.3± 5.5f |
| dp/dtmax | 9108±324 | 8601±224 | 5137± 180 * | 5446±173* |
| dp/dtmin | -8458±235 | -8088±196 | -3452±113* | -4643±149*† |
| Infarct size, % | 0 | 0 | 42.5± 1.4 * | 39.1 ± 1.2 * |
Values are mean ± SE. BW, body weight; HW, heart weight; WLW, wet lung weight; LVEDP, left ventricle end-diastolic pressure; SAP, systolic arterial pressure; MAP, mean arterial pressure; DAP, diastolic arterial pressure; HR, heart rate.
P<0.05 vs. sham+Vehicle.
P<0.05 vs. CHF+ Vehicle.
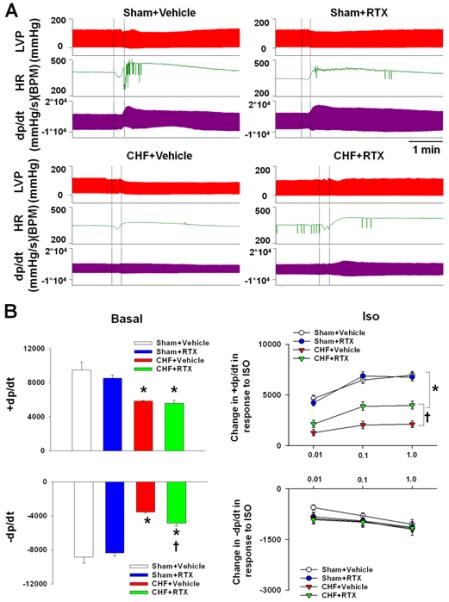
Hemodynamic and echocardiographic measurements point to several other important differences between CHF+vehicle and CHF+RTX rats. CHF+vehicle rats exhibited elevated LVEDP and time-dependent decline in ejection fraction and fractional shortening compared with sham+vehicle rats (Tables 1 and S1), indicating decreased ventricular function. However, the increased LVEDP observed in CHF+vehicle rats was largely prevented in CHF+RTX rats, which is consistent with the finding that lung congestion was also reduced in CHF+RTX rats. MI-induced cardiac dilation in CHF rats, indicated by increased LV systolic and diastolic diameters and volumes, was significantly reduced by RTX treatment compared to sham rats (Table S1). Interestingly, although LVEDP and cardiac dilation were reduced by RTX, there was no significant difference in ejection fraction or fractional shortening 2 weeks and 6 weeks after MI between CHF+RTX and CHF+vehicle rats (Table S1). Dp/dt measurements (Table 1 and Figure 2) further show differences between CHF+vehicle and CHF+RTX rats. Compared to sham+vehicle rats, CHF+vehicle rats exhibited decreased dp/dtmax and dp/dtmin, suggesting both systolic and diastolic dysfunction in the hearts of CHF rats (Table 1). However, the blunted dp/dtmin in CHF+vehicle rats was significantly increased in CHF+RTX rats, potentially indicating improved diastolic function in CHF+RTX rats. Although there was no significant difference in dp/dtmax at rest between CHF+RTX and CHF+vehicle rats, the dp/dtmax response to isoproterenol (ISO) stimulation in CHF+RTX rats was significantly greater compared to CHF+vehicle rats (Figure 2). At the molecular level, the decreased protein expression of β1-AR in myocardium of CHF+vehicle rats was partially reversed in CHF+RTX rats (Figure 3D). There were no significant differences in cardiac morphological, echocardiographic or hemodynamic parameters between sham+vehicle and sham+RTX rats.
Figure 2.
Representative tracings (A) and mean data (B) showing basal dp/dtmax and diastolic dp/dtmin as well as the dose-dependent responses to beta-adrenergic receptor stimulation with ISO (0.01, 0.1 and 1.0 μg/kg, 0.2 ml). Values are mean ± SE. n=6/each group. *P<0.05 vs. sham+vehicle; †, P<0.05 vs. CHF+vehicle.
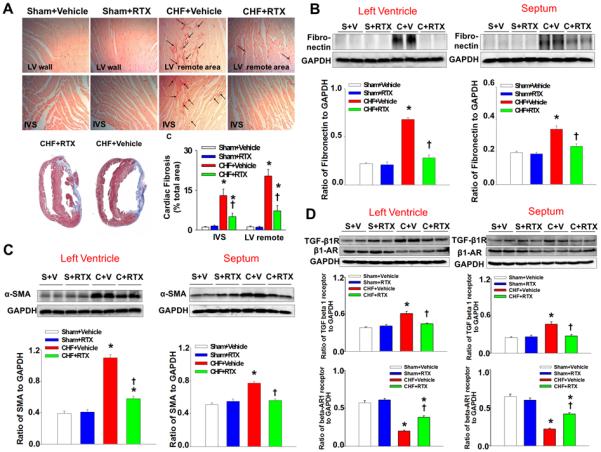
Figure 3.
A, representative sirus red stain (top) and mean data (bottom right) showing that RTX reduces cardiac fibrosis induced by myocardium infarction (MI); bottom left, Masson’s trichrome staining showing similar infarcted size between CHF+Vehicle and CHF+RTX rats in top panel. Black arrows point to the sirius red-positive staining. B-D, western blotting data showing the protein expression of α-smooth muscle actin (α-SMA), fibronectin, transforming growth factor (TGF) beta 1 and β1-adrenergic receptors (β1-AR) in the left ventricle (remote region) and septum in sham+Vehicle, sham+RTX, CHF+Vehicle and CHF+RTX rats. Data are expressed as mean±SE. n=6/each group. *, P<0.05 vs. sham+vehicle, †, P<0.05 vs. CHF+vehicle.
Effects of RTX on cardiac fibrosis in sham and CHF rats
Considering that the RTX-mediated reduction in LVEDP in CHF rats may be related to an improvement in cardiac compliance, we further evaluated cardiac fibrosis using picrosirus red staining and protein expression of two fibrotic markers (α smooth muscle actin and fibronectin) as well as their upstream activator, transforming growth factor (TGF) β receptor I in the non-infarcted LV and inter-ventricular septum (IVS) walls from sham and CHF rats treated with either vehicle or RTX. These data show that picrosirus red staining was increased in CHF + vehicle treated rats compared to sham rats (Figure 3A). Alpha smooth muscle actin, fibronectin and TGF β receptor I were increased in CHF + vehicle rats (Figure 3B-3D). RTX treatment in CHF rats reduced the expression of all these markers in the non-infarcted LV and IVS walls (Figure 3). RTX did not cause any significant change in either picrosirus red staining or protein expressions of fibrotic markers and TGF β receptor I in non-infarcted LV and IVS walls in sham rats (Figure 3).
Effects of RTX on basal sympathetic tone and arterial baroreflex function in sham and CHF rats
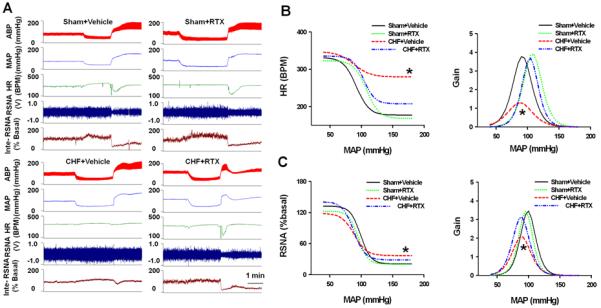
Baseline RSNA and CSNA were significantly higher in CHF+vehicle rats compared to sham+vehicle rats, indicating increased renal and cardiac sympathetic tone in CHF. RTX lowered basal RSNA and CSNA in CHF rats (Figure 1D, Figure S4 and Figure S5). Furthermore, urinary NE excretion was significantly reduced in CHF+RTX rats compared to vehicle treated rats (Figure 1E). In addition, RTX significantly increased the average slope and Gainmax, and decreased the minimum HR and RSNA of the arterial baroreflex control of both HR and RSNA (Figure 4 and Table S2). In contrast to the CHF rats, there were no significant effects on the average slope, Gainmax, and minimum RSNA or HR of arterial baroreflex control in sham rats (Figure 4 and Table S2).
Figure 4.
Original tracing (A) and mean data (B) showing baroreflex curves and gain in sham+vehicle, sham+RTX, CHF+Vehicle and CHF+RTX rats. Data are expressed as mean±SE. n=8-10/each group. * P<0.05 vs. sham+vehicle and sham+RTX and CHF+RTX.
In vivo PV loop analysis in sham and CHF rats treated with vehicle or RTX
Using left ventricular PV loop analysis (Figure 5 and Table S3), basal load-independent contractile function, as measured by the slope of the PRSW relationship and ESPVR, was significantly reduced in CHF+vehicle rats compared to sham+vehicle rats. Load sensitive measures of systolic and diastolic function such as the maximum and minimum rates of pressure change (dP/dtmax and dP/dtmin) and ejection fraction were also significantly lower in CHF+vehicle compared to sham+vehicle rats (Table S3). Furthermore, compared to sham+vehicle rats, higher Tau and LVEDP were found in CHF+vehicle rats, indicating compromised LV active and passive diastolic function. In addition, compared to sham+vehicle rats, CHF+vehicle rats presented with significant LV chamber enlargement, as shown by increased end systolic and diastolic volume (Table S3). These data are consistent with the data obtained by echocardiographic measurements (Table S2). The EDPVR was also significantly elevated in CHF+vehicle rats compared to sham+vehicle rats, suggesting increased ventricular stiffness in CHF (Figure 5 and Table S3). Compared to the CHF+vehicle rats, structural and functional abnormalities were ameliorated by RTX, as shown by a reduction in LV chamber size, increased dP/dtmin, reduced Tau and LVEDP as well as reduced stiffness as shown by the EDPVR (Figure 5 and Table S3). Interestingly, RTX treatment did not have a significant effect on basal contractile function such as dP/dtmax, PRSW and ESPVR in CHF rats. In addition, RTX had no effect on contractile and relaxation parameters in sham rats.
Figure 5.
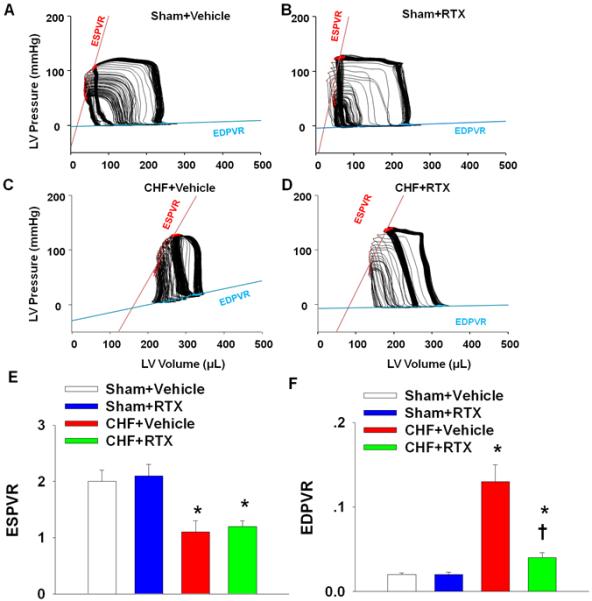
A-D, original 10-sec recordings of steady state P-V loops obtained with a Millar P-V conductance catheter system from sham+vehicle, sham+RTX, CHF+vehicle and CHF+RTX rats. E and F, mean data showing the effect of RTX on the systolic (ESPVR, E) and diastolic (EDPVR, F) function that are independent of systemic vascular resistance in sham and CHF rats. Data are expressed as mean±SE. n=7-8/each group. *, P<0.05 vs. sham+vehicle, †, P<0.05 vs. CHF+vehicle.
Effects of RTX on cardiac apoptosis in sham and CHF rats
Cardiac remodeling includes hypertrophy, fibrosis and apoptosis. Based on the beneficial effects of RTX on cardiac hypertrophy and cardiac fibrosis in CHF rats, we further investigated the effect of RTX on cardiac apoptosis in sham and CHF rats. TUNEL staining was performed and representative photomicrographs are depicted in Figure 6A and Figure S6. We found that compared to sham+vehicle rats, there was a significant increase in TUNEL-positive cells in the LV infarct, LV peri-infarct, LV remote and IVS regions of the myocardium in CHF+vehicle rats (Figure 6B-6E). However, TUNEL-positive cells were markedly reduced in the LV infarct, LV peri-infarct, LV remote and IVS regions of the myocardium from CHF+RTX rats compared to CHF+vehicle rats (Figure 6). There was no significant difference in the number of TUNEL-positive cells in the heart between sham+vehicle and sham+RTX rats. Furthermore, data from immunohistochemical staining demonstrated that there were increased cleaved caspase 3-positive cardiomyocytes in the LV peri-infarct, LV remote and IVS regions of the myocardium in CHF+vehicle rats compared to sham+vehicle rats, which was largely prevented by epicardial application of RTX following coronary artery ligation (Figure S7A). In addition, protein expression of cleaved caspase 3 in LV remote and IVS regions of the myocardium was significantly increased in CHF+vehicle rats compared to sham+vehicle rats whereas RTX treatment prevented the elevated protein expression of cleaved caspase 3 in the myocardium of CHF rats (Figure S7B and S7C).
Figure 6.
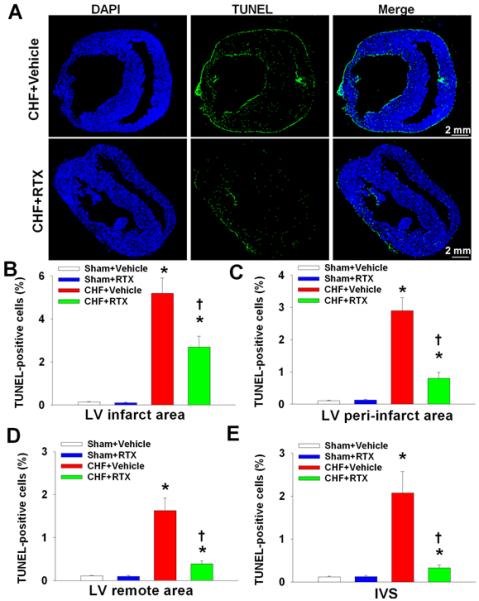
Representative scanning images (A) and mean data (B-D) showing TUNEL-positive staining in LV infarct, LV peri-infarct, LV remote and IVS regions from sham+vehicle, sham+RTX, CHF+Vehicle and CHF+RTX rats. Data are expressed as mean±SE. n=5-6/each group. *, P<0.05 vs. sham+vehicle, †, P<0.05 vs. CHF+vehicle.
DISCUSSION
The results of the current study clearly demonstrate that epicardial application of RTX just prior to the creation of an MI abolished the enhanced CSAR, prevented the exaggerated renal and cardiac sympatho-excitation and increased the blunted baroreflex in CHF rats. Importantly, we found that epicardial application of RTX prevented many of the pathological indices of the CHF state. These included elevated LVEDP, lung edema, cardiac hypertrophy, chamber dilation and contractile reserve in response β-adrenergic receptor stimulation with ISO. RTX also ameliorated cardiac fibrosis and apoptosis in CHF rats. To our knowledge, these are the first data to show that selective cardiac sympathetic afferent deafferentation may offer a clinical benefit to improve cardiac and autonomic function in the CHF state.
Increased cardiac sympathetic nerve activity is a primary characteristic of patients suffering from CHF11, 12. Prolonged stimulation of the β-adrenergic neurohormonal axis has been shown to contribute to the progression of CHF and mortality in both animal models and in humans13, 14. Measurements of cardiac NE spillover in CHF patients reflecting cardiac sympathetic nerve activity, indicate that it is increased earlier and to a greater extent than sympathetic nerve activity to other organs15-17. Data based on direct CSNA recording in the present study also supports the concept that CHF rats have markedly higher basal cardiac sympathetic tone than sham rats. However, the mechanisms underlying this augmented cardiac sympathetic outflow in CHF are not fully understood. One potential explanation is an increased CSAR control of cardiac sympathetic efferent outflow (also called the “cardiac-cardiac reflex”) in CHF. It is well known that myocardial ischemia releases large amounts of metabolites including bradykinin, ATP, prostaglandins and protons that stimulate cardiac sympathetic afferent nerve endings and cause an excitatory response of MAP, HR and sympathetic nerve activity1, 3, 4. Previous studies from this laboratory1, 2 demonstrated that acute epicardial application of lidocaine, which blocked cardiac sympathetic afferent input, decreased baseline MAP, HR and RSNA in anesthetized CHF dogs and rats but not in sham animals, indicating that the CSAR is tonically activated and contributes to the elevated renal sympathetic nerve activity in CHF. We further reported that the sites at which the CSAR is sensitized reside at both the afferent endings18 and in the central nervous system19. Whether the tonic activation of the CSAR also contributes to the elevated cardiac sympathetic nerve activity has not been explored. In the present study, we provide direct evidence that selective cardiac sympathetic afferent deafferentation by epicardial application of RTX prevents local excessive cardiac sympathetic activation in CHF rats. After blocking the CSAR control of cardiac sympathetic activation by RTX, we also observed multiple improvements in the structure and function of the myocardium in CHF rats. These findings highlight a critical contribution of the CSAR to the enhanced cardiac sympathetic tone as well as cardiac remodeling in CHF.
We examined multiple indices of cardiac remodeling and function in CHF rats treated with RTX. Our data show a reduction in chamber size, lower LVEDP, reduced cardiac hypertrophy, and attenuated cardiac fibrosis and apoptosis. Based on these protective effects, we believe the RTX-mediated improvement in cardiac diastolic function in CHF rats to be of primary importance. CHF rats treated with RTX did not exhibit the expected increase in LVEDP and lung congestion compared to CHF+vehicle rats. Our P-V loop data established a direct beneficial effect of RTX on cardiac diastolic function in the failing heart, independent of systemic vascular resistance. Furthermore, our molecular data suggest that RTX attenuated cardiac fibrosis in non-infarcted areas of the LV and IVS of CHF rats by reducing activation of the classic TGF-β receptor fibrotic signaling pathway. Taken together, these data suggest that the lower LVEDP observed in CHF+RTX rats is, at least in part, due to an RTX-mediated improvement of cardiac compliance. Because peripheral venous beds are highly innervated by sympathetic nerves and RTX-induced CSAR attenuation reduced the exaggerated systemic sympatho-excitation in CHF, another possibility is a reduction in venoconstriction and thus reduced cardiac filling, which would contribute to lower LVEDP in RTX-treated CHF rats.
With regard to the effect of RTX on cardiac systolic function in CHF, it is important to emphasize that the favorable effects of RTX on dp/dtmax were not evident in the basal (unstimulated) state, but rather in response to β-adrenergic receptor stimulation with ISO, indicating that the myocardial contractile reserve was substantially increased after RTX treatment. This finding is consistent with the observation that the downregulation of β1-AR in the myocardium of CHF rats was partially prevented by RTX. The underlying mechanisms by which RTX partially restored the β1-AR expression in CHF remain to be identified. A reasonable speculation is that by removing the influence of the CSAR on sympathetic outflow to the heart and thereby reducing excessive myocardial NE release, an upregulation of the β1-AR takes place14. However, this hypothesis awaits confirmation.
RTX treatment affected the remodeling process in other ways. In CHF+RTX rats, LV chamber size was smaller as reflected by reduced LV end-systolic and end-diastolic diameters and volumes compared to CHF+vehicle rats even when ejection fraction and infarct size were similar. It is known that during the progression of CHF persistent ischemia and inflammation in the ischemic zone impair granulation tissue, extracellular matrix formation (ECM) and ischemic myocyte survival, resulting in extension and expansion of the failing myocardium20. The current study provides strong evidence for the fact that epicardial RTX applied at the time of MI creation reduces this vicious cycle and delays progression of the CHF state. In a previous study by White et al.21, it was clearly shown that LV end-systolic volume rather than ejection fraction was the major determinant of survival in patients who recovered from an MI. Therefore, the RTX-mediated reduction of cardiac volume may potentially contribute to enhanced survival for MI patients.
RTX significantly ameliorated cardiac apoptosis in both infarcted and non-infarcted areas of left ventricle in CHF rats. It is well known that cell death is an important determinant of cardiac remodeling because it causes a loss of contractile tissue, compensatory hypertrophy of myocardial cells and reparative fibrosis22. Therefore, the RTX-mediated reduction in cardiac apoptosis may play an important role in these multiple RTX-mediated cardiac protective effects in CHF rats. The mechanisms underlying the reduction of cardiac apoptosis remain unknown. Considering that 1) excessive β-AR stimulation has been reported to induce cardiac myocyte apoptosis both in vivo and in vitro23, 24 and 2) RTX prevents the exaggerated cardiac sympathetic tone in CHF, it is reasonable to speculate that RTX may reduce cardiac apoptosis through the CSNA-β-adrenergic neurohormonal axis. Further studies will be needed to explore the molecular mechanisms underlying the RTX-induced protection against cardiac apoptosis in CHF.
The CSAR is a sympatho-excitatory reflex and contributes to the elevation in sympathetic tone. The latter can antagonize baroreflex function in the CHF state. In a previous study2 we reported that in anesthetized rats with CHF, acute epicardial application of lidocaine decreased baseline RSNA and improved the impaired baroreflex control of RSNA, suggesting that tonic cardiac sympathetic afferent input plays an important role in blunting the baroreflex in CHF. In a more recent study25, we further reported that acute stimulation of cardiac sympathetic afferents by epicardial application of capsaicin significantly decreased basal discharge of barosensitive nucleus of the solitary tract (NTS) neurons in anesthetized rats, indicating that the NTS plays an important role in processing the interactions between the CSAR and the baroreflex. However, because of the acute nature of the experiments in these previous studies, it is not known if the interaction between the CSAR and the baroreflex exists in more chronic pathological conditions such as CHF. In this study, we further found that epicardial application of RTX at the time of MI creation prevents the blunted baroreflex control of HR and RSNA 9-11 weeks post-MI, suggesting a chronic interaction between the CSAR and the baroreflex in CHF. In addition, the effects of lidocaine1, 2 and RTX on the improvement in baroreflex function appear to be specific to the CHF state since in sham-operated rats, we failed to find any change in RSNA and baroreflex parameters following epicardial application of either lidocaine1, 2 or RTX indicating that there is little, if any, tonic cardiac sympathetic afferent input in either the regulation of sympathetic nerve activity or interaction with the baroreflex in the normal state.
In the current study, RTX was selectively delivered to the epicardium to ablate the TRPV1-expressing cardiac afferents. A previous study by Zahner et al.5 showed that sympathetic vasomotor tone is intact after systemic treatment with RTX. Another study by Szolcsanyi et al.26 reported that bradycardia and the fall in blood pressure evoked by electrical stimulation of the peripheral vagus were not impaired after systemic RTX injection, suggesting that cardiac parasympathetic tone remained unaltered after RTX pretreatment. In addition, an earlier study by Barber et al.27 reported that epicardial application of phenol (a non-selective neurotoxin) interrupted sympathetic afferents but not vagal afferents in a canine model, suggesting that vagal afferents are mainly located in deeper left ventricular layers and are less affected by epicardial application of drugs. This finding is important for the current where epicardial application of RTX was used since it makes epicardial application of RTX more specific in targeting cardiac sympathetic afferents, even if a small subpopulation of vagal afferents also express TRPV1 receptors. Therefore, it appears that RTX selectively targets cardiac sympathetic afferents without impairing cardiac sympathetic and parasympathetic efferent nerves and vagal afferent nerves. Using this selective cardiac afferent desensitization strategy, we demonstrated multiple beneficial effects of RTX-induced inhibition of the CSAR on cardiac and autonomic function in CHF rats. In contrast to the current study genetic knockout techniques to systemically delete TRPV1 receptors in mice, have reported conflicting findings28-31 concerning either the beneficial or deleterious effects of global deletion of TRPV1 on cardiac function. There are several major differences between selective TRPV1-expressing cardiac afferent ablation and systemic pharmacological antagonism or genetic deletion of TRPV1 receptor strategies. The greatest difference between the current study and those mentioned above is that RTX not only deletes TRPV1 receptors in cardiac afferents but also damages the TRPV1-expressing nerve endings rather than only blocking TRPV1 receptors. Considering that TRPV1-expressing cardiac sympathetic afferent endings also express many other sensory receptors such as tachykinin and purinergic receptors, RTX also impairs these sensory receptors following damage of TRPV1-expressing cardiac afferent nerve endings. This explains why epicardial RTX application blocks both epicardial capsaicin and bradykinin-induced cardiovascular responses whereas TRPV1 receptor blockade using epicardial ido-RTX only abolishes epicardial capsaicin-induced responses without any effect on the bradykinin-induced cardiovascular response5. In addition, it should be pointed out that knockout animals have systemic TRPV1 deletion from birth, which raises the issue of developmental and compensatory changes. Similar issues may be at play in other models of global TRPV1 deletion such as in a neonatal rat model that was treated with a large dose of capsaicin32. There is clear evidence showing that adult rats that are treated with capsaicin as neonates exhibit reductions in both basal coronary flow and cardiomyocyte size32. This effect may result in an increase in cardiac damage when the heart is exposed to ischemia or pressure overload. Finally, global TRPV1 deletion using either genetic knockout or systemic pharmacological techniques will affect the central and peripheral nervous system as well resulting in the loss of both protective and deleterious effects of TRPV1 activation. Therefore, in our opinion selective ablation of TRPV1-expressing sensory afferents may or may not achieve the same cardiovascular effects in MI rats compared to those studies using pharmacological antagonism or genetic deletion of TRPV1 receptors.
Limitations
In this study, epicardial application of RTX was performed at the time of MI rather than several days or weeks after MI. Therefore, beneficial effects of RTX on cardiac and autonomic function indicate an early protective effect against the progress of deleterious cardiac remodeling in post-MI rats. However, whether epicardial application of RTX several days or weeks after MI has similar beneficial effect on cardiac dysfunction in post-MI rats remains unknown. We speculate that epicardial application of RTX during MI or very early post-MI in rats should have greater protective effects on cardiac dysfunction than during the mid or late post-MI period. For example, with regard to the current finding that epicardial application of RTX during MI largely prevents cardiac fibrosis in post-MI rats, it is less likely that epicardial application of RTX at the mid or late post-MI period would have the same beneficial effect because cardiac fibrosis would already be well developed during that period. A previous study by Judd and Wexler33 reported that myocardial hydroxyproline content (an amino acid primarily found in collagen) was increased by day 7 post MI and increased further by about 2 weeks post MI. This may suggest a potentially ideal therapeutic window for RTX-mediated protective effect against cardiac fibrosis caused by MI. Once myocardial collagen accumulation is well established, the potential protective effect of RTX against cardiac fibrosis observed in the current study may be reduced. Clearly further studies are needed to address this issue.
Another potential limitation of this study relates to potential reinnervation after RTX-induced cardiac afferent ablation. A previous study by Karai et al.34 reported that either subcutaneous or intrathecal injection of RTX (6μg/ml, 30μl) attenuates thermal nociception and inflammatory hyperalgesia in rats. Interestingly, the beneficial effects of subcutaneous RTX injection on inflammatory hyperalgesia only lasted about 20 days whereas the effect of intrathecal injection of RTX lasted much longer (at least 1 year). In the current study, when applying a higher concentration (50 μg/ml) of RTX to the surface of heart, we found that this treatment abolished the CSAR for at least 9-11weeks. However, we did observe a recovery of CSAR sensitivity at 6 months after epicardial application of RTX, indicating that this intervention if carried out in a clinical setting may need to be repeated. On the other hand, based on the study by Karai et al.34, intrathecal RTX injection at thoracic segments to ablate TRPV1-expressing cardiac sympathetic afferent neurons may be an alternative for longer ablation of the CSAR in MI patients. Clearly further studies are needed to address this issue. In addition, we acknowledge a limitation of this study is that all the terminal experiments were performed under anesthesia. Some of hemodynamic data such as basal blood pressure and heart rate should be further confirmed by conscious telemetry recording. Finally, we acknowledge a limitation that the current study did not answer the question of whether epicardial application of RTX during coronary ligation could reduce infarct size because we excluded animals with small MIs in both CHF+Vehicle and CHF+RTX groups in order to rule out a possible contribution of surgical variability to cardiac function. Additional studies should be carried out to address this important issue.
In summary, the present study provides the first evidence demonstrating that selective cardiac sympathetic afferent deafferentation by epicardial application of RTX during MI prevents cardiac and autonomic dysfunction in rats 9-11 weeks post MI. These findings demonstrate the importance and therapeutic potential of targeted deletion of CSAR in ventricular remodeling after MI. Figure S8 summarizes the potential role of the CSAR in the regulation of sympathetic outflow and how interruption of this reflex may be used in sympatho-excitatory disease states.
Perspectives
RTX, an ultrapotent analogue of capsaicin and capable of inducing rapid degeneration of TRPV1-expressing afferent neurons and fibers5-7, has been widely explored for its analgesic properties35-37. The current study reports a novel use of RTX for potential intervention in the cardiac remodeling process in patients following an MI. We believe it is possible to deliver RTX to the surface of the heart using pericardial centesis and thereby selectively ablate cardiac sympathetic afferents in CHF patients. Therefore, our current findings that RTX-induced CSAR deafferentation prevents cardiac remodeling and reduces cardiovascular dysfunction in CHF rats has high translational potential. Ongoing studies from this laboratory are further investigating the potential beneficial effects of epicardial application of RTX on cardiac dysfunction in larger animal models.
Supplementary Material
Acknowledgements
The authors thank Ms Johnnie F. Hackley, Ms Pam Curry, Ms Kaye Talbitzer and Mr. Richard Robinson for their expert technical assistance. We also thank Dr. David Wert as well as the UNMC Tissue Sciences Facility for their help with the histopathology, TUNEL and cleaved caspase 3 staining experiments.
Sources of Funding
This work was supported from The American Heart Association (11GRNT7530022) and in part, by a grant from the National Heart, Lung, and Blood Institute (PO1 HL62222). The authors have no conflict of interest to disclosure with regard to funding or compensation from industry for this study.
Footnotes
Disclosures
None.
References
- (1).Wang W, Zucker IH. Cardiac sympathetic afferent reflex in dogs with congestive heart failure. Am J Physiol. 1996;271:R751–R756. doi: 10.1152/ajpregu.1996.271.3.R751. [DOI] [PubMed] [Google Scholar]
- (2).Gao L, Schultz HD, Patel KP, Zucker IH, Wang W. Augmented input from cardiac sympathetic afferents inhibits baroreflex in rats with heart failure. Hypertension. 2005;45:1173–1181. doi: 10.1161/01.HYP.0000168056.66981.c2. [DOI] [PubMed] [Google Scholar]
- (3).Wang W, Ma R. Cardiac sympathetic afferent reflexes in heart failure. Heart Fail Rev. 2000;5:57–71. doi: 10.1023/A:1009898107964. [DOI] [PubMed] [Google Scholar]
- (4).Longhurst JC, Tjen ALS, Fu LW. Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann N Y Acad Sci. 2001;940:74–95. doi: 10.1111/j.1749-6632.2001.tb03668.x. [DOI] [PubMed] [Google Scholar]
- (5).Zahner MR, Li DP, Chen SR, Pan HL. Cardiac vanilloid receptor 1-expressing afferent nerves and their role in the cardiogenic sympathetic reflex in rats. J Physiol. 2003;551:515–523. doi: 10.1113/jphysiol.2003.048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Szallasi A, Blumberg PM. Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience. 1989;30:515–520. doi: 10.1016/0306-4522(89)90269-8. [DOI] [PubMed] [Google Scholar]
- (7).Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM. Resiniferatoxin. An ultrapotent neurotoxin of capsaicin-sensitive primary afferent neurons. Ann N Y Acad Sci. 1991;632:473–475. doi: 10.1111/j.1749-6632.1991.tb33161.x. [DOI] [PubMed] [Google Scholar]
- (8).Wang HJ, Pan YX, Wang WZ, et al. Exercise training prevents the exaggerated exercise pressor reflex in rats with chronic heart failure. J Appl Physiol. 2010;108:1365–1375. doi: 10.1152/japplphysiol.01273.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang HJ, Li YL, Zucker IH, Wang W. Exercise training prevents skeletal muscle afferent sensitization in rats with chronic heart failure. Am J Physiol Regul Integr Comp Physiol. 2012;320:R1260–R1270. doi: 10.1152/ajpregu.00054.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang HJ, Li YL, Gao L, Zucker IH, Wang W. Alteration in skeletal muscle afferents in rats with chronic heart failure. J Physiol. 2010;588:5033–5047. doi: 10.1113/jphysiol.2010.199562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- (12).Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615–621. doi: 10.1161/01.cir.73.4.615. [DOI] [PubMed] [Google Scholar]
- (13).Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res. 2000;45:713–719. doi: 10.1016/s0008-6363(99)00370-3. [DOI] [PubMed] [Google Scholar]
- (14).Fowler MB, Laser JA, Hopkins GL, Minobe W, Bristow MR. Assessment of the beta-adrenergic receptor pathway in the intact failing human heart: progressive receptor down-regulation and subsensitivity to agonist response. Circulation. 1986;74:1290–1302. doi: 10.1161/01.cir.74.6.1290. [DOI] [PubMed] [Google Scholar]
- (15).Ramchandra R, Hood SG, Denton DA, Woods RL, McKinley MJ, McAllen RM, May CN. Basis for the preferential activation of cardiac sympathetic nerve activity in heart failure. Proc Natl Acad Sci U S A. 2009;106:924–928. doi: 10.1073/pnas.0811929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Meredith IT, Broughton A, Jennings GL, Esler MD. Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med. 1991;325:618–624. doi: 10.1056/NEJM199108293250905. [DOI] [PubMed] [Google Scholar]
- (17).Rundqvist B, Elam M, Bergmann-Sverrisdottir Y, Eisenhofer G, Friberg P. Increased cardiac adrenergic drive precedes generalized sympathetic activation in human heart failure. Circulation. 1997;95:169–175. doi: 10.1161/01.cir.95.1.169. [DOI] [PubMed] [Google Scholar]
- (18).Wang W, Schultz HD, Ma R. Cardiac sympathetic afferent sensitivity is enhanced in heart failure. Am J Physiol. 1999;277:H812–H817. doi: 10.1152/ajpheart.1999.277.2.H812. [DOI] [PubMed] [Google Scholar]
- (19).Ma R, Zucker IH, Wang W. Central gain of the cardiac sympathetic afferent reflex in dogs with heart failure. Am J Physiol. 1997;273:H2664–H2671. doi: 10.1152/ajpheart.1997.273.6.H2664. [DOI] [PubMed] [Google Scholar]
- (20).Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- (21).White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- (22).Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, Uno Y, Minatoguchi S, Fujiwara T, Fujiwara H. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res. 1998;82:1130–1138. doi: 10.1161/01.res.82.11.1130. [DOI] [PubMed] [Google Scholar]
- (23).Goldspink DF, Burniston JG, Ellison GM, Clark WA, Tan LB. Catecholamine-induced apoptosis and necrosis in cardiac and skeletal myocytes of the rat in vivo: the same or separate death pathways? Exp Physiol. 2004;89:407–416. doi: 10.1113/expphysiol.2004.027482. [DOI] [PubMed] [Google Scholar]
- (24).Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- (25).Wang WZ, Gao L, Pan YX, Zucker IH, Wang W. Differential effects of cardiac sympathetic afferent stimulation on neurons in the nucleus tractus solitarius. Neurosci Lett. 2006;409:146–150. doi: 10.1016/j.neulet.2006.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM. Resiniferatoxin: an ultrapotent selective modulator of capsaicin-sensitive primary afferent neurons. J Pharmacol Exp Ther. 1990;255:923–928. [PubMed] [Google Scholar]
- (27).Barber MJ, Mueller TM, Davies BG, Zipes DP. Phenol topically applied to canine left ventricular epicardium interrupts sympathetic but not vagal afferents. Circ Res. 1984;55:532–544. doi: 10.1161/01.res.55.4.532. [DOI] [PubMed] [Google Scholar]
- (28).Thilo F, Liu Y, Schulz N, Gergs U, Neumann J, Loddenkemper C, Gollasch M, Tepel M. Increased transient receptor potential vanilloid type 1 (TRPV1) channel expression in hypertrophic heart. Biochem Biophys Res Commun. 2010;401:98–103. doi: 10.1016/j.bbrc.2010.09.017. [DOI] [PubMed] [Google Scholar]
- (29).Huang W, Rubinstein J, Prieto AR, Thang LV, Wang DH. Transient receptor potential vanilloid gene deletion exacerbates inflammation and atypical cardiac remodeling after myocardial infarction. Hypertension. 2009;53:243–250. doi: 10.1161/HYPERTENSIONAHA.108.118349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Horton JS, Buckley CL, Stokes AJ. Successful TRPV1 antagonist treatment for cardiac hypertrophy and heart failure in mice. Channels (Austin) 2013;7:17–22. doi: 10.4161/chan.23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Buckley CL, Stokes AJ. Mice lacking functional TRPV1 are protected from pressure overload cardiac hypertrophy. Channels (Austin) 2011;5:367–374. doi: 10.4161/chan.5.4.17083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zanesco A, Costa SK, Riado SR, Nathan LP, de Oliveira CF, De Luca IM, Antunes E, De Nucci G. Modulation of coronary flow and cardiomyocyte size by sensory fibers. Hypertension. 1999;34:790–794. doi: 10.1161/01.hyp.34.4.790. [DOI] [PubMed] [Google Scholar]
- (33).Judd JT, Wexler BC. Prolyl hydroxylase and collagen metabolism after experimental mycardial infarction. Am J Physiol. 1975;228:212–216. doi: 10.1152/ajplegacy.1975.228.1.212. [DOI] [PubMed] [Google Scholar]
- (34).Karai L, Brown DC, Mannes AJ, Connelly ST, Brown J, Gandal M, Wellisch OM, Neubert JK, Olah Z, Iadarola MJ. Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J Clin Invest. 2004;113:1344–1352. doi: 10.1172/JCI20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tender GC, Li YY, Cui JG. The role of nerve growth factor in neuropathic pain inhibition produced by resiniferatoxin treatment in the dorsal root ganglia. Neurosurgery. 2013;73:158–165. doi: 10.1227/01.neu.0000429850.37449.c8. [DOI] [PubMed] [Google Scholar]
- (36).Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol. 2013;716:61–76. doi: 10.1016/j.ejphar.2013.03.003. [DOI] [PubMed] [Google Scholar]
- (37).Kissin I, Szallasi A. Therapeutic targeting of TRPV1 by resiniferatoxin, from preclinical studies to clinical trials. Curr Top Med Chem. 2011;11:2159–2170. doi: 10.2174/156802611796904924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.