

Abstract
Mitochondria move, fuse and divide in cells. The dynamic behavior of mitochondria is central to the control of their structure and function. Three conserved mitochondrial dynamin-related GTPases (i.e., mitofusin, Opa1 and Drp1 in mammals and Fzo1, Mgm1 and Dnm1 in yeast) mediate mitochondrial fusion and division. In addition to dynamins, recent studies demonstrated that phospholipids in mitochondria also play key roles in mitochondrial dynamics by interacting with dynamin GTPases and by directly changing the biophysical properties of the mitochondrial membranes. Changes in phospholipid composition also promote mitophagy, which is a selective mitochondrial degradation process that is mechanistically coupled to mitochondrial division. In this review, we will discuss the biogenesis and function of mitochondrial phospholipids.
Keywords: Dynamin-related GTPase, Mitochondrial fission, Mitochondrial fusion, Mitophagy, Apoptosis, Drp1, Mitofusin, Opa1
Introduction
Mitochondria are important for many cellular processes, such as energy production, heat generation, metabolism, cellular proliferation, differentiation and cell death [1, 2]. To accommodate these diverse functions, mitochondria adopt different shapes, sizes, numbers and distributions in different cell types [3–7]. The morphological organization of mitochondria is regulated primarily by organelle fusion and division. Under steady-state conditions, mitochondria fuse and divide constitutively at similar rates, persistently maintaining the overall organelle morphology. In addition to maintaining morphology, fusion mixes the contents of mitochondria, including proteins, lipids and nucleic acids. Division allows cells to make small organelles to facilitate efficient transport during interphase and inheritance during cell division.
In addition, mitochondria modulate fusion and division as part of physiological and signaling mechanisms in response to different stimuli [8, 9]. The induction of mitochondrial division is associated with many types of cellular and organellar stresses [10]. For example, increased division and decreased fusion synergistically fragment mitochondria during apoptosis, which facilitates the efficient release of the proapoptotic factor cytochrome c from mitochondria by severing the mitochondrial outer membrane or remodeling the membrane via hemifusion. Under pathological conditions, such as neurodegenerative diseases and cardiac reperfusion injury after ischemia, mitochondria also become fragmented [11–13]. At the organelle level, mitochondrial damage and dysfunction often result in hyper-division in response to a variety of mitochondrial stresses, such as the loss of membrane potential across the inner membrane, which can inhibit mitochondrial fusion, and oxidative damage, which can facilitate the degradation of mitochondria by mitophagy. Mitochondria also hyper-fuse in response to different types of stress, such as starvation, which induces the degradation of many cellular components by autophagy; elongation allows the mitochondria to escape from degradation and maintain the production of intracellular energy [14, 15]. The hyper-fusion of mitochondria is also observed when cytosolic protein synthesis is inhibited. Under these conditions, hyper-fusion helps maintain the survival of cells by promoting ATP production [16].
Due to their evolutionary origin, mitochondria consist of two membranes: the outer and inner membranes [17]. These two membranes have separate but linked fusion machineries, which are highly conserved from yeast to humans [18, 19]. Outer membrane fusion is controlled by two dynamin-related GTPases: mitofusion (mammals)/Fzo1 (yeast) and Opa1/Mgm1 [20–27]. Mitofusin/Fzo1 is inserted into the outer membrane via two transmembrane domains, with the GTPase domain facing the cytosol. In contrast, Opa1/Mgm1 exhibits two forms: one form contains a transmembrane domain that is inserted into the inner membrane, and the other form lacks a transmembrane domain and is located in the intermembrane space. Mitofusin/Fzo1 and Opa1/Mgm1 form a protein complex that connects the two membranes; therefore, Opa1/Mgm1 also contribute to stable, full fusion of the outer membrane in addition to their role in the fusion of the inner membrane. In yeast, the mitochondrial protein Ugo1 physically connects Fzo1 and Mgm1 and forms the fusion contact site between the two membranes [28, 29]. In contrast to fusion machineries, only the outer membrane-located division machinery has been identified in yeast and mammals. However, algae have separate machineries for the outer and inner membranes [30]. A central component of mitochondrial division is the soluble dynamin-related GTPase Drp1 (mammals)/Dnm1 (yeast), which is assembled onto the surface of mitochondria by separate but potentially collaborative receptor proteins in mammals (i.e., Mff, Fis1 and Mid/MIEF) and by receptor–adapter complexes in yeast (i.e., Fis1-Mdv1 and Num1-Mdm36) [31–40]. Demonstrating the importance of mitochondrial fusion and division in human health, mutations in mitofusin 2, Opa1 and Drp1 can cause different human disorders, such as Charcot–Marie–Tooth type 2A for mitofusin 2, dominant optic atrophy 1 for OPA1 and postneonatal death with neuronal defects for Drp1 [11]. In addition, abnormalities in Drp1 has been linked to a variety of age-related neurodegenerative diseases including Alzheimer’s, Parkinson’s and Huntington’s diseases [11, 41]. These defects are located primarily in central and peripheral nerves, revealing a high demand for proper mitochondrial membrane dynamics in neurons.
The mitochondrial membranes synthesize different phospholipids, such as cardiolipin (CL), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG), after importing precursors such as phosphatidic acid (PA) and phosphatidylserine (PS), which are synthesized in the ER [42]. PE is then exported from mitochondria to the ER and converted to phosphatidylcholine (PC). Therefore, mitochondria are part of the phospholipid biosynthetic network and exchange phospholipids through mitochondria–ER contact sites. A proper composition of phospholipids is necessary for the biogenesis, maintenance, assembly and activity of the dynamin-related GTPases [43]. Phospholipids also signal mitophagy [44]. Finally, phospholipids modulate the biophysical properties of the mitochondrial membranes to facilitate efficient lipid mixing. In this review, we discuss recent findings related to the roles of phospholipids in mitochondrial division, fusion and degradation.
Biosynthesis of mitochondrial phospholipids
Similar to other cellular membranes, such as the plasma membrane, the ER and the Golgi complex, the mitochondrial membrane consists of PC, PE, phosphatidylinositol (PI), PS and PA, with PC and PE as the major components, accounting for a total of 60–80 % of the phospholipids in mitochondria [42, 45, 46]. In addition to these universal phospholipids, mitochondria contain a specific phospholipid [i.e., cardiolipin (CL) (~10 %)] that is synthesized in the mitochondrial inner membrane and remains primarily in mitochondria [42, 47]. CL is also synthesized in bacterial membranes, supporting the symbiotic origin of mitochondria during evolution. The biosynthetic pathways for these phospholipids are well characterized and conserved in yeast and mammals, as described below.
Phosphatidic acid: a major precursor for phospholipids
Although PA is a minor component of biological membranes, PA plays a major role as a precursor lipid in the biosynthesis of phospholipids. In addition, de novo phospholipid synthesis is accomplished primarily by consecutive modifications of this simple phospholipid. The small amount of PA present at steady state may be explained by its rapid consumption for the production of other phospholipids immediately after PA synthesis.
Several PA synthesis pathways exist (Fig. 1). As a major mechanism, glycerol 3-phosphate or dihydroacetone phosphate (DHAP) is acylated in two steps. First, acylation is mediated by the glycerol-3-phosphate/DHAP acyltransferases Sct1p and Gpt2p in the ER in yeast [48, 49]. This step occurs in both the mitochondrial outer membrane (via Gpat1 and Gpat2) and the ER (via Gpat3 and Gpat4) in mammals [50]. The second acylation of the resulting lyso-PA is mediated by lyso-PA acyltransferase (Slc1p and Ale1p in yeast and LPAAT in mammals) [51–53]. PA can also be generated via the removal of the choline headgroup of PC by phospholipase D or via the phosphorylation of diacylglycerol (DAG) by DAG kinase [54–56]. In mammals, another phospholipase D that is located in the mitochondrial outer membrane (mitoPLD) also generates PA from CL [57].
Fig. 1.

Biosynthetic pathways for mitochondrial phospholipids. The major precursor PA is synthesized in the ER and mitochondrial outer membranes. The CDP-DAG pathway is located in the ER and mitochondrial inner membranes and produces a variety of phospholipids including PI, PS, PE, PC and CL. The ER-located Kennedy pathway generates PE and PC
The CDP-DAG pathway
All other phospholipids are produced via two synthetic pathways: the cytidine diphosphate diacylglycerol (CDP-DAG) and Kennedy pathways [43, 58]. In the CDP-DAG pathway, which is located in the ER and in mitochondria, PA is first converted to the intermediate phospholipid CDP-DAG by CDP-DAG synthases (Fig. 1). In yeast, Cds1 and Tam41 convert PA to CDP-DAG in the ER and in the mitochondrial matrix, respectively [59, 60]. Two mammalian Cds1 orthologs (i.e., Cds1 and Cds2) are present in the ER [61]; a mammalian Tam41 ortholog has been identified, but its function remains unclear.
In yeast, CDP-DAG is used to make different phospholipids: PI, PS and the PG precursor phosphatidylglycerolphosphate (PGP) [43]. PI is generated by the PI synthase Pis1, which replaces cytidine monophosphate (CMP) with an inositol group. PS is produced by the PS synthase Cho1, which replaces CMP with serine [62, 63]. Because both Pis1 and Cho1 are located in the ER membrane, PI and PS are imported into mitochondria from the ER. The PGP synthase Pgs1 is located in the mitochondrial matrix and replaces CMP with glycerol 3-phosphate to form PGP [64]. To produce PG, PGP is dephosphorylated by the PGP phosphatases Gep4 in yeast and PTPMT1 in mammals [65, 66]. Using PG and CDP-DAG, the CL synthase Crd1 forms CL by combining these substrates in the mitochondrial inner membrane [67–69]. In mammals, PI and PGP are synthesized via mechanisms similar to those employed in yeast. CL is also produced from PG and CDP-DAG by the CL synthase CLS1 [70, 71]. In contrast, PS is primarily generated from PC and PE but not from CDP-DAG [58]. The mammalian PS synthases PSS1 and PSS2 catalyze the exchange of choline and ethanolamine in PC and PE, respectively, for serine to generate PS [72, 73].
PS is used as a precursor to produce PE and PC (Fig. 1). In yeast, PS is transported from the ER to other organelles and converted to PE via decarboxylation by Psd1 in the mitochondrial inner membrane [74] or by Psd2 in endosomes/Golgi complex [75, 76]. PE is then transported back to the ER and methylated by the phospholipid methyltransferases Cho2 and Opi3 to produce PC [77, 78]. In mammals, a single PS decarboxylase (i.e., Pisd) generates PE in the mitochondrial inner membrane and a PE methyltranferase (i.e., PEMT) subsequently methylates PE to produce PC in the ER and at ER-mitochondria contact sites [79, 80].
The Kennedy pathway
The two most abundant phospholipids (i.e., PC and PE) can be also synthesized de novo via three successive reactions in the ER-located Kennedy pathway (Fig. 1) [81]. The enzymes involved in this pathway are also highly conserved from yeast to humans [82]. First, choline and ethanolamine kinases generate choline-phosphate (for PC) and ethanolamine-phosphate (for PE), respectively. Second, these products are combined with CDP to form CDP-choline and CDP-ethanolamine by choline- and ethanolamine-phosphate cytidylyltransferases, respectively. Finally, to synthesize PC and PE, DAG-cholinephosphotransferase and DAG-ethanolaminephosphotransferase transfer the choline-phosphate and ethanolamine-phosphate head groups, respectively, to DAG, which is generated via the dephosphorylation of PA by the PA phosphatases Pah1 in yeast and Lipin in mammals [81, 83].
Dynamics of mitochondrial phospholipids during biosynthesis
Inter-organellar transfer between the ER and mitochondria
Because phospholipids are synthesized in both the ER and mitochondria, some phospholipids, such as PA, PS and PE, travel between these organelles. These two organelles are physically connected by protein machineries that form inter-organellar contact sites. In mammals, mitofusin 2, which mediates fusion of the mitochondrial outer membrane, also tethers the ER and mitochondria [84, 85]. In yeast, the ERMES protein complex, which consists of the ER membrane protein Mmm1 and four mitochondrial proteins (i.e., Mdm10, Mdm12, Mdm34 and Gme1) connect the two organelles [86–88]. The ER–mitochondria contact sites are important for phospholipid transfer, and cells defective in these contact sites exhibit defects in the biosynthesis of phospholipids.
Intra-mitochondrial transfer between the outer and inner membranes
Because many of the phospholipid biosynthetic enzymes are located in the mitochondrial inner membrane, phospholipids and their precursor lipids also move between the outer and inner membranes in mitochondria. In contrast to ER–mitochondria contact sites, components of the intra-mitochondrial transfer pathway are evolutionarily conserved from yeast to mammals. The key proteins in this pathway are homologous small proteins located in the intermembrane space (i.e., Ups1, 2 and 3) and their binding protein Mdm35 in yeast and their orthologs in mammals (i.e., PRELI for Ups1 and TRIAP1 for Mdm35) [89–92]. Ups1 was originally identified as a protein necessary for the biogenesis of Mgm1, which is a mitochondrial fusion protein; therefore, Ups1 is required for the maintenance of mitochondrial morphology [93]. Later studies demonstrated that Ups1 and its homologous protein Ups2 antagonistically regulate the biosynthesis of CL through the transfer of PA between the outer and inner membranes [89, 94]. The function of Ups proteins has been proposed to directly mediate lipid transfer or to regulate this transfer step [89, 95]. In addition to PA, the Ups proteins regulate the transfer of PS and PE, suggesting that these proteins support a universal mechanism for lipid transfer and biosynthesis in mitochondria [96, 97].
Roles of phospholipids in mitochondrial dynamics
Mitochondrial fusion
Phosphatidic acid (PA)
Phospholipids affect the biophysical properties of bilayer membranes and facilitate membrane curvature. PA has a small, negatively charged head group and can therefore create negative curvature in the membrane (Fig. 2) [45]. This curvature makes biological membranes prone to fusion, and PA has been called a fusogenic phospholipid [98]. For example, SNARE-mediated membrane fusion during exocytosis and endocytosis requires PA [98]. Similarly, the fusion of the mitochondrial outer membrane is facilitated by PA [57]. Outer membrane fusion in mammals is mediated by two homologous GTPases: mitofusin 1 and 2. These two proteins tether two opposing outer membranes prior to fusion [99]. After the tethering of two opposing outer membranes, an outer membrane-localized phospholipase D (i.e., MitoPLD) generates PA from CL to facilitate membrane fusion [57]. PA may change the conformation of trans-mitofusin complexes formed between tethered mitochondria and merge the membranes.
Fig. 2.
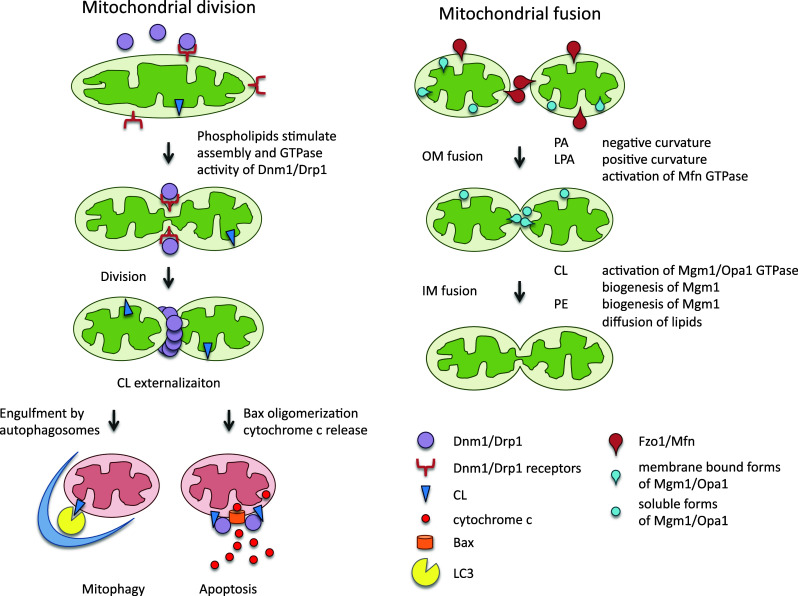
Roles of phospholipids in mitochondrial division, fusion and degradation. During mitochondrial division, phospholipids help assemble Dnm1/Drp1 on the surface of mitochondria together with its receptor proteins. Upon stress, CL is externalized to the outer membrane and can promote mitophagy and apoptosis. During mitochondrial fusion, dynamin-related GTPases and phospholipids work together. While Fzo1/mitofusin along with PA and LPA mediate outer membrane fusion, Mgm1/Opa1, CL and PE control inner membrane fusion
Lysophosphatidic acid (LPA)
LPA is a phospholipid similar in structure to PA, except LPA has only one acyl chain and PA has two. A glycerol-3-phosphate acyltransferase, which is located in the mitochondrial outer membrane, catalyzes the acylation of glycerol 3-phosphate with long-chain fatty acyl CoA to form LPA [50, 100, 101]. Unlike PA, due to its single acyl chain, LPA generates positive curvature in bilayer membranes (Fig. 2). Nonetheless, similar to PA, LPA promotes mitofusin-mediated mitochondrial fusion [100]. Because PA and LPA form opposite membrane curvature, these proteins may function to bend membranes at different steps of mitochondrial fusion or in different regions to biophysically stabilize intermediate structures, such as stalk intermediate and hemifusion or fusion pores between two membranes. LPA may also directly interact with mitofusin and regulate its function, as LPA stimulates the GTPase activity of purified mitofusin in vitro [100].
Cardiolipin (CL)
Like its mammalian homolog Opa1, Mgm1 support both outer membrane fusion and inner membrane fusion [20–24]. Two versions of the Mgm1 protein are generated posttranslationally: one version is embedded in the inner membrane, and the other version is soluble and localizes to the intermembrane space after proteolytic cleavage of the membrane-bound form [102]. CL binds to a soluble form of Mgm1 and stimulates its GTPase activity (Fig. 2) [103, 104]. The CL–Mgm1 interaction is mediated by several conserved positively charged lysines in Mgm1 that may serve as a binding site for negatively charged CL [104]. In contrast, the GTPase activity of the membrane-embedded form of Mgm1 is not stimulated by CL [103]. Interestingly, when soluble Mgm1 is mixed with CL-containing and Mgm1-embedded liposomes, the GTPase activity of soluble Mgm1 is further increased due to interactions with both CL and the membrane form of Mgm1. Interactions of Mgm1 with phospholipids are not specific to CL, as Mgm1 can tubulate and tether liposomes and promote GTP-stimulated hemifusion of liposomes independently of CL [105]. This type of non-selective interaction with phospholipids and the activation of GTPase activity were also observed for Opa1, which is a mammalian ortholog of Mgm1, as discussed below.
In addition to activating the GTPase activity of Mgm1, CL is also important for the biogenesis of Mgm1 (Fig. 2). As described above, Mgm1 exists in two forms as a result of proteolytic cleavage of its transmembrane domain [102, 106, 107]. This cleavage event is coupled to the insertion of the transmembrane domain of Mgm1 into the inner membrane during import [89]. When CL levels are reduced, the translocation of the transmembrane domain of Mgm1p is also reduced due to disassembly of the inner membrane-localized protein translocase and the matrix-localized import motor complex, leading to defects in the proteolytic processing of Mgm1 and defects in subsequent mitochondrial fusion [89, 93].
The mammalian ortholog of Mgm1 (i.e., OPA1) is subjected to alternative mRNA splicing in addition to proteolytic processing, creating at least eight isoforms. Similar to Mgm1, these isoforms can be grouped into two major classes based on the presence of a transmembrane domain [108–110]. Further, a purified soluble form of OPA1 directly binds to liposomes containing acidic phospholipids, such as CL, PA and PS, and can tubulate liposomes [111]. Interactions of OPA1 with these phospholipids also stimulate GTPase activity. Among these three acidic phospholipids, CL is most abundant in the mitochondrial membrane; therefore, CL may be a major physiological stimulator of OPA1. As a possible mechanism for GTPase activation, CL stimulates the oligomerization of OPA1. Many dynamins and dynamin-related GTPase are activated upon the homotypic assembly of this protein [111, 112]. Mutations associated with optic atrophy 1 affect the ability of OPA1 to bind to phospholipids, and these biochemical changes may play a role in the pathogenesis of this disease.
Acyl chains of CL undergo remodeling by tafazzin, a transacylase which replaces saturated acyl chains with unsaturated acyl chains in the CL maturation pathway [113]. This enzyme is defective in Barth syndrome, a severe genetic disorder that causes cardiomyopathy and skeletal myopathy [113]. In addition, ALCAT1 mediates pathological CL remodeling and promotes oxidative damage to acyl chains of CL [114]. ALCAT1-mediated oxidative damage of CL leads to reduced mitofusion 2 levels [115]. Therefore, the intact acyl composition of CL is important for mitofusin-mediated outer membrane fusion.
Phosphatidylethanolamine (PE)
In mammalian cells, PE is more abundant in mitochondria than in other organelles and mitochondrial PE is important for mitochondrial functions and cellular proliferation. Decreasing PE levels by knocking out the mouse phosphatidylserine decarboxylase leads to embryonic lethality and fragmentation of mitochondria [116]. Like CL, PE has conical shape due to a small head group diameter relative to its acyl chains [117]. This produces membrane curvatures; therefore, these lipids alone form micelles, but not bilayers. This structural similarity suggests that PE and CL may share functions in mitochondrial dynamics [45]. Indeed, PE is important for the biogenesis of Mgm1 and subsequent mitochondrial fusion. psd1∆ yeast cells have reduced mitochondrial fusion efficiency and increased numbers of fragmented mitochondria (Fig. 2) [118]. In addition, in vitro fusion assays using liposomes demonstrated that a lack of PE slows the mixing of lipids after fusion, suggesting that relatively high levels of PE may increase lateral movements of lipids in the mitochondrial membranes [118]. Further supporting the hypothesis that CL and PE perform redundant functions, simultaneous depletion of these two phospholipids in the crd1∆psd1∆ yeast mutant caused accelerated defects in mitochondrial fusion and fragmentation of mitochondria; these changes likely occur due to reduced levels of Mgm1 [119].
Mitochondrial division
The roles of phospholipids in mitochondrial division have also been studied extensively using a number of biochemical and structural assays [19]. Although these studies clearly demonstrated the importance of phospholipids, many studies on mitochondrial division examined the effects of liposomes with mixed phospholipids on the mitochondrial division dynamin Drp1/Dnm1 and its binding proteins, leaving the issue of phospholipid specificity unclear.
Drp1/Dnm1 plays a direct role in mitochondrial fission as a mechanochemical enzyme [19, 30]. Drp1 is a cytosolic protein, which assembles onto the surface of mitochondria by associating with its receptor proteins, including the Mff, Fis1 and Mid/MIEF proteins [31–34, 120–124]. In contrast, Dnm1 is recruited to mitochondria by the Fis1–Mdv1 complex and the Mdm36–Num1 complex [35, 38, 40, 125–129]. Similar to the inner membrane fusion dynamin OPA1/Mgm1, the mitochondrial division dynamin Drp1/Dnm1 also binds to liposomes containing mitochondrial phospholipids, including PC, PE, PI, PA, PS and CL (Fig. 2) [130]. The GTPase activity of Dnm1 is stimulated by interactions with liposomes. This enzymatic stimulation likely results from the stimulation of oligomerization on the surface of liposomes, because Dnm1/Drp1 tubulates liposomes upon oligomerization [131–133]. The phospholipids that bind directly to Drp1/Dnm1 remain unknown, although liposomes made of only PG or PS can interact with these proteins [131]. Drp1 also plays important roles in apoptosis. Drp1 facilitates the release of the proapoptotic factor cytochrome c from mitochondria into the cytosol both in vitro and in vivo [134–136]. CL binds to Drp1 and stimulates its apoptotic function [137]. In a key step for cytochrome c release, Bax oligomerizes in the outer membrane, leading to the permeabilization of the mitochondrial outer membrane (Fig. 2). CL binding increases the ability of Drp1 to promote Bax oligomerization [137]. CL–Drp1 interactions depend on the positively charged arginine at position 247 in the GTPase domain of Drp1, but interestingly, these interactions are independent of GTPase activity.
The conserved mitochondrial outer membrane protein Fis1 plays important roles in the recruitment of Drp1 and Dnm1 to the surface of mitochondria; however, the mechanism of recruitment appears to be different in mammals and yeast, as described above. Fis1 contains a C-terminal hydrophobic region, which serves as a transmembrane domain [40]. Interestingly, the cytosolic domain of yeast Fis1 binds to liposomes containing PC and PG, reversibly clustering liposomes and changing their permeability [138]. These activities may contribute to the mechanism of mitochondrial fission or to apoptotic release of cytochrome c.
In addition to Drp1/Dnm1, dynamin-independent mechanisms for mitochondrial division, such as the Parkinson’s disease-associated protein alpha-synuclein, have also been proposed [139]. Overexpression of alpha-synuclein leads to Drp1-independent fragmentation of mitochondria within cells. Purified alpha-synuclein clusters and fragments liposomes containing PC and CL in a CL-dependent manner. The elucidation of the functional relationships between these two division mechanisms is of interest. Similar to Drp1, alpha-synuclein also controls apoptosis through interactions with CL and cytochrome c in Lewy bodies [140]. This complex formation prevents cytochrome c-promoted apoptosis. Comprehensive reviews on the role of phospholipids in apoptosis have been published elsewhere [141–143].
Mitophagy
Cells can recognize damaged mitochondria and selectively eliminate them using a lysosomal degradation process called mitophagy [8]. Mitochondrial division is part of this degradation mechanism because division creates smaller organelles, which helps autophagosomes engulf the mitochondria [144]. The autophagosomes then fuse with the lysosome to deliver the mitochondria to the degradation compartment. In vivo, in postmitotic neurons, a mitochondrial division deficiency enlarges mitochondria and blocks mitophagy, leading to neurodegeneration [145]. In neurons lacking Drp1, oxidative damage accumulates in halted mitophagy intermediates, causing a loss of mitochondrial respiration. The recognition of damaged mitochondria is a key step in this pathway. Cells can identify damaged mitochondria based on a number of changes, such as reductions in the membrane potential across the inner membrane and oxidative damage to mitochondrial components. The loss of membrane potential recruits the Parkinson’s disease-associated ubiquitin E3 ligase parkin to mitochondria and promotes the ubiquitination of mitochondrial outer membrane proteins [146, 147]. This modification signals autophagosomes to engulf mitochondria. Interestingly, parkin also generates small vesicles called mitochondria-derived vesicles from the mitochondrial membranes in response to reactive oxygen species and delivers these vesicles to lysosomes for degradation [148, 149].
In addition to these mitophagy-related recognition and signaling mechanisms, the exposure of CL on the surface of mitochondria is also used to mark damaged mitochondria (Fig. 2) [44]. When neurons are treated with inhibitors of mitochondrial respiration, CL is moved from the mitochondrial inner membrane to the outer leaflet of the outer membrane by phospholipid scramblase 3, which flips CL from the inner leaflet to the outer leaflet of the membrane. The externalization of CL stimulates engulfment of mitochondria via the recruitment of the autophagosomal protein LC3 through direct interactions [44]. This CL-regulated mitophagy pathway appears to be independent of parkin and the mitophagy adapter protein p62. A parkin-independent mitophagy mechanism has been reported in several other experimental systems, including postmitotic neurons lacking Drp1, as described above [145, 150]. The mitophagy intermediates that accumulated in these neurons were highly ubiquitinated in the absence of parkin. Therefore, Drp1-mediated or CL-stimulated mitophagy may use other E3 ligases for mitochondrial ubiquitination.
Conclusions and future prospects
Interactions between proteins and lipids are fundamental reactions that underlie many biological processes. As discussed above, proteins involved in mitochondrial fusion, division and degradation, including dynamin-related GTPases, bind to phospholipids in mitochondria. These interactions control the biochemical activity, assembly and localization of these proteins. Because the spatial pattern of phospholipids is dynamic and because phospholipid biosynthetic pathways are distributed among three different membranes (e.g., the ER membrane and the outer and inner mitochondrial membranes), phospholipid–protein interactions can generate a wide array of mitochondrial responses in different sub-mitochondrial locations in response to intracellular and environmental changes. Improving our understanding of the dynamic mitochondrial signaling network that consists of protein and lipid elements is an important future challenge. Understanding this complex mechanism will significantly advance our understanding of mitochondrial homeostasis and dynamics. At the molecular level, the amino acid sequences of mitochondrial dynamin-related GTPases indicate that these proteins lack known lipid-binding domains, such as the DAG, PI, PH, C2 and BAR domains. This finding is in clear contrast to endocytic dynamins, which contain a PH domain and bind to phosphoinositides. Therefore, the identification of lipid-binding domains and interaction mechanisms in mitochondrial dynamin-related GTPases is important.
Acknowledgments
We are grateful to a number of researchers who advanced our understanding of phospholipid biogenesis and mitochondrial fusion, division and mitophagy, and we apologize that we were not able to include all of the relevant studies due to space limitations. This work was supported by Grants from the NIH (GM084015 to MI and GM089853 and NS084154 to HS).
References
- 1.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19(5):757–766 [DOI] [PMC free article] [PubMed]
- 3.Sesaki H, Adachi Y, Kageyama Y, Itoh K, Iijima M (2013) In vivo functions of Drp1: Lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta. doi:10.1016/j.bbadis.2013.11.024 [DOI] [PMC free article] [PubMed]
- 4.Lackner LL. Determining the shape and cellular distribution of mitochondria: the integration of multiple activities. Curr Opin Cell Biol. 2013;25:471–476. doi: 10.1016/j.ceb.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto K, Shaw JM (2005) Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 39:503–536 [DOI] [PubMed]
- 7.Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 2013;1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shutt TE, McBride HM (2013) Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta 1833(2):417–424 [DOI] [PubMed]
- 10.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho DH, Nakamura T, Lipton SA. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci. 2010;67:3435–3447. doi: 10.1007/s00018-010-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal. 2013;19:400–414. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace DC. Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120267. doi: 10.1098/rstb.2012.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann NY Acad Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 19.Kageyama Y, Zhang Z, Sesaki H. Mitochondrial division: molecular machinery and physiological functions. Curr Opin Cell Biol. 2011;23:427–434. doi: 10.1016/j.ceb.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. 2003;14:2342–2356. doi: 10.1091/mbc.E02-12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 22.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- 24.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 25.Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 26.Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 28.Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol. 2001;152:1123–1134. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoppins S, Horner J, Song C, McCaffery JM, Nunnari J. Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J Cell Biol. 2009;184:569–581. doi: 10.1083/jcb.200809099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura Y, Itoh K, Sesaki H (2011) SnapShot: mitochondrial dynamics. Cell 145:1158, 1158.e151 [DOI] [PMC free article] [PubMed]
- 31.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammermeister M, Schodel K, Westermann B. Mdm36 is a mitochondrial fission-promoting protein in Saccharomyces cerevisiae . Mol Biol Cell. 2010;21:2443–2452. doi: 10.1091/mbc.E10-02-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerveny KL, Studer SL, Jensen RE, Sesaki H. Yeast mitochondrial division and distribution require the cortical num1 protein. Dev Cell. 2007;12:363–375. doi: 10.1016/j.devcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tieu Q, Okreglak V, Naylor K, Nunnari J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol. 2002;158:445–452. doi: 10.1083/jcb.200205031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev. 2011;67:103–118. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res. 2013;52:590–614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Kroon AI, Koorengevel MC, Goerdayal SS, Mulders PC, Janssen MJ, de Kruijff B. Isolation and characterization of highly purified mitochondrial outer membranes of the yeast Saccharomyces cerevisiae (method) Mol Membr Biol. 1999;16:205–211. doi: 10.1080/096876899294670. [DOI] [PubMed] [Google Scholar]
- 47.Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Z, Zou J. The initial step of the glycerolipid pathway: identification of glycerol 3-phosphate/dihydroxyacetone phosphate dual substrate acyltransferases in Saccharomyces cerevisiae. J Biol Chem. 2001;276:41710–41716. doi: 10.1074/jbc.M104749200. [DOI] [PubMed] [Google Scholar]
- 49.Pagac M, Vazquez HM, Bochud A, Roubaty C, Knopfli C, Vionnet C, Conzelmann A. Topology of the microsomal glycerol-3-phosphate acyltransferase Gpt2p/Gat1p of Saccharomyces cerevisiae. Mol Microbiol. 2012;86:1156–1166. doi: 10.1111/mmi.12047. [DOI] [PubMed] [Google Scholar]
- 50.Wendel AA, Lewin TM, Coleman RA. Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim Biophys Acta. 2009;1791:501–506. doi: 10.1016/j.bbalip.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leung DW. The structure and functions of human lysophosphatidic acid acyltransferases. Front Biosci. 2001;6:D944–D953. doi: 10.2741/leung. [DOI] [PubMed] [Google Scholar]
- 52.Benghezal M, Roubaty C, Veepuri V, Knudsen J, Conzelmann A. SLC1 and SLC4 encode partially redundant acyl-coenzyme A 1-acylglycerol-3-phosphate O-acyltransferases of budding yeast. J Biol Chem. 2007;282:30845–30855. doi: 10.1074/jbc.M702719200. [DOI] [PubMed] [Google Scholar]
- 53.Riekhof WR, Wu J, Jones JL, Voelker DR. Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2007;282:28344–28352. doi: 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- 54.Rose K, Rudge SA, Frohman MA, Morris AJ, Engebrecht J. Phospholipase D signaling is essential for meiosis. Proc Natl Acad Sci USA. 1995;92:12151–12155. doi: 10.1073/pnas.92.26.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han GS, O’Hara L, Siniossoglou S, Carman GM. Characterization of the yeast DGK1-encoded CTP-dependent diacylglycerol kinase. J Biol Chem. 2008;283:20443–20453. doi: 10.1074/jbc.M802866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai J, Abramovici H, Gee SH, Topham MK. Diacylglycerol kinases as sources of phosphatidic acid. Biochim Biophys Acta. 2009;1791:942–948. doi: 10.1016/j.bbalip.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 58.Hermansson M, Hokynar K, Somerharju P. Mechanisms of glycerophospholipid homeostasis in mammalian cells. Prog Lipid Res. 2011;50:240–257. doi: 10.1016/j.plipres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Shen H, Dowhan W. Reduction of CDP-diacylglycerol synthase activity results in the excretion of inositol by Saccharomyces cerevisiae. J Biol Chem. 1996;271:29043–29048. doi: 10.1074/jbc.271.46.29043. [DOI] [PubMed] [Google Scholar]
- 60.Tamura Y, Harada Y, Nishikawa S, Yamano K, Kamiya M, Shiota T, Kuroda T, Kuge O, Sesaki H, Imai K, et al. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013;17:709–718. doi: 10.1016/j.cmet.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inglis-Broadgate SL, Ocaka L, Banerjee R, Gaasenbeek M, Chapple JP, Cheetham ME, Clark BJ, Hunt DM, Halford S. Isolation and characterization of murine Cds (CDP-diacylglycerol synthase) 1 and 2. Gene. 2005;356:19–31. doi: 10.1016/j.gene.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 62.Nikawa J, Yamashita S. Molecular cloning of the gene encoding CDPdiacylglycerol-inositol 3-phosphatidyl transferase in Saccharomyces cerevisiae. Eur J Biochem. 1984;143:251–256. doi: 10.1111/j.1432-1033.1984.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 63.Letts VA, Klig LS, Bae-Lee M, Carman GM, Henry SA. Isolation of the yeast structural gene for the membrane-associated enzyme phosphatidylserine synthase. Proc Natl Acad Sci USA. 1983;80:7279–7283. doi: 10.1073/pnas.80.23.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang SC, Heacock PN, Clancey CJ, Dowhan W. The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J Biol Chem. 1998;273:9829–9836. doi: 10.1074/jbc.273.16.9829. [DOI] [PubMed] [Google Scholar]
- 65.Osman C, Haag M, Wieland FT, Brugger B, Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 2010;29:1976–1987. doi: 10.1038/emboj.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Houtkooper RH, Akbari H, van Lenthe H, Kulik W, Wanders RJ, Frentzen M, Vaz FM. Identification and characterization of human cardiolipin synthase. FEBS Lett. 2006;580:3059–3064. doi: 10.1016/j.febslet.2006.04.054. [DOI] [PubMed] [Google Scholar]
- 68.Chang SC, Heacock PN, Mileykovskaya E, Voelker DR, Dowhan W. Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem. 1998;273:14933–14941. doi: 10.1074/jbc.273.24.14933. [DOI] [PubMed] [Google Scholar]
- 69.Tuller G, Hrastnik C, Achleitner G, Schiefthaler U, Klein F, Daum G. YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae . FEBS Lett. 1998;421:15–18. doi: 10.1016/s0014-5793(97)01525-1. [DOI] [PubMed] [Google Scholar]
- 70.Chen D, Zhang XY, Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J. 2006;398:169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu B, Xu FY, Jiang YJ, Choy PC, Hatch GM, Grunfeld C, Feingold KR. Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1) J Lipid Res. 2006;47:1140–1145. doi: 10.1194/jlr.C600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 72.Tomohiro S, Kawaguti A, Kawabe Y, Kitada S, Kuge O. Purification and characterization of human phosphatidylserine synthases 1 and 2. Biochem J. 2009;418:421–429. doi: 10.1042/BJ20081597. [DOI] [PubMed] [Google Scholar]
- 73.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 74.Clancey CJ, Chang SC, Dowhan W. Cloning of a gene (PSD1) encoding phosphatidylserine decarboxylase from Saccharomyces cerevisiae by complementation of an Escherichia coli mutant. J Biol Chem. 1993;268:24580–24590. [PubMed] [Google Scholar]
- 75.Trotter PJ, Voelker DR. Identification of a non-mitochondrial phosphatidylserine decarboxylase activity (PSD2) in the yeast Saccharomyces cerevisiae . J Biol Chem. 1995;270:6062–6070. doi: 10.1074/jbc.270.11.6062. [DOI] [PubMed] [Google Scholar]
- 76.Gulshan K, Shahi P, Moye-Rowley WS. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol Biol Cell. 2010;21:443–455. doi: 10.1091/mbc.E09-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kodaki T, Yamashita S. Yeast phosphatidylethanolamine methylation pathway. Cloning and characterization of two distinct methyltransferase genes. J Biol Chem. 1987;262:15428–15435. [PubMed] [Google Scholar]
- 78.Kodaki T, Yamashita S. Characterization of the methyltransferases in the yeast phosphatidylethanolamine methylation pathway by selective gene disruption. Eur J Biochem. 1989;185:243–251. doi: 10.1111/j.1432-1033.1989.tb15109.x. [DOI] [PubMed] [Google Scholar]
- 79.Schuiki I, Daum G. Phosphatidylserine decarboxylases, key enzymes of lipid metabolism. IUBMB Life. 2009;61:151–162. doi: 10.1002/iub.159. [DOI] [PubMed] [Google Scholar]
- 80.Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2013;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 81.Gibellini F, Smith TK. The Kennedy pathway––De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010;62:414–428. doi: 10.1002/iub.337. [DOI] [PubMed] [Google Scholar]
- 82.Lykidis A. Comparative genomics and evolution of eukaryotic phospholipid biosynthesis. Prog Lipid Res. 2007;46:171–199. doi: 10.1016/j.plipres.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 83.Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J Biol Chem. 2006;281:9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal. 2008;10:621–633. doi: 10.1089/ars.2007.1934. [DOI] [PubMed] [Google Scholar]
- 85.Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, et al. MITOL regulates endoplasmic reticulum–mitochondria contacts via Mitofusin2. Mol Cell. 2013;51:20–34. doi: 10.1016/j.molcel.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 86.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum–mitochondria connections. Proc Natl Acad Sci USA. 2011;108:14151–14156. doi: 10.1073/pnas.1111314108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nguyen T, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA, Shaw JM. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13:880–890. doi: 10.1111/j.1600-0854.2012.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tamura Y, Endo T, Iijima M, Sesaki H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol. 2009;185:1029–1045. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tamura Y, Iijima M, Sesaki H. Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J. 2010;29:2875–2887. doi: 10.1038/emboj.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Potting C, Wilmes C, Engmann T, Osman C, Langer T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010;29:2888–2898. doi: 10.1038/emboj.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Potting C, Tatsuta T, Konig T, Haag M, Wai T, Aaltonen MJ, Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013;18:287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 93.Sesaki H, Dunn CD, Iijima M, Shepard KA, Yaffe MP, Machamer CE, Jensen RE. Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol. 2006;173:651–658. doi: 10.1083/jcb.200603092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol. 2009;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science. 2012;338:815–818. doi: 10.1126/science.1225625. [DOI] [PubMed] [Google Scholar]
- 96.Tamura Y, Onguka O, Hobbs AE, Jensen RE, Iijima M, Claypool SM, Sesaki H. Role for two conserved intermembrane space proteins, Ups1p and Ups2p, [corrected] in intra-mitochondrial phospholipid trafficking. J Biol Chem. 2012;287:15205–15218. doi: 10.1074/jbc.M111.338665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tamura Y, Onguka O, Itoh K, Endo T, Iijima M, Claypool SM, Sesaki H. Phosphatidylethanolamine Biosynthesis in Mitochondria: phosphatidylserine (PS) trafficking is independent of a PS decarboxylase and intermembrane space proteins UPS1P and UPS2P. J Biol Chem. 2012;287:43961–43971. doi: 10.1074/jbc.M112.390997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ammar MR, Kassas N, Chasserot-Golaz S, Bader MF, Vitale N. Lipids in regulated exocytosis: what are they doing? Front Endocrinol (Lausanne) 2013;4:125. doi: 10.3389/fendo.2013.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 100.Ohba Y, Sakuragi T, Kage-Nakadai E, Tomioka NH, Kono N, Imae R, Inoue A, Aoki J, Ishihara N, Inoue T, et al. Mitochondria-type GPAT is required for mitochondrial fusion. EMBO J. 2013;32:1265–1279. doi: 10.1038/emboj.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gimeno RE, Cao J. Thematic review series: glycerolipids. Mammalian glycerol-3-phosphate acyltransferases: new genes for an old activity. J Lipid Res. 2008;49:2079–2088. doi: 10.1194/jlr.R800013-JLR200. [DOI] [PubMed] [Google Scholar]
- 102.Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol. 2004;165:167–173. doi: 10.1083/jcb.200403022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rujiviphat J, Meglei G, Rubinstein JL, McQuibban GA. Phospholipid association is essential for dynamin-related protein Mgm1 to function in mitochondrial membrane fusion. J Biol Chem. 2009;284:28682–28686. doi: 10.1074/jbc.M109.044933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abutbul-Ionita I, Rujiviphat J, Nir I, McQuibban GA, Danino D. Membrane tethering and nucleotide-dependent conformational changes drive mitochondrial genome maintenance (Mgm1) protein-mediated membrane fusion. J Biol Chem. 2012;287:36634–36638. doi: 10.1074/jbc.C112.406769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sesaki H, Southard SM, Hobbs AE, Jensen RE. Cells lacking Pcp1p/Ugo2p, a rhomboid-like protease required for Mgm1p processing, lose mtDNA and mitochondrial structure in a Dnm1p-dependent manner, but remain competent for mitochondrial fusion. Biochem Biophys Res Commun. 2003;308:276–283. doi: 10.1016/s0006-291x(03)01348-2. [DOI] [PubMed] [Google Scholar]
- 107.McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature. 2003;423:537–541. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- 108.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19:2113–2122. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19:2113–2122. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, Liu X, Jefferson LS, Xiong J, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li J, Liu X, Wang H, Zhang W, Chan DC, Shi Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc Natl Acad Sci USA. 2012;109:6975–6980. doi: 10.1073/pnas.1120043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kung C. A possible unifying principle for mechanosensation. Nature. 2005;436:647–654. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 118.Chan EY, McQuibban GA. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1) J Biol Chem. 2012;287:40131–40139. doi: 10.1074/jbc.M112.399428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae . J Biol Chem. 2012;287:17589–17597. doi: 10.1074/jbc.M111.330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49 and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24(5):659–667 [DOI] [PMC free article] [PubMed]
- 122.Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, Sesaki H, Kvansakul M, Ryan MT. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol. 2014;204:477–486. doi: 10.1083/jcb.201311014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA. 2013;110:E1342–E1351. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. 2014;22:367–377. doi: 10.1016/j.str.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lackner LL, Ping H, Graef M, Murley A, Nunnari J. Endoplasmic reticulum-associated mitochondria–cortex tether functions in the distribution and inheritance of mitochondria. Proc Natl Acad Sci USA. 2013;110:E458–E467. doi: 10.1073/pnas.1215232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cerveny KL, Studer SL, Jensen RE, Sesaki H. Yeast mitochondrial division and distribution require the cortical num1 protein. Dev Cell. 2007;12:363–375. doi: 10.1016/j.devcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 128.Hammermeister M, Schodel K, Westermann B. Mdm36 is a mitochondrial fission-promoting protein in Saccharomyces cerevisiae . Mol Biol Cell. 2010;21:2443–2452. doi: 10.1091/mbc.E10-02-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cerveny KL, McCaffery JM, Jensen RE (2001) Division of mitochondria requires a noverl DNM1-interacting protien, Net2p. Mol Biol Cell 12(2):309–321 [DOI] [PMC free article] [PubMed]
- 130.Lackner LL, Horner JS, Nunnari J. Mechanistic analysis of a dynamin effector. Science. 2009;325:874–877. doi: 10.1126/science.1176921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yoon Y, Pitts KR, McNiven MA. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell. 2001;12:2894–2905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 136.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wells RC, Hill RB. The cytosolic domain of Fis1 binds and reversibly clusters lipid vesicles. PLoS One. 2011;6:e21384. doi: 10.1371/journal.pone.0021384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bayir H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, Zhao Q, Belikova NA, Vlasova II, Maeda A, et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome c: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem. 2009;284:15951–15969. doi: 10.1074/jbc.M900418200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cosentino K, Garcia-Saez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014;181C:62–75. doi: 10.1016/j.chemphyslip.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 142.Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 143.Zhang T, Saghatelian A. Emerging roles of lipids in BCL-2 family-regulated apoptosis. Biochim Biophys Acta. 2013;1831:1542–1554. doi: 10.1016/j.bbalip.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 144.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol CB. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 150.Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci USA. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]