

Abstract
Objective
Indirect evidence suggests that lateralized changes in motoneuron behavior post-stroke are potentially due to a depolarizing supraspinal drive to the motoneuron pool, but the pathways responsible are unknown. In this study, we assessed vestibular evoked myogenic potentials (VEMPs) in the neck muscles of hemispheric stroke survivors with contralesional spasticity to quantify the relative levels of vestibular drive to the spastic-paretic and contralateral motoneuron pools.
Methods
VEMPs were recorded from each sternocleidomastoid muscle in chronic stroke survivors. Side-to-side differences in cVEMP amplitude were calculated and expressed as an asymmetry ratio, a proxy for the relative amount of vestibular drive to each side.
Results
Spastic-paretic VEMPs were larger than contralateral VEMPs in 13/16 subjects. There was a strong positive relationship between the degree of asymmetry and the severity of spasticity in this subset of subjects. Remaining subjects had larger contralateral responses.
Conclusion
Vestibular drive to cervical motoneurons is asymmetric in spastic stroke survivors, supporting our hypothesis that there is an imbalance in descending vestibular drive to motoneuron pools post-stroke. We speculate this imbalance is a consequence of the unilateral disruption of inhibitory corticobulbar projections to the vestibular nuclei.
Significance
This study sheds new light on the underlying mechanisms of post-stroke spasticity.
Keywords: Hemispheric stroke, post-stroke spasticity, chronic stroke subjects, vestibulospinal, human, motoneuron, vestibular evoked myogenic potentials
1. Introduction
Spastic hypertonia or “spasticity” is a frequent and often disabling sequel to hemispheric stroke (Watkins et al., 2002, Urban et al., 2010). It is a motor disorder, manifesting as a sharply lateralized muscular hypertonia on the contralesional side with exaggerated phasic and tonic stretch reflex activity (Lance, 1980). Clinically, spasticity presents as an increase in the resistance of a passive limb to externally applied joint motion and is commonly associated with deficits in both motor and functional performance (Bohannon et al., 1987, O'Dwyer et al., 1996, Watkins et al., 2002, Sommerfeld et al., 2004). Emerging evidence implicates changes in motoneuron excitability as central to the genesis of post-stroke spasticity, however the physiological basis underlying the lateralized hyperexcitability of spastic-paretic motoneurons is incompletely understood (Katz and Rymer, 1989), due in part to the lack of a widely accepted animal model of hemispheric stroke (Wright and Rang, 1990). However, one plausible explanation for the sharply lateralized nature of spasticity is that spastic-paretic motoneurons receive a tonic increase in excitatory synaptic drive generated by a sharply lateralized supraspinal pathway released from cortical suppression following a stroke mediated disruption of inhibitory corticobulbar projections (Katz and Rymer, 1989).
Previous studies provide indirect evidence suggesting that post-stroke changes in spastic-paretic motoneuron behavior are likely due to a depolarizing supraspinal drive to the resting spastic-paretic motoneuron pool (Burke and Ashby, 1972, Burke et al., 1972, Burne et al., 2005, Mottram et al., 2009). Mottram et al. (2009) showed that there was a greater incidence of co-modulation in the spontaneous discharge of spastic-paretic motor units that was not seen on the clinically spared side. In addition, they found a lack of initial acceleration in firing rate following motor unit recruitment in low threshold spastic-paretic motor units, a potential marker of a depolarized baseline membrane potential. While not definitive, both findings are highly suggestive of a common, low-level depolarizing drive to the spastic-paretic motoneuron pool. However, the specific supraspinal pathways that mediate these changes are unknown. The candidate supraspinal pathway or pathways mediating these changes should be sharply lateralized, project to the antigravity muscle groups, and be able to strongly modulate motoneuron excitability.
In humans, the major supraspinal pathways that influence motoneuron excitability arise from the brainstem, and are the reticulospinal, rubrospinal, and vestibulospinal pathways. The reticulospinal pathways are largely bilateral in both their anatomical spinal distribution (Nyberg-Hansen, 1965, Nathan et al., 1996) and synaptic action (Schepens and Drew, 2006, Davidson et al., 2007). Rubrospinal pathways are believed to be rudimentary in humans, with few fibers reaching the spinal cord (Nathan and Smith, 1982). Alternatively, the unilateral nature of the vestibular pathways, especially the lateral vestibulospinal tract is consistent with the sharply lateralized nature of spasticity (Nyberg-Hansen and Mascitti, 1964).
Vestibulospinal pathways have been long implicated as a prime driver of decerebrate rigidity, which occurs following transection of the brainstem between the red nucleus and vestibular nuclei. In quadrupeds, a hallmark of decerebrate rigidity is rigid extension of the limbs (decerebrate posturing) accompanied by hyperactive reflexes that appear to be driven by unopposed vestibulospinal drive to the antigravity motoneuron pools (Fulton et al., 1930, Bach and Magoun, 1947). While not a precise analog for decerebrate posturing, the antigravity limb posturing that follows an upper motoneuron lesion is modified though postural changes and abolished following VIII nerve transection (Denny-Brown, 1964, 1965), supporting a potential contributing role for vestibulospinal projections. Vestibulospinal drive is also an important facilitator of the tonic vibration reflex (Gillies et al., 1971a, b), a useful analog for the tonic stretch reflex (Kimura, 2013). Surgical transection of the vestibular nerve or interruption of central vestibular pathways results in a marked decrease in the tonic vibration reflex (Gillies et al., 1971a, b), motoneuron excitability (Molina-Negro et al., 1980) and antigravity muscle tone (Bach and Magoun, 1947), highlighting the potentially unique contributions of the vestibular nuclei.
While there are known deficits in vestibular functioning subsequent to stroke (Marsden et al., 2005), the role descending vestibular pathways play in generating lateralized spasticity in humans has yet to be comprehensively investigated. For these reasons, the aim of the current study was to quantify the relative levels of descending vestibular drive to spastic-paretic and clinically-spared or contralateral motoneuron pools in chronic stroke survivors with demonstrable spasticity. Specifically, we hypothesized that there would be asymmetries in the amplitudes of the cervical vestibular evoked myogenic potentials (cVEMPs) elicited in the spastic-paretic and contralateral sternocleidomastoid muscles, due to a disruption of inhibitory corticobulbar projections to the contralesional vestibular nuclei. The degree of asymmetry in cVEMP amplitude between the two sides was used as a proxy for the relative amount of vestibular drive impinging onto the spastic-paretic and contralateral motoneuron pools. We propose that the lateralized disruption of corticobulbar projections causes an imbalance in descending vestibular drive to the motoneuron pools that sets the baseline membrane potential of spastic-paretic motoneurons closer to neuron activation threshold.
2. Methodology
In order to differentiate between the two sides, the sternocleidomastoid muscle on the clinically affected side will be classified as spastic-paretic and the sternocleidomastoid muscle on the clinically spared side will be classified as contralateral. Additionally, the vestibular nuclear complex on the spastic-paretic side will be referred to as the contralesional vestibular nuclei while the vestibular nuclear complex on the contralateral side will be referred to as the ipsilesional vestibular nuclei.
2.1 Subject population
Informed, written consent was obtained prior to experimentation. The Northwestern University Institutional Review Board approved all experimental procedures. We enrolled 17 chronic stroke survivors (115.7 ± 105.9 months post stroke) ranging in age from 44-71 (56.5 ± 7.0 years) with a single brain lesion that resulted in lateralized spasticity in at least one limb. Clinical assessments for each subject were performed by a dedicated research physical therapist. Spasticity was assessed in both the elbow flexors and plantar flexors using the modified Ashworth scale (MAS), a 6-point rating scale used to measure passive muscle resistance (Bohannon and Smith, 1987). For each subject, we calculated an antigravity spasticity index (AGSI) by dividing the sum of the MAS in the elbow flexors and plantar flexors by eight; the maximum possible attainable MAS score. Subject demographic and clinical information is detailed in Table 1.
Table 1.
Subject demographic and clinical information from 17 chronic stroke survivors.
| Subject ID | Sex | Age, yr | Months Post Stroke | Antigravity Spasticity Index | Lesion Location | Paretic Side/Hand Dominance |
|---|---|---|---|---|---|---|
| 2 | F | 52 | 33 | 0.125 | L BG | R / R |
| 3 | F | 52 | 302 | 0.750 | R MCA | L / R |
| 4 | M | 56 | 68 | 0.375 | L BG - lateral thalamic | R / R |
| 5 | F | 62 | 226 | 0.375 | R BG | L / R |
| 6 | M | 52 | 45 | 0.500 | R Carotid dissection | L / L |
| 7 | M | 51 | 39 | 0.188 | R MCA | L / L |
| 8 | M | 66 | 99 | 0.625 | R Periatrial WM - L Medial Temporal | L / R |
| 9 | M | 48 | 16 | 0.313 | L IC - BG - centrum semi-ovale | R / L |
| 10 | M | 62 | 225 | 0.375 | L MCA | R / R |
| 11 | F | 54 | 84 | 0.750 | R BG | L / R |
| 12 | F | 61 | 154 | 0.375 | R IC | L / R |
| 13 | M | 56 | 35 | 0.563 | R IC - corona radiata | L / R |
| 14 | F | 58 | 301 | 0.438 | L Carotid artery | R / R |
| 15 | F | 64 | 24 | 0.625 | R BG | L / R |
| 16 | M | 52 | 35 | 0.625 | L Thalamic | R / L |
| 17 | F | 71 | 265 | 0.375 | L BG - centrum semiovale | R / R |
| 18 | F | 44 | 16 | 0.438 | R IC - thalamic - corona radiata | L / R |
Abbreviations: MCA, middle cerebral artery; BG, basal ganglia; WM, white matter; IC, internal capsule.
Notes: Subject 1 was excluded because of comorbid conditions. Notch filtering was used on both sides of subject 13 due to 60 Hz noise.
It should be noted that while subject 8 had a chronic focal lesion in the right periatrial white matter and a chronic microhemmorhage in the left medial temporal lobe, this subject presented with lateralized spasticity in both the upper and lower limbs on the left side. The left sided spasticity was presumably an outcome of the right-sided white matter lesion.
At the time of the study, no subject was taking any vestibular suppressants or medications that would influence motoneuron excitability, such as SSRIs, benzodiazepines, or GABA analogues. To exclude significant hearing loss, hearing sensitivity was assessed at 500 Hz using pure-tone audiometry as the cVEMP amplitude is dependent upon ossicle integrity. Subjects were excluded if they had greater than a 10 dB difference between the left and right ears. Additionally, all subjects reported a negative history of vestibular disorders or prior neurological dysfunction.
2.2 Data collection
Surface electromyographic (EMG) activity was recorded from the spastic-paretic and contralateral sternocleidomastoid muscles. Prior to electrode placement, the sternocleidomastoid muscles and forehead were prepped using an abrasive alcohol pad. Disposable silver/silver chloride electrodes were placed symmetrically over the middle third of each sternocleidomastoid muscle (Sheykholeslami et al., 2001) and at the sternoclavicular notch. A single ground electrode was placed in the center of the forehead. EMG was sampled over a 150 ms epoch, from 50 ms prior to 100 ms after stimulus onset (DAM 50 Differential Amplifier, World Precision Instruments, Sarasota, FL). EMG data were amplified ×1000, band-pass filtered 10-1,000 Hz and sampled at 7936.5 Hz using a Power 1401 analog to digital converter linked to a PC running Spike 2 software (Cambridge Electrode Design, Cambridge, UK). Data sets were analyzed offline using customized software programs (Spike 2; Matlab, Mathworks Inc., Natick, MA).
Subjects lay supine on an examination table with their torso centered on a foam wedge that elevated the torso. Figure 1 depicts the subject positioning and electrode placement. Each subject was instructed to maintain a moderate level contraction in the sternocleidomastoid muscle by raising their head against gravity while a train of auditory stimuli was delivered. In some cases, in order to match the level of EMG activation between the spastic-paretic and contralateral sides, it was necessary to instruct the subject to lift and turn their head slightly in the direction opposite (towards) to the side being stimulated. This allowed the subject to generate comparable levels of background EMG between the two sides without compromising the cVEMP amplitude (Ito et al., 2007). Subjects were given brief rest periods between trials in order to reduce fatigue which could influence the cVEMP threshold. Testing occurred in one session that lasted approximately two hours.
Figure 1.
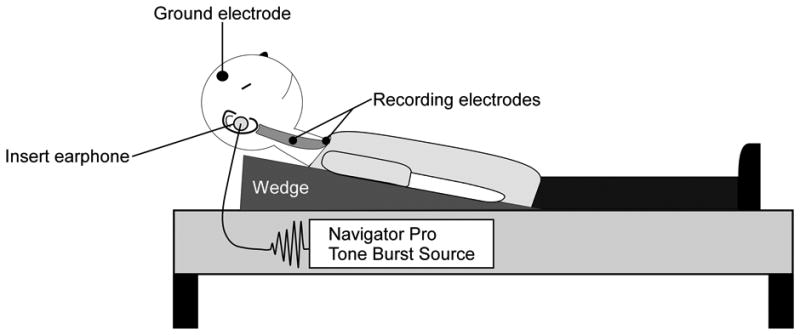
Cervical VEMPs were recorded from the tonically active sternocleidomastoid muscles in 17 chronic stroke subjects. Subjects lay supine with surface electrodes placed bilaterally over the middle third of the sternocleidomastoid muscles and at the sternoclavicular notch. A ground electrode was placed on the forehead. To activate their sternocleidomastoid, subjects lifted their head against gravity while monaural acoustic stimuli were presented through insert earphones placed snugly in the ear canal.
Acoustic stimuli were generated using the Biologic Navigator Pro Auditory Evoked Potential system (Natus Medical, San Carlos, CA). Each trial consisted of a train of 128 short tone bursts delivered monaurally at a repetition rate of 5/s with an impulse intensity of 95 dB nHL (130 dB SPL) via standard foam EA-3 insert earphones. Each 500 Hz short tone burst was presented with rarefaction polarity and Blackman ramping (2 ms plateau and 1 ms rise and fall times). Previous studies have demonstrated that the above stimulus parameters produce robust and reliable cVEMPs (Young, 2006).
2.3 Data and statistical analysis
The cVEMP is a vestibular-dependent biphasic surface potential representing a modulation of sternocleidomastoid motoneuron discharge (Colebatch and Rothwell, 2004). The interpeak amplitude (μV), peak latencies (ms), and the interpeak interval (ms) were measured from the unrectified EMG stimulus triggered waveform average ipsilateral to the stimulated ear (Fig. 2). The peak latencies were calculated with respect to stimulus onset and were defined as p13 and n23. A peak was labeled p13 if it occurred between 10 and 20 ms and exceeded two standard deviation above baseline based on the pre-stimulus EMG during a 50 ms period. The subsequent peak of opposite polarity immediately following p13 was designated as n23. The interpeak amplitude was calculated as the difference been the p13 and n23 peaks. The interpeak interval was calculated by subtracting the latency of p13 from n23.
Figure 2.
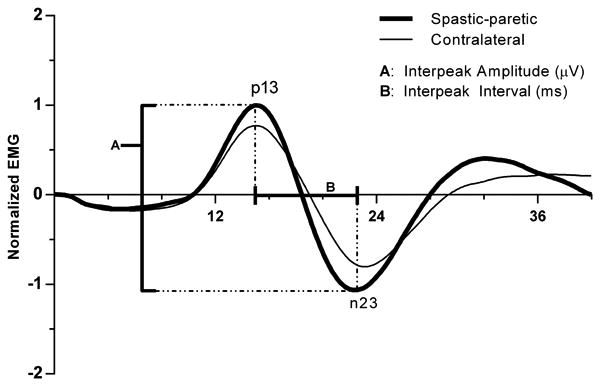
Population spastic-paretic and contralateral cVEMPs recorded in response to monaural acoustic stimulation (128 short tone bursts; 500 Hz; 95 dB nHL; 1 ms rise/fall; 2 ms plateau; 5/s). A peak was labeled p13 if it occurred between 10 and 20 ms and exceeded two standard deviations above baseline based on the pre-stimulus unrectified EMG (50 ms). The subsequent peak of opposite polarity immediately following p13 was designated as n23. The interpeak amplitude (A) was calculated as the difference been the p13 and n23 peaks. The interpeak interval (B) was calculated by subtracting the latency of p13 from n23. To normalize, the interpeak amplitude was divided by the mean rectified pre-stimulus EMG (50 ms).
At least two waveforms were obtained from each spastic-paretic and contralateral sternocleidomastoid muscle in response to ipsilateral acoustic stimulation, with each waveform constituted a single trial consisting of 128 short tone bursts. For each subject, one trial was selected from each side that minimized the percent difference in mean pre-stimulus rectified EMG (50ms) between the two sides. As the relationship between background EMG level and the interpeak amplitude is linear (Colebatch et al., 1994), normalized corrected interpeak amplitudes were calculated by dividing the interpeak amplitude by the mean pre-stimulus rectified EMG (50ms) which corrected for differences in muscle activation between trials.
Side-to-side differences in descending vestibular drive were quantified by expressing the spastic-paretic and contralateral corrected interpeak amplitudes (CA) as an asymmetry ratio (AR, %). The asymmetry ratio was defined as 100 × ((spastic CA − contralateral CA) ÷ (spastic CA + contralateral CA)). Positive asymmetry ratios indicate larger cVEMPs and thus greater drive to the spastic motoneuron pool while negative values indicate larger cVEMPs on the contralateral side. Finally, we calculated the Con/Spa ratio, a measure of the contralateral corrected interpeak amplitude relative to the spastic corrected interpeak amplitude.
Data are expressed in terms of mean ± standard deviation, unless otherwise noted. The corrected interpeak amplitude, peak latencies, and interpeak interval from each subject were averaged to calculate population means. A paired two-tailed Student's t-test was used to analyze any significant differences in the spastic-paretic and contralateral corrected interpeak amplitudes, the spastic-paretic and contralateral peak latencies, and the spastic-paretic and contralateral interpeak intervals. The relation between (1) the asymmetry ratio and antigravity spasticity index and the relation between (2) the Con/Spa ratio and antigravity spasticity index were calculated using least-squares linear regression analysis. To compare the spastic-paretic and contralateral background EMG levels, we used the Wilcoxon matched-pairs signed rank test. For statistical comparisons, a p-value ≤ 0.05 was considered to be significant. Statistical analysis was performed using Prism 6 (Graph Pad Software, Inc., La Jolla, CA USA).
3. Results
Descending vestibular drive to the spastic-paretic and contralateral sternocleidoma stoid motoneuron pools was asymmetric and in the majority of subjects, the degree of asymmetry between the two sides was strongly correlated with the severity of spasticity. Monaural acoustic stimulation elicited cVEMPs on both the spastic-paretic and contralateral sides in response to ipsilateral stimulation in 16/17 (94%) subjects (Fig. 3). Of the 16 subjects that had bilateral cVEMPs, 13 subjects had cVEMPs that were larger on the spastic-paretic side whereas 3 subjects had cVEMPs that were larger on the contralateral side. The remaining subject (subject 7: age 51, 0.188 AGSI, R MCA stroke) lacked consistent responses on the contralateral side and was excluded from any further statistical analysis.
Figure 3.
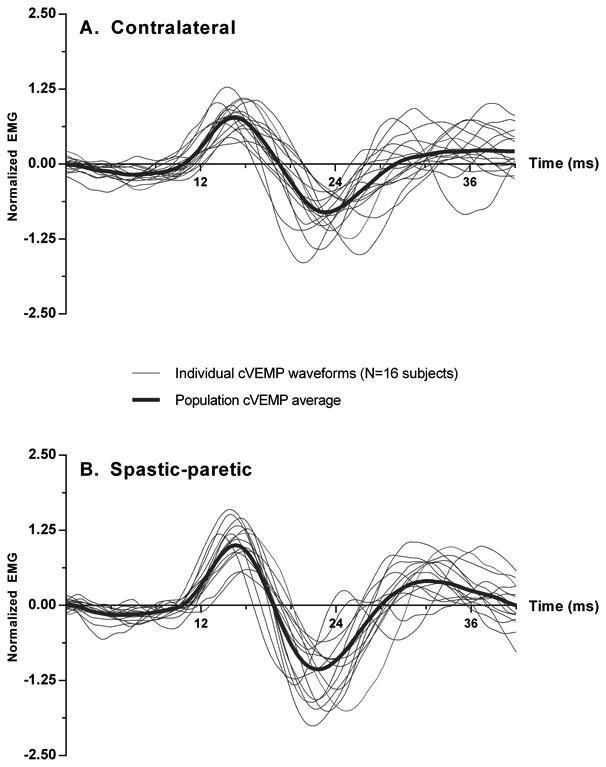
Individual normalized cVEMPs (thin lines) obtained from the contralateral (A) and spastic-paretic (B) sternocleidomastoid muscles of sixteen chronic stroke subjects. Each individual trace represents an average over 128 consecutive short tone burst repetitions. The spastic-paretic and contralateral population averages are shown (thick line).
The individual cVEMPs elicited on the spastic-paretic and contralateral sides from the 16 subjects with bilateral responses are superimposed and depicted in Figure 3. Morphologically, the cVEMPs were similar in shape, latency and duration to those previously reported in the literature (Basta et al., 2005). Comparisons of the mean corrected interpeak amplitudes revealed significant differences between the contralateral and spastic-paretic sides. The population cVEMPs for the contralateral and spastic-paretic cVEMPs are depicted in Figure 2. The mean corrected interpeak amplitudes for the contralateral and spastic-paretic sides were 1.83 ± 0.58 (range: 0.84-2.93; 95% confidence interval [CI]: 1.52-2.13) and 2.31 ± 0.81 (range: 0.80-3.60; 95% CI: 1.88-2.74), respectively (Fig. 4). Overall, the population spastic-paretic corrected interpeak amplitude was significantly larger than the population contralateral corrected interpeak amplitude (n=16, 2-tailed paired t-test, p=0.0480).
Figure 4.
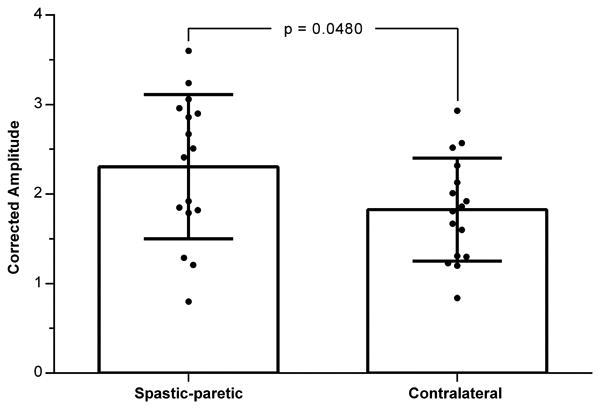
Spastic-paretic and contralateral averaged corrected amplitudes from 16 subjects. Mean and standard deviation are given expressed as the ratio of the interpeak amplitude divided by the mean rectified prestimulus EMG (50 ms). The individual spastic-paretic and contralateral corrected amplitudes for each subject are represented by a filled circle. Significant results are marked with an asterisk.
To illustrate the relationship between the vestibular reflex asymmetries and the degree of spasticity, the subjects were broken into four groups based on the magnitude of their antigravity spasticity index - minimum, mild, moderate, and severe. Representative responses from each group are shown in Figure 5. Next, the individual asymmetry ratios from the 13 subjects with positive asymmetry ratios were plotted as a function of the antigravity spasticity index (Fig. 6). We found that the asymmetry ratio was significantly and positively related to the antigravity spasticity index, a measure of spasticity severity (linear regression: p=0.0034; R2=0.5570). The linear regression equation that describes the data set is indicated in the upper left-hand portion of the figure.
Figure 5.
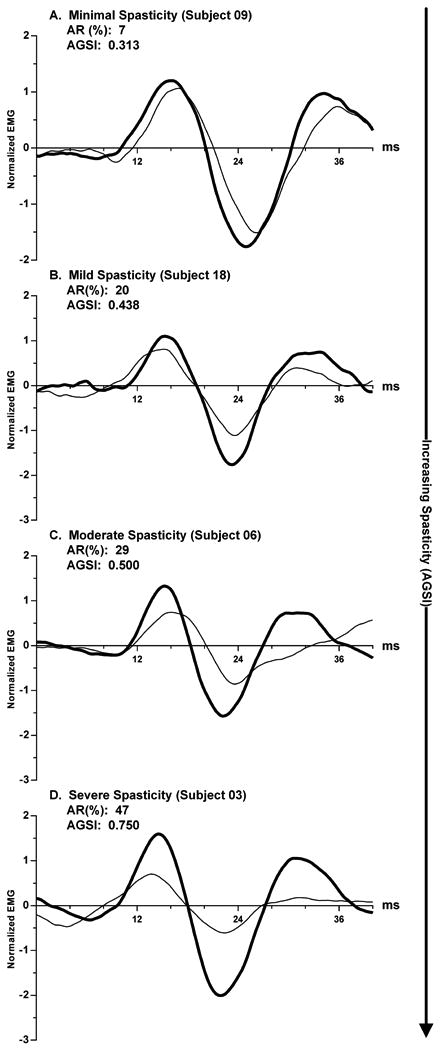
Representative spastic-paretic (bold) and contralateral (thin) cVEMPs from 4 subjects are ranked ordered as a function of increasing severity of spasticity (minimal, mild, moderate and severe). Asymmetry ratios (AR) and the antigravity spasticity index (AGSI) are indicated for each subject.
Figure 6.
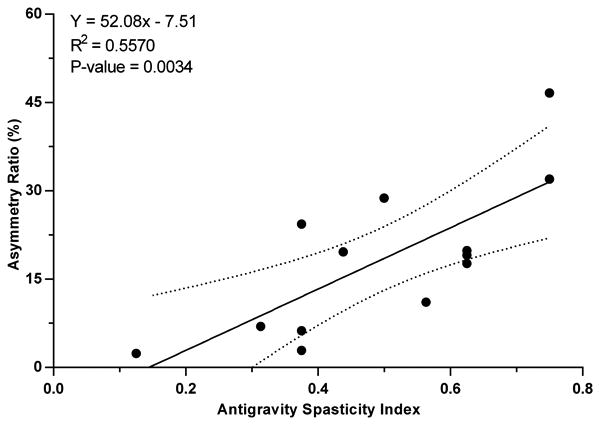
For the 13 subjects with positive asymmetry ratios there is a significant increase in the degree of cVEMP amplitude asymmetry between the spastic-paretic and contralateral sides with increasing severity of spasticity, indicated by the antigravity spasticity index. The linear regression line and ± 95% confidence intervals (dotted lines) are shown. The regression equation, coefficient of determination, and p-value are indicated in the upper left of the figure.
The mean Con/Spa ratio for the 13 subjects with positive asymmetry ratios was 0.71 ± 0.18 (range: 0.36-0.95; 95% CI: 0.60-0.82). The Con/Spa ratio was plotted as a function of the antigravity spasticity index (Fig. 7) for each of the subjects with a positive asymmetry ratio. There was a significant negative relationship between the Con/Spa ratio and the antigravity spasticity index (linear regression: p=0.0023; R2=0.5866). In this subset of subjects, with increasing severity of spasticity the relative size of the contralateral corrected interpeak amplitude decreased with respect to the spastic-paretic corrected interpeak amplitude. The linear regression equation that describes the data set is indicated in the upper right-hand portion of the figure.
Figure 7.
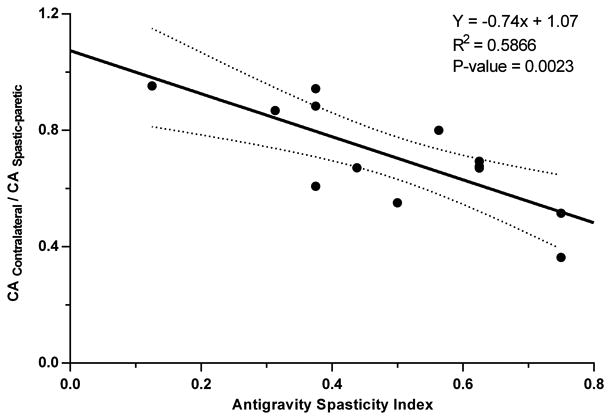
The relationship between the Con-Spa ratio and the severity of spasticity quantified using the AGSI. For the 13 subjects with positive asymmetry ratios, as the severity of spasticity increases there is a significant decrease in the Con-Spa ratio, a measure of the contralateral corrected amplitude relative to the spastic-paretic corrected amplitude. The linear regression line and ± 95% confidence intervals (dotted lines) are shown. The regression equation, coefficient of determination, and p-values are indicated in the upper right of the figure.
The spastic-paretic and contralateral mean prestimulus background EMG values (N=16 subjects) were 125.40 ± 66.90 μV (range: 38.52-347.65 μV; 95% CI: 89.75-161.05 μV) and 138.13 ± 74.89 μV (range: 39.98-340.23 μV; 95% CI: 98.22-178.03 μV) respectively. The mean spastic-paretic background EMG level was significantly smaller than the mean contralateral background EMG level (Wilcoxon matched-pairs signed rank test, p-value=0.0131). However, as the interpeak amplitude is linearly related to the tonic background activity, by normalizing the interpeak amplitude a valid inter-subject comparison of cVEMP amplitudes was possible (Colebatch et al., 1994). Within individual subjects, the tonic background EMG levels between the spastic-paretic and contralateral sternocleidomastoid muscles were closely matched. Additionally, we selected trials that minimized the percent difference in background EMG activity between the spastic-paretic and contralateral sides. The mean percent difference between the spastic-paretic and contralateral pre-stimulus rectified EMG (50ms) was 9.84 ± 14.40 % (range: 0.10-56.10; 95% CI: 2.19-17.50). Three subjects had percent differences (PD) that were greater than 15 percent (subj. 3 (PD: 25.5%; AR: 46.6), subj. 8 (PD: 56.1%; AR: 17.7), and subj. 14 (PD: 16.4%; AR: -39)). However, there was no relationship between the value of the asymmetry ratio and the percent difference between the spastic-paretic and contralateral sides (n=16; linear regression: Y=0.36x + 6.68; R2=0.0606; p-value=0.3581).
There was no significant difference in p13 peak latency between the two sides (n=16, 2-tailed t-test, p=0.8994), however the spastic-paretic n23 peak latency was significantly shorter than the contralateral n23 peak latency (n=16, paired 2-tailed t-test, p=0.0105). Additionally, the cVEMP on the spastic-paretic side had a significantly shorter interpeak interval than the contralateral cVEMP (n=16, 2-tailed paired t-test, p=0.0065). The mean p13 and n23 peak latencies of cVEMPs elicited on the contralateral side were 15.21 ± 0.97 ms (range: 13.50-16.91 ms; 95% CI: 14.70-15.73 ms) and 23.50 ± 1.91 ms (range: 20.94-27.94 ms; 95% CI: 22.49-24.52 ms) respectively. The mean p13 and n23 latencies of cVEMPs elicited on the spastic-paretic side were 15.20 ± 0.76 ms (range: 13.63-16.15 ms; 95% CI: 14.79-15.60 ms) and 22.47 ± 1.43 ms (range: 20.31-24.84 ms; 95% CI: 21.71-23.23 ms) respectively. The mean interpeak interval on the contralateral side was 8.29 ± 1.62 ms (range: 6.30-12.98 ms; 95% CI: 7.43-9.15 ms) and the mean interpeak interval on the spastic-paretic side was 7.28 ± 0.97 ms (range: 5.67-8.69 ms; 95% CI: 6.76-7.79 ms).
4. Discussion
The aim of the present study was to quantify the relative distribution of descending vestibular drive to the spastic-paretic and contralateral sternocleidomastoid motoneuron pools in chronic stroke survivors. The main finding of this study was that there were significant differences in the relative amounts of vestibular drive impinging upon the spastic-paretic and contralateral motoneuron pools (Figs. 2, 4). In the majority of subjects, the spastic-paretic corrected interpeak amplitude was larger than the contralateral corrected interpeak amplitude, a difference reflected in the finding of a positive asymmetry ratio. Additionally, in subjects with positive asymmetry ratios there was a strong positive correlation between the degree of asymmetry and the severity of spasticity as measured by the Ashworth scale (Figs. 5, 6). These results lend support to our hypothesis that following hemispheric stroke there is an asymmetry in descending vestibular drive to cervical motoneuron pools in spastic stroke survivors.
4.1 Corticobular projections to the vestibular nuclei
Direct corticobulbar projections from premotor cortices to the contralateral caudal medial and lateral vestibular nuclei in primates have been demonstrated using retrograde tracers injected into the vestibular nuclear complex (Akbarian et al., 1993, 1994). These projections could descend in close proximity to the corticospinal tract (Terao et al., 2000, Marsden et al., 2005) or could be collaterals of the corticospinal axons (Keizer and Kuypers, 1984, Marsden et al., 2005); we are unable to distinguish these alternatives currently. From a functional perspective, it is likely that these corticobulbar fibers play a dominant role in the suppression of descending vestibular output (Akbarian et al., 1994). Electrophysiological studies of corticovestibular modulation do provide evidence for polysynaptic inhibition of vestibulospinal neurons (Wilson et al., 1999b) but it remains unclear as to whether these suppressive effects are mediated directly by inhibitory synaptic projections, or via regional inhibitory interneuronal pathways.
Indirect evidence suggesting that these corticobulbar projections inhibit descending vestibular output comes from early studies that investigated decerebrate rigidity in the feline (Sherrington, 1898, Fulton et al., 1930, Bach and Magoun, 1947). Cortical modulation of vestibular output pathways is supported by the striking differences between alert animals and preparations involving intercollicular transection. Immediately following transection, there is an exaggeration of postural reflexes characterized by increased extensor tone (Sherrington, 1898), that is reduced sharply following ablation of the lateral vestibular nucleus (Fulton et al., 1930, Bach and Magoun, 1947). Although not a precise analog for human stroke, the loss of descending corticobulbar fibers following intercollicular transection in the feline model parallels many of the changes that occur following hemispheric stroke in humans.
4.2 Quantification of vestibular drive to the spastic-paretic and contralateral motoneuron pools
Despite the fact that the p13 component of the cVEMP represents a brief inhibition of tonic motoneuron discharge (Colebatch and Rothwell, 2004), the corrected interpeak amplitudes and asymmetry ratio were used as a measure of the relative level of descending vestibular drive. We propose that the spastic-paretic and contralateral cVEMP interpeak amplitudes are proportional to the relative amounts of descending vestibular drive to the respective sternocleidomastoid motoneuron pools, irrespective of the sign of such drive. In other more distal spinal motoneuron pools, release of lateral vestibular neurons would presumably induce increased excitation. We propose that when the background EMG levels between the two sides are matched, the corrected interpeak amplitude is proportional to the amount of descending sacculocollic drive to the sternocleidomastoid motoneuron pool and is not an indicator of motoneuron excitability. This assumption was based on studies which showed that in stroke survivors, side-to-side differences in reflex amplitude disappear when background EMG levels are matched (Burne et al., 2005). It follows that differences in corrected interpeak amplitude between the spastic-paretic and contralateral sides could be due to the amount of vestibular drive impinging upon the motoneuron pool rather than to differences in motoneuron excitability.
4.3 Inhibitory interneurons reciprocally interconnect the medial vestibular nuclei
Interestingly, as the severity of spasticity increased, the ratio of the contralateral corrected interpeak amplitude to the spastic-paretic corrected interpeak amplitude decreased (Fig. 7). In order to better understand how this could arise, we need to briefly review the cVEMP pathway. Previous research has demonstrated that high intensity acoustic stimulation activates irregular primary saccular afferents (McCue and Guinan, 1994, 1997). In reduced animal preparations, saccular nerve stimulation evokes inhibitory postsynaptic potentials in ipsilateral sternocleidomastoid motoneurons via a disynaptic pathway (Kushiro et al., 1999) that descends in the medial longitudinal fasciculus as part of the medial vestibulospinal tract (Kushiro et al., 1999, Kim et al., 2010). The individual medial vestibular nuclei, from which the medial vestibulospinal tracts arise, are reciprocally interconnected through an extensive network of inhibitory commissural pathways (Carleton and Carpenter, 1983, Buttner-Ennever, 1992, Furuya et al., 1992). These pathways allow for the synchronous operation of the left and right medial vestibular nuclei in a push-pull manner, whereby an increase in activity on one-side results in a simultaneous decrease in activity on the other side. It follows that a corticobulbar lesion that increases the activity in the contralesional medial vestibular nucleus would also indirectly cause a decrease in the activity of the ipsilesional medial vestibular nucleus. Potentially, this could explain our results whereby with increasing spasticity, neurons in the contralesional medial vestibular nucleus are increasingly disinhibited and perched closer to threshold. In parallel, there is a subsequent decrease in activity in the ipsilesional medial vestibular nucleus that places these primary vestibular neurons farther away from their thresholds.
4.4 Peak latencies
While there were marginal differences between the spastic-paretic and contralateral p13 peak latencies, the mean spastic-paretic n23 peak latency was significantly shorter than the mean contralateral n23 peak latency. We propose three possible explanations that may account for the differences seen in peak latencies between the two sides. First, contralesional primary medial vestibular neurons may be tonically depolarized and thus closer to threshold due to a lack of cortical inhibition. If we assume that the intrinsic excitability (i.e. gain, EPSP rise time) of the primary vestibular neurons has not changed, differences in latency (<1ms) may arise from contralesional primary vestibular neurons being perched closer to threshold. Second, following stroke there are changes in the molecular phenotype of muscle fibers, shifting towards an increased proportion of fast-twitch type II myosin heavy chain isoforms (De Deyne et al., 2004). The peak latencies may be shorter on the spastic-paretic side because of the increased proportion of type II muscle fibers which have a faster conduction velocity relative to the type I muscle fibers (Kupa et al., 1995). Third, differences could have arisen due to the experimental protocol whereby to closely match EMG levels between the two sides, a subset of subjects were asked to rotate their head slightly to the left or right to increase or decrease the background EMG activity. We may have inadvertently shortened one sternocleidomastoid muscle while lengthening the other. Previous studies have shown that while the corrected interpeak amplitude is not affected by head position, the peak latencies are (Ito et al., 2007). We carefully controlled for background EMG levels because our main measure, the interpeak amplitude, is largely determined by the stimulus intensity and background EMG activity (Lim et al., 1995, Akin et al., 2004) while the peak latencies are not.
4.5 Negative asymmetry ratios were found in a small subset of subjects
Despite probable damage to the corticobulbar pathways that resulted in mild to moderate spasticity, three subjects had consistently larger corrected interpeak amplitudes on their contralateral side (Table 1: subjects 4, 14, and 17). We suggest three possible explanations for why these subjects, whose MAS scores indicated mild to moderate spasticity, had negative asymmetry ratios. First, given the inherent variability in both lesion size and site, it is not surprising that a small subset of subjects had negative asymmetry ratios. Compared to subjects with more focal lesions, these subjects tended to have diffuse lesions (subject 17) involving multiple areas and likely impacted various ascending and descending pathways. Second, the course of recovery and reorganization of cortical areas following stroke is dependent on intrinsic mechanisms that vary as a function of the time post-stroke as well as the site and extent of the lesion (Teasell et al., 2005). It is plausible that over the relatively long course of the natural recovery following stroke, these subjects may have differed in the reorganization of various cortical areas and pathways involved in vestibular modulation and compensation. Third, it is conceivable that the Ashworth measurements were confounded by contracture (Patrick and Ada, 2006), a scenario where the increased resistance is due to peripheral changes in muscle properties rather than changes in stretch reflex excitability (O'Dwyer et al., 1996). We quantified the deep tendon reflexes, a measure of phasic reflex excitability and found that these subjects tended to have hypoactive to normal reflexes in the upper limbs. Additionally, these subjects were at least 68 months post stroke (range: 68- 301 months post stroke) which would give ample time for soft-tissue changes and contracture to set in (O'Dwyer et al., 1996).
4.6 Alternative mechanisms
While asymmetries in descending vestibular drive to the spastic-paretic and contralateral motoneuron pools is the most parsimonious explanation for our findings, other explanations are certainly conceivable. Spastic-paretic motoneuron hyperexcitability could result from an increase in the intrinsic excitability of motoneurons themselves (Katz and Rymer, 1989). Augmented persistent inward currents (PICs), which are voltage-sensitive conductances that are subject to powerful modulation by descending monoaminergic drive (Hounsgaard et al., 1988), could increase the likelihood of motoneuron discharge through amplification of group 1a afferent or other sensory input (Lee and Heckman, 2000). While there is evidence for the role of PICs in spinal forms of spasticity (Barbeau et al., 1981, Li et al., 2004), there is sparse evidence that supports a role for enhanced PICs and monoamines in cerebral forms of spasticity (Mottram et al., 2009). Additionally, the bilateral and diffuse nature of brainstem monoaminergic projections (Björklund and Skagerberg, 1982) does not readily explain the sharply lateralized nature of stroke-induced spasticity.
It is probable that reticulospinal pathways, which mediate the acoustic startle reflex (Leitner et al., 1980), are disrupted following stroke. Indeed, previous research shows a lateralized increased startle reflex amplitude in about 25% of stroke subjects (Jankelowitz and Colebatch, 2004a). However, cortical involvement is questioned, due in part to the bilateral distribution of corticoreticular projections (Matsuyama and Drew, 1997, Rho et al., 1997) and the finding that spinal cord injured patients also exhibited increased startle, which indicates a potential change at the segmental level (Jankelowitz and Colebatch, 2004a). Additionally, reticulospinal pathways cannot easily explain the sharply lateralized nature of spasticity as they are largely bilateral in both their anatomical spinal distribution (Nyberg-Hansen, 1965, Nathan et al., 1996) and synaptic action (Schepens and Drew, 2006, Davidson et al., 2007). However, we cannot completely dismiss reticular pathways given the extensive interconnection of the vestibular and the reticular complexes (Ladpli and Brodal, 1968, Carleton and Carpenter, 1983).
At the segmental level, enhanced fusimotor drive or disruption of descending pathways that mediate presynaptic inhibition could contribute to our findings. While there is no evidence for a post-stroke enhancement of spindle afferent sensitivity (Wilson et al., 1999a), there is evidence for a reduction in presynaptic inhibition (Lamy et al., 2009) although this is bilaterally distributed within the spinal cord and correlated poorly with the severity of spasticity. Finally, changes in spinal reflex circuitry may occur following stroke but as Burke et al. (2013) noted, current research has yielded conflicting results that often correlate poorly with spasticity.
4.7 Previous studies
Apparent discrepancies between our results and the results of a previous study that quantified vestibular reflexes post-stroke may be due to differences in study design. Jankelowitz and Colebatch (2004b) evaluated vestibulospinal and vestibulocollic reflexes by means of transmastoid galvanic vestibular stimulation in stroke survivors. They found no significant differences in either the cVEMP amplitudes or latencies between the clinically affected and clinically spared sides (Jankelowitz and Colebatch, 2004b). This earlier study differed from the present study design in three ways: (1) The study was not designed to assess the contribution of descending pathways to post-stroke spasticity; (2) The subject population consisted of both chronic (> 6 months post-stroke) and acute (< 6 months post-stroke) subjects that presented with a variety of lesion locations, both cortical and subcortical; and (3) they utilized transmastoid galvanic vestibular stimulation which activates a population of vestibular afferents distinct from those excited by acoustical stimulation (Bacsi et al., 2003).
In contrast, we primarily recruited chronic spastic stroke survivors with lesions in the internal capsule or basal ganglia, areas where corticobulbar projection fibers would most likely be damaged. Additionally, while transmastoid galvanic stimulation bilaterally activates a wide range of canal and otolith irregular vestibular afferents, monaural acoustic stimulation allows both selective and independent activation of ipsilesional and contralesional saccular afferents.
4.8 Clinical implications and conclusion
In summary, our current observations provide evidence for asymmetries in descending vestibular drive following stroke that result in spastic hypertonia. We propose that the asymmetric vestibular drive is a source of an uncompensated low-level ionotropic drive to the motoneuron pool that keeps spinal motoneurons on the spastic-paretic side closer to their activation threshold and thus more readily activated. However, more research will need to be done to evaluate the effect of vestibular drive on spinal motoneuron excitability post-stroke. If vestibular projections are definitively shown to be involved in maintaining heightened spinal motoneuron excitability post-stroke, therapies could be developed that target the tonic synaptic depolarizing drive to the spastic-paretic motoneuron pool. Modulation of descending vestibular activity through electrical or acoustical stimuli represents a novel therapeutic modality. These potential therapeutic targets will allow for a more specific and effective treatment of spasticity in hemiparetic stroke survivors by addressing the underlying causation of the increased motoneuronal activity.
Highlights.
Spastic stroke survivors display significant asymmetries in vestibulocollic drive to clinically affected and clinically spared motoneuron pools innervating cervical muscles.
There is a strong correlation between the degree of asymmetry in vestibular mediated reflex amplitudes elicited on the clinically affected and clinically spared sides, and the severity of spasticity.
Vestibular drive is a likely source of ionotropic excitation that places spastic-paretic motoneurons in a hyperexcitable state.
Acknowledgments
This research was supported by a predoctoral fellowship from the American Heart Association and a National Institutes of Health T32 Training Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbarian S, Grusser OJ, Guldin WO. Corticofugal projections to the vestibular nuclei in squirrel monkeys: further evidence of multiple cortical vestibular fields. J Comp Neurol. 1993;332:89–104. doi: 10.1002/cne.903320107. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Grusser OJ, Guldin WO. Corticofugal connections between the cerebral cortex and brainstem vestibular nuclei in the macaque monkey. J Comp Neurol. 1994;339:421–437. doi: 10.1002/cne.903390309. [DOI] [PubMed] [Google Scholar]
- Akin FW, Murnane OD, Panus PC, Caruthers SK, Wilkinson AE, Proffitt TM. The influence of voluntary tonic EMG level on the vestibular-evoked myogenic potential. J Rehabil Res Dev. 2004;41:473–480. doi: 10.1682/jrrd.2003.04.0060. [DOI] [PubMed] [Google Scholar]
- Bach LM, Magoun HW. The vestibular nuclei as an excitatory mechanism for the cord. J Neurophysiol. 1947;10:331–337. doi: 10.1152/jn.1947.10.5.331. [DOI] [PubMed] [Google Scholar]
- Bacsi AM, Watson SR, Colebatch JG. Galvanic and acoustic vestibular stimulation activate different populations of vestibular afferents. Clin Neurophysiol. 2003;114:359–365. doi: 10.1016/s1388-2457(02)00376-0. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Filion M, Bedard P. Effects of agonists and antagonists of serotonin on spontaneous hindlimb EMG activity in chronic spinal rats. Neuropharmacology. 1981;20:99–107. doi: 10.1016/0028-3908(81)90191-x. [DOI] [PubMed] [Google Scholar]
- Basta D, Todt I, Ernst A. Normative data for P1/N1-latencies of vestibular evoked myogenic potentials induced by air- or bone-conducted tone bursts. Clin Neurophysiol. 2005;116:2216–2219. doi: 10.1016/j.clinph.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Björklund A, Skagerberg G. Descending monoaminergic projections to the spinal cord. In: Sjolund B, Björklund A, editors. Brain Stem Control of Spinal Mechanisms. Amsterdam: Elsevier Biomedical Press; 1982. pp. 55–88. [Google Scholar]
- Bohannon RW, Larkin PA, Smith MB, Horton MG. Relationship between static muscle strength deficits and spasticity in stroke patients with hemiparesis. Phys Ther. 1987;67:1068–1071. doi: 10.1093/ptj/67.7.1068. [DOI] [PubMed] [Google Scholar]
- Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67:206–207. doi: 10.1093/ptj/67.2.206. [DOI] [PubMed] [Google Scholar]
- Burke D, Ashby P. Are spinal “presynaptic” inhibitory mechanisms suppressed in spasticity? J Neurol Sci. 1972;15:321–326. doi: 10.1016/0022-510x(72)90073-1. [DOI] [PubMed] [Google Scholar]
- Burke D, Knowles L, Andrews C, Ashby P. Spasticity, decerebrate rigidity and the claspknife phenomenon: an experimental study in the cat. Brain. 1972;95:31–48. doi: 10.1093/brain/95.1.31. [DOI] [PubMed] [Google Scholar]
- Burke D, Wissel J, Donnan GA. Pathophysiology of spasticity in stroke. Neurology. 2013;80:S20–26. doi: 10.1212/WNL.0b013e31827624a7. [DOI] [PubMed] [Google Scholar]
- Burne JA, Carleton VL, O'Dwyer NJ. The spasticity paradox: movement disorder or disorder of resting limbs? J Neurol Neurosurg Psychiatry. 2005;76:47–54. doi: 10.1136/jnnp.2003.034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner-Ennever JA. Patterns of connectivity in the vestibular nuclei. Ann N Y Acad Sci. 1992;656:363–378. doi: 10.1111/j.1749-6632.1992.tb25222.x. [DOI] [PubMed] [Google Scholar]
- Carleton SC, Carpenter MB. Afferent and efferent connections of the medial, inferior and lateral vestibular nuclei in the cat and monkey. Brain Res. 1983;278:29–51. doi: 10.1016/0006-8993(83)90223-8. [DOI] [PubMed] [Google Scholar]
- Colebatch JG, Halmagyi GM, Skuse NF. Myogenic potentials generated by a clickevoked vestibulocollic reflex. J Neurol Neurosurg Psychiatry. 1994;57:190–197. doi: 10.1136/jnnp.57.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colebatch JG, Rothwell JC. Motor unit excitability changes mediating vestibulocollic reflexes in the sternocleidomastoid muscle. Clin Neurophysiol. 2004;115:2567–2573. doi: 10.1016/j.clinph.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Davidson AG, Schieber MH, Buford JA. Bilateral spike-triggered average effects in arm and shoulder muscles from the monkey pontomedullary reticular formation. J Neurosci. 2007;27:8053–8058. doi: 10.1523/JNEUROSCI.0040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Deyne PG, Hafer-Macko CE, Ivey FM, Ryan AS, Macko RF. Muscle molecular phenotype after stroke is associated with gait speed. Muscle Nerve. 2004;30:209–215. doi: 10.1002/mus.20085. [DOI] [PubMed] [Google Scholar]
- Denny-Brown D. The extrapyramidal system and postural mechanisms. Clin Pharmacol Ther. 1964;5:812–827. [Google Scholar]
- Denny-Brown D. The Nature of Dystonia. Bulletin of the New York Academy of Medicine. 1965;41:858–869. [PMC free article] [PubMed] [Google Scholar]
- Fulton JF, Liddell EGT, Mck Rioch D. The influence of unilateral destruction of the vestibular nuclei upon posture and the knee jerk. Brain. 1930;53:327–343. [Google Scholar]
- Furuya N, Yabe T, Koizumi T. Neurotransmitters in the vestibular commissural system of the cat. Ann N Y Acad Sci. 1992;656:594–601. doi: 10.1111/j.1749-6632.1992.tb25238.x. [DOI] [PubMed] [Google Scholar]
- Gillies JD, Burke DJ, Lance JW. Supraspinal control of tonic vibration reflex. J Neurophysiol. 1971a;34:302–309. doi: 10.1152/jn.1971.34.2.302. [DOI] [PubMed] [Google Scholar]
- Gillies JD, Burke DJ, Lance JW. Tonic vibration reflex in the cat. J Neurophysiol. 1971b;34:252–262. doi: 10.1152/jn.1971.34.2.252. [DOI] [PubMed] [Google Scholar]
- Hounsgaard J, Hultborn H, Jespersen B, Kiehn O. Bistability of alpha-motoneurones in the decerebrate cat and in the acute spinal cat after intravenous 5-hydroxytryptophan. J Physiol. 1988;405:345–367. doi: 10.1113/jphysiol.1988.sp017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Karino S, Murofushi T. Effect of head position on vestibular evoked myogenic potentials with toneburst stimuli. Acta Otolaryngol. 2007;127:57–61. doi: 10.1080/00016480600740597. [DOI] [PubMed] [Google Scholar]
- Jankelowitz SK, Colebatch JG. The acoustic startle reflex in ischemic stroke. Neurology. 2004a;62:114–116. doi: 10.1212/01.wnl.0000101711.48946.35. [DOI] [PubMed] [Google Scholar]
- Jankelowitz SK, Colebatch JG. Galvanic evoked vestibulospinal and vestibulocollic reflexes in stroke. Clin Neurophysiol. 2004b;115:1796–1801. doi: 10.1016/j.clinph.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Katz RT, Rymer WZ. Spastic hypertonia: mechanisms and measurement. Arch Phys Med Rehabil. 1989;70:144–155. [PubMed] [Google Scholar]
- Keizer K, Kuypers HG. Distribution of corticospinal neurons with collaterals to lower brain stem reticular formation in cat. Exp Brain Res. 1984;54:107–120. doi: 10.1007/BF00235823. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee HS, Kim JS. Medial vestibulospinal tract lesions impair sacculo-collic reflexes. J Neurol. 2010;257:825–832. doi: 10.1007/s00415-009-5427-5. [DOI] [PubMed] [Google Scholar]
- Kimura J. Electrodiagnosis in diseases of nerve and muscle : principles and practice. 4th. New York: Oxford University Press; 2013. p. 219. [Google Scholar]
- Kupa EJ, Roy SH, Kandarian SC, De Luca CJ. Effects of muscle fiber type and size on EMG median frequency and conduction velocity. J Appl Physiol. 1995;79:23–32. doi: 10.1152/jappl.1995.79.1.23. [DOI] [PubMed] [Google Scholar]
- Kushiro K, Zakir M, Ogawa Y, Sato H, Uchino Y. Saccular and utricular inputs to sternocleidomastoid motoneurons of decerebrate cats. Exp Brain Res. 1999;126:410–416. doi: 10.1007/s002210050747. [DOI] [PubMed] [Google Scholar]
- Ladpli R, Brodal A. Experimental studies of commissural and reticular formation projections from the vestibular nuclei in the cat. Brain Res. 1968;8:65–96. doi: 10.1016/0006-8993(68)90173-x. [DOI] [PubMed] [Google Scholar]
- Lamy JC, Wargon I, Mazevet D, Ghanim Z, Pradat-Diehl P, Katz R. Impaired efficacy of spinal presynaptic mechanisms in spastic stroke patients. Brain. 2009;132:734–748. doi: 10.1093/brain/awn310. [DOI] [PubMed] [Google Scholar]
- Lance JW. Pathophysiology of spasticity and clinical experience with baclofen. In: Feldman RGRRY, Koella WW, editors. Spasticity: Disordered Motor Control. Year Book Medical Publishers; Chicago: 1980. pp. 185–203. Miami. [Google Scholar]
- Lee RH, Heckman CJ. Adjustable amplification of synaptic input in the dendrites of spinal motoneurons in vivo. J Neurosci. 2000;20:6734–6740. doi: 10.1523/JNEUROSCI.20-17-06734.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner DS, Powers AS, Hoffman HS. The neural substrate of the startle response. Physiol Behav. 1980;25:291–297. doi: 10.1016/0031-9384(80)90219-x. [DOI] [PubMed] [Google Scholar]
- Li Y, Harvey PJ, Li X, Bennett DJ. Spastic long-lasting reflexes of the chronic spinal rat studied in vitro. J Neurophysiol. 2004;91:2236–2246. doi: 10.1152/jn.01010.2003. [DOI] [PubMed] [Google Scholar]
- Lim CL, Clouston P, Sheean G, Yiannikas C. The influence of voluntary EMG activity and click intensity on the vestibular click evoked myogenic potential. Muscle Nerve. 1995;18:1210–1213. doi: 10.1002/mus.880181021. [DOI] [PubMed] [Google Scholar]
- Marsden JF, Playford DE, Day BL. The vestibular control of balance after stroke. J Neurol Neurosurg Psychiatry. 2005;76:670–678. doi: 10.1136/jnnp.2004.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama K, Drew T. Organization of the projections from the pericruciate cortex to the pontomedullary brainstem of the cat: a study using the anterograde tracer Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1997;389:617–641. doi: 10.1002/(sici)1096-9861(19971229)389:4<617::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- McCue MP, Guinan JJ., Jr Acoustically responsive fibers in the vestibular nerve of the cat. J Neurosci. 1994;14:6058–6070. doi: 10.1523/JNEUROSCI.14-10-06058.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue MP, Guinan JJ., Jr Sound-evoked activity in primary afferent neurons of a mammalian vestibular system. Am J Otol. 1997;18:355–360. [PubMed] [Google Scholar]
- Molina-Negro P, Bertrand RA, Martin E, Gioani Y. The role of the vestibular system in relation to muscle tone and postural reflexes in man. Acta Otolaryngol. 1980;89:524–533. doi: 10.3109/00016488009127170. [DOI] [PubMed] [Google Scholar]
- Mottram CJ, Suresh NL, Heckman CJ, Gorassini MA, Rymer WZ. Origins of abnormal excitability in biceps brachii motoneurons of spastic-paretic stroke survivors. J Neurophysiol. 2009;102:2026–2038. doi: 10.1152/jn.00151.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan PW, Smith M, Deacon P. Vestibulospinal, reticulospinal and descending propriospinal nerve fibres in man. Brain. 1996;119:1809–1833. doi: 10.1093/brain/119.6.1809. [DOI] [PubMed] [Google Scholar]
- Nathan PW, Smith MC. The rubrospinal and central tegmental tracts in man. Brain. 1982;105:223–269. doi: 10.1093/brain/105.2.223. [DOI] [PubMed] [Google Scholar]
- Nyberg-Hansen R. Sites and Mode of Termination of Reticulo-Spinal Fibers in the Cat. An Experimental Study with Silver Impregnation Methods. J Comp Neurol. 1965;124:71–99. doi: 10.1002/cne.901240107. [DOI] [PubMed] [Google Scholar]
- Nyberg-Hansen R, Mascitti TA. Sites and Mode of Termination of Fibers of the Vestibulospinal Tract in the Cat. An Experimental Study with Silver Impregnation Methods. J Comp Neurol. 1964;122:369–383. doi: 10.1002/cne.901220307. [DOI] [PubMed] [Google Scholar]
- O'Dwyer NJ, Ada L, Neilson PD. Spasticity and muscle contracture following stroke. Brain. 1996;119:1737–1749. doi: 10.1093/brain/119.5.1737. [DOI] [PubMed] [Google Scholar]
- Patrick E, Ada L. The Tardieu Scale differentiates contracture from spasticity whereas the Ashworth Scale is confounded by it. Clin Rehabil. 2006;20:173–182. doi: 10.1191/0269215506cr922oa. [DOI] [PubMed] [Google Scholar]
- Rho MJ, Cabana T, Drew T. Organization of the projections from the pericruciate cortex to the pontomedullary reticular formation of the cat: a quantitative retrograde tracing study. J Comp Neurol. 1997;388:228–249. doi: 10.1002/(sici)1096-9861(19971117)388:2<228::aid-cne4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Schepens B, Drew T. Descending signals from the pontomedullary reticular formation are bilateral, asymmetric, and gated during reaching movements in the cat. J Neurophysiol. 2006;96:2229–2252. doi: 10.1152/jn.00342.2006. [DOI] [PubMed] [Google Scholar]
- Sherrington CS. Decerebrate Rigidity, and Reflex Coordination of Movements. J Physiol. 1898;22:319–332. doi: 10.1113/jphysiol.1898.sp000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheykholeslami K, Murofushi T, Kaga K. The effect of sternocleidomastoid electrode location on vestibular evoked myogenic potential. Auris Nasus Larynx. 2001;28:41–43. doi: 10.1016/s0385-8146(00)00091-2. [DOI] [PubMed] [Google Scholar]
- Sommerfeld DK, Eek EU, Svensson AK, Holmqvist LW, von Arbin MH. Spasticity after stroke: its occurrence and association with motor impairments and activity limitations. Stroke. 2004;35:134–139. doi: 10.1161/01.STR.0000105386.05173.5E. [DOI] [PubMed] [Google Scholar]
- Teasell R, Bayona NA, Bitensky J. Plasticity and reorganization of the brain post stroke. Top Stroke Rehabil. 2005;12:11–26. doi: 10.1310/6AUM-ETYW-Q8XV-8XAC. [DOI] [PubMed] [Google Scholar]
- Terao S, Miura N, Takeda A, Takahashi A, Mitsuma T, Sobue G. Course and distribution of facial corticobulbar tract fibres in the lower brain stem. J Neurol Neurosurg Psychiatry. 2000;69:262–265. doi: 10.1136/jnnp.69.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban PP, Wolf T, Uebele M, Marx JJ, Vogt T, Stoeter P, et al. Occurence and clinical predictors of spasticity after ischemic stroke. Stroke. 2010;41:2016–2020. doi: 10.1161/STROKEAHA.110.581991. [DOI] [PubMed] [Google Scholar]
- Watkins CL, Leathley MJ, Gregson JM, Moore AP, Smith TL, Sharma AK. Prevalence of spasticity post stroke. Clin Rehabil. 2002;16:515–522. doi: 10.1191/0269215502cr512oa. [DOI] [PubMed] [Google Scholar]
- Wilson LR, Gandevia SC, Inglis JT, Gracies J, Burke D. Muscle spindle activity in the affected upper limb after a unilateral stroke. Brain. 1999a;122:2079–2088. doi: 10.1093/brain/122.11.2079. [DOI] [PubMed] [Google Scholar]
- Wilson VJ, Zarzecki P, Schor RH, Isu N, Rose PK, Sato H, et al. Cortical influences on the vestibular nuclei of the cat. Exp Brain Res. 1999b;125:1–13. doi: 10.1007/s002210050651. [DOI] [PubMed] [Google Scholar]
- Wright J, Rang M. The spastic mouse and the search for an animal model of spasticity in human beings. Clin Orthop Relat Res. 1990:12–19. [PubMed] [Google Scholar]
- Young YH. Vestibular evoked myogenic potentials: optimal stimulation and clinical application. J Biomed Sci. 2006;13:745–751. doi: 10.1007/s11373-006-9106-6. [DOI] [PubMed] [Google Scholar]