

Abstract
Epidithiodioxopiperazine alkaloids possess an astonishing array of molecular architecture and generally exhibit potent biological activity. Nearly twenty distinct families have been isolated and characterized since the seminal discovery of gliotoxin in 1936. Numerous biosynthetic investigations offer a glimpse at the relative ease with which Nature is able to assemble this class of molecules, while providing synthetic chemists inspiration for the development of more efficient syntheses. Herein, we discuss the isolation and characterization, proposed fungal biogeneses, and total syntheses of epidithiodioxopiperazines.
1. Introduction
Nearly twenty distinct families of epidithiodioxopiperazine fungal metabolites have been reported since the seminal discovery of gliotoxin in 1936. This unique class of natural products is characterized by a sulfur-bridged dioxopiperazine (1), a feature generally requisite for the potent biological activity prevalent among the class.1-5
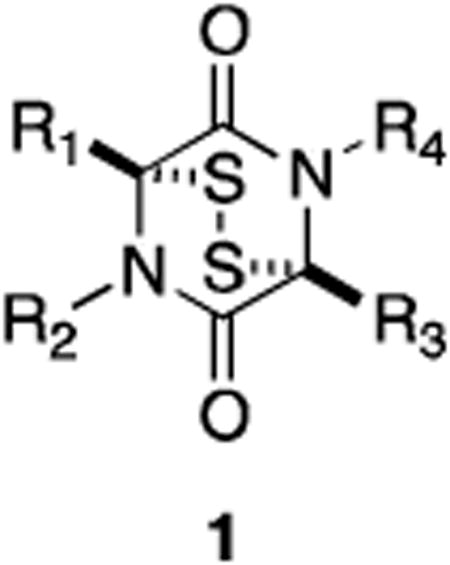
All natural epidithiodioxopiperazines discovered to date contain (or are derived from) at least one aromatic amino acid. Representative molecules from each family to be discussed are shown in Figure 1 (tyrosine- and/or phenylalanine-derived) and Figure 2 (tryptophan-derived). In Sections 2-4, we present an overview of the structures of naturally occurring epidithiodioxopiperazines, relevant physiological properties, and some of the more interesting of the proposed fungal biogeneses.
Figure 1.

Representative epidithiodioxopiperazines derived from tyrosine and/or phenylalanine.
Figure 2.

Representative tryptophan-derived epidithiodioxopiperazines.
2. Epidithiodioxopiperazines Derived from Phenylalanine or Tyrosine
In 1936, a novel substance with substantial antifungal and antiviral activity was isolated from the wood fungus Gliocladium fimbriatum by Weindling and Emerson.6 A putative structure (16, Figure 3) was proposed for the metabolite based on degradation studies. This structure, however, could not account for some experimental observations, leading Johnson and Woodward to propose a revised structure for gliotoxin (2) in 1958.7 Absolute stereochemistry was later determined by X-ray analysis.8 Gliotoxin and related metabolites (Figure 3) have since been isolated from a variety of fungi—including several Penicillium and Aspergillus species, Gliocladium, Thermoascus, and Candida—and have been the focus of numerous synthetic and biosynthetic studies that have formed the basis for much of the research discussed herein.2,3
Figure 3.

Gliotoxins.
The initial interest in the chemotherapeutic potential of gliotoxin as an antifungal or antiviral agent waned as in vivo studies revealed gliotoxin to be generally toxic.9 Moreover, gliotoxin has been implicated as a virulence factor of Aspergillus fumigatus, the main source of invasive aspergillosis and a leading cause of death in immunocompromised patients.10 However, interest in the molecule was renewed when it was discovered that gliotoxin displayed selective toxicity to cells of the hematopoietic system.11,12 Specifically, gliotoxin exhibits antiproliferative activity against T and B cells and inhibits phagocytic activity with considerable selectivity toward immune system cells, leading to promising studies that have demonstrated that gliotoxin prevents graft-versus-host disease after bone marrow transplantation.13
Generally, the toxicity of gliotoxin and other epidithiodioxopiperazines can be attributed to two mechanisms: generation of reactive oxygen species (Figure 4) and mixed disulfide formation (Figure 5). In the presence of a suitable reducing agent such as glutathione or dithiothreitol, epidithiodioxopiperazines are reduced to the corresponding dithiols (23). Autooxidation back to the disulfide (1) occurs with the production of superoxide ions and hydrogen peroxide, known to cause oxidative damage to cells.14,15 Gliotoxin has specifically been shown to induce single- and double-stranded DNA damage in the presence of glutathione, presumably by hydroxyl radicals generated in this redox process.16
Figure 4.

Redox cycling of epidithiodioxopiperazines.
Figure 5.

Mixed disulfide formation.
Evidence suggests that gliotoxin and other epidithiodioxopiperazines are also capable of forming mixed disulfides with free thiol groups in cells.17,18 Following incubation with radiolabelled gliotoxin, cells were shown to contain protein-bound [35S]-gliotoxin (24).3 This result could be reversed if cells were treated with both gliotoxin and excess reducing agent (dithiothreitol), suggesting a covalent interaction. Moreover, the antiviral activity of gliotoxin is lost in the presence of excess dithiothreitol. This evidence supports a mechanism of toxicity resulting from mixed disulfide formation between gliotoxin and protein.19
The toxicity of epidithiodioxopiperazines appears to be directly related to the presence of an intact disulfide ring or the reduced dithiol. Indeed, dethiogliotoxin lacks the antibacterial activity of gliotoxin. Reduction and methylation of the disulfide bridge (bisdethiodi(methylthio)gliotoxin, 19) also results in a loss of antiviral activity.4,20
Not surprisingly, the simple disulfide 25 and dithiol 26 exhibit potent biological activity, highlighting the importance of the fragment to the observed toxicity of epidithiodioxopiperazines (Figure 6).21 The great structural diversity in epidithiodioxopiperazines may have evolved only to mask the core disulfide moiety to prevent degradation by target organisms, rather than to impart any sort of selectivity or increased toxicity.2
Figure 6.

Simple biologically active epidithiodioxopiperazines.
Like gliotoxin, the hyalodendrins (Figure 7) are derived biosynthetically from phenylalanine and serine. Hyalodendrin (3) was originally isolated by Strunz in 1974 from Hyalodendron sp.22 The same fungus was later shown to produce the bis(methylthio) derivative (29)23 and epitetrasulfide 28.24 Epitrithiohyalodendrin (27) has only been observed as a product of the unidentified fungus NRRL 3888, along with 3 and 29.25 Not surprisingly, hyalodendrin and the epitri- and epitetrasulfide derivatives exhibit antibacterial activity, while the bisdethiodi(methylthio) analogue is inactive against fungi and bacteria and relatively non-toxic to mice.22-25 Interestingly, it was observed that hyalodendrin could be converted to epitetrasulfide 28 in the presence of HCl with heating in methanol and the culture medium. Racemic tetrasulfide was isolated when HCl was omitted from the same conditions.
Figure 7.

Hyalodendrins and related compounds.
Enantiomers of the hyalodendrins (except for epitrisulfide 27) have been isolated from both terrestrial and marine sources. Gliovictin (32) was first isolated from Helminthosporium victoriae in 1974,26 the same year that researchers at Eli Lilly reported the isolation of the same structure (named A26771E) along with the disulfide (A26771A, 30) and epitetrasulfide (A26771C, 31) from Penicillium turbatum.27 Fenical has also isolated gliovictin from the marine deuteromycete Asteromyces cruciatus.28 Predictably, 30 and 31 both showed antiviral and antibacterial activity, while gliovictin–lacking sulfur atoms capable of redox cycling or mixed disulfide formation–was inactive.27
Kawahara and coworkers reported the isolation of dithiosilvatin (4) and silvathione (33) in 1987 from Aspergillus silvaticus (Figure 8).29 Dioxopiperazinethiones such as 33 are rare and are possible intermediates in the formation of trioxopiperazines from epidithiodioxopiperazines. The authors reported the conversion of 4 to the bisdethiodi(methylthio) derivative (34) by reductive methylation (NaBH4, MeI), a compound previously isolated by Hanson and O'Leary from Gliocladium deliquescens.30
Figure 8.
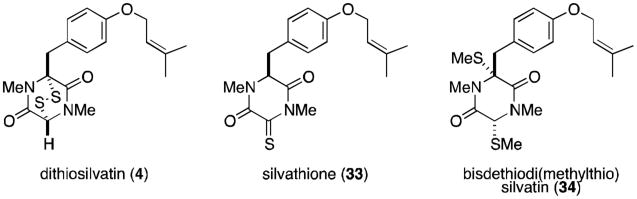
Silvatins.
The next subgroup of epidithiodioxopiperazines to be discussed is characterized by the presence of at least one seven-membered dihydrooxepine ring, and includes the aranotins (Figure 9), emethallicins (Figure 10), and emestrins (Figure 11). Aranotin (5) and acetylaranotin (35) have been isolated from Arachniotus aureus and Aspergillus terreus and exhibit antiviral activity that is apparently selective for RNA viruses such as the polio, Coxsackie (A21), rhino-, and parainfluenza viruses through inhibition of viral RNA synthesis.31-37 Structurally, the aranotins are related to gliotoxin, with the similarities most evident in apoaranotin (37). Apoaranotin can be considered a chimera of gliotoxin and aranotin, containing the cyclohexadienol of gliotoxin and the dihydrooxepine ring of aranotin. Asteroxepin (39) was the first monooxepine derivative to be isolated and is further unique in that it contains one unsubstituted amide. This dioxopiperazine may provide evidence for the sequence of steps in the biosynthesis of the more complex aranotins.
Figure 9.

Aranotins.
Figure 10.

Emethallicins.
Figure 11.

Emestrins and related metabolites.
Closely related to the aranotins are the emethallicins (Figure 10). Both families share the same absolute stereochemistry, and emethallicin A (6) differs from apoaranotin (37) only in the ester moieties. This observation was experimentally confirmed by hydrolysis of 6 followed by acetylation to afford compound 37, identical to naturally occurring apoaranotin.38 Emethallicins B, D, and F (40, 42, and 44) share this same monooxepine core and differ from apoaranotin only in ester substitution and sulfur content of the epipolythiodioxopiperazine ring. Emethallicin C (41) is symmetrical and the only emethallicin to contain two dihydrooxepine rings, more closely resembling aranotin and acetylaranotin.
Emethallicin A (6) was first isolated from Emericella heterothallica in 1989 by Kawai and coworkers, who later reported the isolation of emethallicins B-F (40-44) from the same fungus.38-41 Interestingly, Kawai was unable to convert synthetic emethallicin D monoacetate (42, R=Ac) to the naturally occurring metabolite (42, R=H). Basic hydrolytic conditions only succeeded at forming the disulfide and tetrasulfide monoacetates of 6 and 40, respectively.39 This disproportionation of the trisulfide is similar to a result obtained by Waring and coworkers, who similarly converted trisulfide gliotoxin E (21) to the disulfide, gliotoxin (2), and the tetrasulfide, gliotoxin G (22).42
All of the emethallicins exhibit fairly strong inhibitory activities upon histamine release from mast cells, with IC50 values ranging from 1.0 × 10−6 to 2.0 × 10−8 M. Generally, activity is stronger for the original emethallicins than for the acetate derivatives. Micromolar inhibition of 5-lipoxygenase has also been reported.43
The final class of known epipolythiodioxopiperazine metabolites known to contain at least one dihydrooxepine ring are the macrocyclic emestrins (Figure 11). Emestrin (7) was isolated in 1985 from the fungus Emericella striata, and later from E. quadrilineata, E. foveolata, E. acristata, and E. parvathecia.44-46 Trisulfide emestrin B (45), piperazinethione aurantioemestrin (47), and trioxopiperazine dethiosecoemestrin (48) were later isolated from E. striata.46-49 It has been postulated that the latter two compounds are derived biosynthetically from emestrin. Emestrin displays potent antifungal and antibacterial activity, but is also very toxic to mammals.
Recently, Kanda and coworkers reported the isolation of MPC1001 (46) and its analogues (not shown) from Cladorrhinum sp. KY4922, contributing eight new members to the emestrin family of natural products.50 MPC1001 contains a methoxy group rather than the free phenol found in emestrin, but is otherwise structurally and stereochemically identical. MPC1001 and its epipolysulfide analogues all showed antiproliferative activity in the DU145 human prostate cancer cell line.50
Epicorazines (Figure 12) have been isolated from several organisms, including Epicoccum nigrum (epicorazine A and B, 8 and 49),51-53 E. purpurascens (epicorazine B),54 and Stereum hirsutum (epicorazine C, 50).55 The only difference between 8 and 49 is the absolute stereochemistry at C6. This cis-configuration is shared between 49 and 50.
Figure 12.

Epicorazines.
Epicorazine A, B, and C display only marginal activity against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecalis (VRE), and are inactive against Gram-negative organisms. All three exhibit antiproliferative effects against L929 mouse fibroblast cells and K562 human leukemia cells, as well as cytotoxicity toward the HeLa human cervical carcinoma cell line.55
The scabrosin esters were originally isolated from the lichen Xanthoparmelia scabrosa in 1978, but it was not until 1999 that the correct structures were determined (Figure 13).56,57 The isolation of these compounds was of particular interest, as this report marked the first epidithiodioxopiperazine to be isolated from lichenized fungi. Submicromolar activity, similar to that of gliotoxin (2), was observed for the scabrosins against the murine P815 mastocytomia cell line, as well as low nanomolar activity in MCF7 human breast carcinoma cell line. Scabrosin esters have also been shown to induce apoptosis concomitantly with a large increase in mitochondrial membrane potential and significant decrease in total cellular ATP. Mitochondrial ATP synthase is the proposed cellular target of the scabrosins.58
Figure 13.

Scabrosin esters.
The sirodesmins (Figure 14) were first discovered in 1977 as metabolites of Sirodesmium diversum 59 and later from the unrelated fungus Phoma lingam.60 Sirodesmins A-H (10, 56-60) are characterized by a spirofused tetrahydrofuran cyclopentylpyrrolidine skeleton. Sirodesmins A-C are epimers of G and H at the spirocenter. Notably, sirodesmin H was the first example of a naturally occurring epimonosulfide.
Figure 14.

Sirodesmins.
Sirodesmins are potent antiviral agents, particularly against the rhinovirus.59 Sirodesmin G (originally named sirodesmin PL, 58) has specifically been shown to exhibit activity against Gram-positive bacteria,61 although it has also been implicated as the causative agent of blackleg disease in canola crops (along with phomalirazine, 61). Metabolites 58 and 61 have both been isolated from the ascomycetous fungus Leptosphaeria maculans, the organism known to be responsible for blackleg disease.62
3 Tryptophan–Derived Epidithiodioxopiperazines
The remaining epidithiodioxopiperazine alkaloids to be discussed are all derived from tryptophan (Figure 2). Of this subset, the sporidesmins (Figure 15) are the most densely functionalized and the only members to contain a substituted aromatic ring. Sporidesmin was discovered by researchers investigating the source of the disease facial eczema that plagued sheep in New Zealand and Australia. The disease caused extensive liver damage in infected sheep and ultimately resulted in death. Thornton and Percival eventually established that ingestion of pasture grasses on which the fungus Pithomyces chartarum (previously known as Sporidesmium bakeri) was growing was the cause of this serious disease.63,64 Sporidesmin (11) was isolated and implicated as the main toxic agent produced by P. chartarum.65 The structure and absolute configuration were subsequently determined by crystallographic means.8,66,67
Figure 15.
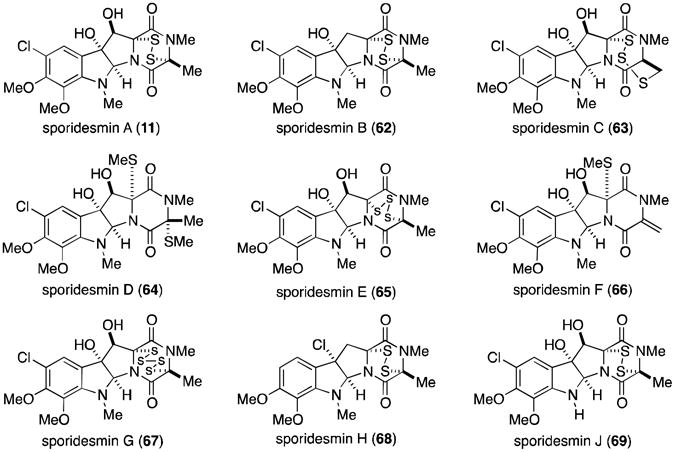
Sporidesmins.
As an interesting aside, veterinarians discovered that zinc sulfate doses gave sheep protection from the effects of sporidesmin.68 Metals such as zinc are now known to inhibit generation of the superoxide anion radical, with epidithiodioxopiperazines shown to form a 2:1 complex with zinc ion.69,70
Extensive amounts of research have focused on the sporidesmins, producing the complete characterization of all nine derivatives (11, 62-69). All contain a densely functionalized, tryptophan–derived pyrroloindoline core coupled to an alanine residue. Sporidesmin C (63)71 is the most unusual, containing a novel trisulfide [4.3.3] ring system.
A great deal of chemistry applicable to most of the epipolythiodioxopiperazines was discovered through investigations of the sporidesmins. For example, the trisulfide sporidesmin E (65) is readily converted to the disulfide (11) upon treatment with triphenyl phosphine. Alternatively, di- and trisulfides can be converted to tetrasulfides, by using hydrogen polysulfide or dihydrogen disulfide. This was demonstrated by the conversion of sporidesmins A (11) and E (65) into sporidesmin G (67).
In 1944, Waksman and Bugie reported the isolation of a new antibiotic metabolite of the fungus Chaetomium cochliodes that they named chetomin.72 It was not until thirty years later that Walter and co-workers determined the structure of chetomin (12), revealing a nearly dimeric core likely formed from two molecules each of tryptophan and serine.73 The two fragments are joined by a bond between the β-pyrrolidinoindoline carbon and the indole nitrogen, a common feature of all five chaetocochins (Figure 16).74 Chetomin is the only molecule in the family to contain a disulfide bridge within both dioxopiperazine rings. Dethio-tetra(methylthio)chetomin (70) and chaetocochin C (73) differ only in the oxidation state of the sulfurs, while chaetocochins A (71) and B (72) are macrocyclic analogues of 70 and 73, each containing a novel 14-membered ring.
Figure 16.

Metabolites of the fungus Chaetomium cochliodes.
Chetomin was recently isolated from Chaetomium seminudum by Fujimoto and coworkers in 2004, along with three new metabolites named the chetoseminudins (Figure 17).75 Chetoseminudin A (74) is merely the pentasulfide homolog of chetomin. From a biosynthetic viewpoint, the more interesting discoveries are chetoseminudins B-D (75-77), monomeric bisdethiodi(methylthio) structures that potentially provide insight as to the biosynthetic sequence that produces chetomin, the chaetocochins, and other related epipolythiodioxopiperazines derived from tryptophan and serine.76
Figure 17.
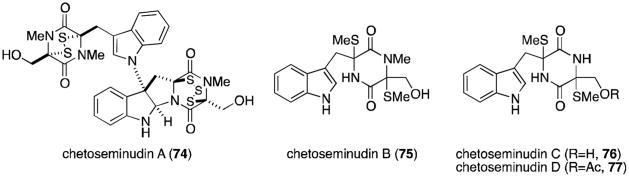
Chetoseminudins.
Chetomin (12) has an unprecedented mechanism of action as a cancer chemotherapeutic agent. Solid tumors must adapt to oxygen deprivation through induction of the heterodimeric transcription factor hypoxia-inducible factor 1 (HIF-1) in order to survive. Overexpression of HIF-1 is associated with radioresistance in tumors, increased risk of metastasis, and a poor prognosis for patients.77-79 In normal cells, the α-subunit of HIF-1 (HIF-1α) is hydroxylated and degraded by vHL proteosome. As oxygen levels decrease and become the rate-limiting reagent in the hydroxylation reaction, HIF-1α accumulates and binds to transcriptional coactivators p300 and CREB binding protein (CBP). Consequent to this binding is the transcription of proteins requisite to the survival of hypoxic cancer cells, facilitating tumor growth and progression.80
Chetomin has been shown to inhibit the interaction between HIF-1 and p300 both in vitro and in cells, despite extensive surface interactions between the two proteins. Specifically, Kung and co-workers have shown that chetomin disrupts the tertiary structure of p300, inhibiting the transcriptional activity of HIF-1.77 No other small molecule has been identified to mediate an antitumor response through this mechanism of action. More recently, Hilton and coworkers proposed that chetomin and other epidithiodioxopiperazines bind zinc at the CH1 domain of p300, ultimately resulting in ejection of a stable zinc–epidithiodioxopiperazine complex. Loss of zinc from the CH1 domain causes the previously observed disruption of the p300 tertiary structure.1,81
Chaetocin A (13, Figure 18) is a dimeric epidithiodioxopiperazine also derived from two molecules each of tryptophan and serine. It was isolated from Chaetomium minutum in 1970.82,83 Fifteen years passed before the penta- and hexasulfide homologs chaetocin B and C (78 and 79) were isolated from Chaetomium spp., along with the novel octasulfide chetracin A (80).84 In 2012, several related metabolites were isolated from Oidiodendron truncatum, including the tetra-, penta- and hexasulfide homologs melinacidin IV, chetracins B and C (82, 83, and 84), and the dethiotetra(methylthio) derivative chetracin D (85).85
Figure 18.

Chaetocins and related metabolites.
The three chaetocin metabolites (and likely the chetracins) can be interconverted through either desulfurization of 78 and 79 with triphenyl phosphine to generate chaetocin (13), or by sulfurization of chaetocin with phosphorus pentasulfide in carbon disulfide to afford a mixture of chaetocins B and C (78 and 79).84
Recently, chaetocin A (13) was identified as the first known inhibitor of lysine-specific histone methyltransferases.86 Histone methylation is an important process in controlling gene expression patterns, especially during cellular differentiation and embryonic development. The activity of histone methyltransferases is disregulated in some tumors, making chaetocin an attractive tool for the study of the molecular mechanism of histone methylation.86 Additionally, melinacidin IV and chetracin B (82 and 83) display nanomolar (3 – 54 nM) activity against five human cancer cell lines (HCT-8, Bel-7402, BGC-823, A549, and A2780).85
Several metabolites related to the chaetocins have recently been reported, possessing a C3-C3′ linkage to indole rather than the additional monomer found in the chaetocins (Figure 19). T988 A, B, and C (86-88) were originally isolated from Tilachlidium sp., although recently it was shown that Oidiodendron truncatum also produces the same metabolites, in addition to oidioperazine A (89) and the chetracins (83-85).75,87 Chetoseminudin C (76) was also isolated from O. truncatum, suggesting that it may be a common intermediate to all of the tryptophan- and serine-derived epipolythiodioxopiperazines discussed thus far. T988 A and B are cytotoxic to P388 leukemia cells.87
Figure 19.
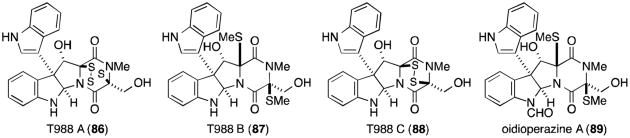
Fungal metabolites related to the chaetocins.
Verticillin A (14, Figure 20) is quite similar to chaetocin A (13), derived from two molecules of alanine rather than serine. Only verticillins A, D and E (14, 92 and 93) are symmetrical, while the two tryptophan residues of the remaining verticillins are coupled to different amino acids on the two halves (either alanine, serine, or tyrosine). Verticillin B (90), for instance, contains alanine and serine residues on the northern and southern halves of the molecule, respectively. Verticillins A-C were isolated from Verticillium sp., while the remaining compounds in Figure 20 are produced by Gliocladium sp.88-90
Figure 20.
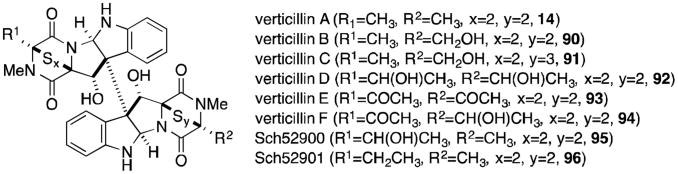
Verticillin A and related metabolites.
Gliocladines A-E (Figure 21, 97-101) were isolated in 2005 from Gliocladium roseum, along with verticillin A, Sch52900, and Sch52901 (14, 95, and 96).91 Gliocladines A and B are simply the penta- and hexasulfide homologs of verticillin A, thus it is not surprising that the same organism produces all three compounds. The structures of gliocladines C and D (99 and 100) should also look familiar, as they are the alanine derivatives of T988 A and C (Figure 19). Bionectra byssicola F120 has also been shown to produce bionectins A and B (102, 103), the glycine and tyrosine derivatives of T988C.92
Figure 21.

Verticillin-type epipolythiodioxopiperazines.
The dimeric subset of epipolythiodioxopiperazines increased greatly in number with the discovery of the leptosins (Figure 22) from a strain of Leptosphaeria sp. attached to the marine alga Sargassum tortile.93-95 Leptosins A-K (104-118) all contain at least one valine residue, a feature unique among all families of epipolythiodioxopiperazines. Leptosins A-C, G, H, and K (104-113) share the 12,12′-dihydroxylated, octacyclic core of the verticillins, gliocladines, and chetracins. The C3-C3′ linkage to indole is once again produced in leptosins D-F (114-116), the valine derivatives of the T988s, gliocladines C-E, and the bionectins. In 1994, two remarkable additions to this family were discovered, the epimers leptosin I and J (117 and 118).95 These two compounds are characterized by a C12-C11′ ether linkage, introducing an additional ring to the structure that reduces the degrees of freedom of the molecule. Leptosins are generally toxic to P388 leukemia cells.
Figure 22.

Leptosins.
In summary, over 100 epipolythiodioxopiperazine alkaloids have been isolated and characterized to date. While this was intended to be a comprehensive review of all known members of this class of natural products, some were undoubtedly and inadvertently omitted. One cannot help but marvel at the remarkable biological activity of the class as a whole, although it is unlikely that any epipolythiodioxopiperazine will ever achieve therapeutic utility in the clinic due to the general toxicity inherent in the disulfide through mechanisms discussed above. Certainly others have and will continue to argue otherwise, but it is our opinion that the true contribution to medicine will be realized by employing epipolythiodioxopiperazines as tools for the study of novel biological pathways. Natural products such as chetomin (12), gliotoxin (2), and chaetocin A (13) have already contributed to our understanding of the respective biological targets of these molecules.
Many of the families (particularly those containing a C3-C3′ indoline linkage) share a great deal of structural similarity beyond the epipolythiodioxopiperazine moiety that defines the class. It is likely that hundreds of additional variants exist in nature and have yet to be discovered. While numerous biosynthetic studies have been conducted, we still have only a limited understanding of the ease with which nature is able to assemble epipolythiodioxopiperazines, molecules that have for over fifty years taunted synthetic chemists. In the next section of this review, we present the notable biosynthetic discoveries reported to date.
4. Biosynthetic Investigations of Epidithiodioxopiperazines
Researchers have investigated the biosynthesis of gliotoxin (2) forr nearly forty-five years, but still very little experimental evidence exists to support the numerous proposals. In 1958 and 1960, Suhadolnik reported the only undisputed incorporation study, demonstrating that Trichodermaviride incorporates isotopically labeled phenylalanine (119) and serine (120) into gliotoxin (Scheme 1).96,97 Walsh recently implicated the nonribosomal peptide synthetase GliP in the catalysis of the peptide coupling and dioxopiperazine cyclization reactions.98 Some debate has occurred as to whether cyclo-Phe-Ser (121) is a biosynthetic intermediate to gliotoxin. Although doubly labeled 121 is incorporated into gliotoxin by T. viride, the fungus Penicillium terlikowskii poorly incorporates the same compound.99-101 Walsh observed that release of the dioxopiperazine from the enzyme is slow, allowing for some speculation that further transformations may occur to the enzyme-bound intermediate prior to release.
Scheme 1.

Proposed biosynthesis of gliotoxin.
In 1968, Neuss proposed arene oxide 128 as a biosynthetic intermediate to both the gliotoxins and aranotins. Cyclization at the ortho position would provide the tricyclic core of gliotoxin (see 127) whereas electrocyclic ring opening of the arene oxide could give the oxepine ring (129) characteristic of the aranotins (Scheme 2).33,34 However, structures of metabolites recently isolated from both gliI and gliG deletion mutants of Aspergillus fumigatus suggest that this epoxidation occurs after epidithiol formation in the biosynthesis of the gliotoxins and aranotins.103,104
Scheme 2.
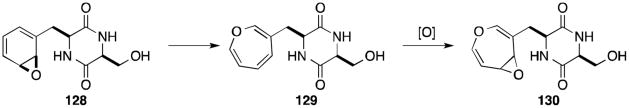
Proposed oxepine ring formation.
The incorporation of sulfur into epidithiodioxopiperazines was until recently poorly understood. Sulfur transfer readily occurs among various potential donors, including methionine, cysteine, and sodium sulfate, complicating [35S] feeding studies.2,102 In 2011, two groups independently provided evidence supporting a GliC–mediated bishydroxylation of a dioxopiperazine (121 to 122).103,104 Elimination of water to generate diiminium 123 could be followed by nucleophilic attack of the cysteine thiolate residues of two glutathione (GSH) molecules, catalyzed by the specialized glutathione S-transferase GliG.103-105 Sequential GliJ peptidase and GliI C–S lyase activity is proposed to reveal the free dithiol (125).103 N-methylation (126), epoxidation to the arene oxide and cyclization would complete the tricyclic core (127). Finally, Scharf and coworkers have unequivocally shown that GliT catalyzes the oxidation to the disulfide to complete the biosynthesis of gliotoxin (2).106 The order of tailoring events is up for debate, but recent advances in genomics and proteomics tools to study and manipulate secondary metabolite production will certainly produce much more accurate insight as to the formation of gliotoxin in nature.
Recall that the toxicity of epidithiodioxopiperazines generally arises from redox cycling or mixed disulfide formation (Figures 4 and 5). How is it then that producing fungi, such as Aspergillus fumigatus, are immune to toxicity inherent in the structure of gliotoxin? Several studies published in the last two years have provided convincing evidence that GliT is responsible for the self-resistance of A. fumigatus to gliotoxin.106,107 Deletion of the gliT gene renders A. fumigatus mutants highly sensitive to gliotoxin, toxicity that can be reversed by addition of glutathione. Moreover, concentrations of reduced gliotoxin increase with the concomitant depletion of intracellular glutathione levels. It is likely that alterations of the cellular redox status and mixed disulfide formation contributes to the growth inhibition observed in the absence of gliT.
Perhaps the cytotoxicity of gliotoxin is an indirect consequence of the true role of gliotoxin and other epidithiodioxopiperazines produced by fungi. The same redox cycling that produces deleterious consequences in naïve organisms may, in Aspergillus fumigatus, serve as a buffer against cellular oxidative stress.107 Gliotoxin and other epidithiodioxopiperazines may have evolved as simple antioxidants, with the host protected from unwanted redox cycling and mixed disulfide formation by GliT.
Sirodesmin PL (58) has also been extensively studied from a biosynthetic perspective. Putative biosynthetic gene clusters for sirodesmin and gliotoxin identified from Leptosphaeria maculans and Aspergillus fumigatus, respectively, are given in Figure 23.108-110 Based on recent advances to our understanding of gliotoxin biosynthesis and the identification of homologs in the sirodesmin gene cluster, we are able to predict many of the experimentally unverified steps in the biosynthesis of sirodesmin PL (Scheme 3).
Figure 23.
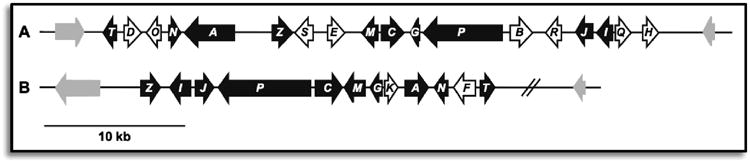
Putative epidithiodioxopiperazine gene clusters for sirodemsin PL (A) and gliotoxin (B).110
Scheme 3.

Proposed biosynthetic pathway of sirodesmin PL.
Feeding studies have shown that Leptosphaeria maculans incorporate labelled tyrosine and serine into both sirodesmin PL (58) and presumed intermediate phomamide (132).59,111-113 We propose the incorporation of sulfur to proceed analogously to the gliotoxin biosynthesis, through dihydroxylation (133), imine formation and glutathione addition (134). Unmasking of the dithiol (135) and oxidation to the disulfide (136) could be followed by oxidation to the arene oxide,Claisen rearrangementand cyclization to give 137. Subsequent cyclization and epoxide opening, addition of water and oxidation to the enone would provide known L. maculans metabolite phomalirazine (61).59,112 Oxidative spirorearrangement, N-methylation, and ketone reduction would lead to another known metabolite, desacetyl-sirodemin PL (59). Acetylation by SirH would complete the biosynthesis of sirodesmin PL (58).2,65,108,110
Wang and coworkers recently utilized a targeted gene-deletion approach to identify nine clustered genes necessary for the biosynthesis of acetylaranotin in Aspergillus terreus (Scheme 4).114 AtaP, a nonribosomal peptide synthetase homologous to GliP and SirP, is responsible for the coupling of two molecules of phenylalanine to dioxopiperazine 138. Bishydroxylation of 138, glutathione conjugation, conversion to dithiol139 and oxidation to the disulfide (140) is proposed to proceed as described above in the homologous pathways. Epoxidation of 140 could be catalyzed by the cytochrome P450 AtaF to give intermediate 141, which could undergo nucleophilic attack of the amide nitrogens to afford diol 142. AtaH–mediated bisacetylation to 143 and two subsequent ring expansions to the dihydrooxepins would complete the biosynthesis of acetylaranotin (35).
Scheme 4.

Proposed biosynthetic pathway for acetylaranotin.
Alternatively, dithiol 139 could undergo bismethylation to 144 (Scheme 5).114 Epoxidation, cyclization, ring expansion and acetylation as described above would complete the biosynthesis of bisdethiodi(methylthio)acetylaranotin (36).
Scheme 5.

Proposed biosynthetic pathway for bisdethiodi(methylthio)acetylaranotin.
Epidithiodioxopiperazine alkaloids possess an astonishing array of molecular architecture that has for decades challenged and inspired synthetic chemists. Biosynthetic studies presented in this section provide a glimpse at the efficiency and elegance with which Nature is able to assemble these compounds. Synthetic chemists strive to mimic and in turn better understand the mechanisms by which microorganisms are able to produce such complexity, hoping to channel some of Nature's efficiency into novel synthetic pathways. In the next section, we present many of the synthetic advances made toward several of the epidithiodioxopiperazines presented above.
5. Introduction to Synthetic Studies
Over 100 naturally occurring epidithiodioxopiperazines have been isolated. However, relatively few have succumbed to total synthesis despite decades of effort, highlighting the challenging synthetic nature of the class of molecules. Initial interest in the biological activity of epidithiodioxopiperazines sparked the synthetic interest of several groups, with six different naturally occurring members of the class yielding to synthetic chemists between 1973 and 1981. Three decades passed before renewed interest in epidithiodioxopiperazines arose, sparked by isolation reports of new metabolites, exciting results from the biological community detailing novel mechanisms of action in cells, and advances in genomics that invigorated interest in elucidating the biosynthesis of these secondary metabolites.115 Between 2009 and 2013 alone, syntheses of twelve additional epidithiodioxo-piperazine alkaloids have been reported. A casual glance through Sections 2 and 3 is enough to impress upon the reader the great degree of structural similarity shared among and between families of epidithiodioxopiperazines. We anticipate that in the coming decade total syntheses of entire families of this class of fungal metabolites will be completed concomitantly with great advances in synthetic methods and biosynthetic understanding. In Sections 6 and 7, we present a comprehensive review of total syntheses of epidithiodioxopiperazines.
6. Early Epidithiodioxopiperazine Syntheses (1973-1981)
The total synthesis of sporidesmin A was completed by Kishi and coworkers in 1973. In a series of communications, Kishi described a novel strategy for the synthesis of epidithiodioxopiperazines using a dithioacetal moiety as a protecting group for the disulfide bridge.116-118 Thus protected, the dithioacetal is stable to acidic, basic, and reducing conditions, allowing for the introduction of thiol groups at an early stage in a total synthesis. Synthesis of the sporidesmins began with the treatment of dioxopiperazine 147 with the dithiane derivative of p-anisaldehyde in the presence of acid to afford dithioacetal-protected dioxopiperazine 148 (Scheme 6).116 Condensation with acid chloride 149 and subsequent methoxymethyl deprotection gave compound 150. Treatment of ketone 150 with DIBAL-H at -78 °C resulted in stereoselective reduction to the alcohol, which was then converted into acetate 151 in 80% yield. Cyclization to the diacetate (152) proceeded upon addition of iodosobenzene diacetate, and hydrolysis of the acetates gave the corresponding diol. Treatment of the diol with m-chloroperbenzoic acid (mCPBA) afforded an intermediate sulfoxide, which decomposed to the disulfide upon exposure to strong Lewis acid, revealing (±)-sporidesmin A (11).
Scheme 6.

Total synthesis of (±)-sporidesmin A.
Intermediate 151 was also used by Kishi in a total synthesis of (±)-sporidesmin B (Scheme 7).119 Reduction of the acetate gave the methylene (153), which underwent an oxidative cyclization similar to that reported in the sporidesmin A synthesis to benzoate 154. The disulfide was revealed as described above, completing the synthesis of (±)-sporidesmin B (62).
Scheme 7.

Synthesis of (±)-sporidesmin B.
Fukuyama and Kishi reported the first synthesis of dehydrogliotoxin (18, Scheme 8) in 1973.117 Benzoic acid derivative 156 was coupled to dioxopiperazine 155 and treated with diazomethane to give methyl ester157. Radical bromination with NBS and benzoyl peroxide was followed by treatment with potassium thioacetate to give dithioacetate 158. Cleavage of the thioacetates and addition of p-methoxybenzaldehyde and boron trifluoride etherate gave a 1:1 diasteromeric mixture of thioacetals 159 in good yield. This masked disulfide is stable to a variety of conditions that the disulfide would not otherwise survive, allowing Kishi to introduce sulfur at an early stage in the synthesis.
Scheme 8.

Synthesis of (±)-dehydrogliotoxin.
A three-step conversion to the primary chloride (160) was followed by addition of phenyllithium to affect cyclization on the desired bridgehead carbon in 79% yield (161). Addition of phenyllithium with benzyl chloromethyl ether efficiently installed the benzyl protected serine side chain (162).121 Cleavage of the benzyl and methyl ethers and conversion to the epidithiodioxopiperazine gave (±)-dehydrogliotoxin (18).
The biosynthesis of gliotoxin (2) is believed to proceed through the intramolecular nucleophilic ring-opening of a phenylalanine-derived arene oxide (122) and has been the subject of considerable speculation and interest. Kishi and coworkers drew inspiration from this biogenetic hypothesis in devising a brilliant total synthesis of gliotoxin. The total synthesis of (±)-gliotoxin was completed in 1976 utilizing the same disulfide protecting strategy as employed above for the sporidesmins, and was re-engineered in 1981 by the same route starting from optically pure dithioacetal 163 obtained from resolution (Scheme 9).120,121 Coupling of 163 with t-butoxy arene oxide 164 in the presence of triton B afforded 165 and 166 in a 2:1 ratio. Acetylation, deprotection, mixed anhydride formation, and reduction gave alcohol 167 in 77% yield from 165. Primary alcohol 167 was converted to the chloride following mesylation, and the secondary ester deprotected to reveal alcohol 168. The key stereoselective cyclization-alkylation reaction was achieved upon addition of phenyllithium to 168 and benzoxymethyl chloride, affording cycloadduct 169 in modest yield (53%). The primary alcohol was revealed upon removal of the benzyl group, and the thioacetal oxidatively removed to afford either (±)- or (+)-gliotoxin (2).
Scheme 9.

Synthesis of (+)-gliotoxin.
Strunz and Kakushima followed the precedent set by Kishi and completed a total synthesis of hyalodendrin (3, Scheme 10).122 Known thioacetal 170 was alkylated sequentially with chloromethyl methyl ether and benzyl bromide, then deprotected to reveal hyalodendrin (3).
Scheme 10.
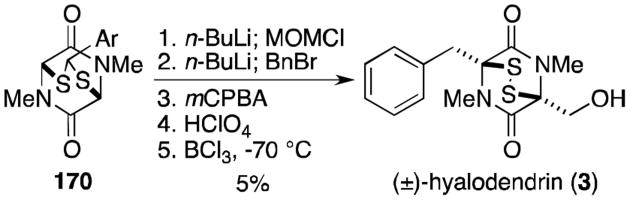
Synthesis of (±)-hyalodendrin.
In 1979 and 1980, Williams and Rastetter reported the total synthesis of gliovictin (32, Scheme 11).123,124 Sarcosine anhydride (171) was converted in three steps to silyl enol ether 173. Alkylation with benzyl bromide and concomitant sulfenylation and deprotection gave methyl sulfide 175. Additional sulfenylation gave a 3:1 mixture of diastereomers (176), favoring the undesired anti isomer. Reduction of the mixture of aldehydes completed the total synthesis of gliovictin (32) and epi-gliovictin (177).
Scheme 11.

Total synthesis of (±)-gliovictin and epi-gliovictin.
A synthesis of hyalodendrin (3, Scheme 12) was achieved from silyl enol ether 174, used previously in the gliovictin synthesis.123 Addition of monoclinic sulfur to the enolate of 174 followed by reductive workup provided thiol 178. Conversion to the enolic methyl disulfide, deprotection of the silyl group, and sulfenylation with triphenylmethyl chlorodisulfide gave a mixture of diastereomers, unfortunately favoring the undesired anti isomer (180). Reduction and oxidation of 180 gave hyalodendrin (3) in 29% yield.
Scheme 12.

Williams and Rastetter's total synthesis of (±)-hyalodendrin.
In 1993, Williams and Miknis reported a total synthesis of aspirochlorine, the structure of which contains a unique seven-membered bicyclo[3.2.2] disulfide ring system.125 4-chloro-resorcinol (181) was converted to the benzaldehyde via a Gatterman formylation, protected as the MOM ether, and cyclized to coumarilic acid derivative (182, Scheme 13). Schotten–Baumann coupling of the glycine derivative was followed by protection of the phenol to afford 183 in excellent yield. After conversion of 183 to the hydroxamic ester, tricyclic compound 184 was formed upon treatment with NBS. The bromide was converted to the methyl ether, the photolabile o-nitrobenzyl group removed, the dioxopiperazine ring oxidized, and the acetate protecting group cleaved to afford a mixture of diastereomers (185). The methyl ethers were converted to the bisthioacetates (186) in good yield and themixture of diastereomers treated with excess methoxyamine and camphor sulfonic acid to give (±)-aspirochlorine (187).
Scheme 13.

Total synthesis of (±)-aspirochlorine.
7. Recent Epidithiodioxopiperazine Syntheses (2009-2013)
Strangely, synthetic activity in the field of epidithiodioxopiperazines remained relatively dormant for several decades following the landmark work of Kishi and the few other laboratories noted above. Beginning in about 2009, a resurgence of activity burst onto the chemical scene and several remarkable total syntheses of epidithiodioxopiperazines have been reported in the last four years. Movassaghi and coworkers were the first to publish in this modern synthetic revival, reporting the synthesis of (+)-11,11′-dideoxyverticillin A (196, Scheme 14).126 The authors showed that the amine resulting from cleavage of the N-Boc carbamate of 188 was readily cyclized to dioxopiperazine 189 in morpholine. Exposure of 189 to bromine produced 3-bromopyrroloindoline 190, and the amides were subsequently methylated upon treatment with iodomethane. Addition of tris(triphenylphosphine)cobalt chloride gave the desired dimeric intermediate 191. After extensive investigation searching for appropriate stereoselective hydroxylation conditions, the dimer was eventually oxidized with bis(pyridine)silver(I) permanganate to a fragile octacyclic tetraol (192). Exposure of 192 to Fu's (R)-(+)-4-pyrrolidinopyridinyl(pentamethyl-cyclopentadienyl)iron (PPY) catalyst with t-butyldimethylsilyl chloride (TBSCl) gave selectively the alanine-derived protected hemiaminals (193). Removal of the benzenesulfonyl groups with sodium amalgam revealed diaminodiol 194. Treatment of 194 with K2CS3 followed by ethanolamine gave diaminotetrathiol 195, which readily oxidized to (+)-11,11′-dideoxyverticillin A (196) when partitioned between aqueous hydrochloric acid and dichloromethane and treated with potassium triiodide.
Scheme 14.

Biomimetic total synthesis of (+)-11,11′-dideoxyverticillin A.
Chaetocin (13, Scheme 15) is quite similar in structure to 11,11′-dideoxyverticillin A, derived from two molecules of serine rather than alanine. Substituted dioxopiperazine 199 was prepared in five steps from N-Me,Cbz-D-Ser (197) and D-Trp-OMe (198).127,128 Bromocyclization gave tetracycle 200, converted to the tribromide (201) under radical conditions and hemiaminal 202 following addition of water. Movassaghi's reductive dimerization conditions using a Co(I) complex afforded the desired dimer (203) in modest yield. Addition of 203 to condensed hydrogen sulfide and BF3· OEt2 formed the tetrathiol, oxidized to the bis(disulfide) upon addition of iodine to complete the synthesis of (+)-chaetocin (13).
Scheme 15.

Sodeoka's total synthesis of (+)-chaetocin.
Sodeoka originally planned to form the tetraol from the product resulting from dimerization of 200. It was noted that this dimer was not stable to the radical conditions employed above, so the synthetic plan was altered to that shown in Scheme 15 to circumvent this problem. Recall that Movassaghi had similar difficulties in forming related tetraol 192 and eventually settled on the mild oxidation discussed above.
Mere months after Sodeoka reported the first total synthesis of chaetocin, Movassaghi published the synthesis of (+)-chaetocin (13, Scheme 16), the hexasulfide (+)-chaetocin C (79), and the octasulfide (+)-12,12′-dideoxychetracin A (81, Scheme 17), all from the natural amino acids l-serine and l-tryptophan.129
Scheme 16.

Movassaghi's total synthesis of (+)-chaetocin.
Scheme 17.

Total syntheses of chaetocin C and 12,12′-dideoxychetracin A.
Bromocyclization of dioxopiperazine 204 gave endo-tetracyclic bromide 205. N-methylation, protecting group exchange, and key reductive radical dimerization afforded the dimeric octacycle (206). Selective tetrahydroxylation was achieved using Py2AgMnO4. Tetraol 207 is analogous to 203 used in Sodeoka's synthesis, but interestingly the acetate allows for differentiation of the hemiaminals, resulting in regioselective substitution of hydrogen sulfide. Protection of resulting thiohemiaminal as the dithioisobutyrate (208) served both to prevent opening of the hemiaminal under polar protic conditions and to activate the hemiaminal to mild ionization in future steps. Mild deprotection of the sulfonyl group was followed by chemoselective hydrazinolysis and addition of triphenylmethanesulfenyl chloride to give disulfane 209. Ionization of the isobutyrates and cyclization to the epidithiodioxopiperazines with concomitant loss of a triphenylmethyl cation was followed by removal of the acetates using Otera's catalyst to afford (+)-chaetocin (13).
Chaetocin C (79) and 12,12′-dideoxychetracin A (81), the epitri- and epitetrathiodioxopiperazine analogues of chaetocin, were similarly synthesized from a common intermediate (Scheme 17).129
Hydrazinolysis of diaminodithioisobutyrate 210 was followed by treatment with the corresponding sulfur source to give 211 or 212. It was necessary to protect the indoline nitrogens as the trifluoroacetates before addition of Lewis acid to form the polysulfide bridges (213 and 214), presumably to prevent decomposition pathways encountered over longer reactions times necessary to the formation of the larger polysulfide bridges. Global deprotection gave either (+)-chaetocin C (79) or (+)-12,12′-dideoxychetracin A (81).
In 2011, Overman and coworkers reported the total synthesis of (+)-gliocladine C (99, Scheme 18).130 Known 2-indolinone 215 (prepared from isatin and indole) was reduced and Boc-protected to afford compound 216. Conversion to the oxindole ester (217) proceeded efficiently upon treatment with 2,2,2-trichloro-1,1-dimethylethyl chloroformate, triethyl amine, and 10 mol % of Fu's (S)-(–)-4-pyrrolidinopyrindinyl-(pentamethylcyclopentadienyl)iron catalyst. The oxindole was elaborated to indoline 218 over four steps in 54% yield. Aldol condensation with the lithium enolate of the piperazinedione provided exclusively the Z isomer of 219, which readily cyclized to hexacycle 220 upon treatment with BF3·OEt2. Grignard addition, silyl protection of the resultant alcohol, asymmetric dihydroxylation, and acetylation gave advanced intermediate 221. Addition of the epimeric mixture of silyl ethers to condensed hydrogen sulfide and BF3·OEt2 gave the dithiol, oxidized to the disulfide upon exposure to oxygen. Removal of the acetate revealed (+)-gliocladine C (99).
Scheme 18.

Synthesis of (+)-gliocladine C.
This approach was later shown in 2013 to be generally applicable to several epidithiodioxopiperazine natural products characterized by a hydroxyl group adjacent to the quaternary stereocenter (Scheme 19).131 Overman and coworkers reported that trioxopiperazine 220 could be converted to (+)-leptosin D (114) through an analogous pathway as for gliocladine C, substituting MeMgCl for i-PrMgCl in the Grignard addition. Alternatively, if methyl Grignard addition to 220 is followed by elimination of the resultant alcohol, dihydroxylation and TBS protection, the same general method can then be employed to furnish (+)-T988C (88) from 223. Bionectins A and C, the analogous glycine derivatives, were also prepared from 220. Stereoselective reduction of the secondary amide replaced previous Grignard additions, with the resulting alcohol elaborated to protected triol 224. The dithiol intermediate formed upon addition of H2S and BF3·OEt2 was converted either to the epidisulfide, (+)-bionectin A(102), or the dithioether, (+)-bionectin C(225).
Scheme 19.

Divergent syntheses of (+)-leptosin D, (+)-T988C, (+)-bionectin A and (+)-bionectin C.
Boyer and Movassaghi completed the total synthesis of two structurally related compounds in 2012. The most direct route to the first, (+)-gliocladin B (230), is detailed in Scheme 20.132 Bromocyclization proceeded as above, providing tetracycle 227 in good yield with excellent stereoselectivity (97:3 endo:exo). A Friedel–Crafts type coupling of N-TIPS-5-bromoindole with 227 formed the desired 3-3′ bond. Several indole derivatives were screened, and the 5-bromo derivative was found to give the best yield and selectivity. The bromine and silyl protecting group were removed and the resulting compound oxidized to diol 229. Exposure of this diol to sodium thiomethoxide and trifluoracetic acid gave the dithioether derivative. (+)-Gliocladin B (230) was revealed upon removal of the benzenesulfonyl group.
Scheme 20.

Total synthesis of (+)-gliocladin B.
Although it has not to date been isolated from a natural source, it is worth noting that the authors were able to convert diol 229 into the disulfide (+)-12-deoxybionectin A (233) using conditions analogous to those discussed above in the chaetocin syntheses (Scheme 21).132 Disulfide 233 is also easily converted to (+)-gliocladin B (230) by reduction of the disulfide with sodium borohydride in the presence of iodomethane.
Scheme 21.

Synthesis of (+)-12-deoxybionectin A and (+)-gliocladin B.
In 2013, Movassaghi and coworkers expanded on this work with a synthesis of the C12-hydroxylated analogues, (+)-bionectin A (102) and (+)-bionectin C (225, Scheme 22).133 Anti-aldol reaction of aldehyde 234 and ethyl iminogylcinate 235 gave alcohol 236. Subsequent TBS protection of the alcohol and hydrolysis of the Schiff base revealed β-hydroxy amino ester 237. Coupling with sarcosine, deprotection and concomitant formation of the dioxopiperazine, and diastereoselective bromocyclization gave bromopyrroloindoline 238. Unfortunately, the presence of the C12-hydroxyl group prevented the successful application of an intermolecular Friedel–Crafts reaction as in the gliocladin B synthesis. The C3-C3′ bond was instead formed through a silver-mediated intramolecular Friedel–Crafts reaction of 239 to 240. Removal of the bridging silyl group revealed the desired core structure (241) in moderate yield. Following Boc protection of the alcohol, treatment with bis(pyridine)silver(I) permanganate gave the monohydroxylated trioxopiperazine rather than the expected dihydroxylated dioxopiperazine. While this unexpected product, analogous to trioxopiperazine 220 prepared by Overman and coworkers above, could likely be converted to a number of naturally occurring analogues, the authors only demonstrate its conversion to the bionectins. Sodium borohydride reduction gave the desired diol, converted to dipivaloate 242 upon treatment with pivaloyl chloride and DMAP. Treatment of 242 with 4-mercaptobutan-2-one and trifluoroacetic acid resulted in concomitant formation of the bisthioethers with deprotection of the Boc groups. The benzensulfonyl group was removed, the dithiols revealed and oxidized with KI3 to afford the epidisulfide, (+)-bionectin A (102). Reductive methylation gave the dithioether, (+)-bionectin C (225).
Scheme 22.

Total synthesis of (+)-bionectin A and (+)-bionectin C.
Epiccocin G (250) lacks a disulfide bridge and was thus not discussed in Section 2. Nonetheless, the structure is remarkably similar to that of epicorazine A (8), and a discussion of Nicolaou's recent total synthesis is certainly relevant in this context.134 Hydroxy enone 243 (prepared in two steps from N-Boc-tyrosine) was converted in four steps to hydroxy methyl ester 244 (Scheme 23). Separate deprotections to either the amine or carboxylic acid gave two derivatives that were coupled to form amide 245. Deprotection and cyclization to the dioxopiperazine was followed by conversion to the bistrifluoroacetate (246), which eliminated to bisdiene 247 upon exposure to Pd(PPh3)4 catalyst. Treatment with S8 and NaHMDS gave a mixture of oligosulfenylated compounds (248) that were readily converted to the bisdimethylthio compound (249) upon reduction with sodium borohydride and addition of iodomethane. Bisendoperoxide 251 was generated on reaction with singlet oxygen, and addition of DBU induced a Kornblum–DeLaMare rearrangement to give a bishydroxy enone. Reduction to the ketone completed the total synthesis of epicoccin G (250).
Scheme 23.

Total synthesis of epicoccin G.
The next epidithiodioxopiperazine total synthesis to be discussed was reported recently by the Reisman group, who completed an elegant synthesis of (–)-acetylaranotin (35).135 Pyrrolidine 254 was synthesized with >98% ee by (1,3)-dipolar cycloaddition of cinnamaldimine 253 to t-butyl acrylate (252) and subsequent cleavage of the t-butyl ester (Scheme 24). Protection of the amine and ozonolytic cleavage of the alkene provided lactone 255 in good yield. Ethynylmagnesium bromide was added to the hydroxylactone to form the hydroxy acid, which upon addition of triphenylphosphine and DIAD underwent Mitsunobu lactonization to 256. Reduction to the diol (257) was followed by bis-TBS protection of the alcohols and selective deprotection of the primary alcohol. The aldehyde obtained following oxidation with Dess–Martin periodinane was efficiently converted to chlorohydrin 258.
Scheme 24.

Key pyrrolidine synthesis.
Formation of the dihydrooxepine (259) was realized upon treatment of alkyne 258 with catalytic [Rh(cod)-Cl]2 and tris(4-fluorophenyl)phosphine (Scheme 25).135 Elimination of HCl and hydrolysis of the ester gave acid 260, while deprotection of the Teoc group and HCl elimination of the same compound gave amine 261. BOP-Cl–mediated coupling of 260 and 261 delivered amide 262, which readily cyclized to dioxopiperazine 263 after TBAF·(t-BuOH)4–induced desilylation. Synthesis of (–)-acetylaranotin (35) was completed following formation of tetrasulfide 264, bisacetylation, mild reduction to the dithiol, and oxidation to the natural disulfide. This publication marked the first total synthesis of a dihydrooxepine-containing epidithiodioxopiperazine natural product.
Scheme 25.

Completion of the total synthesis of (–)-acetylaranotin.
Tokuyama and coworkers completed an elegant total synthesis of acetylaranotin that intercepts an advanced intermediate from the Reisman synthesis (Scheme 26).136 Known β,γ-unsaturated ketone 265 was converted to the α,β-unsaturated ketone with catalytic DBU, then treated with basic hydrogen peroxide to give epoxyketone 266. Enone 267 was prepared from 266 through Wharton rearrangement and Dess–Martin oxidation. This enone was converted to the corresponding dienol silyl ether on treatment with TMSOTf, then oxidized to an epoxide that opened to form the desired γ-hydroxyenone on acidic workup with silica gel. Following protection as the silylether (268), a Baeyer–Villiger oxidation gave enol lactone 269. Dihydrooxepine 270 was revealed through formation of the enol triflate and reduction. Coupling and cyclization of two equivalents of 270 to dioxopiperazine 271 proceeded over four steps, analogous to Reisman's synthesis. Oxidation of the diol with nor-AZADO was followed by sodium borohydride reduction with CeCl3 to afford the correct diastereomer (263), thus intercepting Reisman and coworker's synthesis. The final three steps were completed as discussed above, albeit in lower yield, to complete the synthesis of (–)-acetylaranotin (35).
Scheme 26.

Tokuyama's formal synthesis of (–)-acetylaranotin.
8. Conclusions
Epidithiodioxopiperazine alkaloids possess an astonishing array of molecular architecture and, with that, corresponding synthetic challenges to construct such substances. The biosynthesis of many of the natural metabolites touched on in Section 4 has certainly inspired many of the chemists cited in this review, as several have sought to exploit insights from Nature's strategic bond constructions in a synthetic laboratory context.
Many new and exciting biological targets have recently been discovered for this family of natural substances, yet their intrinsic chemical and biochemical reactivities and toxicity have thus far precluded the development of any natural member of this family or synthetic analogs as serious therapeutic candidates. Nonetheless, this family of alkaloids will almost certainly continue to inspire and challenge synthetic chemists, and new and exciting chapters in the biogenesis and chemical biology of these agents are yet to be written.
Acknowledgments
This review is dedicated to Professor Yoshito Kishi of Harvard University on the occasion of his 77th birthday (kiju) and is also dedicated to Tohru Fukuyama on the occasion of his retirement from the University of Tokyo. Financial support was generously provided by the National Institutes of Health (grant RO1 CA070375).
Footnotes
Notes: The authors declare no competing financial interest.
References
- 1.Cook KM, Hilton ST, Mecinovic J, Motherwell WB, Figg WD, Schofield CJ. J Biol Chem. 2009;284:26831–26838. doi: 10.1074/jbc.M109.009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 3.Waring P, Beaver J. Gen Pharmacol. 1996;27:1311–1316. doi: 10.1016/s0306-3623(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 4.Waring P, Eichner RD, Mullbacher A. Med Res Rev. 1988;8:499–524. doi: 10.1002/med.2610080404. [DOI] [PubMed] [Google Scholar]
- 5.Iwasa E, Hamashima Y, Sodeoka M. Isr J Chem. 2011;51:420–433. [Google Scholar]
- 6.Weindling R, Emerson OH. Phytopathology. 1936;26:1068–1070. [Google Scholar]
- 7.Bell MR, Johnson JR, Wildi BS, Woodward RB. J Am Chem Soc. 1958;80:1001–1001. [Google Scholar]
- 8.Beecham AF, Fridrichsons J, Mathieson AM. Tetrahedron Lett. 1966:3131. doi: 10.1016/s0040-4039(01)99927-7. [DOI] [PubMed] [Google Scholar]
- 9.Jordan TW, Cordiner SJ. Trends Pharmacol Sci. 1987;8:144–149. [Google Scholar]
- 10.Kwon-Chung KJ, Sugui JA. Med Mycol. 2009;47 Suppl 1:S97–103. doi: 10.1080/13693780802056012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullbacher A, Eichner RD. Proc Natl Acad Sci Biol. 1984;81:3835–3837. doi: 10.1073/pnas.81.12.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullbacher A, Waring P, Eichner RD. J Gen Microbiol. 1985;131:1251–1258. doi: 10.1099/00221287-131-5-1251. [DOI] [PubMed] [Google Scholar]
- 13.Mullbacher A, Moreland AF, Waring P, Sjaarda A, Eichner RD. Transplantation. 1988;46:120–125. doi: 10.1097/00007890-198807000-00022. [DOI] [PubMed] [Google Scholar]
- 14.Munday R. Chem-Biol Interact. 1982;41:361–374. doi: 10.1016/0009-2797(82)90112-0. [DOI] [PubMed] [Google Scholar]
- 15.Chai CL, Waring P. Redox Rep. 2000;5:257–264. doi: 10.1179/135100000101535799. [DOI] [PubMed] [Google Scholar]
- 16.Eichner RD, Waring P, Geue AM, Braithwaite AW, Mullbacher A. J Biol Chem. 1988;263:3772–3777. [PubMed] [Google Scholar]
- 17.Cordiner SJ, Jordan TW. Biochem J. 1983;212:197–204. doi: 10.1042/bj2120197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mason JW, Kidd JG. J Immunol. 1951;66:99–106. [PubMed] [Google Scholar]
- 19.Hurne AM, Chai CL, Waring P. J Biol Chem. 2000;275:25202–25206. doi: 10.1074/jbc.M002278200. [DOI] [PubMed] [Google Scholar]
- 20.Trown PW, Bilello JA. Antimicrob Agents Chemother. 1972;2:261–266. doi: 10.1128/aac.2.4.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullbacher A, Waring P, Tiwari-Palni U, Eichner RD. Mol Immunol. 1986;23:231–235. doi: 10.1016/0161-5890(86)90047-7. [DOI] [PubMed] [Google Scholar]
- 22.Stillwell MA, Magasi LP, Strunz GM. Can J Microbiol. 1974;20:759–764. doi: 10.1139/m74-116. [DOI] [PubMed] [Google Scholar]
- 23.Strunz GM, Heissner CJ, Kakushima M, Stillwell MA. Can J Chem. 1974;52:325–326. [Google Scholar]
- 24.Strunz GM, Kakushima M, Stillwell MA. Can J Chem. 1975;53:295–297. [Google Scholar]
- 25.DeVault RL, Rosenbrook W., Jr J Antibiot. 1973;26:532–534. doi: 10.7164/antibiotics.26.532. [DOI] [PubMed] [Google Scholar]
- 26.Dorn F, Arigoni D. Experientia. 1974;30:134–135. [Google Scholar]
- 27.Michel KH, Chaney MO, Jones ND, Hoehn MM, Nagarajan R. J Antibiot. 1974;27:57–64. doi: 10.7164/antibiotics.27.57. [DOI] [PubMed] [Google Scholar]
- 28.Shin JH, Fenical W. Phytochemistry. 1987;26:3347. [Google Scholar]
- 29.Kawahara N, Nozawa K, Nakajima S, Kawai K. J Chem Soc Perkin Trans. 1987;1:2099–2101. [Google Scholar]
- 30.Hanson JR, Oleary MA. J Chem Soc Perkin Trans. 1981;1:218–220. [Google Scholar]
- 31.Nagarajan R, Huckstep LL, Lively DH, Delong DC, Marsh MM, Neuss N. J Am Chem Soc. 1968;90:2980. [Google Scholar]
- 32.Nagarajan R, Neuss N, Marsh MM. J Am Chem Soc. 1968;90:6518. doi: 10.1021/ja01025a053. [DOI] [PubMed] [Google Scholar]
- 33.Neuss N, Boeck LD, Brannon DR, Cline JC, DeLong DC, Gorman M, Huckstep LL, Lively DH, Mabe J, Marsh MM, Molloy BB, Nagarajan R, Nelson JD, Stark WM. Antimicrob Agents Chemother. 1968;8:213–219. [PubMed] [Google Scholar]
- 34.Neuss N, Nagarajan R, Molloy BB, Huckstep LL. Tetrahedron Lett. 1968:4467. [Google Scholar]
- 35.Trown PW, Lindh HF, Milstrey KP, Gallo VM, Mayberry BR, Lindsay HL, Miller PA. Antimicrob Agents Chemother. 1968;8:225–228. [PubMed] [Google Scholar]
- 36.Cosulich DB, Nelson NR, Van den Hende JH. J Am Chem Soc. 1968;90:6519. [Google Scholar]
- 37.Murdock KC. J Med Chem. 1974;17:827–835. doi: 10.1021/jm00254a010. [DOI] [PubMed] [Google Scholar]
- 38.Kawahara N, Nakajima S, Yamazaki M, Kawai K. Chem Pharm Bull. 1989;37:2592–2595. doi: 10.1248/cpb.37.2592. [DOI] [PubMed] [Google Scholar]
- 39.Kawahara N, Nozawa K, Yamazaki M, Nakajima S, Kawai K. Chem Pharm Bull. 1990;38:73–78. doi: 10.1248/cpb.38.73. [DOI] [PubMed] [Google Scholar]
- 40.Kawahara N, Nozawa K, Nakajima S, Kawai K, Yamazaki M. J Chem Soc Chem Commun. 1989:951–952. [Google Scholar]
- 41.Kawahara N, Nozawa K, Yamazaki M, Nakajima S, Kawai K. Heterocycles. 1990;30:507–515. [Google Scholar]
- 42.Waring P, Eichner RD, Tiwaripalni U, Mullbacher A. Aust J Chem. 1987;40:991–997. [Google Scholar]
- 43.Ueno Y, Umemori K, Niimi E, Tanuma S, Nagata S, Sugamata M, Ihara T, Sekijima M, Kawai K, Ueno I, et al. Nat Toxins. 1995;3:129–137. doi: 10.1002/nt.2620030303. [DOI] [PubMed] [Google Scholar]
- 44.Seya H, Nozawa K, Nakajima S, Kawai KI, Udagawa SI. J Chem Soc Perkin Trans. 1986;1:109–116. [Google Scholar]
- 45.Seya H, Nakajima S, Kawai K, Udagawa S. J Chem Soc Chem Commun. 1985:657–658. [Google Scholar]
- 46.Kawai K, Nozawa K, Seya H, Kawahara N, Udagawa S, Nakajima S. Heterocycles. 1987;26:475–479. [Google Scholar]
- 47.Kawahara N, Nozawa K, Nakajima S, Kawai K. J Chem Soc Chem Commun. 1986:1495–1496. [Google Scholar]
- 48.Seya H, Nozawa K, Udagawa S, Nakajima S, Kawai K. Chem Pharm Bull. 1986;34:2411–2416. doi: 10.1248/cpb.34.2411. [DOI] [PubMed] [Google Scholar]
- 49.Nozawa K, Udagawa SI, Nakajima S, Kawai KI. Chem Pharm Bull. 1987;35:3460–3463. [Google Scholar]
- 50.Onodera H, Hasegawa A, Tsumagari N, Nakai R, Ogawa T, Kanda Y. Org Lett. 2004;6:4101–4104. doi: 10.1021/ol048202d. [DOI] [PubMed] [Google Scholar]
- 51.Deffieux G, Gadret M, Leger JM, Carpy A. Acta Crystallogr B. 1977;33:1474–1478. [Google Scholar]
- 52.Deffieux G, Baute MA, Baute R, Filleau MJ. J Antibiot. 1978;31:1102–1105. doi: 10.7164/antibiotics.31.1102. [DOI] [PubMed] [Google Scholar]
- 53.Deffieux G, Filleau MJ, Baute R. J Antibiot. 1978;31:1106–1109. doi: 10.7164/antibiotics.31.1106. [DOI] [PubMed] [Google Scholar]
- 54.Brown AE, Finlay R, Ward JS. Soil Biol Biochem. 1987;19:657–664. [Google Scholar]
- 55.Kleinwachter P, Dahse HM, Luhmann U, Schlegel B, Dornberger K. J Antibiot. 2001;54:521–525. doi: 10.7164/antibiotics.54.521. [DOI] [PubMed] [Google Scholar]
- 56.Begg WR, Elix JA, Jones AJ. Tetrahedron Lett. 1978:1047–1050. [Google Scholar]
- 57.Ernst-Russell MA, Chai CLL, Hurne AM, Waring P, Hockless DCR, Elix JA. Aust J Chem. 1999;52:279–283. [Google Scholar]
- 58.Moerman KL, Chai CLL, Waring P. Toxicol Appl Pharmacol. 2003;190:232–240. doi: 10.1016/s0041-008x(03)00189-3. [DOI] [PubMed] [Google Scholar]
- 59.Curtis PJ, Greatbanks D, Hesp B, Cameron AF, Freer AA. J Chem Soc Perkin Trans. 1977;1:180–189. doi: 10.1039/p19770000180. [DOI] [PubMed] [Google Scholar]
- 60.Ferezou JP, Riche C, Quesneauthierry A, Pascardbilly C, Barbier M, Bousquet JF, Boudart G. Nouv J Chim. 1977;1:327–334. [Google Scholar]
- 61.Boudart G. Appl Environ Microbiol. 1989;55:1555–1559. doi: 10.1128/aem.55.6.1555-1559.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elliott CE, Gardiner DM, Thomas G, Cozijnsen A, Van De Wouw A, Howlett BJ. Mol Plant Pathol. 2007;8:791–802. doi: 10.1111/j.1364-3703.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 63.Thornton RH, Sinclair DP. Nature. 1959;184:1327–1328. doi: 10.1038/1841327b0. [DOI] [PubMed] [Google Scholar]
- 64.Thornton RH, Percival JC. Nature. 1959;183:63. doi: 10.1038/183063a0. [DOI] [PubMed] [Google Scholar]
- 65.Synge RLM, White EP. Chem Ind. 1959:1546–1547. [Google Scholar]
- 66.Fridrichsons J, Mathieson AM. Acta Crystallogr. 1965;18:1043. [Google Scholar]
- 67.Fridrichsons J, Mathieson AM. Tetrahedron Lett. 1962:1265–1268. [Google Scholar]
- 68.Smith BL, Embling PP, Towers NR, Wright DE, Payne E. New Zealand veterinary journal. 1977;25:124–127. doi: 10.1080/00480169.1977.34379. [DOI] [PubMed] [Google Scholar]
- 69.Waring P, Egan M, Braithwaite A, Mullbacher A, Sjaarda A. Int J Immunopharmacol. 1990;12:445–457. doi: 10.1016/0192-0561(90)90028-l. [DOI] [PubMed] [Google Scholar]
- 70.Munday R. J Appl Toxicol. 1984;4:182–186. doi: 10.1002/jat.2550040405. [DOI] [PubMed] [Google Scholar]
- 71.Hodges R, Shannon JS. Aust J Chem. 1966;19:1059. [Google Scholar]
- 72.Waksman SA, Bugie E. J Bacteriol. 1944;48:527–530. doi: 10.1128/jb.48.5.527-530.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mcinnes AG, Taylor A, Walter JA. J Am Chem Soc. 1976;98:6741–6741. doi: 10.1021/ja00437a074. [DOI] [PubMed] [Google Scholar]
- 74.Li GY, Li BG, Yang T, Yan JF, Liu GY, Zhang GL. J Nat Prod. 2006;69:1374–1376. doi: 10.1021/np0602970. [DOI] [PubMed] [Google Scholar]
- 75.Fujimoto H, Sumino M, Okuyama E, Ishibashi M. J Nat Prod. 2004;67:98–102. doi: 10.1021/np0302201. [DOI] [PubMed] [Google Scholar]
- 76.Welch TR, Williams RM. Tetrahedron. 2013;69:770–773. doi: 10.1016/j.tet.2012.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 78.Staab A, Loeffler J, Said HM, Diehlmann D, Katzer A, Beyer M, Fleischer M, Schwab F, Baier K, Einsele H, Flentje M, Vordermark D. BMC Cancer. 2007;7:213. doi: 10.1186/1471-2407-7-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mohammed KA, Jadulco RC, Bugni TS, Harper MK, Sturdy M, Ireland CM. J Med Chem. 2008;51:1402–1405. doi: 10.1021/jm7010854. [DOI] [PubMed] [Google Scholar]
- 80.Melillo G. Methods Enzymol. 2007;435:385–402. doi: 10.1016/S0076-6879(07)35020-9. [DOI] [PubMed] [Google Scholar]
- 81.Block KM, Wang H, Szabo LZ, Polaske NW, Henchey LK, Dubey R, Kushal S, Laszlo CF, Makhoul J, Song Z, Meuillet EJ, Olenyuk BZ. J Am Chem Soc. 2009;131:18078–18088. doi: 10.1021/ja807601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hauser D, Weber HP, Sigg HP. Helv Chim Acta. 1970;53:1061–1073. doi: 10.1002/hlca.19700530521. [DOI] [PubMed] [Google Scholar]
- 83.Weber HP. Acta Crystallogr B. 1972;B 28:2945. [Google Scholar]
- 84.Saito T, Suzuki Y, Koyama K, Natori S, Iitaka Y, Kinoshita T. Chem Pharm Bull. 1988;36:1942–1956. [Google Scholar]
- 85.Li L, Li D, Luan Y, Gu Q, Zhu T. J Nat Prod. 2012;75:920–927. doi: 10.1021/np3000443. [DOI] [PubMed] [Google Scholar]
- 86.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 87.Feng Y, Blunt JW, Cole AL, Munro MH. J Nat Prod. 2004;67:2090–2092. doi: 10.1021/np030326w. [DOI] [PubMed] [Google Scholar]
- 88.Minato H, Matsumoto M, Katayama T. J Chem Soc Perkin Trans. 1973;117:1819–1825. doi: 10.1039/p19730001819. [DOI] [PubMed] [Google Scholar]
- 89.Joshi BK, Gloer JB, Wicklow DT. J Nat Prod. 1999;62:730–733. doi: 10.1021/np980530x. [DOI] [PubMed] [Google Scholar]
- 90.Chu M, Truumees I, Rothofsky ML, Patel MG, Gentile F, Das PR, Puar MS, Lin SL. J Antibiot. 1995;48:1440–1445. doi: 10.7164/antibiotics.48.1440. [DOI] [PubMed] [Google Scholar]
- 91.Dong JY, He HP, Shen YM, Zhang KQ. J Nat Prod. 2005;68:1510–1513. doi: 10.1021/np0502241. [DOI] [PubMed] [Google Scholar]
- 92.Zheng CJ, Kim CJ, Bae KS, Kim YH, Kim WG. J Nat Prod. 2006;69:1816–1819. doi: 10.1021/np060348t. [DOI] [PubMed] [Google Scholar]
- 93.Takahashi C, Minoura K, Yamada T, Numata A, Kushida K, Shingu T, Hagishita S, Nakai H, Sato T, Harada H. Tetrahedron. 1995;51:3483–3498. [Google Scholar]
- 94.Takahashi C, Numata A, Ito Y, Matsumura E, Araki H, Iwaki H, Kushida K. J Chem Soc Perkin Trans. 1994;1:1859–1864. [Google Scholar]
- 95.Takahashi C, Numata A, Matsumura E, Minoura K, Eto H, Shingu T, Ito T, Hasegawa T. J Antibiot. 1994;47:1242–1249. doi: 10.7164/antibiotics.47.1242. [DOI] [PubMed] [Google Scholar]
- 96.Winstead JA, Suhadolnik RJ. J Am Chem Soc. 1960;82:1644–1647. [Google Scholar]
- 97.Suhadolnik RJ, Chenoweth RG. J Am Chem Soc. 1958;80:4391–4392. [Google Scholar]
- 98.Balibar CJ, Walsh CT. Biochemistry. 2006;45:15029–15038. doi: 10.1021/bi061845b. [DOI] [PubMed] [Google Scholar]
- 99.Bulock JD, Leigh C. J Chem Soc Chem Commun. 1975:628–629. [Google Scholar]
- 100.Kirby GW, Patrick GL, Robins DJ. J Chem Soc Perkin Trans. 1978;1:1336–1338. [Google Scholar]
- 101.Macdonald JC, Slater GP. Can J Biochem. 1975;53:475–478. doi: 10.1139/o75-066. [DOI] [PubMed] [Google Scholar]
- 102.Kirby GW, Rao GV, Robins DJ. J Chem Soc Perkin Trans. 1988;1:301–304. [Google Scholar]
- 103.Davis C, Carberry S, Schrettl M, Singh I, Stephens JC, Barry SM, Kavanagh K, Challis GL, Brougham D, Doyle S. Chem Biol. 2011;18:542–552. doi: 10.1016/j.chembiol.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 104.Scharf DH, Remme N, Habel A, Chankhamjon P, Scherlach K, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. J Am Chem Soc. 2011;133:12322–12325. doi: 10.1021/ja201311d. [DOI] [PubMed] [Google Scholar]
- 105.Scharf DH, Heinekamp T, Remme N, Hortschansky P, Brakhage AA, Hertweck C. Appl Microbiol Biotechnol. 2012;93:467–472. doi: 10.1007/s00253-011-3689-1. [DOI] [PubMed] [Google Scholar]
- 106.Scharf DH, Remme N, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. J Am Chem Soc. 2010;132:10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- 107.Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O'Brien J, Nolan A, Stephens J, Fenelon O, Doyle S. PLoS Pathog. 2010;6:e1000952. doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gardiner DM, Cozijnsen AJ, Wilson LM, Pedras MS, Howlett BJ. Mol Microbiol. 2004;53:1307–1318. doi: 10.1111/j.1365-2958.2004.04215.x. [DOI] [PubMed] [Google Scholar]
- 109.Gardiner DM, Howlett BJ. FEMS Microbiol Lett. 2005;248:241–248. doi: 10.1016/j.femsle.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 110.Fox EM, Howlett BJ. Mycol Res. 2008;112:162–169. doi: 10.1016/j.mycres.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 111.Ferezou JP, Quesneauthierry A, Barbier M, Kollmann A, Bousquet JF. J Chem Soc Perkin Trans. 1980;1:113–115. [Google Scholar]
- 112.Bulock JD, Clough LE. Aust J Chem. 1992;45:39–45. [Google Scholar]
- 113.Kremer A, Li SM. Microbiology. 2010;156:278–286. doi: 10.1099/mic.0.033886-0. [DOI] [PubMed] [Google Scholar]
- 114.Guo CJ, Yeh HH, Chiang YM, Sanchez JF, Chang SL, Bruno KS, Wang CC. J Am Chem Soc. 2013;135:7205–7213. doi: 10.1021/ja3123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Welch TR, Williams RM. In: Biomimetic Organic Synthesis. Poupon E, Nay B, editors. Wiley-VCH; 2011. pp. 117–148. [Google Scholar]
- 116.Kishi Y, Nakatsuka S, Fukuyama T, Havel M. J Am Chem Soc. 1973;95:6493–6495. doi: 10.1021/ja00800a079. [DOI] [PubMed] [Google Scholar]
- 117.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6492–6493. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]
- 118.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6490–6492. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]
- 119.Nakatsuka S, Fukuyama T, Kishi Y. Tetrahedron Lett. 1974:1549–1552. [Google Scholar]
- 120.Fukuyama T, Kishi Y. J Am Chem Soc. 1976;98:6723–6724. doi: 10.1021/ja00437a063. [DOI] [PubMed] [Google Scholar]
- 121.Fukuyama T, Nakatsuka S, Kishi Y. Tetrahedron. 1981;37:2045–2078. [Google Scholar]
- 122.Strunz GM, Kakushima M. Experientia. 1974;30:719–720. doi: 10.1007/BF01924146. [DOI] [PubMed] [Google Scholar]
- 123.Williams RM, Rastetter WH. J Org Chem. 1980;45:2625–2631. [Google Scholar]
- 124.Williams RM, Rastetter WH. Tetrahedron Lett. 1979:1187–1190. [Google Scholar]
- 125.Miknis GF, Williams RM. J Am Chem Soc. 1993;115:536–547. [Google Scholar]
- 126.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]
- 128.Iwasa E, Hamashima Y, Fujishiro S, Hashizume D, Sodeoka M. Tetrahedron. 2011;67:6587–6599. [Google Scholar]
- 129.Kim J, Movassaghi M. J Am Chem Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.DeLorbe JE, Horne D, Jove R, Mennen SM, Nam S, Zhang FL, Overman LE. J Am Chem Soc. 2013;135:4117–4128. doi: 10.1021/ja400315y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Movassaghi M, Boyer N. Chem Sci. 2012;3:1798–1803. doi: 10.1039/C2SC20270K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coste A, Kim J, Adams TC, Movassaghi M. Chem Sci. 2013;4:3191–3197. doi: 10.1039/C3SC51150B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nicolaou KC, Totokotsopoulos S, Giguere D, Sun YP, Sarlah D. J Am Chem Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Codelli JA, Puchlopek ALA, Reisman SE. J Am Chem Soc. 2012;134:1930–1933. doi: 10.1021/ja209354e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fujiwara H, Kurogi T, Okaya S, Okano K, Tokuyama H. Angew Chem Int Ed. 2012;51:13062–13065. doi: 10.1002/anie.201207307. [DOI] [PubMed] [Google Scholar]