

Abstract
Flavin-dependent oxidoreductases are increasingly recognized as important biocatalysts for various industrial applications. In order to identify novel activities and to improve these enzymes in engineering approaches, suitable screening methods are necessary. We developed novel microtiter-plate-based assays for flavin-dependent oxidases and dehydrogenases using redox dyes as electron acceptors for these enzymes. 2,6-dichlorophenol-indophenol, methylene green, and thionine show absorption changes between their oxidized and reduced forms in the visible range, making it easy to judge visually changes in activity. A sample set of enzymes containing both flavoprotein oxidases and dehydrogenases – pyranose 2-oxidase, pyranose dehydrogenase, cellobiose dehydrogenase, d-amino acid oxidase, and l-lactate oxidase – was selected. Assays for these enzymes are based on a direct enzymatic reduction of the redox dyes and not on the coupled detection of a reaction product as in the frequently used assays based on hydrogen peroxide formation. The different flavoproteins show low Michaelis constants with these electron acceptor substrates, and therefore these dyes need to be added in only low concentrations to assure substrate saturation. In conclusion, these electron acceptors are useful in selective, reliable and cheap MTP-based screening assays for a range of flavin-dependent oxidoreductases, and offer a robust method for library screening, which could find applications in enzyme engineering programs.
Keywords: 2,6-dichlorophenol-indophenol; flavin-dependent oxidoreductases; high-throughput screening; methylene green; thionine
1 Introduction
Oxidation of various organic substrates by enzymes is an area in biocatalysis that has attracted considerable interest, since the chemical industry is in search of greener alternatives to those currently used industrial processes. A limited number of enzymes can be used for these biocatalytic oxidation reactions, including dehydrogenases, oxidases, peroxidases, and monooxygenases [1, 2]. Flavin-dependent oxidoreductases are of significant importance in this area, and several enzymes of this group are currently employed as biocatalysts [3]. This is partly because of the ability of flavin-dependent oxidoreductases to react directly with molecular oxygen thus avoiding other cofactors, cosubstrates or metal catalyst for oxidation reactions, but also for the enormous catalytic versatilities that have been shown by flavin oxidoreductases. An additional area of interest for flavoenzymes is electrochemistry, where suitable enzymes can be applied in biosensors or for biofuel cell applications. Here, dehydrogenases that lack oxygen reactivity are favorable since a futile reaction of the electrode-bound enzymes with oxygen will result in hydrogen peroxide formation, which can impede stability of the biocatalyst. Recently, several new flavin-containing oxidoreductases have been introduced to this area in addition to commonly used glucose oxidases, yet there is wide interest in the identification of novel enzymes for these above-mentioned applications [4, 5]. For example, it has been proposed that, based on genome sequence data, many attractive and so far unknown flavoproteins can still be obtained from nature [3].
Flavin-dependent oxidoreductases show a reaction mechanism consisting typically of two half-reactions [6]. In the reductive half-reaction an electron donor substrate (e.g. a sugar or an amino acid) is oxidized, while the flavin is reduced (reactions 1 or 2).
| (1) |
| (2) |
In the ensuing oxidative half-reaction the reduced flavin is re-oxidized by the second substrate oxygen, yielding the oxidized prosthetic group and H2O2 (shown for reduced FADH2 in reaction 3), or by alternative electron acceptors such as quinones, redox dyes, or chelated metal ions [7]. The two-electron redox systems of the redox dyes DCPIP, thionine (Thi), and methylene green (MG) are shown in reactions 4–6, with the phenothiazines MG and Thi showing an oxidized cationic dye-form and a colorless reduced leuco-form [8, 9]
| (3) |
| (4) |
| (5) |
| (6) |
It was the aim of our work to test whether redox dyes that are frequently used in biosensor applications to transport electrons from the prosthetic group of an oxidoreductase can also be used as colorimetric substrates in microtiter plate (MTP)-based active assays for flavin-dependent oxidoreductases.
2 Materials and methods
2.1 Chemicals
All chemicals were of the highest purity commercially available. Lactose monohydrate, α-d-glucose monohydrate and dipotassium hydrogen phosphate anhydrous were purchased from Lactan (Graz, Austria). 2,6-dichlorophenol-indophenol (DCPIP), MG, Thi, sodium dithionite, d-methionine, l-(+)-lactic acid, and all other chemicals were obtained from Sigma-Aldrich (Vienna, Austria) unless otherwise stated.
2.2 Enzymes
Pyranose 2-oxidase (POx, EC 1.1.3.10) from Trametes multicolor, pyranose dehydrogenase (PDH, EC 1.1.99.29) from Agaricus meleagris, cellobiose dehydrogenase (CDH, EC 1.1.99.18) from Neurospora crassa, d-amino acid oxidase (DAAO, EC 1.4.3.3) from Trigonopsis variabilis, and l-lactate oxidase (LOx, EC 1.13.12.4) from Aerococcus viridans were prepared and purified to apparent homogeneity as described previously [7, 10–13].
2.3 Screening for electron acceptors
The following electron acceptors were tested for their reactivity with 0.1 U TmPOx and 100 mM d-glucose in 50 mM phosphate buffer pH 6.5: DCPIP (150 μM), methylene blue (15 μM), MG (25 μM), celestine blue (75 μM), Meldola's blue (50 μM), Thi (20 μM), toluidine blue (15 μM), reactive blue 2 (160 μM), direct red 81 (80 μM), acid orange 74 (150 μM), sudan orange (100 μM), malachite oxalate green (8 μM), K3[Cr(CN)6] (12 mM), and K3[Fe(CN)6] (1 mM). The different reactions were followed on a U-3000 spectrophotometer (Hitachi, Tokyo, Japan).
2.4 Spectral properties of redox dyes
Electron acceptor stock solutions were prepared as follows: DCPIP was dissolved in a small amount of ethanol over low heat and then adjusted to the desired concentration (3 mM) with reverse osmosis water; MG and Thi stock solutions (0.5 and 1 mM, respectively) were prepared in reverse osmosis water. Spectra of the selected redox dyes DCPIP (70 μM), MG (25 μM), and Thi (20 μM) were measured in 50 mM phosphate buffer (pH 6.5), and these dyes were reduced by adding sodium dithionite (Na2S2O4). The changes in the absorption spectra were recorded with a U-3000 spectrophotometer (Hitachi) in the range from 400 to 800 nm. To investigate the pH dependence of the spectral properties of the electron acceptors DCPIP, MG, and Thi, absorbance spectra were recorded in the range of pH 3.0–9.0 in steps of 1.0 pH unit by using the following buffers: 50 mM citrate buffer (pH 3.0–5.0), 50 mM phosphate buffer (pH 5.0–8.0), and 50 mM Tris buffer (8.0–9.0). The concentration of the redox dyes was 70 μM for DCPIP, 25 μM for MG, and 20 μM for Thi.
2.5 Microtiter plate activity assays
MTP assays were performed in standard 96-well plates. Each well contained 100 μL of the assay mixture, which was composed of the electron acceptor (450 μM DCPIP, 75 μM MG, or 100 μM Thi), varying activities of purified POx, PDH, CDH, DAAO, or LOx (0.2, 0.02, or 0.002 U), 50 mM phosphate buffer and the appropriate substrate (d-glucose for POx and PDH, lactose for CDH, d-methionine for DAAO, and l-lactic acid for LOx). The individual composition of the reaction mixtures for the respective wells is shown in Fig. 1, with the first column of the 96-well plate representing the negative control (no enzyme added). Electron acceptor reduction was followed at room temperature (22°C) at 520 nm for DCPIP, 655 nm for MG, and 600 nm for Thi using the Microplate reader Sunrisea (Tecan, Austria). The relative absorption change was calculated according to Eq. (7).
Figure 1.

Compositions of the reaction mixtures used in the microtiter plate assays shown in Fig. 3, each well contained additionally 450 μM DCPIP, 75 μM MG, or 100 μM Thi as indicated. These dye concentrations were selected so that changes in the absorption can be detected by eye, which is not possible when higher concentrations are used. The first column represents the negative control (no enzyme added). Line A: POx, pyranose 2-oxidase from Trametes multicolor; B: PDH, pyranose dehydrogenase from Agaricus meleagris; C: CDH, cellobiose dehydrogenase from Neurospora crassa; D: DAAO, D-amino acid oxidase from Trigonopsis variabilis; E: LOx, L-lactate oxidase from Aerococcus viridans. PPB, phosphate buffer; d-Gluc, d-glucose; d-MET, d-methionine; LLA, l-lactic acid.
| (7) |
where A0 is the initial electron acceptor absorption and At is the absorption measured at the time point t. The absorption change in percentage was plotted versus time.
In addition, realistic enzyme samples were employed in the MTP-based assays, using two different samples and expression systems. Crude, intracellular extracts of Escherichia coli were used as a source of POx, and culture supernatants of Pichia pastoris were the source of recombinant CDH. POx was recombinantly produced in E. coli as reported in ref. [14], while CDH was obtained from shaken flask cultures of P. pastoris basically as described by Sygmund [15], however, omitting the trace element solution from the medium. The following reaction mixtures were analyzed as negative controls: (i) cell lysates of E. coli BL21 Star DE3 harboring the pET21d+ expression vector without the POx gene, d-glucose and dye; (ii) dye and substrate without enzyme; (iii) dye and enzyme without substrate. The MTP set-up was as described above, and the total volume per well was increased to maximally 200 μL, depending on the amount/activity of protein applied.
2.6 Steady-state kinetic measurements
Apparent steady-state kinetic constants for the two-electron, two-proton acceptor DCPIP and the 2-e–, 1-H+ acceptors MG and Thi were determined at room temperature (22°C) for POx, PDH, CDH, DAAO, and LOx. Initial rates of the DCPIP assay were recorded at 520 nm, where the pH dependence of the spectral properties of DCPIP is least pronounced. A molar extinction coefficient ε520 nm of 6.8 mM–1 cm–1 was used for pH values below 6.5, whereas a value of 6.6 mM–1 cm–1 was used when the pH was above 6.5 [16]. The reaction in the MG assay was followed at 655 nm and the corresponding molar extinction coefficient ε655 was 46.6 mM–1 cm–1. Initial rates of the reduction of Thi were recorded at 600 nm and a molar extinction coefficient ε600 of 55.4 mM–1 cm–1 was applied. Although Thi does not strictly obey Lambert–Beer's law [17], for the small concentration range that was employed in these experiments the absorption is linearly dependent on the Thi concentration. The molar extinction coefficients for MG and Thi were determined experimentally at pH 6.5 from the slope of the regression line of various electron acceptor concentrations versus their absorption intensity at 655 and 600 nm for MG and Thi, respectively (data not shown).
Kinetic constants were calculated by nonlinear least-square regression, fitting the data to the Henri-Michaelis-Menten or Hill equation (Sigma Plot 11, Systat Software). The path length of the quartz cuvettes used was varied between 3 and 10 mm depending on the electron acceptor concentration used so that the initial absorption value did not exceed a value of 2.0.
2.6.1 2, 6-dichlorophenol-indophenol
Initial rates of reduction of DCPIP were followed at 520 nm. The reaction mixtures for the determination of the apparent steady-state kinetic constants contained varied concentrations of DCPIP (0.0075 to maximally 1.2 mM), substrate in saturating concentrations (100 mM d-glucose for POx, 200 mM d-glucose for PDH, 30 mM lactose for CDH, 200 mM d-methionine for DAAO, and 200 mM l-lactic acid for LOx), 50 mM phosphate buffer (pH 6.5 for POx and LOx, pH 7.5 for PDH, pH 6.0 for CDH, and pH 8.0 DAAO) and a suitable amount of the respective enzyme (POx, PDH, CDH, DAAO, and LOx).
2.6.2 Methylene green
Initial rates of the reduction of MG were recorded at 655 nm, and the assay mixtures contained varied MG concentrations (0.003–0.2 mM), while all other conditions were as for DCPIP.
2.6.3 Thionine
For the determination of initial rates of the reduction of Thi absorption changes were recorded at 600 nm. Thi concentrations were varied from 0.001 to maximally 0.3 mM. All other conditions were as with DCPIP.
3 Results
3.1 Screening for suitable electron acceptors
We performed an initial screening for suitable electron acceptors that show a visible color change in reasonable time, using the flavoenzyme POx since it was previously shown that this enzyme reduces a wide range of different quinones, redox dyes, and complexed metal ions [7]. The electron acceptors that were tested in this screening included DCPIP, methylene blue, MG, celestine blue, Meldola's blue, Thi, toluidine blue, reactive blue 2, direct red 81, acid orange 74, sudan orange, malachite oxalate green, K3[Cr(CN)6], and K3[Fe(CN)6]. Quinones were not included because of the possibility of polymerization at higher pH values [18]. Some of these potential electron acceptors did not show any reaction in the POx-catalyzed reaction (e.g. reactive blue 2, direct red 81 or K3[Cr(CN)6]) while other showed very slow color changes under the initial screening conditions (e.g. malachite oxalate green). Based on this initial screening we selected DCPIP, MG, and Thi as the most promising electron acceptors (redox dyes) since they reacted directly with POx, and showed distinct color changes that are clearly visible even by eye within a short reaction period.
3.2 Spectral properties of the selected redox dyes
Absorption changes of DCPIP, MG, and Thi are shown in Fig. 2 for the reduction by Na2S2O4 at pH 6.5. For DCPIP and Thi the maximal change in absorption between the oxidized and reduced states was recorded at 600 nm, and a color shift from blue and purple (oxidized state) to colorless (completely reduced state) could be observed for DCPIP and Thi, respectively. MG is turquoise-colored in the oxidized form and shows an absorption maximum at 655 nm, while the reduced form is again colorless. To investigate the dependence of the DCPIP, MG, and Thi spectra on the pH value, their absorption profiles were recorded in the range of pH 4.0–9.0. The absorbance of Thi and MG are pH-independent (data not shown), while the spectral properties of DCPIP show a strong pH dependency [16]. Since the lowest effect of the pH was observed at 520 nm, this wavelength was used for further experiments.
Figure 2.
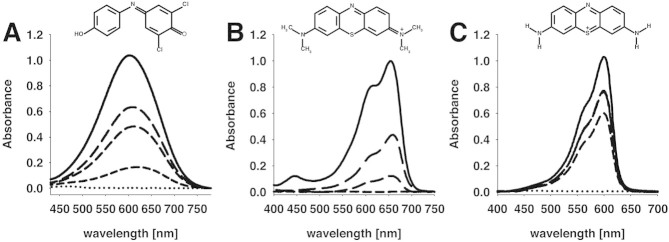
Absorption spectra of (A) 70 μM DCPIP, (B) 25 μM MG and (C) 20 μM thionine in 50 mM phosphate buffer (pH 6.5) of the oxidized state (solid line) and the (partly) reduced state (discontinuous lines). Sodium dithionite was added stepwise until complete reduction was attained. Spectra were obtained in 10-mm quartz cuvettes, and the concentration of the dyes was chosen so that the maximum absorption value was 1.
3.3 Microtiter plate assay
The reduction of DCPIP, MG, and Thi by the sample enzymes in the presence of a suitable electron-donor substrate is shown in Fig. 3 by characteristic color changes. Each well of the 96-well MTP contained the respective electron acceptor (DCPIP, MG, or Thi), varying activities of one of the flavoenzymes POx, PDH, CDH, DAAO, or LOx plus the appropriate electron donor substrate. The different assay conditions are shown in Fig. 1, in which the first column of the 96-well MTP represents the negative control (no enzyme added). All five tested flavin-dependent enzymes show discoloration of the redox dye within 1 h, even with the lowest activities tested. This color change and differences in intensities can be judged easily by eye. Some of these colorimetric changes could be observed rapidly within the first minutes of the reaction, even when using only low activities of 0.002 U per well, e.g. for CDH and LOx.
Figure 3.
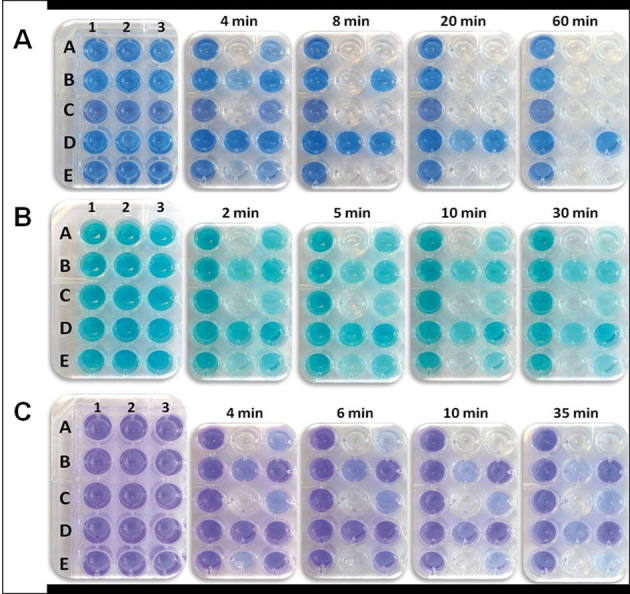
Microtiter-plate-based activity assays using (A) DCPIP, (B) MG, or (C) Thi as indicators for the activity of various flavoenzymes. The composition of the individual assay mixtures per well is as given in Fig 1. The plates to the far left show the beginning of the reaction at 0 min.
The absorbance changes over time, calculated according to Eq. (7), for the reduction of DCPIP, MG, and Thi are shown in Fig. 4. Again, the absorbance changes resulting from the reduction of the redox dyes by the tested flavoenzymes can be conveniently measured on a microtiter plate reader and give significant results for the activity ranges chosen within a reasonable time of less than 60 min. After approximately 1 h the slow re-oxidation of MG by air could be observed for the system POx/glucose, presumably because glucose was completely oxidized at this point. This slight re-oxidation of the reduced form by oxygen is also known from the blue bottle experiment using methylene blue, a compound that is structurally related to MG [19, 20]. This re-oxidation was not observed for the other two redox dyes used.
Figure 4.
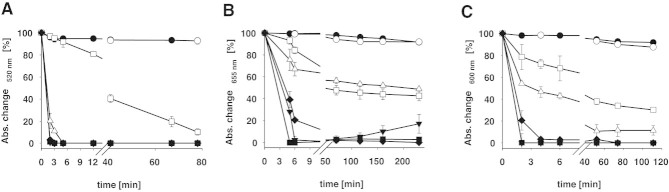
Time course of the absorption change of (A) DCPIP, (B) methylene green, or (C) thionine when reacting with different flavoenzymes and the appropriate enzyme substrate. The activity stated is the total activity added per well. Absorbance values were measured directly in 96-well plates by using a microplate reader. Data presented are the mean of duplicate independent measurements ± the standard deviation shown by the error bars.Symbols: •, negative control I, no enzyme added; ○, negative control II, no substrate added; ▾, pyranose 2-oxidase from Trametes multicolor (0.2 U); ▵, pyranose dehydrogenase from Agaricus meleagris (0.2 U); ▪, cellobiose dehydrogenase from Neurospora crassa (0.02 U); □, d-amino acid oxidase from Trigonopsis variabilis (0.2 U); ♦, l-lactate oxidase from Aerococcus viridans (0.02 U).
In addition, we used two different realistic enzyme samples as typically employed in enzyme engineering approaches, cell lysates of E. coli overexpressing POx from T. multicolor and culture supernatants of P. pastoris cultivations, in which CDH from N. crassa was extracellularly expressed (Fig. 5). Even low activities of only 2 and 10 mU per well gave absorbance changes over time that could be clearly distinguished from the reaction blanks. When using these low enzyme activities, DCPIP and MG gave unambiguous results with 3 h, while Thi proved to be less sensitive and only the higher of the two enzyme activities (10 mU per well) could be clearly identified within the reaction time of 3 h (Fig. 5).
Figure 5.

Time course of the absorption change of (A) DCPIP, (B) methylene green, or (C) thionine when reacting with cell lysates of E. coli expressing POx or clear supernatant of P. pastoris expressing CDH; d-glucose and lactose were used as the respective substrates. The activities stated are the total activity added per well. Data presented are the mean of duplicate independent measurements ± the standard deviation shown by the error bars.Symbols: •, negative control I, cell lysates of E. coli carrying the pET21d+ expression vector without the POx gene; ○, negative control II, no enzyme added; ▾, negative control III, clear P. pastoris supernatant containing 0.01 U cellobiose dehydrogenase from Neurospora crassa without addition of the electron donor (sugar) substrate; ▵ and ▪, cell lysates of E. coli expressing pyranose 2-oxidase from Trametes multicolor (0.002 and 0.01 U); □ and ♦, clear supernatant of P. pastoris containing cellobiose dehydrogenase from Neurospora crassa (0.002 and 0.01 U).
3.4 Catalytic constants
The apparent steady-state kinetic constants were measured for the 2-e–, 2-H+ acceptor DCPIP (used in varying concentrations of 7.5 μM to maximal 1.2 mM while the second electron-donor substrate was held constant in saturating concentrations) and the 2-e–, 1-H+ acceptors MG (3.0–200 μM) and Thi (1.0–300 μM). Kinetic data are summarized in Table 1. The five investigated flavoproteins show very low apparent Michaelis constants mostly in the micromolar range for these redox dyes. The five flavoenzymes differ significantly in their catalytic constants kcat and catalytic efficiencies kcat/Km for these electron acceptors, with the lowest efficiencies found for DAAO and PDH. This is also evident from Fig. 3 and 4, where the slowest change in color/decrease in absorbance is observed for these two enzymes.
Table 1.
Kinetic properties of TmPOx, AmPDH, NcCDH, TvDAAO, and AvLOx with DCPIP, methylene green and thionine as electron acceptors and d-glucose as saturating substrate for POx and PDH, lactose for CDH, d-methionine for DAAO and l-lactic acid for LOxa)
| Enzyme | DCPIP | Methylene green | Thionine | Oxygen | Refs. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KM (mM) | kcat (s–1) | kcat/KM (mM–1 s–1) | KM (mM) | kcat (s–1) | kcat/KM (mM–1 s–1) | KM (mM) | kcat (s–1) | kcat/KM (mM–1 s–1) | KM (mM) | kcat (s–1) | kcat/KM (mM–1 s–1) | ||
| POx | 1.88 ± 0.15 | 70 ± 4 | 38 | 0.12 ± 0.01 | 1.63 ± 0.10 | 13 | 0.20 ± 0.01 | 4.2 ± 0.1 | 24 | 0.090 | 71 | 790 | [7] |
| PDH | 0.66 ± 0.07 | 2.7 ± 0.13 | 4.1 | 0.14 ± 0.01 | 0.34 ± 0.08 | 2.5 | 0.12 ± 0.01 | 0.25 ± 0.01 | 2.1 | nd | nd | nd | – |
| CDH | 0.037 ± 0.001 | 3.7 ± 0.04 | 99 | 0.042 ± 0.003 | 15.1 ± 0.4 | 360 | 0.014 ± 0.001 | 7.2 ± 0.1 | 511 | nd | nd | nd | – |
| DAAO | 0.031 ± 0.002 | 0.033 ± 0.001 | 1.0 | 0.062 ± 0.006 | 0.190 ± 0.008 | 3.0 | 0.032 ± 0.005 | 0.161 ± 0.004 | 5.0 | 0.755 ± 0.076 | 229 ± 12 | 303 | [35] |
| LOx | 1.60 ± 0.15 | 189 ± 11 | 118 | 0.14 ± 0.01 | 3.0 ± 0.2 | 22 | 0.11 ± 0.003 | 1.30 ± 0.05 | 12 | 0.83 ± 0.03 | 140 ± 1 | 167 | [13] |
Published data for the kinetic constants with oxygen are given, for the flavoprotein dehydrogenases these data are not available. Data presented are the mean of triplicate independent measurements ± the standard deviation.
4 Discussion
Rapid, convenient, and reliable colorimetric assays are a prerequisite for different approaches in enzyme development, ranging from screening of mutational libraries to the identification of novel enzymes from metagenomic libraries. Typically, large numbers of samples have to be evaluated by activity-based assays in these approaches, and microassays are preferred for this purpose because they facilitate rapid screening of numerous samples and substantially reduce reagent consumption. When screening for flavin-dependent oxidases the second reaction product, hydrogen peroxide, is frequently detected in a second, enzyme-coupled reaction. Here, chromogenic substrates such as 4-aminoantipyrine together with a suitable phenolic compound [21], o-dianisidine [22], or 2,2'-azinobis(3-ethylbenzthiazoline)-6-sulfonic acid (ABTS) [23, 24] are used together with horseradish peroxidase or another suitable peroxidase. Obviously, these approaches cannot be used for flavin-dependent dehydrogenases since these only show negligible activity with oxygen. Various quinones are often used directly as an electron acceptor by a number of flavin-dependent oxidoreductases. A drawback for the use of quinones in screening assays, however, is that the redox reaction can only be followed in the UV range, which makes the use of special and rather expensive MTP necessary. Furthermore, quinones can give confounded results due to their spontaneous oxidative side reactions in the alkaline pH range [18]. Recently, ferricyanide was used in a high-throughput screening assay that is based on the formation of Prussian Blue, but it was shown that this assay is highly specific for CDH while other flavin-dependent carbohydrate oxidoreductases (glucose oxidase, PDH) do not show similar reactions [25].
It was the aim of our work to test whether mediators that are used in enzyme-based biosensors [26] can also be used as convenient colorimetric substrates in MTP-based activity assays used for high-throughput screening of flavin-dependent oxidoreductases. To this end, we initially tested 14 different compounds that are often employed as redox mediators in biosensors, mainly various phenothiazine derivatives, for their ability to directly react with the flavin adenine dinucleotide (FAD)-dependent enzyme POx. Based on rapid direct enzyme-catalyzed reduction of the mediators and clear color changes we selected DCPIP, MG, and Thi, which show distinct absorption changes between their oxidized and reduced forms in the visible range, as the most promising substrates in these assays for further characterization. These three redox dyes are direct substrates (electron acceptors) for various flavin-dependent oxidoreductases, both oxidases and true dehydrogenases. These studied flavin-dependent oxidoreductases belong to three different enzyme superfamily of flavoproteins (POx, PDH, and CDH are members of the glucose-methanol-choline (GMC) oxidoreductase family; DAAO belongs to the glutathione reductase family [27] and l-LOx to the l-α-hydroxy acid oxidase family [28]). They all contain covalently or non-covalently bound FAD or flavin mononucleotide (FMN): monomeric PDH and each subunit of the homotetramer POx bind one FAD covalently [29, 30], homodimeric DAAO [31], and the flavin domain of the monomeric flavocytochrome CDH [32] contain one non-covalently bound FAD, whereas each subunit of the homotetramer LOx binds one FMN non-covalently [33].
All five tested enzymes (POx from T. multicolor, PDH from A. meleagris, CDH from N. crassa, DAAO from T. variabilis, LOx from A. viridans) reacted rapidly and efficiently with these electron acceptors so that decolorization of the dye in the MTP was visible within few minutes. This is also expressed by the high apparent catalytic constants kcat determined for these electron acceptors in presence of an excess of the respective electron donor substrates, which were found to range from 0.033 to 189 s–1. This indicates that the selected redox dyes might be useful for activity assays of a wide range of different flavin-dependent oxidoreductases. Furthermore, the apparent Michaelis constants of all tested enzymes were typically found to be in the micromolar range for these three electron acceptors, indicating that only low concentrations have to be used to the assays to assure substrate saturation and hence maximum reaction velocities. Another clear advantage of these dyes for activity assays is that they react directly with the enzymes to be tested in contrast to frequently used assays that are based on a second, enzyme-coupled assay such as the ones that are frequently used for the detection of the reaction product hydrogen peroxide as this will also reduced costs for these assays; these dyes are cheap and there are no extra costs for additional reagents and enzymes. The reduced forms of the redox dyes were stable in the presence of oxygen in these assays, and only MG showed some slight re-oxidation by oxygen after a prolonged incubation period and once the electron donor substrate was exhausted from the reaction mixture. This should however not interfere with MTP-based screening. Furthermore, MG and Thi did not show a pH dependence of their absorption properties, and this will make their application in enzyme activity assays possible even over a wide pH range. The proposed method might be useful in the primary screening for desired enzyme activities of libraries. To avoid a potential bias in the screening, i.e. identification of variants showing improved activities only with these specific electron acceptors and not with the specific electron donor substrates that often are of interest in an enzyme improvement program (“you get what you screen for” [34]), two or more electron acceptors can be used in the library screening for specific activities, which will be possible because of the ease of application. In conclusion, the activity assays based on the electron acceptors DCPIP, MG, and Thi offer a rapid, robust and cheap method for screening of a large number of samples, which could find applications in enzyme engineering programs of flavin-dependent oxidoreductases or identification of novel enzyme activities from metagenomic libraries, to name a few examples.
Acknowledgments
This work was supported by BOKU University of Natural Resources and Life Sciences (BOKU Doc Program) and by the Austrian Science Fund (FWF), Doctoral Programme BioToP–Biomolecular Technology of Proteins (FWF W1224) and individual project P22094 (to CKP), which is gratefully acknowledged. We thank Wolfgang Graschopf for his help and support.
The authors declare no financial or commercial conflict of interest.
Glossary
Abbreviations:
- CDH
cellobiose dehydrogenase
- DAAO
d-amino acid oxidase
- DCPIP
2,6-dichlorophenol-indophenol
- FAD
flavin adenine dinucleotide
- LOx
l-lactate oxidase
- MG
methylene green
- MTP
microtiter plates
- Thi
thionine
- PDH
pyranose dehydrogenase
- POx
pyranose 2-oxidase
References
- 1.Hollmann F, Arends IWCE, Bühler K, Schallmey A, Bühler B. Enzyme-mediated oxidations for the chemist. Green Chem. 2011;13:226–265. [Google Scholar]
- 2.Monti D, Ottolina G, Carrea G, Riva S. Redox reactions catalyzed by isolated enzymes. Chem. Rev. 2011;111:4111–4140. doi: 10.1021/cr100334x. [DOI] [PubMed] [Google Scholar]
- 3.Winter RT, Fraaije MW. Applications of flavoprotein oxidases in organic synthesis: Novel reactivities that go beyond amine and alcohol oxidations. Curr. Org. Chem. 2012;16:2542–2550. [Google Scholar]
- 4.Shao M, Zafar MN, Falk M, Ludwig R, et al. Optimization of a membraneless glucose/oxygen enzymatic fuel cell based on a bioanode with high coulombic efficiency and current density. ChemPhysChem. 2013;14:2260–2269. doi: 10.1002/cphc.201300046. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig R, Ortiz R, Schulz C, Harreither W, et al. Cellobiose dehydrogenase modified electrodes: Advances by materials science and biochemical engineering. Anal. Bioanal. Chem. 2013;405:3637–3658. doi: 10.1007/s00216-012-6627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dijkman WP, de Gonzalo G, Mattevi A, Fraaije MW. Flavoprotein oxidases: Classification and applications. Appl. Microbiol. Biotechnol. 2013;97:5177–5188. doi: 10.1007/s00253-013-4925-7. [DOI] [PubMed] [Google Scholar]
- 7.Leitner C, Volc J, Haltrich D. Purification and characterization of pyranose oxidase from the white rot fungus Trametes multicolor. Appl. Environ. Microbiol. 2001;67:3636–3644. doi: 10.1128/AEM.67.8.3636-3644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akkermans RP, Roberts SL, Marken F, Coles BA, et al. Methylene green voltammetry in aqueous solution: Studies using thermal, microwave, laser, or ultrasonic activation at platinum electrodes. J. Phys. Chem. B. 1999;103:9987–9995. [Google Scholar]
- 9.Wondrak GT. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apoptosis. Free Rad. Biol. Med. 2007;43:178–190. doi: 10.1016/j.freeradbiomed.2007.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sygmund C, Kittl R, Volc J, Halada P, et al. Characterization of pyranose dehydrogenase from Agaricus meleagris and its application in the C-2 specific conversion of d-galactose. J. Biotechnol. 2008;133:334–342. doi: 10.1016/j.jbiotec.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Nidetzky B. Stability and stabilization of d-amino acid oxidase from the yeast Trigonopsis variabilis. Biochem. Soc. Trans. 2007;35:1588–1592. doi: 10.1042/BST0351588. [DOI] [PubMed] [Google Scholar]
- 12.Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, et al. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 2012;78:6161–6171. doi: 10.1128/AEM.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unterweger B, Stoisser T, Leitgeb S, Birner-Grünberger R, Nidetzky B. Engineering of Aerococcus viridansl-lactate oxidase for site-specific PEGylation: Characterization and selective bioorthogonal modification of a S218C mutant. Bioconj. Chem. 2012;23:1406–1414. doi: 10.1021/bc2006847. [DOI] [PubMed] [Google Scholar]
- 14.Spadiut O, Radakovits K, Pisanelli I, Salaheddin C, et al. A thermostable triple mutant of pyranose 2-oxidase from Trametes multicolor with improved properties for biotechnological applications. Biotechnol. J. 2009;4:525–534. doi: 10.1002/biot.200800260. [DOI] [PubMed] [Google Scholar]
- 15.Sygmund C, Santner P, Krondorfer I, Peterbauer CK, et al. Semi-rational engineering of cellobiose dehydrogenase for improved hydrogen peroxide production. Microb. Cell Fact. 2013;12:38. doi: 10.1186/1475-2859-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baminger U, Subramaniam SS, Renganathan V, Haltrich D. Purification and characterization of cellobiose dehydrogenase from the plant pathogen SclerotiumAtheliarolfsii. Appl. Environ. Microbiol. 2001;67:1766–1774. doi: 10.1128/AEM.67.4.1766-1774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabinowitch E, Epstein LF. Polymerization of dyestuffs in solution: Thionine and methylene blue. J. Am. Chem. Soc. 1941;63:69–78. [Google Scholar]
- 18.Frébortová J, Fraaije MW, Galuszka P, Sebela M, et al. Catalytic reaction of cytokinin dehydrogenase: Preference for quinones as electron acceptors. Biochem. J. 2004;380:121–130. doi: 10.1042/BJ20031813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook AG, Toliver RM, Willams JE. The blue bottle experiment revisited: How blue? How sweet? J. Chem. Educ. 1994;71:160–161. [Google Scholar]
- 20.Azmat R, Ahmed S, Qureshi S, Vali Mohammed F, Uddin F. Aerobic oxidation of d-glucose by methylene green in alkaline aqueous solution by visible spectrophotometry. J. Appl. Sci. 2006;6:2784–2788. [Google Scholar]
- 21.Ribitsch D, Winkler S, Gruber K, Karl W, et al. Engineering of choline oxidase from Arthrobacter nicotianae for potential use as biological bleach in detergents. Appl. Microbiol. Biotechnol. 2010;87:1743–1752. doi: 10.1007/s00253-010-2637-9. [DOI] [PubMed] [Google Scholar]
- 22.Gabler M, Hensel M, Fischer L. Detection and substrate selectivity of new microbial d-amino acid oxidases. Enzyme Microb. Technol. 2000;27:605–611. doi: 10.1016/s0141-0229(00)00262-3. [DOI] [PubMed] [Google Scholar]
- 23.Spadiut O, Brugger D, Coman V, Haltrich D, et al. Engineered pyranose 2-oxidase: Efficiently turning sugars into electrical energy. Electroanalysis. 2010;22:813–820. [Google Scholar]
- 24.Salaheddin C, Spadiut O, Ludwig R, Tan TC, et al. Probing active-site residues of pyranose 2-oxidase from Trametes multicolor by semi-rational protein design. Biotechnol. J. 2009;4:535–543. doi: 10.1002/biot.200800265. [DOI] [PubMed] [Google Scholar]
- 25.Vasilchenko LG, Ludwig R, Yershevich OP, Haltrich D, et al. High-throughput screening for cellobiose dehydrogenases by Prussian Blue in situ formation. Biotechnol. J. 2012;7:919–930. doi: 10.1002/biot.201100480. [DOI] [PubMed] [Google Scholar]
- 26.Privett BJ, Shim JH, Schoenfisch MH. Electrochemical sensors. Anal. Chem. 2008;80:4499–4571. doi: 10.1021/ac8007219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caldinelli L, Iametti S, Barbiroli A, Bonomi F, et al. Unfolding intermediate in the peroxisomal flavoprotein d-amino acid oxidase. J. Biol. Chem. 2004;279:28426–28434. doi: 10.1074/jbc.M403489200. [DOI] [PubMed] [Google Scholar]
- 28.Maeda-Yorita K, Aki K, Sagai H, Misaki H, et al. l-Lactate oxidase and l-lactate monooxygenase: Mechanistic variations on a common structural theme. Biochimie. 1995;77:631–642. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 29.Tan TC, Spadiut O, Wongnate T, Sucharitakul J, et al. The 1.6-Å crystal structure of pyranose dehydrogenase from Agaricus meleagris rationalizes substrate specificity and reveals a flavin intermediate. PLoS ONE. 2013;8:e53567. doi: 10.1371/journal.pone.0053567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hallberg MB, Leitner C, Haltrich D, Divne C. Crystal structure of the 270 kDa homotetrameric lignin-degrading enzyme pyranose 2-oxidase. J. Mol. Biol. 2004;341:781–796. doi: 10.1016/j.jmb.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 31.Pilone MS. d-Amino acid oxidase: New findings. Cell. Mol. Life Sci. 2000;57:1732–1747. doi: 10.1007/PL00000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zamocky M, Ludwig R, Peterbauer C, Hallberg BM, et al. Cellobiose dehydrogenase – a flavocytochrome from wood-degrading, phytopathogenic and saprotropic fungi. Curr. Protein Pept. Sci. 2006;7:255–280. doi: 10.2174/138920306777452367. [DOI] [PubMed] [Google Scholar]
- 33.Li SJ, Umena Y, Yorita K, Matsuoka T, et al. Crystallographic study on the interaction of l-lactate oxidase with pyruvate at 1.9 Å resolution. Biochem. Biophys. Res. Comm. 2007;358:1002–1007. doi: 10.1016/j.bbrc.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt-Dannert C, Arnold FH. Directed evolution of industrial enzymes. Trends Biotechnol. 1999;17:135–136. doi: 10.1016/s0167-7799(98)01283-9. [DOI] [PubMed] [Google Scholar]
- 35.Trampitsch C, Slavica A, Riethorst W, Nidetzky B. Reaction of Trigonopsis variabilisd-amino acid oxidase with 2,6-dichloroindophenol: Kinetic characterisation and development of an oxygen-independent assay of the enzyme activity. J. Mol. Cat. B: Enzymatic. 2005;32:271–278. [Google Scholar]