

Abstract
In 2001, with-no-lysine (WNK) kinases were identified as the genes responsible for the human hereditary hypertensive disease pseudohypoaldosteronism type II (PHAII). It took a further 6 years to clarify that WNK kinases participate in a signaling cascade with oxidative stress-responsive gene 1 (OSR1), Ste20-related proline-alanine-rich kinase (SPAK), and thiazide-sensitive NaCl cotransporter (NCC) in the kidney and the constitutive activation of this signaling cascade is the molecular basis of PHAII. Since this discovery, the WNK–OSR1/SPAK–NCC signaling cascade has been shown to be involved not only in PHAII but also in the regulation of blood pressure under normal and pathogenic conditions, such as hyperinsulinemia. However, the molecular mechanisms of WNK kinase regulation by dietary and hormonal factors and by PHAII-causing mutations remain poorly understood. In 2012, two additional genes responsible for PHAII, Kelch-like 3 (KLHL3) and Cullin3, were identified. At the time of their discovery, the molecular mechanisms underlying the interaction between these genes and their involvement in PHAII were unknown. Here we review the pathophysiological roles of the WNK signaling cascade clarified to date and introduce a new mechanism of WNK kinase regulation by KLHL3 and Cullin3, which provides insight on previously unknown mechanisms of WNK kinase regulation.
Keywords: Hypertension, Kidney, Kinases, Membrane transport, Ubiquitin
With-no-lysine kinases and pseudohypoaldosteronism type II
Polymerase chain reaction (PCR)-based homology cloning of mitogen-activated protein kinases (MAP) and MEK kinase initially identified WNK1 kinase (Xu et al., 2000). Subsequently, a database search revealed the existence of homologous kinase genes in mammals and in other species: four homologues (WNK1–4) were discovered in mammals, one in Drosophila melanogaster, one in Caenorhabditis elegans, and eight in Arabidopsis thaliana, but none was discovered in yeast (Verissimo and Jordan, 2001). The kinases were named “with-no-lysine” (WNK) kinases because the lysine (K) residue present in subdomain II of most kinases was not conserved in WNK kinases but instead replaced with a cysteine residue. As shown in Fig. 1, a kinase domain exists at the N-terminus of WNK kinases, followed by an autoinhibitory domain (Xu et al., 2002) and a coiled-coil domain. Another coiled-coil domain is present at the C-terminus.
Figure 1. Structures of WNK, OSR1, and SPAK kinases.
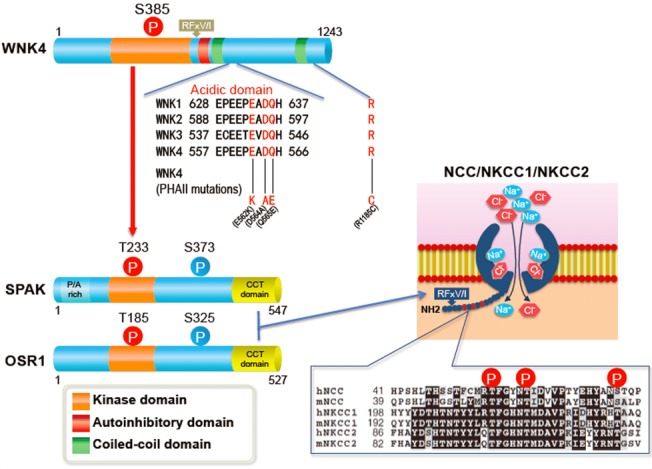
Acidic domains are located downstream of the first coiled-coil domain and conserved in all WNK kinases. Three of four pseudohypoaldosteronism type II-causing mutations in WNK4 are located in the acidic domain. WNK kinases activate OSR1 and SPAK by phosphorylating threonine residues in their kinase domains (T185 and T233). Serine residues (S325 in OSR1 and S373 in SPAK) in the S motif are also phosphorylated by WNK kinases, but their phosphorylation is not involved in the activation of the kinases. Conserved C-terminal domains in OSR1 and SPAK (shown in yellow) bind to the RFx[V/I] motif in WNK and solute carrier family 12 transporters. The N-terminal regions of NCC, NKCC1, and NKCC2 around the sites phosphorylated by OSR1 and SPAK are highly conserved.
In 2001, WNK1 and WNK4 were identified as the genes responsible for the autosomal dominant hereditary hypertensive disease pseudohypoaldosteronism type II (PHAII; Wilson et al., 2001). In addition to hypertension, PHAII is characterised by hyperkalemia, metabolic acidosis and thiazide sensitivity (Gordon, 1986). Thiazide is widely used as an anti-hypertensive drug: It induces salt excretion into the urine as it is a specific inhibitor for NaCl cotransporter (NCC) in the distal tubules of the kidney. NCC is responsible for the reabsorption of approximately 5%–10% of filtered NaCl in the glomeruli. At the time of this discovery, a substrate for WNK kinases was yet to be identified, but it was expected that NCC was regulated by WNK1 and WNK4 because the activation of NCC was considered the major pathogenesis of PHAII.
The mutations found in the WNK1 gene comprised large deletions in intron 1, which were considered to increase its transcription based on reverse transcription PCR analysis of WNK1 mRNA levels in the leukocytes of patients with PHAII (Wilson et al., 2001). However, after the initial report, the existence of two isoforms in WNK1, full-length WNK1 and a kidney-specific WNK1 lacking the kinase domain, was clarified (Delaloy et al., 2003; O'Reilly et al., 2003). Exactly which isoform is increased in patients with PHAII, and whether WNK1 expression is indeed increased in the human kidney, remains undetermined (Delaloy et al., 2008). In the case of WNK4, four missense mutations were identified in patients with PHAII, three of which are clustered within a distance of four amino acids in a region termed the “acidic domain” (Wilson et al., 2001). As shown in Fig. 1, this domain is well conserved in all WNK kinase isoforms.
Discovery of the WNK–oxidative stress-responsive gene 1/Ste20-related proline–alanine-rich kinase–solute carrier family 12a transporter signaling cascade
After the identification of WNK1 and WNK4 as the genes underlying PHAII, numerous investigations of the effects of coexpressing WNK1 and WNK4 with transporters, including NCC, were published (Kahle et al., 2003; Wilson et al., 2003; Yang et al., 2003; Kahle et al., 2004; Yamauchi et al., 2004, 2005; Cai et al., 2006; Gamba, 2006; Garzon-Muvdi et al., 2007; Ring et al., 2007; Yang et al., 2007a). In most studies, WNK4 was demonstrated to exert an inhibitory effect on the transporters. However, the detailed mechanisms of this regulation, in particular the intracellular signaling cascades involved, were poorly understood. Then, in 2005, two groups identified that oxidative stress-responsive gene 1 (OSR1) and Ste20-related proline-alanine-rich kinase (SPAK) were substrates of WNK1 and WNK4 (Moriguchi et al., 2005; Vitari et al., 2005). OSR1 and SPAK are related serine–threonine kinases that possess an N-terminal catalytic domain similar to those of other members of the Ste20 kinase subfamily, and two conserved regions known as the serine motif (S motif) and conserved C-terminal (CCT) domain. SPAK also possesses a unique 48-amino-acid N-terminal extension that primarily consists of alanine and proline. The CCT domains of OSR1 and SPAK were shown to interact with the RFv[V/I] motif in WNK kinases and solute carrier family 12 (SLC12) transporters (Fig. 1). OSR1 and SPAK were already identified as regulators of the SLC12A2 [also known as Na-K-2Cl-cotransporter 1 (NKCC1)] cotransporter (Flemmer et al., 2002; Piechotta et al., 2002; Dowd and Forbush, 2003; Piechotta et al., 2003): through in vitro experiments, Moriguchi et al. (2005) demonstrated that SLC12A3 (also known as NCC) and SLC12A1 [also known as Na-K-2Cl-cotransporter 2 (NKCC2)], which belong to the same transporter family as NKCC1, could also be substrates of OSR1 and SPAK. To prove this notion in the kidney in vivo, Yang et al. (2007c) generated anti-phosphorylated NCC (pNCC) antibodies that recognised potential serine and threonine phosphorylation sites deduced from sequence alignment with NKCC1. They also generated a mouse model of PHAII: a knock-in mouse carrying a PHAII-causing missense mutation of WNK4 (D561A), corresponding to the D574A mutation in patients with PHAII (Yang et al., 2007c). Wnk4D561A/+ mice exhibited a PHAII phenotype, including increased thiazide sensitivity, indicating that NCC is activated in the kidneys of the mutant mice. Using anti-pNCC antibodies, Yang et al. (2007c) demonstrated that NCC phosphorylation at three sites (Thr53, Thr58 and Ser71 in mouse NCC) was significantly increased in the kidneys of PHAII model mice, and that pNCC was concentrated on the apical plasma membranes of the distal convoluted tubules. Phosphorylation of SPAK and OSR1 was also increased in Wnk4D561A/+ mice, suggesting that WNK–OSR1/SPAK–NCC signaling was present in the kidney and activated by the PHAII-causing WNK4 mutation. Subsequently, by crossing Wnk4D561A/+ mice with SPAK and OSR1 knock-in mice, in which the T-loop Thr residues in SPAK (Thr243) and OSR1 (Thr185) were mutated to Ala to prevent activation by WNK kinases, Chiga et al. (2011) demonstrated that NCC phosphorylation and PHAII phenotypes in Wnk4D561A/+ mice were dependent on WNK–OSR1/SPAK signaling. Thus, the WNK–OSR1/SPAK–NCC signaling cascade in the kidney was established, and its activation was shown to be the pathogenic mechanism underlying PHAII. The WNK kinase responsible for NCC phosphorylation in the kidney was later identified as WNK4 through the analysis of WNK1, WNK3 and WNK4 knockout mice (Ohta et al., 2009; Oi et al., 2012; Castaneda-Bueno et al., 2012; Susa et al., 2012).
The mechanism of NCC activation by phosphorylation was initially investigated by Pacheco-Alvarez et al. (2006) using the Xenopus laevis oocyte expression system. Phosphorylation-incompetent mutant NCC molecules were present on the plasma membrane, but their transport activity was significantly decreased, suggesting that phosphorylation of NCC is important for its transport activity. As previously mentioned, analysis of NCC phosphorylation in the kidney in vivo clarified that phosphorylated NCC was exclusively present on the apical plasma membranes of the distal convoluted tubules (Yang et al., 2007c; Pedersen et al., 2010; Lee et al., 2013), suggesting that phosphorylation regulates the plasma membrane expression of NCC. Hossain Kahn et al. (2012) found that phosphorylation of NCC decreased its ubiquitination: decreased endocytosis and/or degradation may underlie the increased phosphorylated NCC accumulation evident in the apical plasma membranes of the distal convoluted tubules.
Regulators of WNK signaling
After the discovery of the WNK–OSR1/SPAK–NCC signaling cascade in the kidney and its involvement in PHAII, its pathophysiological roles outside of PHAII were investigated (Fig. 2). Salt intake regulates this cascade, partly through aldosterone (Chiga et al., 2008; Vallon et al., 2009). High and low salt intake decreased and increased the phosphorylation of OSR1/SPAK and NCC in the kidney, respectively, adjusting the excretion of NaCl according to its intake. This regulation was abolished in Wnk4D561A/+ mice (Chiga et al., 2008): A high-salt diet did not down-regulate WNK–OSR1/SPAK–NCC signaling in PHAII model mice. Elucidation of the mechanism of this dysregulation was one of the important unanswered questions in the molecular pathogenesis of PHAII. Potassium intake also regulates this cascade; high and low potassium intake decreased and increased WNK–OSR1/SPAK–NCC signaling, respectively (Vallon et al., 2009; Sorensen et al., 2013; van der Lubbe et al., 2013). As the initial phenotype of PHAII is hyperkalemia rather than hypertension, WNK–OSR1/SPAK–NCC signaling must also regulate potassium homeostasis in the body. In this regard, it is reasonable to predict that this signaling cascade is regulated by potassium intake. Although Naito et al. (2011) reported that extracellular potassium levels directly regulated WNK1 activity in cultured cells; the mechanisms of WNK kinase regulation by dietary potassium remain unclear. Hormonal factors also regulate WNK signaling. In addition to aldosterone, angiotensin II (San-Cristobal et al., 2009; Talati et al., 2010; van der Lubbe et al., 2011; Castaneda-Bueno et al., 2012; Castaneda-Bueno and Gamba, 2012) and vasopressin (Mutig et al., 2010; Pedersen et al., 2010; Rieg et al., 2013; Saritas et al., 2013) reportedly activated this signaling cascade. However, the details of intracellular signaling from these hormones to WNK kinases are poorly understood. Recently, insulin was identified as a powerful activator of this signaling cascade, and the phosphatidylinositol 3-kinase/Akt pathway was shown to mediate the signal from insulin to WNK4 (Sohara et al., 2011; Nishida et al., 2012; Chavez-Canales et al., 2013). Constitutive activation of this cascade caused by hyperinsulinemia may underlie the pathogenesis of salt-sensitive hypertension in metabolic syndrome (Nishida et al., 2012; Komers et al., 2012).
Figure 2. Regulators and effectors of WNK–OSR1/SPAK kinase signaling.
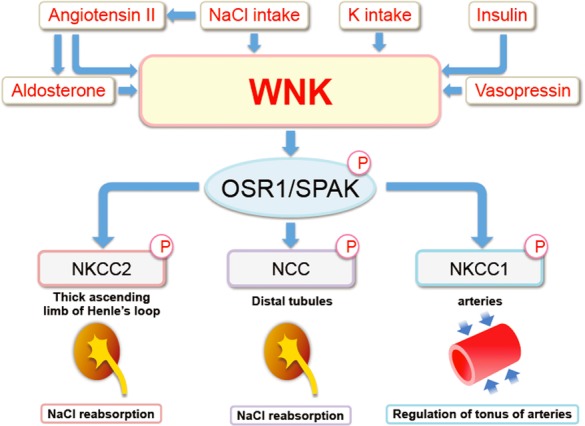
NaCl and K intakes regulate WNK kinase–OSR1/SPAK-NCC signaling in the kidney. Angiotensin II, aldosterone, vasopressin, and insulin also regulate WNK–OSR1/SPAK–NCC signaling in the kidney. A WNK1/WNK3–SPAK–Na-K-2Cl-cotransporter 1 cascade regulates arterial tonus.
Extrarenal roles of WNK–OSR1/SPAK kinase signaling
In addition to NaCl and K homeostasis in the kidney, WNK–OSR1/SPAK signaling has been shown to be involved in the regulation of arterial tonus. In this context, the transporter involved is not NCC but NKCC1. SPAK knockout mice showed a decreased response to phenylephrine and decreased phosphorylation of NKCC1 (Yang et al., 2010). Similarly, heterozygous WNK1 knockout mice exhibited reduced phosphorylation of NKCC1 and reduced arterial tonus (Bergaya et al., 2011; Susa et al., 2012). Zeniya et al. (2013) reported the existence of WNK3–SPAK–NKCC1 signaling in vascular smooth muscle cells, which was regulated by salt intake through angiotensin II. Thus, WNK–OSR1/SPAK signaling is involved in the regulation of blood pressure by modulating both NaCl excretion in the kidney and vascular tonus in the arteries (Fig. 2).
In addition, mutation of the WNK1 gene was shown to be responsible for human neuropathy (Shekarabi et al., 2008). WNK kinases were also shown to regulate KCl cotransporters (KCC family; Kahle et al., 2005; de de Los Heros et al., 2006; Garzon-Muvdi et al., 2007; Rinehart et al., 2009). The reciprocal regulation of NKCC1 and SLC12A5 (also known as KCC2) by WNK kinases is postulated to regulate intracellular chloride concentration, thereby regulating the excitability of neuronal cells (Kahle et al., 2006). Although data supporting this idea are accumulating, further validation by in vivo experiments is necessary.
Discovery of Kelch-like 3 and Cullin3 as pseudohypoaldosteronism type II causing genes
Although several upstream regulators of this cascade have been identified (Fig. 2), exactly how these regulators regulate WNK kinase activity remains largely unknown. Similarly, how PHAII-causing mutations of WNK4 activate the cascade remained unelucidated. Recently, two new genes [Kelch-like protein 3 (KLHL3) and Cullin3] were identified as genes responsible for causing PHAII (Boyden et al., 2012; Louis-Dit-Picard et al., 2012). However, how these genes were involved in causing PHAII was unknown. Determining how these genes (WNKs, KLHL3 and Cullin3) interact and how their mutation causes a common hypertensive disease would contribute to the understanding of the molecular pathogenesis of human hypertension, and also to the identification of new targets for anti-hypertensive drugs.
KLHL3 is a member of the Kelch-like protein family, which consists of 42 members (Dhanoa et al., 2013). Kelch-like ECH-associated protein 1 (Keap1), known as the E3 ligase to NRF2, also belongs to the KLHL family and is designated KLHL19 (Dhanoa et al., 2013). In general, KLHL proteins contain one BTB domain, one BTB and C-terminal Kelch (BACK) domain, and five to six Kelch domains (Fig. 3). The BTB domain was named based on a homologous, 115-amino-acid domain present in D. melanogaster bric a brac 1, tramtrack, and broad complex proteins and facilitates the protein–protein interaction (Zollman et al., 1994). The Kelch domain forms one blade of a β-propeller structure, as shown in Fig. 3. This domain is also involved in the protein–protein interaction. Kelch domain-containing proteins have been shown to participate in many cellular functions, such as the regulation of cell morphology and gene expression (Adams et al., 2000). Mutations in KLHL genes reportedly cause multiple human diseases. KLHL7 mutations cause autosomal dominant retinitis pigmentosa (Friedman et al., 2009; Kigoshi et al., 2011), and a missense mutation in KLHL9 causes distal myopathy (Cirak et al., 2010). Mutations in KLHL16 are linked to human giant axonal neuropathy (Bomont et al., 2000). In investigations of the molecular pathogenesis of these diseases, Kigoshi et al. (2011) clarified that KLHL7 assembles with Cullin3 and exerts E3 ligase activity. Likewise, KLHL20 was also reported to function as an E3 ligase in combination with Cullin3 on death-associated protein kinase (Lee et al., 2010), PDZ-Rho guanine nucleotide exchange factor (Lin et al., 2011) and promyelocytic leukemia protein (Yuan et al., 2011). KLHL7 and KLHL20 proteins bind to Cullin3 via their BTB domains and capture their substrates with their Kelch repeats. Therefore, it has been speculated that the KLHL3-Cullin3 complex also acts an E3 ligase on an unknown target protein.
Figure 3. Structure of Kelch-like proteins.
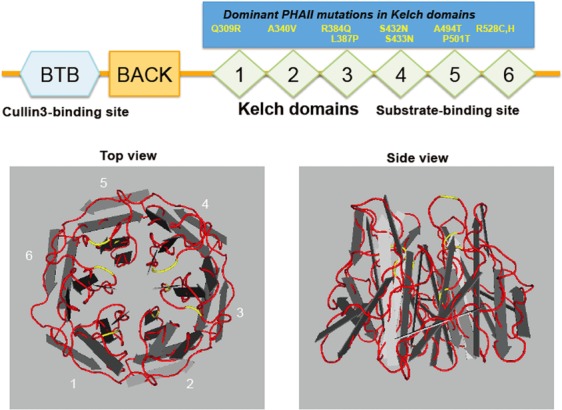
The upper panel shows the structure of Kelch-like (KLHL) proteins with N-terminal BTB and BACK domains and five to six C-terminal Kelch domains, and most autosomal dominant mutations causing pseudohypoaldosteronism type II (PHAII). The BTB domain is a binding site for Cullin 3 and Kelch repeats constitute a propeller structure, as shown in the lower panels, and capture a substrate. Each Kelch domain forms a blade, and most PHAII-causing mutations (shown in yellow lines) are located in the loop regions linking each blade, which may be involved in substrate binding.
WNK kinases are substrates of Kelch-like protein 3-Cullin3 E3 ligase
As mutations in WNK4, KLHL3 and Cullin3 cause the same disease, PHAII, and the activation of WNK–OSR1/SPAK–NCC signaling underlies its pathogenesis, it is reasonable to speculate that components of this signaling cascade, in particular WNK4, could be the substrate of KLHL3-Cullin3 E3 ligase. A French group reported that KLHL3 was able to bind to NCC and regulate its intracellular localisation (Louis-Dit-Picard et al., 2012). They did not investigate whether NCC was ubiquitinated by KLHL3. Then, Ohta et al. (2013) and Wakabayashi et al. (2013) reported that WNK1 and WNK4 were substrates of KLHL3-Cullin3 E3 ligase, respectively. In both studies, the binding of KLHL3 to NCC was not reproduced. Subsequently, two further reports (Shibata et al., 2013; Wu and Peng, 2013) supported WNK4 as a target of KLHL3-Cullin3 E3 ligase.
Analyses of PHAII-causing mutations in WNK4, KLHL3 and Cullin 3 also clearly disclosed how these three proteins interact. As previously mentioned, PHAII-causing mutations in WNK4 were clustered in the acidic domain, which is highly conserved in all WNK kinases (Fig. 1). Wakabayashi et al. (2013) and Mori et al. (2013) showed via fluorescent correlation spectroscopy that binding of KLHL3 to WNK4 was abolished by PHAII-causing mutations in WNK4, indicating that the acidic domain is involved in binding KLHL3. In contrast to WNK4, mutations in KLHL3 were not confined to a single domain, but present in the BTB, BACK and Kelch domains. Mutations in the BTB and BACK domains affected the ability of KLHL3 to bind Cullin3, whereas mutations in the Kelch domains affected the ability of KLHL3 to bind WNK1 and WNK4 (Mori et al., 2013). Impaired binding of KLHL3 to either Cullin3 or WNK4 decreased the ubiquitination of WNK4, resulting in increased WNK4 within cells. PHAII-causing Cullin3 mutations are clustered around the splice donor and acceptor sites of exon 9. Boyden et al. (2012) showed via experiments in cultured cells that these mutations resulted in the skipping of exon 9. Osawa et al. (2013) and Tsuji et al. (2013) verified that exon 9 was skipped in the leukocytes of patients with PHAII. Mutant Cullin3 lacking a portion of exon 9 did not show reduced binding to KLHL3, but E3 ligase activity towards WNK4 was significantly decreased (Wakabayashi et al., 2013). Thus, all PHAII-causing mutations in WNK4, KLHL3 and Cullin3 resulted in a common consequence: reduced ubiquitination of WNK4 and increased WNK4 protein within cells (Fig. 4). This increase in WNK4 protein was confirmed in the kidneys of Wnk4D561A/+ mice (Wakabayashi et al., 2013).
Figure 4. Molecular pathogenesis of pseudohypoaldosteronism type II.
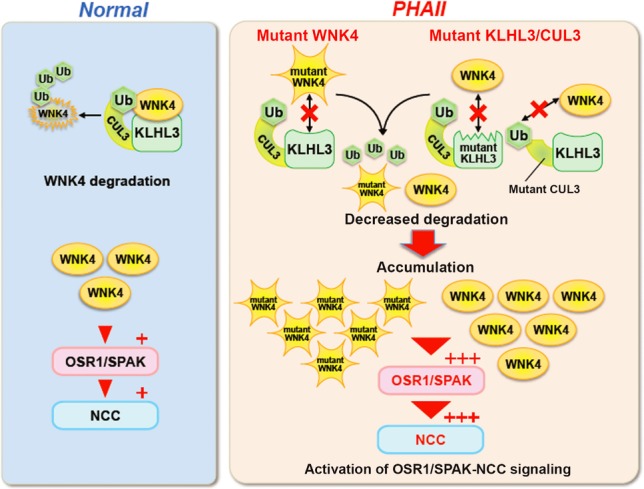
Under normal conditions, WNK 4 protein within cells are maintained by appropriate degradation after ubiquitination by KLHL3-Cullin3 E3 ligase. However, PHAII causing-mutations in the acidic domain of WNK4 and in the Kelch domains of KLHL3 affect their binding, thereby reducing the ubiquitination and degradation of WNK4. PHAII-causing mutant Cullin3 lacking the portion corresponding to exon 9 exhibits lower E3 ligase activity in combination with KLHL3 toward WNK4. Thus, PHAII-causing mutations in three different genes have a common consequence: decreased ubiquitination and increased WNK4 protein levels within cells. The increase in WNK4 protein was confirmed in the kidneys of Wnk4D561A/+ PHAII model mice. Furthermore, increased WNK4 protein levels in the kidneys of WNK4 transgenic mice activated OSR1/SPAK–NCC signaling. Although WNK4 is the major WNK kinase regulating NCC in the kidney, other WNKs normally expressed at low levels could also be increased in kidneys with PHAII caused by KLHL3 and Cullin3 mutations, thereby contributing to the more severe phenotypes resulting from these mutations compared with those resulting from WNK1 or WNK4 mutations alone.
Increased WNK4 in kidney activates OSR1/SPAK–NCC signaling and causes PHAII
Long-standing controversy exists about the influence of WNK4 on NCC function (McCormick and Ellison, 2011). Initially, WNK4 overexpression experiments in X. laevis oocytes showed that WNK4 is a negative regulator of NCC (Wilson et al., 2003; Yang et al. 2003). Further analyses by Yang et al. (2005, 2007b) showed that the inhibitory effect of WNK4 on NCC was not kinase activity dependent. Therefore, this inhibitory effect cannot be mediated by OSR1/SPAK–NCC signaling. Casteneda-Bueno et al. (2012) reported that WNK4 knockout mice exhibit a phenotype reminiscent of Gitelman syndrome (Gitelman syndrome is caused by the loss of function of NCC), indicating that WNK4 is a positive regulator of NCC in vivo. In fact, NCC phosphorylation, and even NCC protein abundance, was markedly decreased in the kidneys of WNK4 knockout mice. Thus, it is barely possible that a decrease in WNK4 levels activate NCC, and there is little evidence that WNK4 is a negative regulator of NCC in vivo, except that WNK4 BAC transgenic mice harboring a single copy of the wild-type WNK4 transgene exhibited a Gitelman syndrome-like phenotype (Lalioti et al., 2006). The results of this transgenic mouse study were obtained through analysis of a single line of wild-type WNK4 transgenic mice, and whether WNK4 protein abundance was indeed increased in the kidney was not shown. Data from transgenic mouse studies should be interpreted with caution, as there is no guarantee that transgenes are expressed in the same manner as endogenous genes. Sometimes, transgenes disrupt endogenous genes by homologous recombination. To circumvent the problems inherent in transgenic mouse studies, analysis of multiple lines of transgenic mice with different copy numbers is necessary. Proof that an observed phenotype is dependent on the level of the protein overexpressed is very important to draw a definite conclusion. Wakabayashi et al. (2013) reproduced the method of transgenic mouse generation used by Lalioti et al. (2006) to generate several lines of WNK4 BAC transgenic mice. They showed that, as WNK4 protein levels in the kidney increased, phosphorylation of OSR1, SPAK and NCC robustly increased. Furthermore, their WNK4 transgenic mice mimicked the phenotype of PHAII model mice. These results indicate that increased wild-type WNK4 in the kidney activates the OSR1/SPAK–NCC signaling cascade and causes PHAII.
Thus, impaired ubiquitination and a consequent increase in WNK4 protein was established as the molecular pathogenesis of PHAII caused by mutations in WNK4, KLHL3 and Cullin3 (Fig. 4). However, WNK kinases other than WNK4 may also be regulated by the KLHL3-Cullin3 complex. The amino acid sequence of the KLHL3 binding site in WNK4 is highly conserved in other WNK kinases (Fig. 1), and both the WNK1 and WNK4 proteins were shown to be regulated by KLHL3-Cullin3 (Ohta et al., 2013; Wakabayashi et al., 2013). Therefore, levels of both WNK1 and WNK4 may be increased in the kidneys of patients with PHAII carrying the KLHL3 and Cullin3 mutations, further contributing to the activation of OSR1/SPAK–NCC signaling and explaining the more severe PHAII phenotypes evident with Cullin3 and KLHL3 mutations than with WNK1 and WNK4 mutations (Boyden et al., 2012). PHAII-causing mutations in WNK1 consist of large deletions in intron 1 (Wilson et al., 2001): This deletion was recently discovered to increase full-length WNK1 transcription in the kidneys of a mouse model of the WNK1 mutation (Vidal-Petiot et al., 2013). The mechanism elucidated in this study may not be directly related to the pathogenesis of PHAII caused by WNK1 mutations. However, PHAII should be considered a disease caused by increased WNK kinase caused by the dysregulation of either transcription or the ubiquitination of WNK kinases.
Future perspectives
Analyses of PHAII pathogenesis suggest that the regulation of levels of WNK kinase protein is an important regulatory mechanism of WNK–OSR1/SPAK–SLC12 signaling. In addition to WNK1 and WNK4, it is hypothesised that other WNKs, such as WNK2 and WNK3, could be substrates of KLHL3-Cullin3 E3 ligase because the KLHL3-binding domain of WNK4 (the acidic domain) is highly conserved in all WNK isoforms. Furthermore, KLHL2 is the closest homolog to KLHL3 among KLHL proteins, and it is also the closest homolog to D. melanogaster Kelch (63% homology; (Soltysik-Espanola et al., 1999). Kelch repeats in these three proteins are highly conserved. KLHL2 shares almost perfect homology (98%) with KLHL3 in the loop regions of the Kelch repeats connecting each blade, in which most of the PHAII-causing KLHL3 mutations cluster (Boyden et al., 2012; Louis-Dit-Picard et al., 2012). The high degree of homology between KLHL2 and KLHL3 is not evident between KLHL3 and other Kelch-like proteins (Prag and Adams, 2003). The function of the loops connecting the blades of the Kelch repeats has not yet been evaluated in KLHL3, but given that these loops form the top face of the β-propeller (Fig. 3) and that this face is considered the substrate-binding pocket, extensive homology in these loop domains between KLHL2 and KLHL3 supports the theory of shared substrate specificity between KLHL2 and KLHL3. Takahashi et al. (2013) verified that KLHL2 in combination with Cullin3 could function as an E3 ligase for all WNK isoforms. These data suggest that all WNK kinases could be regulated by KLHL2 and KLHL3 in multiple cell types. Regulation of WNK kinases by KLHL2 and KLHL3 could be involved in PHAII and in other contexts where WNK kinases are regulated. The hormones and diets known to regulate WNK–OSR1/SPAK signaling (Fig. 2) may not directly regulate WNK but rather regulate KLHLs, thereby regulating WNK kinase. In addition, the binding of WNKs to KLHL2 and KLHL3 could be regulated by external stimuli, such as the phosphorylation of serine and threonine residues in Kelch domains. Further analyses focusing on these points are necessary, in addition to the confirmation of PHAII pathogenesis in vivo in PHAII model mice carrying KLHL3 and Cullin3 mutations.
Conclusions
Why PHAII-causing missense mutations in WNK4 are clustered and how these mutations activate downstream signaling to NCC remained undetermined. Recent advancements in genetics, in particular whole-exome sequencing, revealed two additional genes responsible for causing PHAII, and their discovery helped to construct a complete picture of the molecular pathogenesis of PHAII. Levels of WNK kinases within cells, regulated via ubiquitination by KLHL proteins, are important determinants of the activity of the WNK–OSR1/SPAK–SLC12A signaling cascade. Consequently, KLHL2 and KLHL3 could represent new targets for drug discovery to regulate WNK kinase activity.
Glossary
- KLHL3
Kelch-like 3
- NCC
NaCl cotransporter
- OSR1
oxidative stress-responsive gene 1
- PHAII
pseudohypoaldosteronism type II
- SPAK
Ste20-related proline-alanine rich kinase
- WNK
with-no-lysine
Conflict of interest
The authors have declared no conflict of interest.
References
- Adams J, Kelso R, Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 2000;10:17–24. doi: 10.1016/s0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- Bergaya S, Faure S, Baudrie V, Rio M, Escoubet B, Bonnin P, Henrion D, Loirand G, Achard JM, Jeunemaitre X, Hadchouel J. WNK1 regulates vasoconstriction and blood pressure response to alpha 1-adrenergic stimulation in mice. Hypertension. 2011;58:439–445. doi: 10.1161/HYPERTENSIONAHA.111.172429. [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Valimaki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Cebotaru V, Wang YH, Zhang XM, Cebotaru L, Guggino SE, Guggino WB. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 2006;69:2162–2170. doi: 10.1038/sj.ki.5000333. [DOI] [PubMed] [Google Scholar]
- Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl– cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda-Bueno M, Gamba G. Mechanisms of sodium-chloride cotransporter modulation by angiotensin II. Curr. Opin. Nephrol. Hypertens. 2012;21:516–522. doi: 10.1097/MNH.0b013e32835571a4. [DOI] [PubMed] [Google Scholar]
- Chavez-Canales M, Arroyo JP, Ko B, Vazquez N, Bautista R, Castaneda-Bueno M, Bobadilla NA, Hoover RS, Gamba G. Insulin increases the functional activity of the renal NaCl cotransporter. J. Hypertens. 2013;31:303–311. doi: 10.1097/HJH.0b013e32835bbb83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiga M, Rafiqi FH, Alessi DR, Sohara E, Ohta A, Rai T, Sasaki S, Uchida S. Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J. Cell Sci. 2011;124:1391–1395. doi: 10.1242/jcs.084111. [DOI] [PubMed] [Google Scholar]
- Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int. 2008;74:1403–1409. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- Cirak S, von Deimling F, Sachdev S, Errington WJ, Herrmann R, Bonnemann C, Brockmann K, Hinderlich S, Lindner TH, Steinbrecher A, Hoffmann K, Prive GG, Hannink M, Nurnberg P, Voit T. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain. 2010;133:2123–2135. doi: 10.1093/brain/awq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Los Heros P, Kahle KT, Rinehart J, Bobadilla NA, Vazquez N, San Cristobal P, Mount DB, Lifton RP, Hebert SC, Gamba G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1976–1981. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaloy C, Elvira-Matelot E, Clemessy M, Zhou XO, Imbert-Teboul M, Houot AM, Jeunemaitre X, Hadchouel J. Deletion of WNK1 first intron results in misregulation of both isoforms in renal and extrarenal tissues. Hypertension. 2008;52:1149–1154. doi: 10.1161/HYPERTENSIONAHA.108.120899. [DOI] [PubMed] [Google Scholar]
- Delaloy C, Lu J, Houot AM, Disse-Nicodeme S, Gasc JM, Corvol P, Jeunemaitre X. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol. Cell Biol. 2003;23:9208–9221. doi: 10.1128/MCB.23.24.9208-9221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS. Update on the Kelch-like (KLHL) gene family. Hum. Genomics. 2013;7:13. doi: 10.1186/1479-7364-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J. Biol. Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G. TRPV4: a new target for the hypertension-related kinases WNK1 and WNK4. Am. J. Physiol. Renal. Physiol. 2006;290:F1303–F1304. doi: 10.1152/ajprenal.00030.2006. [DOI] [PubMed] [Google Scholar]
- Garzon-Muvdi T, Pacheco-Alvarez D, Gagnon KB, Vazquez N, Ponce-Coria J, Moreno E, Delpire E, Gamba G. WNK4 kinase is a negative regulator of K+-Cl- cotransporters. Am. J. Physiol. Renal. Physiol. 2007;292:F1197–1207. doi: 10.1152/ajprenal.00335.2006. [DOI] [PubMed] [Google Scholar]
- Gordon RD. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension. 1986;8:93–102. doi: 10.1161/01.hyp.8.2.93. [DOI] [PubMed] [Google Scholar]
- Hossain Khan MZ, Sohara E, Ohta A, Chiga M, Inoue Y, Isobe K, Wakabayashi M, Oi K, Rai T, Sasaki S, Uchida S. Phosphorylation of Na-Cl cotransporter by OSR1 and SPAK kinases regulates its ubiquitination. Biochem. Biophys. Res. Commun. 2012;425:456–461. doi: 10.1016/j.bbrc.2012.07.124. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Macgregor GG, Wilson FH, Van Hoek AN, Brown D, Ardito T, Kashgarian M, Giebisch G, Hebert SC, Boulpaep EL, Lifton RP. Paracellular Cl- permeability is regulated by WNK4 kinase: insight into normal physiology and hypertension. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14877–14882. doi: 10.1073/pnas.0406172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16783–16788. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Ring A, Gimenez I, Gamba G, Hebert SC, Lifton RP. WNK protein kinases modulate cellular Cl- flux by altering the phosphorylation state of the Na-K-Cl and K-Cl cotransporters. Physiology (Bethesda) 2006;21:326–335. doi: 10.1152/physiol.00015.2006. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Wilson FH, Leng Q, Lalioti MD, O'Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- Kigoshi Y, Tsuruta F, Chiba T. Ubiquitin ligase activity of Cul3-KLHL7 protein is attenuated by autosomal dominant retinitis pigmentosa causative mutation. J. Biol. Chem. 2011;286:33613–33621. doi: 10.1074/jbc.M111.245126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komers R, Rogers S, Oyama TT, Xu B, Yang CL, McCormick J, Ellison DH. Enhanced phosphorylation of Na(+)-Cl- co-transporter in experimental metabolic syndrome: role of insulin. Clin. Sci. (Lond.) 2012;123:635–647. doi: 10.1042/CS20120003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat. Genet. 2006;38:1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- Lee DH, Maunsbach AB, Riquier-Brison AD, Nguyen MT, Fenton RA, Bachmann S, Yu AS, McDonough AA. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am. J. Physiol. Cell Physiol. 2013;304:C147–C163. doi: 10.1152/ajpcell.00287.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM, Chen RH. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010;29:1748–1761. doi: 10.1038/emboj.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Lin YM, Kao TC, Chuang HH, Chen RH. PDZ-RhoGEF ubiquitination by Cullin3-KLHL20 controls neurotrophin-induced neurite outgrowth. J. Cell Biol. 2011;193:985–994. doi: 10.1083/jcb.201103015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, Beaurain G, Bonnefond A, Sand O, Simian C, Vidal-Petiot E, Soukaseum C, Mandet C, Broux F, Chabre O, Delahousse M, Esnault V, Fiquet B, Houillier P, Bagnis CI, Koenig J, Konrad M, Landais P, Mourani C, Niaudet P, Probst V, Thauvin C, Unwin RJ, Soroka SD, Ehret G, Ossowski S, Caulfield M, Bruneval P, Estivill X, Froguel P, Hadchouel J, Schott JJ, Jeunemaitre X. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012;44:456–460. doi: 10.1038/ng.2218. S451–S453. [DOI] [PubMed] [Google Scholar]
- McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011;91:177–219. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Wakabayashi M, Mori T, Araki Y, Sohara E, Rai T, Sasaki S, Uchida S. Decrease of WNK4 ubiquitination by disease-causing mutations of KLHL3 through different molecular mechanisms. Biochem. Biophys. Res. Commun. 2013;439:30–34. doi: 10.1016/j.bbrc.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 2005;280:42685–42693. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- Mutig K, Saritas T, Uchida S, Kahl T, Borowski T, Paliege A, Bohlick A, Bleich M, Shan Q, Bachmann S. Short-term stimulation of the thiazide-sensitive Na+-Cl– cotransporter by vasopressin involves phosphorylation and membrane translocation. Am. J. Physiol. Renal. Physiol. 2010;298:F502–509. doi: 10.1152/ajprenal.00476.2009. [DOI] [PubMed] [Google Scholar]
- Naito S, Ohta A, Sohara E, Ohta E, Rai T, Sasaki S, Uchida S. Regulation of WNK1 kinase by extracellular potassium. Clin. Exp. Nephrol. 2011;15:195–202. doi: 10.1007/s10157-010-0378-9. [DOI] [PubMed] [Google Scholar]
- Nishida H, Sohara E, Nomura N, Chiga M, Alessi DR, Rai T, Sasaki S, Uchida S. Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension. 2012;60:981–990. doi: 10.1161/HYPERTENSIONAHA.112.201509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly M, Marshall E, Speirs HJ, Brown RW. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J. Am. Soc. Nephrol. 2003;14:2447–2456. doi: 10.1097/01.asn.0000089830.97681.3b. [DOI] [PubMed] [Google Scholar]
- Ohta A, Rai T, Yui N, Chiga M, Yang SS, Lin SH, Sohara E, Sasaki S, Uchida S. Targeted disruption of the Wnk4 gene decreases phosphorylation of Na-Cl cotransporter, increases Na excretion and lowers blood pressure. Hum. Mol. Genet. 2009;18:3978–3986. doi: 10.1093/hmg/ddp344. [DOI] [PubMed] [Google Scholar]
- Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon's hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 2013;451:111–122. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oi K, Sohara E, Rai T, Misawa M, Chiga M, Alessi D, Sasaki S, Uchida S. A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo. Biology Open. 2012;1:120–127. doi: 10.1242/bio.2011048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Ogura Y, Isobe K, Uchida S, Nonoyama S, Kawaguchi H. CUL3 gene analysis enables early intervention for pediatric pseudohypoaldosteronism type II in infancy. Pediatr. Nephrol. 2013;28:1881–1884. doi: 10.1007/s00467-013-2496-6. [DOI] [PubMed] [Google Scholar]
- Pacheco-Alvarez D, Cristobal PS, Meade P, Moreno E, Vazquez N, Munoz E, Diaz A, Juarez ME, Gimenez I, Gamba G. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 2006;281:28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- Pedersen NB, Hofmeister MV, Rosenbaek LL, Nielsen J, Fenton RA. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 2010;78:160–169. doi: 10.1038/ki.2010.130. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Garbarini N, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl- cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J. Biol. Chem. 2003;278:52848–52856. doi: 10.1074/jbc.M309436200. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J. Biol. Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42. doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieg T, Tang T, Uchida S, Hammond HK, Fenton RA, Vallon V. Adenylyl cyclase 6 enhances NKCC2 expression and mediates vasopressin-induced phosphorylation of NKCC2 and NCC. Am. J. Pathol. 2013;182:96–106. doi: 10.1016/j.ajpath.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, Volkman HM, Wilson FH, Hebert SC, Lifton RP. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2007;104:4020–4024. doi: 10.1073/pnas.0611727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, Mutig K. SPAK differentially mediates vasopressin effects on sodium cotransporters. J. Am. Soc. Nephrol. 2013;24:407–418. doi: 10.1681/ASN.2012040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekarabi M, Girard N, Riviere JB, Dion P, Houle M, Toulouse A, Lafreniere RG, Vercauteren F, Hince P, Laganiere J, Rochefort D, Faivre L, Samuels M, Rouleau GA. Mutations in the nervous system–specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J. Clin. Invest. 2008;118:2496–2505. doi: 10.1172/JCI34088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. U. S. A. 2013;110:7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohara E, Rai T, Yang SS, Ohta A, Naito S, Chiga M, Nomura N, Lin SH, Vandewalle A, Ohta E, Sasaki S, Uchida S. Acute insulin stimulation induces phosphorylation of the Na-Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. PLoS One. 2011;6:e24277. doi: 10.1371/journal.pone.0024277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltysik-Espanola M, Rogers RA, Jiang S, Kim TA, Gaedigk R, White RA, Avraham H, Avraham S. Characterization of Mayven, a novel actin-binding protein predominantly expressed in brain. Mol. Biol. Cell. 1999;10:2361–2375. doi: 10.1091/mbc.10.7.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83:811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- Susa K, Kita S, Iwamoto T, Yang SS, Lin SH, Ohta A, Sohara E, Rai T, Sasaki S, Alessi DR, Uchida S. Effect of heterozygous deletion of WNK1 on the WNK-OSR1/SPAK-NCC/NKCC1/NKCC2 signal cascade in the kidney and blood vessels. Clin. Exp. Nephrol. 2012;16:530–538. doi: 10.1007/s10157-012-0590-x. [DOI] [PubMed] [Google Scholar]
- Takahashi D, Mori T, Wakabayashi M, Mori Y, Susa K, Zeniya M, Sohara E, Rai T, Sasaki S, Uchida S. KLHL2 interacts with and ubiquitinates WNK kinases. Biochem. Biophys. Res. Commun. 2013;437:457–462. doi: 10.1016/j.bbrc.2013.06.104. [DOI] [PubMed] [Google Scholar]
- Talati G, Ohta A, Rai T, Sohara E, Naito S, Vandewalle A, Sasaki S, Uchida S. Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem. Biophys. Res. Commun. 2010;393:844–848. doi: 10.1016/j.bbrc.2010.02.096. [DOI] [PubMed] [Google Scholar]
- Tsuji S, Yamashita M, Unishi G, Takewa R, Kimata T, Isobe K, Chiga M, Uchida S, Kaneko K. A young child with pseudohypoaldosteronism type II by a mutation of Cullin 3. BMC Nephrol. 2013;14:166. doi: 10.1186/1471-2369-14-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Renal. Physiol. 2009;297:F704–712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. Potassium-induced natriuresis is preserved during sodium depletion and accompanied by inhibition of the sodium chloride cotransporter. Am. J. Physiol. Renal. Physiol. 2013 doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- Verissimo F, Jordan P. WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene. 2001;20:5562–5569. doi: 10.1038/sj.onc.1204726. [DOI] [PubMed] [Google Scholar]
- Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc. Natl. Acad. Sci. U. S. A. 2013;110:14366–14371. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013;3:858–868. doi: 10.1016/j.celrep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc. Natl. Acad. Sci. U. S. A. 2003;100:680–684. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Peng JB. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett. 2013;587:1717–1722. doi: 10.1016/j.febslet.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- Xu BE, Min X, Stippec S, Lee BH, Goldsmith EJ, Cobb MH. Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J. Biol. Chem. 2002;277:48456–48462. doi: 10.1074/jbc.M207917200. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4690–4694. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K, Yang SS, Ohta A, Sohara E, Rai T, Sasaki S, Uchida S. Apical localization of renal K channel was not altered in mutant WNK4 transgenic mice. Biochem. Biophys. Res. Commun. 2005;332:750–755. doi: 10.1016/j.bbrc.2005.04.169. [DOI] [PubMed] [Google Scholar]
- Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Invest. 2003;111:1039–1045. doi: 10.1172/JCI17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CL, Liu X, Paliege A, Zhu X, Bachmann S, Dawson DC, Ellison DH. WNK1 and WNK4 modulate CFTR activity. Biochem. Biophys. Res. Commun. 2007a;353:535–540. doi: 10.1016/j.bbrc.2006.11.151. [DOI] [PubMed] [Google Scholar]
- Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J. Clin. Invest. 2007b;117:3403–3411. doi: 10.1172/JCI32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CL, Zhu X, Wang Z, Subramanya AR, Ellison DH. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J. Clin. Invest. 2005;115:1379–1387. doi: 10.1172/JCI22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010;21:1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 2007c;5:331–344. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Yuan WC, Lee YR, Huang SF, Lin YM, Chen TY, Chung HC, Tsai CH, Chen HY, Chiang CT, Lai CK, Lu LT, Chen CH, Gu DL, Pu YS, Jou YS, Lu KP, Hsiao PW, Shih HM, Chen RH. A Cullin3-KLHL20 ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell. 2011;20:214–228. doi: 10.1016/j.ccr.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, Oi K, Chiga M, Takahashi D, Yang SS, Lin SH, Rai T, Sasaki S, Uchida S. Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension. 2013;62:872–878. doi: 10.1161/HYPERTENSIONAHA.113.01543. [DOI] [PubMed] [Google Scholar]
- Zollman S, Godt D, Prive GG, Couderc JL, Laski FA. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 1994;91:10717–10721. doi: 10.1073/pnas.91.22.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]