

Abstract
The development of an angiosarcomatous component in germ cell tumors (GCTs) is rare. Here we studied 12 cases of mediastinal GCTs with an angiosarcomatous component. All patients were men with a mean age of 34 years (range, 24 to 49 years). No patient had a documented testicular GCT. The mean size of mediastinal tumors was 12.9 cm (range, 5.5 to 16.0 cm). Grossly, the tumors were cystic with variegated hemorrhagic, mucinous and fleshy solid areas. Microscopically, all tumors were composed of GCT. The most common GCT component was teratoma (n=10), and other GCT components included seminoma (n=3), yolk sac tumor (n=3), embryonal carcinoma (n=2) and choriocarcinoma (n=1). The angiosarcomatous component was present in primary mediastinal tumors (n=6), metastasis (n=3), or both primary mediastinal tumor and metastasis (n=3). The angiosarcomatous component accounted for an average of 30% (range, 5% to 95%) of the primary mediastinal tumor. In addition, other non–germ cell components, including rhabdomyosarcoma (n = 3), leiomyosarcoma (n = 1), and poorly differentiated carcinoma (n = 1), were also present in the tumors. Of the 10 patients with follow-up available, all patients developed metastasis (n=8) or local recurrence (n=2); seven died of disease at a mean of 33 months (range, 21 to 75 months), and three patients were alive at a mean of 75 months (range, 5 to 120 months). Our findings suggest that the presence of an angiosarcomatous component in mediastinal GCT, even in a small amount, is associated with a poor clinical outcome.
Keywords: germ cell tumor, mediastinum, sarcomatoid transformation, angiosarcoma
INTRODUCTION
Germ cell tumors (GCTs) are the most common tumors in men aged 15 to 35 years. While the majority of GCTs arise from the gonads, some GCTs develop outside the gonads. Most extragonadal GCTs are concentrated in the midline of the body, including the mediastinum, retroperitoneum, pineal gland, and sacrococcygeal region.1-3 Of these locations, the mediastinum is the most common for extragonadal GCTs. In fact, GCTs account for approximately 15% and 25% of mediastinal tumors in adults and children, respectively.4 Like gonadal GCTs, mediastinal GCTs demonstrate a wide spectrum of histologic phenotypes. At the current time, mediastinal GCTs are classified using the same system as that proposed for gonadal GCTs.4
Occasionally, GCTs develop a non-germ cell or somatic component. The somatic components can be epithelial, mesenchymal, hematopoietic, neurogenic malignancies or a combination of them, with sarcomatous malignancies being the most commonly observed.5-9 Although this phenomenon is uncommon in gonadal GCTs, its frequency seems disproportionally high in mediastinal GCTs, considering the fact that mediastinal GCTs account for less than 5 % of all GCTs but up to 50% of GCTs with somatic components.5 The presence of somatic components, particularly sarcomatous components, in the GCTs often leads to a poor response to a conventional cisplatin-based chemotherapeutic regimen.10
While rhabdomyosarcoma accounts for the majority of sarcomatous components in mediastinal GCTs,5-7 angiosarcoma is rare with most cases being reported as case reports or parts of large series of GCTs with sarcomatous components.5-7,9,11 Accordingly, the presence of an angiosarcomatous component in mediastinal GCT is not well understood. In this study, we examined 12 cases of mediastinal GCTs with an angiosarcomatous component, the largest series we are aware of, to further elucidate the clinicopathologic features and outcomes associated with this rare phenomenon.
MATERIALS AND METHODS
With approval from our Institutional Review Board, we retrospectively searched the database in the Department of Pathology and identified 12 cases of mediastinal GCT with an angiosarcomatous component in from 1987 to 2008. Our inclusion criterion was an expansile angiosarcomatous tumor growth of at least one low-power field (4 × objective) replacing the GCT component. None of the 12 patients had any other malignancies documented before their mediastinal GCTs.
The reviewed specimens included biopsies (n=9) and resections (n=12) of the primary mediastinal tumor, and biopsies (n=7) of the metastatic tumor. All specimens were adequately sampled for histologic examination. The reviewed pathologic parameters included tumor size, gross appearance, stage, GCT components, angiosarcomatous components and other somatic components. In some cases, immunohistochemical staining was performed using the avidin-biotin-peroxidase complex method in a Dako AutoStainer (Dako, Carpinteria, CA). The primary antibodies used were antibodies to CD31 (Dako, 1:20 dilution), CD34 (Dako, 1:50 dilution), and factor VIII (Dako, 1:100 dilution). Clinical data, including demographic, presenting symptoms, radiographic studies, treatment and follow-up information, were obtained from the medical records. Overall survival times were measured in months from the date of initial diagnosis to death or last available follow-up.
RESULTS
All 12 patients were men with a mean age of 34 years (median, 34 years; range, 24 to 49 years) (Table 1). No patient had a documented testicular GCT. Presenting symptoms were available for 8 patients and included chest pain (n = 4), shoulder pain (n = 2), cough (n = 2), and shortness of breath (n = 1). In 1 patient, a mediastinal tumor was found incidentally during a workup after an automobile accident. In all patients, a mediastinal tumor was demonstrated by radiographic studies. Nine patients underwent core needle biopsies, which demonstrated GCT only (no sarcomatous component; n = 6), GCT with an angiosarcomatous component (n = 1), angiosarcoma only (n = 1), and undifferentiated sarcoma only (n = 1).
Table 1.
Clinical and Pathologic Features of Mediastinal Germ Cell Tumors with an Angiosarcoma Component
| Case | Age (years) | Presenting Symptom | Mediastinal Tumor Histology | Tumor Stage† | Metastasis Site | Metastasis Histology | Therapy | Follow-up (months) | Status |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 36 | Cough | MT, IT, R, A (10)* | III | Brain | N/B | Su, Ch, Ra | 25 | DOD |
| 2 | 26 | N/A | IT, S, L | III | Bone | A (100) | Su | 49 | DOD |
| 3 | 32 | Chest pain | MT, A (50) | III | Bone | N/B | Su | 27 | DOD |
| 4 | 43 | N/A | IT, S, A (‡) | III | N/A | N/A | Su | 75 | DOD |
| 5 | 40 | Shoulder pain | MT, Ca, A (45) | III | Liver, bone | A (100) | Su, Ch+ | 101 | FOD |
| 6 | 29 | N/A | MT | III | Liver, RPLN | MT, R, A (50) | Su | 21 | DOD |
| 7 | 49 | N/A | MT, YS, EC, SCC, A (45) | III | N/A | N/A | Su | N/A | N/A |
| 8 | 24 | Trauma workup | EC, YS, A (10) | III | Liver, spleen, bone | R, US, A (60) | Su, Ch | 33 | DOD |
| 9 | 35 | Chest pain | IT, MT, A (10) | II | Brain, bone | A (100) | Su, Ch+ | 49 | DOD |
| 10 | 34 | Chest and shoulder pain | MT, A (5) | II | Recurrences | N/B | Su, Ch+ | 120 | FOD |
| 11 | 33 | Cough | S, A (95) | III | N/A | N/A | Su, Ch | N/A | N/A |
| 12 | 24 | Chest pain, SOB | MT, YS | II | Recurrence | MT, A (5) | Su, Ch | 5 | FOD |
A, angiosarcoma; C, choriocarcinoma; Ca, carcinoma; Ch, cisplatin-based chemotherapy; Ch+, cisplatin-based chemotherapy plus doxorubicin; DOD, died of disease; EC, embryonal carcinoma; FOD, free of disease; IT, immature teratoma; MT, mature teratoma; L, leiomyosarcoma; N/A, not available; N/B, not biopsied; R, rhabdomyosarcoma; Ra, radiation therapy; RPLN, retroperitoneal lymph node; S, seminoma; SOB, shortness of breath; Su, surgery; US, undifferentiated sarcoma; YS, yolk sac tumor.
Angiosarcoma component in bold and percentage of angiosarcomatous component in parenthesis.
Tumor stage was assessed at the time of mediastinal tumor resection using the Moran and Suster staging system. 19
The percentage of angiosarcomatous component could not be assessed in this case, because only representative (not all) slides were available for review.
In all patients, the mediastinal tumor was resected. The mean size of tumor was 12.9 cm in the largest dimension (median, 14.5 cm; range, 5.5 to 16.0 cm). Grossly, the tumors were cystic with variegated hemorrhagic, mucinous, and fleshy solid areas. Microscopically, the most common GCT components were mature (n=8) and immature (n=6) teratomas. Other GCT components, including seminoma (n=3), yolk sac tumor (n=3) and embryonal carcinoma (n=2), were also observed (Table 1). The angiosarcomatous component was present in the primary mediastinal tumor (n=9), in metastasis (n=3), and in both the primary mediastinal tumor and metastasis (n=3). In the primary mediastinal tumor, the angiosarcomatous component accounted for an average of 30% (range, 5% to 95%) of the tumor, and was often admixed with GCT components (Fig. 1). The angiosarcomatous components varied in appearance, ranging from clearly vasoformative to poorly differentiated areas in which the vascular nature was not readily recognizable. In most areas, the angiosarcoma components were characterized by irregular vascular spaces that freely anastomosed and branched in a sinusoidal fashion (Fig. 2A). The cells lining the vascular spaces ranged from spindle to epithelioid, and often displayed high-grade cellular atypia with pleomorphic and hyperchromatic nuclei (Fig. 3). On immunostaining (n=5), the lining cells were positive for endothelial markers CD 31 (Fig. 2B), CD 34 (Fig. 2C) and Factor VIII (Fig 2D). In focal areas, the angiosarcoma was composed of closely packed epithelioid cells with poorly formed vascular spaces (Fig. 4). In addition, focal areas of rhabdomyosarcoma (n = 1), poorly differentiated carcinoma (n = 1), or leiomyosarcoma (n = 1) were also observed in 3 cases.
Figure 1.
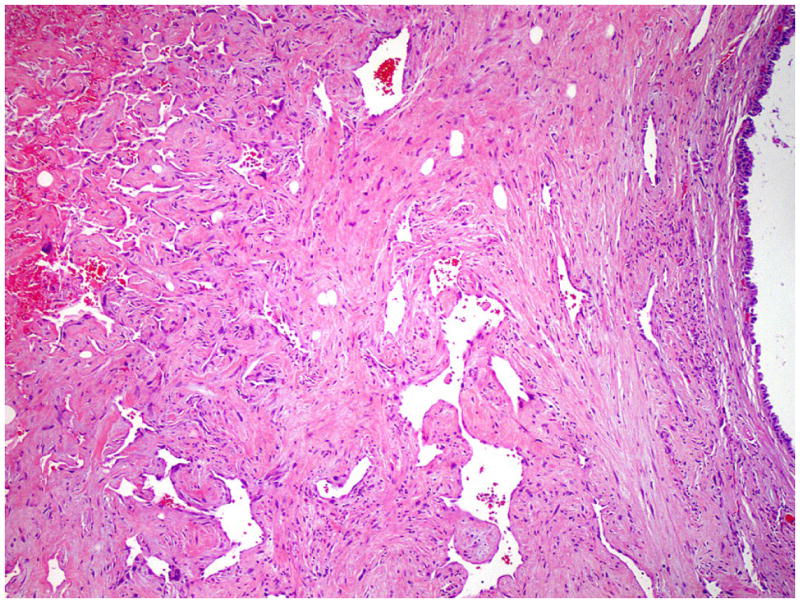
Angiosarcoma admixed with teratoma in a mediastinal GCT. The angiosarcoma component (left) shows irregular vascular spaces and the teratoma component (right) is composed of well differentiated glandular epithelium.
Figure 2.
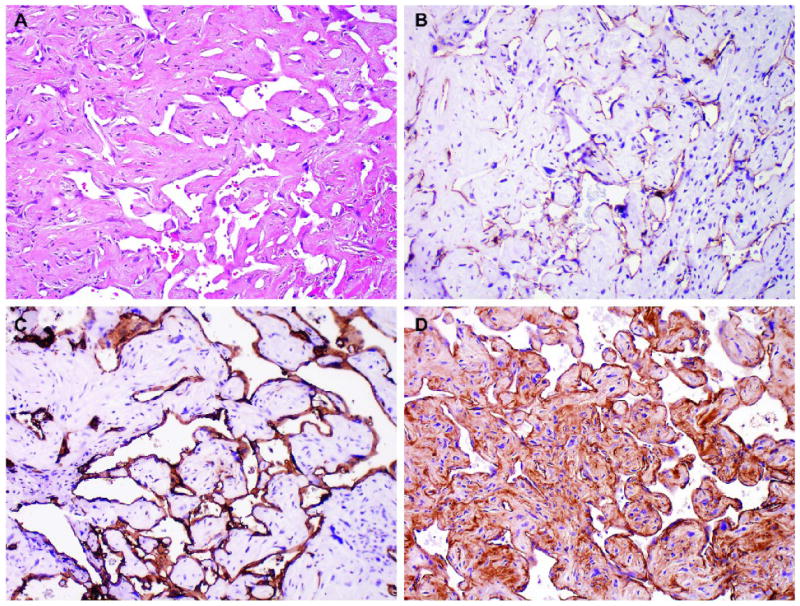
Angiosarcoma showing irregular vascular spaces that anastomose and branch in a sinusoidal fashion (A). The atypical endothelial cells are immunoreactive for CD 31 (B), CD 34 (C) and Factor VIII (D).
Figure 3.
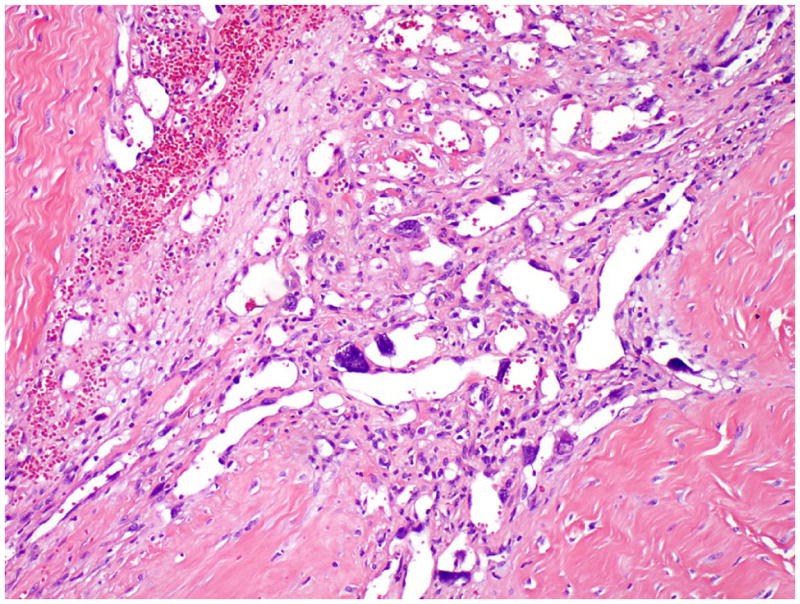
Angiosarcoma showing high grade cellular atypia with pleomorphic and hyperchromatic nuclei.
Figure 4.
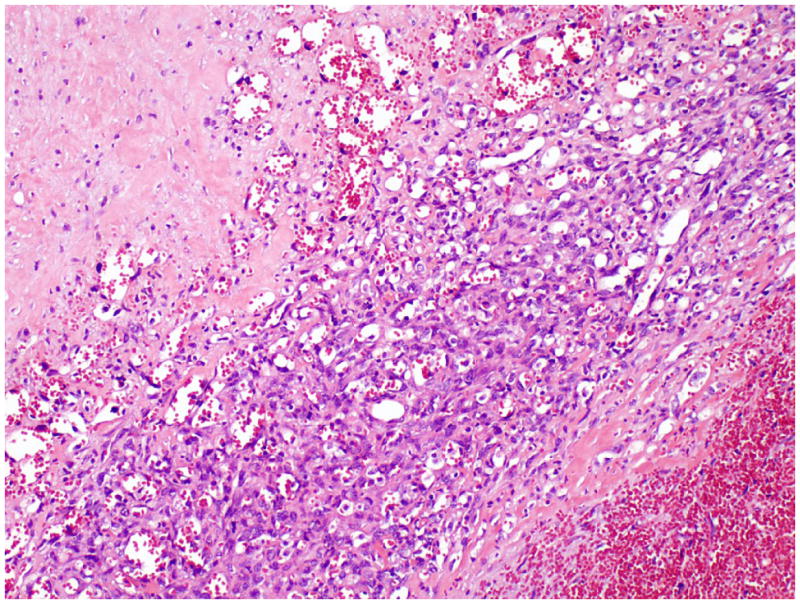
Angiosarcoma showing various differentiations. The well differentiated area shows easily recognizable vascular spaces lined by spindle endothelial cells. The poorly differentiated area is composed of closely packed epithelioid cells with rudimentary formation of vascular spaces.
Follow-up information was available for 10 patients with a mean follow-up of 51 months (median, 41 months; range, 5 to 120 months) (Table 1). Seven patients received cisplatin-based chemotherapy for GCTs, and three of them also received doxorubicin, a sarcoma-oriented drug. In addition, one patient received radiation therapy. In spite of multimodality therapies, patients developed metastasis (n=8) or local recurrence (n=2). The most common metastatic sites were the bones (n = 5), liver (n = 3), and brain (n = 2). In 6 cases, the tumor also involved the lungs, as a result of direct tumor extension or metastasis. The metastatic tumor was biopsied in 6 cases, and angiosarcoma was present in all cases. Seven patients died of disease at a mean of 33 months after the diagnosis (median, 40 months; range, 21 to 75 months). The other 3 patients were alive at a mean of 75 months (median, 101 months; range, 5 to 120 months); 2 of them had received doxorubicin. The clinical outcome was related to the tumor stage, but it was not associated with the amount of angiosarcomatous component in the mediastinal GCT.
DISCUSSION
The origin of mediastinal GCTs remains unclear. Although a small mediastinal GCT may be entirely encased in the thymus, there is no histogenetic relationship between mediastinal GCT and true thymoma.12 Recent studies have suggested that a mediastinal GCT may arise from residual primordial germ cells in the dorsal midline of the body.13,14 During embryogenesis, most primordial germ cells migrate laterally to the genital ridges to become spermatogenetic or ovogenetic stem cells; the cells remaining in the midline usually undergo apoptosis.13 However, this apoptotic process may be disrupted when the ectopic germ cells are exposed to carcinogenic agents. A recent study found that the absence of Bax, a proapoptotic Bcl-2 family member, prevented germ cells from undergoing apoptosis, resulting in large numbers of extragonadal germ cells.14
Mediastinal GCTs demonstrate histologic and serologic characteristics similar to those of gonadal GCTs.15 In addition, most mediastinal GCTs possess an isochromosome of 12p, a characteristic cytogenetic alteration that is consistently present in testicular GCTs.16,17 Therefore, the possibility of the mediastinal GCT representing a metastasis from a gonadal primary GCT should be considered. Whereas the presence of a single tumor in the anterior upper mediastinum with no retroperitoneal involvement would favor a mediastinal primary, the presence of a coexisting or preexisting gonadal GCT would suggest that the mediastinal GCT represents a metastasis. However, it may be difficult to recognize metastasis from a “burnt-out” gonadal GCT, in which the primary tumor has regressed in the gonadal gland.18
The staging system for mediastinal GCT largely follows the TNM classification system used for soft tissue tumors.4 Recently, Moran and Suster19 proposed a different clinical staging system in which mediastinal GCTs are divided into three stages: in stage I, tumors are confined to the mediastinum; in stage II, tumors invade the surrounding structures, such as the pleura, pericardium, or large vessels; and in stage III, tumors show evidence of intrathoracic or extrathoracic metastases.19 According to this classification system, 9 patients in our study had a stage III disease and 3 patients had a stage II disease. Six of the 9 patients at stage III died within 21-75 months, one patient survived and two were lost to followup. Two of the 3 patients at stage II were alive at 5 and 120 months, and one died at 75 months. Our results suggest that this staging system is also useful for predicting the clinical outcome of mediastinal GCTs with an angiosarcomatous component.
There are several hypotheses regarding the origin of an angiosarcomatous component in GCTs.9 In our study, the majority of GCTs (10/12) contained a teratomatous component, suggesting that the angiosarcomatous component may represent a malignant transformation of teratomatous component. Nonetheless, two cases of GCT lacked a teratomatous component, raising the possibility that the angiosarcomatous component may also develop from the dedifferentiation of non-teratomatous GCT components. Cytogenetic studies have demonstrated the presence of i(12p) abnormalities in somatic components.7 Since i(12p) is a specific genetic marker present in 80% of GCTs,16 its prevalence in the somatic components supports the idea that the somatic component arises from GCT cells. Others have suggested that the angiosarcomatous component may also develop in the mediastinal GCTs as a consequence of radiation therapy.9 In our study, only one patient had a documented history of radiation therapy, but he had an angiosarcomatous component in the mediastinal tumor before radiation therapy. Therefore, the development of the angiosarcomatous component was not related to radiation therapy in this patient.
The angiosarcomatous component in mediastinal GCT is characterized by freely anastomosing vascular spaces lined by atypical endothelial cells. Interestingly, there are also some atypical cells in the stroma that do not line the vascular spaces. These atypical stromal cells lack immunoreactivity for endothelial markers (CD 31, CD 34 or Factor VIII). When the atypical stromal cells are scattered and do not form a pure nodule, we consider them to be a part of the stroma that is associated with a teratoma. Only when the atypical stromal cells form a pure nodule of substantial size (larger than a half field viewed with a 4 x objective), they are considered to represent a sarcomatoid transformation. Using this definition, we found that 2 patients in the current study also developed other sarcomatoid transformations, including rhabdomyosarcoma and leiomyosarcoma, and 1 patient developed a poorly differentiated carcinoma.
The angiosarcomatous component may develop in a primary mediastinal GCT or in metastasis. In this study, nine patients showed an angiosarcomatous component in the primary mediastinal GCT, while six patients had an angiosarcomatous component in metastasis. Interestingly, three patients did not have an angiosarcomatous component in the primary mediastinal tumor, but subsequently developed an angiosarcomatous component in metastasis. Although this discrepancy could be due to inadequate sampling of the primary mediastinal tumors, an alternative explanation is the emergence of an angiosarcomatous component in metastasis due to drug selection. Patients with advanced mediastinal GCT often receive cisplatin-based chemotherapy, which is highly effective against conventional GCT components. However, sarcomatous components are notoriously resistant to cisplatin-based chemotherapy, which allows a growth advantage of sarcomatous components over conventional GCT components.
Previous studies have found that the presence of a sarcomatous component in a mediastinal GCT carries a dismal prognosis.5-7 In the 25 cases of mediastinal GCT with sarcomatous components that Malagón et al.5 reported, 16 patients died of tumor at a mean of 13 months, 3 patients were alive at a mean of 25 months, and 5 patients were lost in the follow-up after diagnosis. Likewise, patients with an angiosarcomatous component in mediastinal GCTs also have a poor clinical outcome. Manivel et al.6 reported 3 cases of mediastinal GCT with angiosarcomatous components and all patients died of disease within 9 months. In the current study, despite surgical treatment and chemotherapy, all 10 patients who had follow-up information either developed metastasis or local recurrence, and 7 died of disease. Of the 7 expired patients, 4 had a biopsy of the metastasis. An angiosarcomatous component was present in all 4 cases. In addition, other components, including mature teratoma, rhabdomyosarcoma and undifferentiated sarcoma, were also present in 2 of these cases. Although the angiosarcomatous component is a plausible cause of death in the expired patients, it cannot be ruled out that some patients might die of other sarcomas or even progressive germ cell tumor of the usual type. The poor clinical outcome is indicated whether the angiosarcomatous component is present in a primary mediastinal GCT or in its metastasis. Furthermore, the same outcome is observed in mediastinal GCTs with even a small amount of angiosarcomatous component, such as 5% of the tumor.
The presence of a somatic component in GCT is a major challenge in disease management. Although most GCTs are highly sensitive to cisplatin-based chemotherapy, somatic, particularly sarcomatous, components are resistant to this treatment. Recent studies found that patients with a single histologic type of somatic component could achieve long-term survival when they were treated with a chemotherapeutic regimen based on the specific histologic type of somatic component.20,21 In our study, 3 patients received chemotherapy containing doxorubicin, a drug that has been successfully used to treat sarcomas,22 and 2 achieved long-term survival. Therefore, our limited retrospective study suggests that patients with an angiosarcomatous component may benefit from sarcoma-oriented drugs, although this should be verified in large studies.
In summary, our study represents the largest series of mediastinal GCTs with an angiosarcomatous component to date. The angiosarcomatous component may develop in a primary mediastinal GCT or in its metastasis. The high prevalence of a teratomatous component in the mediastinal GCTs with an angiosarcomatous component suggests that the angiosarcomatous component may arise from malignant transformation of a teratomatous component, although alternative origins are also possible. The presence of an angiosarcomatous component in mediastinal GCT, even in a small amount, is associated with metastasis and local recurrence, and carries a poor clinical outcome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bokemeyer C, Nichols CR, Droz JP, et al. Extragonadal germ cell tumors of the mediastinum and retroperitoneum: results from an international analysis. J Clin Oncol. 2002;20:1864–1873. doi: 10.1200/JCO.2002.07.062. [DOI] [PubMed] [Google Scholar]
- 2.Gonzales-Crussi F, Winkler RF, Mirkin DL. Sacrococcygeal teratomas in infants and children. Arch Pathol Lab Med. 1978;102:420–425. [PubMed] [Google Scholar]
- 3.Parwani AV, Baisden BL, Erozan YS, et al. Pineal gland lesions: a cytopathologic study of 20 specimens. Cancer. 2005;105:80–86. doi: 10.1002/cncr.20849. [DOI] [PubMed] [Google Scholar]
- 4.Travis WD, Brambilla E, Muller-Hermelink HK, editors. World Health Organization Classification of Tumours, Pathology and Genetics: Tumours of the Lung, Pleura, Thymus and Heart. Lyon, France: IARC Press; 2004. [Google Scholar]
- 5.Malagón HD, Valdez AM, Moran CA, et al. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol. 2007;31:1356–1362. doi: 10.1097/PAS.0b013e318033c7c4. [DOI] [PubMed] [Google Scholar]
- 6.Manivel C, Wick MR, Abenoza P, et al. The occurrence of sarcomatous components in primary mediastinal germ cell tumors. Am J Surg Pathol. 1986;10:711–717. doi: 10.1097/00000478-198610000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, Amsterdam A, Prieto V, et al. Teratoma with malignant transformation: diverse malignant histologies arising in men with germ cell tumors. J Urol. 1998;159:133–138. doi: 10.1016/s0022-5347(01)64035-7. [DOI] [PubMed] [Google Scholar]
- 8.True LD, Otis CN, Delprado W, et al. Spermatocytic seminoma of testis with sarcomatous transformation. A report of five cases. Am J Surg Pathol. 1988;12:75–82. doi: 10.1097/00000478-198802000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Ulbright TM, Clark SA, Einhorn LH. Angiosarcoma associated with germ cell tumors. Hum Pathol. 1985;16:268–272. doi: 10.1016/s0046-8177(85)80013-7. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Vela JL, Savage PD, Manivel JC, et al. Poor prognosis of mediastinal germ cell cancers containing sarcomatous components. Cancer. 1990;66:1114–1116. doi: 10.1002/1097-0142(19900915)66:6<1114::aid-cncr2820660606>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Saito A, Watanabe K, Kusakabe T, et al. Mediastinal mature teratoma with coexistence of angiosarcoma, granulocytic sarcoma and a hematopoietic region in the tumor: a rare case of association between hematological malignancy and mediastinal germ cell tumor. Pathol Int. 1998;48:749–753. doi: 10.1111/j.1440-1827.1998.tb03977.x. [DOI] [PubMed] [Google Scholar]
- 12.Rosai J, Parkash V, Reuter VE. The origin of mediastinal germ cell tumors in men. Int J Surg Pathol. 1994;2:73–78. [Google Scholar]
- 13.Molyneaux KA, Stallock J, Schaible K, et al. Time-lapse analysis of living mouse germ cell migration. Dev Biol. 2001;240:488–498. doi: 10.1006/dbio.2001.0436. [DOI] [PubMed] [Google Scholar]
- 14.Runyan C, Gu Y, Shoemaker A, et al. The distribution and behavior of extragonadal primordial germ cells in Bax mutant mice suggest a novel origin for sacrococcygeal germ cell tumors. Int J Dev Biol. 2008;52:333–44. doi: 10.1387/ijdb.072486cr. [DOI] [PubMed] [Google Scholar]
- 15.Malagón HD, Montiel DP. Mediastinal germ cell tumors. Semin Diagn Path. 2005;22:230–240. doi: 10.1053/j.semdp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Bosl GJ, Ilson DH, Rodriguez E, et al. Clinical relevance of the i(12p) marker chromosome in germ cell tumors. J Natl Cancer Inst. 1994;86:349–355. doi: 10.1093/jnci/86.5.349. [DOI] [PubMed] [Google Scholar]
- 17.Schneider DT, Schuster AE, Fritsch MK, et al. Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosomes Cancer. 2002;34:115–125. doi: 10.1002/gcc.10053. [DOI] [PubMed] [Google Scholar]
- 18.Balzer BL, Ulbright TM. Spontaneous regression of testicular germ cell tumors: an analysis of 42 cases. Am J Surg Pathol. 2006;30:858–865. doi: 10.1097/01.pas.0000209831.24230.56. [DOI] [PubMed] [Google Scholar]
- 19.Moran CA, Suster S. Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer. 1997;80:681–690. [PubMed] [Google Scholar]
- 20.Donadio AC, Motzer RJ, Bajorin DF, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol. 2003;21:4285–4291. doi: 10.1200/JCO.2003.01.019. [DOI] [PubMed] [Google Scholar]
- 21.El Mesbahi O, Terrier-Lacombe MJ, Rebischung C, et al. Chemotherapy in patients with teratoma with malignant transformation. Eur Urol. 2007;51:1306–1311. doi: 10.1016/j.eururo.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647–1654. [PubMed] [Google Scholar]