

Summary
Misfolded proteins of the endoplasmic reticulum (ER) are retro-translocated into the cytosol, poly-ubiquitinated, and degraded by the proteasome, a process called ER-associated protein degradation (ERAD). Here, we have used purified components from Saccharomyces cerevisiae to analyze the mechanism of retro-translocation of luminal substrates (ERAD-L), recapitulating key steps in a basic process where the ubiquitin ligase Hrd1p is the only required membrane protein. We show that Hrd1p interacts with substrate through its membrane-spanning domain, discriminating misfolded from folded polypeptides. Both Hrd1p and substrate are poly-ubiquitinated, resulting in the binding of the Cdc48p ATPase complex. Subsequently, ATP hydrolysis by Cdc48p releases substrate from Hrd1p. Finally, ubiquitin chains are trimmed by the de-ubiquitinating enzyme Otu1p, which is recruited and activated by the Cdc48p complex. Cdc48p-dependent membrane extraction of poly-ubiquitinated proteins can be reproduced with reconstituted proteoliposomes. Our results suggest a model for retro-translocation in which Hrd1p forms a membrane conduit for misfolded proteins.
Introduction
Protein homeostasis in the ER is maintained by a quality control system. When a protein misfolds, it is retained in the ER and ultimately retro-translocated into the cytosol, poly-ubiquitinated, and degraded by the proteasome. This pathway is referred to as ER-associated protein degradation (ERAD) (for review, see (Bagola et al., 2011; Brodsky, 2012)). It alleviates cytotoxic stress imposed by protein misfolding and is implicated in numerous diseases (Guerriero and Brodsky, 2012). ERAD is found in all eukaryotic cells, but is best understood in S. cerevisiae. Here, substrates use three ERAD-pathways (ERAD-L, -M, or –C), depending on whether the misfolded domain is localized in the ER lumen, inside the ER membrane, or at the cytosolic side of the ER membrane (Carvalho et al., 2006; Huyer et al., 2004; Vashist and Ng, 2004). The pathways use distinct ubiquitin ligase complexes. ERAD-L requires a hetero-tetrameric membrane protein complex, the Hrd1p complex, comprised of the ubiquitin ligase Hrd1p and three additional membrane proteins (Hrd3p, Usa1p, and Der1p). ERAD-M also requires Hrd1p, but only a subset of the other components, and ERAD-C uses the ubiquitin ligase Doa10p. On the cytosolic side of the ER membrane, all pathways require an ATPase complex, which includes the ATPase Cdc48p and the cofactors Ufd1p and Npl4p.
Among the ERAD-pathways, ERAD-L is arguably most complex, as polypeptides have to be inserted into and moved across the ER membrane. ERAD-L begins with the recognition of a misfolded protein in the ER lumen, which is best understood for misfolded glycoproteins (for review, see (Xie and Ng, 2010)). The N-linked glycan of these proteins is trimmed to generate a terminal α-1,6-mannose residue, which is recognized by the luminal protein Yos9p. Yos9p binds to the luminal domain of Hrd3p, which also binds a misfolded segment around the glycan-attachment site of the substrate. Once a segment of the substrate emerges on the cytoplasmic side of the ER membrane, it is poly-ubiquitinated by the RING finger domain of Hrd1p (Bays et al., 2001a; Bordallo et al., 1998). The major ubiquitin-conjugating enzyme participating in this reaction is Ubc7p, which also requires the activator Cue1p (Bays et al., 2001a; Biederer et al., 1997). The recruitment of the Cdc48p complex to the Hrd1p complex involves recognition of a poly-ubiquitin chain by the cofactor Ufd1p/Npl4p (Meyer et al., 2002). However, it is unclear whether the recruitment requires the ubiquitin chain to be attached to the substrate, to Hrd1p, or to an unknown component. The binding of Cdc48p to the membrane might be facilitated by the adaptor protein Ubx2p (Neuber et al., 2005; Schuberth and Buchberger, 2005). Cdc48p is then thought to pull on the poly-ubiquitinated polypeptide substrate to move it into the cytosol (Bays et al., 2001b; Jarosch et al., 2002; Rabinovich et al., 2002; Ye et al., 2001). Recent experiments have also implicated de-ubiquitinating enzymes (DUBs) in ERAD (for review, see (Liu and Ye, 2012)). Several DUBs associate with Cdc48p or its mammalian homolog p97, and the overexpression of dominant-negative forms blocks ERAD in mammalian cells. However, it remains unclear how DUBs participate in ERAD.
The events of ERAD-L inside the ER membrane are less well understood. Hrd1p seems to be a central component, as its overexpression bypasses the need for the other membrane components, as well as for the luminal protein Yos9p (Carvalho et al., 2010; Denic et al., 2006; Garza et al., 2009; Plemper et al., 1999). Under these conditions, glycan trimming is not required, and both glycosylated and non-glycosylated proteins are degraded. All downstream cytosolic components, including the Cdc48p complex, are still required. Hrd1p overexpression makes Hrd1p unstable and slows, but does not abolish, cell growth. These results suggest that Hrd1p is the only membrane component required for a basic ERAD-L process. The minimal components required for retro-translocation therefore comprise Hrd1p, Ubc7p, Cue1p, and the Cdc48p complex. Because Hrd1p is a multi-spanning membrane protein, it is a good candidate to be part of a retro-translocation channel. In fact, crosslinking experiments show that a retro-translocating substrate interacts with Hrd1p (Carvalho et al., 2010). However, the exact function of Hrd1p during retro-translocation remains unclear.
Our knowledge on ERAD-L comes from genetics and biochemical experiments in intact cells. In vitro reconstitution of ERAD-L using purified components is critical to demonstrate that all ERAD components have been identified. Even more importantly, reconstitution experiments are instrumental in addressing the molecular mechanism of ERAD. Here, we have recapitulated crucial reactions of a basic ERAD-L process with purified S. cerevisiae components in both detergent and reconstituted proteoliposomes. Our results suggest a mechanistic model for how misfolded luminal ER proteins associate with Hrd1p inside the ER membrane and how they are extracted by Cdc48p on the cytoplasmic side of the membrane.
Results
Direct interaction of ERAD-L substrates with Hrd1p
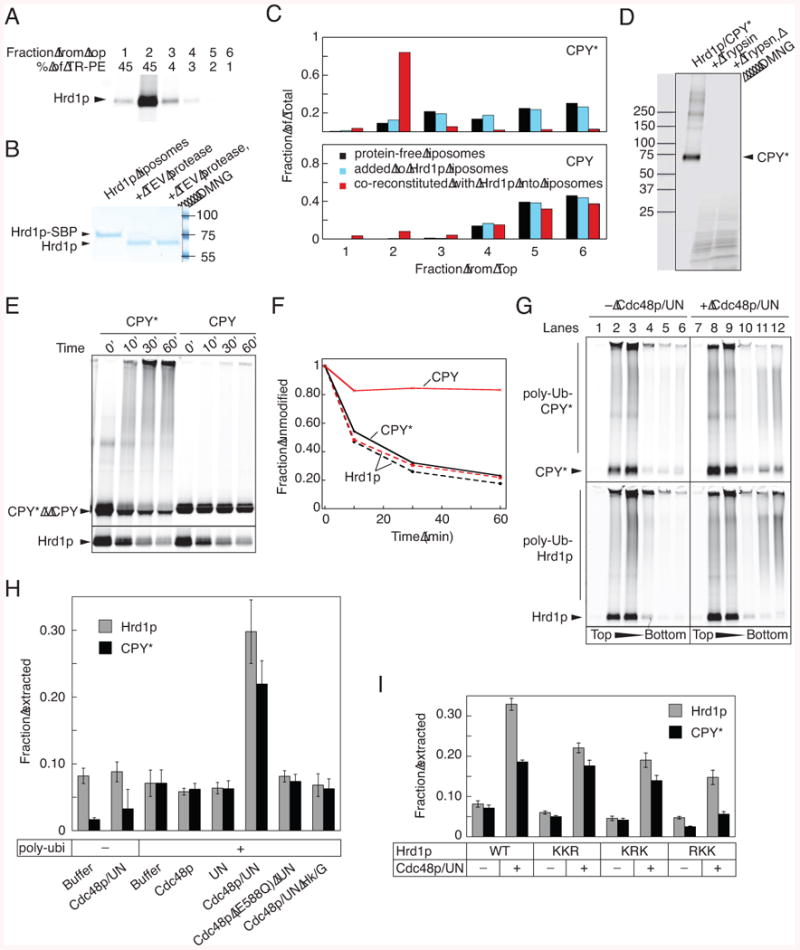
In vivo experiments suggested that Hrd1p is the only membrane protein required for a basic ERAD-L process (Carvalho et al., 2010). We therefore tested whether purified Hrd1p directly interacts with purified misfolded proteins. We used a well-characterized ERAD-L substrate, misfolded pro-carboxypeptidase Y (CPY*), which differs from the native protein by a single point mutation (Finger et al., 1993). His-tagged CPY* was purified in urea after expression in S. cerevisiae, but it remained soluble after removal of urea (Figure S1A, available online). To facilitate detection, CPY* was labeled with a fluorescent dye (Figure S1B).
Hrd1p with a C-terminal streptavidin-binding peptide (SBP) tag (Hrd1p-SBP) was purified from S. cerevisiae in the detergent decylmaltose neopentyl glycol (DMNG). Although Hrd1p appeared pure by SDS-PAGE (Figure S1C), it was heterogeneous in size by gel filtration (Figure S1D), consistent with it forming homo-oligomers in vivo (Carvalho et al., 2010).
For binding experiments, we mixed labeled CPY* with increasing concentrations of immobilized Hrd1p. Quantification of the bound and non-bound fractions gave an apparent affinity of ∼30 nM (Figures 1A, B). Wild type pro-CPY (CPY) did not bind to Hrd1p (Figure 1B; see Figures S1A and S1B for purified and labeled protein, respectively). Even when CPY was treated like CPY*, i.e. denatured in urea followed by removal of urea, only a small fraction was able to bind (Figure 1B). This suggests that Hrd1p can discriminate misfolded from folded polypeptides.
Figure 1. Substrate Interaction with Hrd1p.
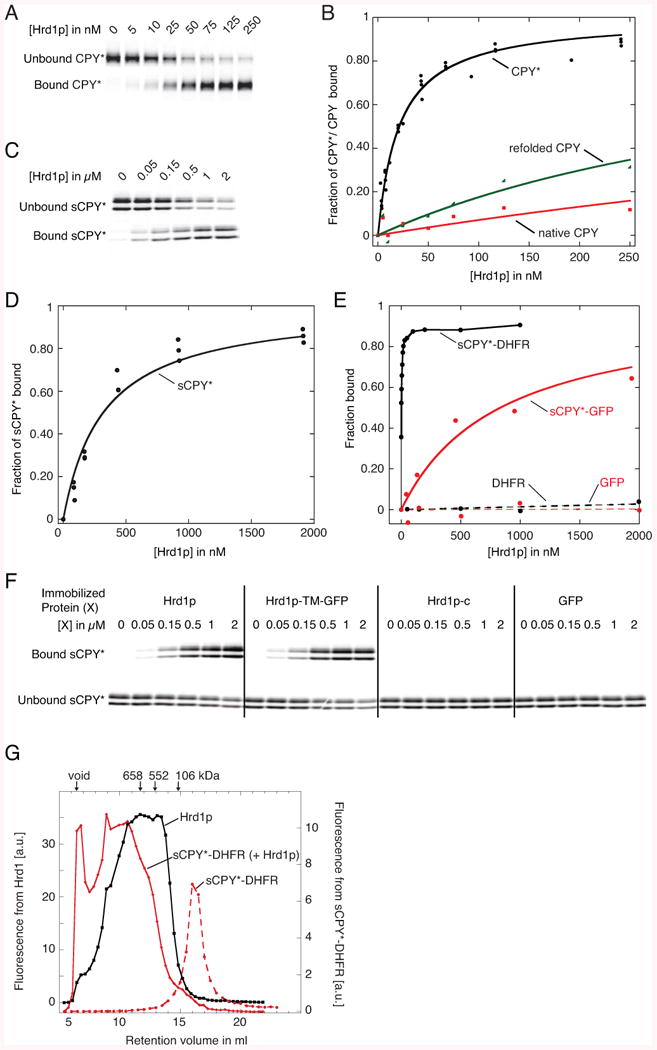
(A) Fluorescently labeled CPY* (10 nM) was incubated with increasing concentrations of bead-immobilized SBP-tagged Hrd1p (Hrd1p). The bound and unbound fractions were analyzed by SDS-PAGE and fluorescence scanning.
(B) Quantification of four different experiments as in (A). Fitting of the data points gives an apparent dissociation constant of 30 nM. Also shown are experiments with wild type CPY, either purified as a native protein or after unfolding and refolding, as done with CPY*.
(C) As in (A), but with sCPY* (100 nM).
(D) Quantification of three different experiments as in (C). The apparent dissociation constant is ∼300 nM.
(E) Quantification of binding experiments of wild type Hrd1p with fluorescently labeled sCPY*-DHFR (10 nM), sCPY*-GFP (100 nM), DHFR (100 nM), or GFP (100 nM).
(F) As in (C), but sCPY* was incubated with either wild type Hrd1p, a fusion of the TMs of Hrd1p with GFP (Hrd1p-TM-GFP), the cytoplasmic domain of Hrd1p (Hrd1p-c), or GFP.
(G) sCPY*-DHFR (200 nM) labeled with DyLight800 was incubated with a mixture of unlabeled Hrd1p (20 μM) and Hrd1p (200 nM) labeled with DyLight680. The sample was subjected to gel filtration in a buffer containing 120 μM DMNG and fractions were analyzed in two fluorescence channels. A control was performed with labeled sCPY*-DHFR alone. The arrows indicate the void volume, and the retention volume of size standards.
See also Figure S1.
Hrd1p also bound other misfolded CPY variants, such as a C-terminal fragment of CPY* (sCPY*), which is an ERAD-L substrate in vivo (Carvalho et al., 2010). sCPY* was purified as a mixture of glycosylated and non-glycosylated species (as shown by treatment with peptide N-glycosidase F (Figure S1A)), and was labeled with a fluorescent dye at a single C-terminal Cys-residue (Figure S1B). sCPY* bound to Hrd1p with significantly lower affinity than CPY* (Figure 1C, D; ∼300 nM). Glycosylated and non-glycosylated sCPY* bound equally well to Hrd1p, consistent with the fact that other ERAD components are required for their discrimination and with both species being substrates in Hrd1p-overexpressing cells (Denic et al., 2006). A fusion of sCPY* with dihydrofolate reductase (DHFR) bound more tightly than sCPY*, while a fusion with GFP (sCPY*-GFP) bound more weakly (Figure 1E; purity shown in Figures S1A and S1B). These differences correlate with the tendency of these proteins to aggregate; the concentration of urea required to keep these proteins in solution was lowest for sCPY*-GFP, intermediate for sCPY*, and highest for sCPY*-DHFR (data not shown). Purified GFP or DHFR alone, or bovine serum albumin, did not bind (Figure 1E; and data not shown; purified DHFR and GFP are shown in Figures S1 and S1B, respectively). Thus, Hrd1p binds selectively to unfolded polypeptides.
The membrane-embedded domain of Hrd1p is necessary and sufficient for substrate interaction, as the C-terminal cytoplasmic domain (Hrd1p-c) did not bind sCPY* (Figure 1F), and a fusion of the membrane-embedded domain of Hrd1p with GFP (Hrd1p-TM-GFP) bound substrate with the same affinity as wild type Hrd1p (Figure 1F; purity of the proteins shown in Figure S1C). A folded state of the TMs seems to be required for substrate interaction, as full-length Hrd1p was unstable in detergents other than DMNG, and this correlated with the loss of substrate binding (data not shown).
We used the high-affinity substrate sCPY*-DHFR to test whether substrate binds equally well to different oligomeric states of Hrd1p. Labeled sCPY*-DHFR alone behaved in gel filtration as a homogeneous, low-molecular weight species (Figure 1G), as the low concentration and the presence of detergent prevented its aggregation. When labeled sCPY*-DHFR was mixed with a 100-fold excess of Hrd1p, most substrate molecules migrated at very high molecular weight, where few Hrd1p molecules were found, and vice versa, the smallest-sized Hrd1p species did not contain bound substrate. Because sCPY*-DHFR is much smaller than the Hrd1p oligomers (∼40 kDa versus >250 kDa), the size shift of the substrate indicates that it preferentially binds to high-molecular weight Hrd1p oligomers.
Substrate poly-ubiquitination by Hrd1p
Next we tested whether Hrd1p poly-ubiquitinates bound substrate. To this end, we purified the ubiquitin-conjugating enzyme Ubc7p and its activator Cue1p (Figure S1C). Cue1p is a single-spanning membrane protein (Biederer et al., 1997), but in our experiments the full-length protein and a truncated version containing only the cytosolic domain (Cue1p-c) behaved identically (data not shown). When Hrd1p was incubated with fluorescently labeled CPY*, Ubc7p, Cue1p-c, purified Uba1p (Figure S1C), ubiquitin, and ATP, poly-ubiquitinated CPY* was generated (Figure 2A). The reaction rate depended mostly on the concentrations of Hrd1p and Ubc7p and was complete within 60 min. No poly-ubiquitination was observed if ATP, Uba1p, Ubc7p, or Hrd1p were omitted, or if an inactive Hrd1p mutant (Hrd1p C399S) was used (Bordallo and Wolf, 1999), and little modification was seen in the absence of Cue1p-c (Figure 2A; for purity of Hrd1p (C399S), see Figure S1C). Efficient poly-ubiquitination was also observed with sCPY* and sCPY*-DHFR (shown for sCPY* in Figure S2A). The experiments with sCPY* showed that the glycosylated and non-glycosylated species were equally modified. Most of the poly-ubiquitin chains on sCPY* and CPY* are linked through Lys48 in ubiquitin, as indicated by the much shorter chains generated with a Lys48Arg ubiquitin mutant (Figure S2B). When ubiquitin was replaced with methylated ubiquitin, which permits the attachment of a single ubiquitin molecule but prevents the synthesis of ubiquitin chains, several modified bands appeared, indicating that the substrates were modified at several different Lys-residues (Figure S2B).
Figure 2. Poly-ubiquitination by Hrd1p.
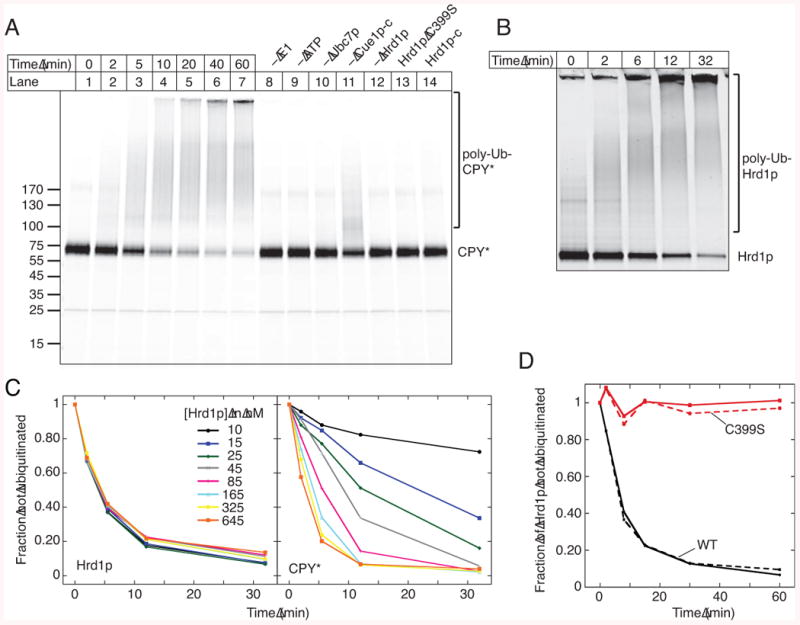
(A) Time-course of ubiquitination of CPY* labeled with DyLight800. Some reactions were analyzed after 60 min with the indicated components omitted. Where indicated, wild type Hrd1p (100 nM) was replaced with 100 nM of an inactive Hrd1 p mutant (C399S) or 1 μM of the cytoplasmic domain of Hrd1 p (Hrd1p-c).
(B) Time-course of auto-ubiquitination of Hrd1p labeled with DyLight680.
(C) The time-course of auto- and substrate-ubiquitination was determined in parallel. The concentration of labeled Hrd1p was kept constant, while that of unlabeled Hrd1p was varied.
(D) The time-course of auto-ubiquitination was determined with 50 nM of labeled wild type (WT) Hrd1p or C399S mutant in the absence or presence of a 10-fold excess of unlabeled WT Hrd1p (solid and broken curves, respectively).
See also Figure S2.
A purified cytoplasmic fragment of Hrd1p (Hrd1p-c) did not ubiquitinate CPY* (Figure 2A, lane 14) or sCPY* (Figure S2A, lane 12), even when added at 10-fold higher concentrations, although it could generate poly-ubiquitin chains (Figure S2C). Thus, substrate needs to bind to the membrane-embedded domain of Hrd1p to become poly-ubiquitinated.
Auto-ubiquitination of Hrd1p
Hrd1p itself was also poly-ubiquitinated. To study Hrd1p auto-ubiquitination in more detail, we attached by sortase labeling a fluorescent dye to the C-terminus of Hrd1p, allowing the simultaneous detection of Hrd1p and substrate. Hrd1p poly-ubiquitination was very efficient (Figure 2B). Experiments with methylated ubiquitin showed that modification of Hrd1p occurs at several different Lys-residues and that Hrd1p molecules often contain two ubiquitin chains (Figure S2C, last lane). Mass spectrometry confirmed the modification of several Lys residues (positions 126, 143, 282, 325, 387, 407, 511, 518, 539, 540, 546), but replacement of single Lys residues with Arg did not drastically reduce auto-ubiquitination (data not shown). Because a Hrd1p mutant in which all 27 Lys residues were changed to Arg did not express, we generated mutants, in which Lys residues were replaced in the trans-membrane domain (residues 1-301), the RING finger domain (302-407), or the C-terminal tail (408-551) (RKK, KRK, and KKR mutants, respectively). The RKK had significantly reduced levels of both auto- and substrate-ubiquitination, whereas the KKR mutant was specifically affected in auto-ubiquitination (Figure S2D). The KRK mutant showed an intermediate phenotype. In none of the mutants, auto-ubiquitination was completely abolished. The cytoplasmic fragment of Hrd1p (Hrd1p-c) showed little or no auto-ubiquitination; much of Hrd1p-c remained unmodified, and no modified protein was seen with methylated ubiquitin (Figure S2C). Thus, the trans-membrane domain is required for efficient auto-ubiquitination.
Hrd1p seems to modify itself by an intra-molecular reaction, since the rate of Hrd1p poly-ubiquitination was independent of the Hrd1p concentration, in contrast to substrate poly-ubiquitination (Figure 2C). Furthermore, when wild type Hrd1p was mixed with an inactive Hrd1p mutant (Hrd1p C399S), only the wild type, and not the mutant, was modified (Figure 2D), even though the two proteins bind each other (Figure S2E).
We found no conditions where only substrate ubiquitination was observed. For example, reducing the concentrations of Ubc7p or Cue1p-c, did not favor substrate- over auto-modification (Figure S2F). Auto-ubiquitination was observed even under conditions where Hrd1p was saturated with substrate, although the rate was somewhat reduced (Figure S2G). Thus, auto-ubiquitination of Hrd1p appears to be an integral part of the poly-ubiquitination reaction.
Poly-ubiquitinated Hrd1p recruits the Cdc48p ATPase complex
Next we tested whether Hrd1p poly-ubiquitination leads to recruitment of the Cdc48p ATPase complex. We first separately purified hexameric Cdc48p and a complex of the heterodimeric cofactor Ufd1p/Npl4p from E. coli (Figure S1C). The individual components assembled into the Cdc48p complex, as shown by gel filtration (Figure S3A). Next, we treated beads containing poly-ubiquitinated Hrd1p with Cdc48p in the presence or absence of Ufd1p/Npl4p. Binding of Cdc48p was observed in the presence of the cofactor at low or physiological salt concentrations (Figure 3A, lane 4 versus 1, and lane 9 versus 6), but not at high salt concentrations (lane 14). Ufd1p/Npl4p alone also bound to ubiquitinated Hrd1p (lanes 2 and 7), indicating that the cofactor is responsible for the recruitment of the Cdc48p complex. When Hrd1p was not pre-incubated with the ubiquitination machinery, Cdc48p binding was drastically reduced (Figure 3C); the residual binding is likely due to purified Hrd1p carrying some ubiquitin chains (shown by mass spectroscopy).
Figure 3. Recruitment of Cdc48p to Ubiquitinated Hrd1p.
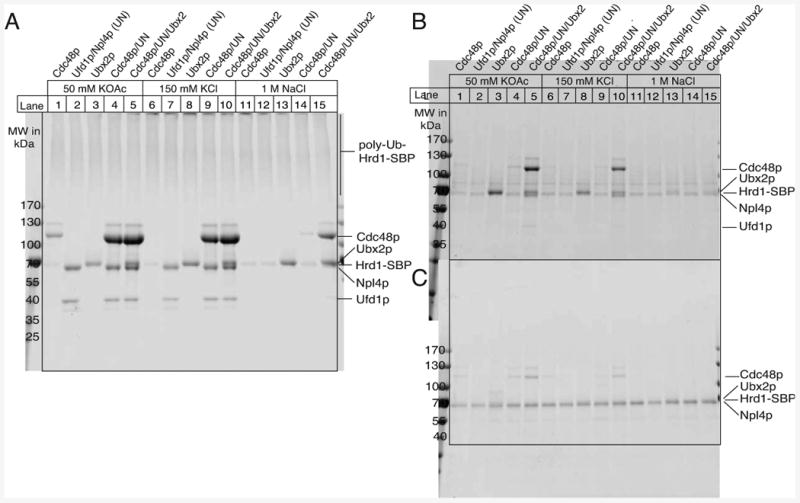
(A) Bead-immobilized Hrd1p (1 μM) was ubiquitinated and incubated at different salt conditions with 2 μM of the indicated components (UN, Ufd1p/Npl4p) in the presence of 250 μM ATPγS. The bound material was analyzed by SDS-PAGE and staining with IRDye Blue.
(B) As in (A), but bead-immobilized Hrd1p was modified with methylated ubiquitin.
(C) As in (A), but with non-ubiquitinated Hrd1p.
See also Figure S3.
We also tested the role of Ubx2p, a protein that interacts with Cdc48p and ubiquitin and is involved in ERAD (Neuber et al., 2005; Schuberth and Buchberger, 2005). Full-length Ubx2p with an N-terminal His-tag was purified in the detergent DMNG by Ni-NTA chromatography, followed by gel filtration (Figure S1C). Ubx2p bound to ubiquitinated Hrd1p (Figure 3A, lanes 3, 8, 13), but not unmodified Hrd1p (Figure 3C). Ubx2p increased the binding affinity of the Cdc48p complex for poly-ubiquitinated Hrd1p, as indicated by the resistance of the interaction to high salt concentrations (Figure 3A, lane 15). In contrast to Cdc48p complex alone, a complex with Ubx2p also bound to mono-ubiquitinated Hrd1p generated with methylated ubiquitin (Figure 3B, lanes 5 and 10). Even in the presence of Ubx2p, the Ufd1p/Npl4p complex was essential for the recruitment of Cdc48p (not shown), suggesting that Ubx2p interacts only weakly with Cdc48p itself. Indeed, gel filtration showed an increased association of Ubx2p's UBX domain (Ubx2p-c) with Cdc48p in the presence of Ufd1p/Npl4p (Figure S3A and B; see Figure S1C for purity of the protein). A similar hierarchy has been observed with another UBX domain containing protein (Hanzelmann et al., 2011). Taken together, our results show that poly-ubiquitinated Hrd1p recruits the Cdc48p complex. Ubx2p stimulates or stabilizes the association, but is not essential, consistent with in vivo data (Neuber et al., 2005; Schuberth and Buchberger, 2005).
Cdc48p-dependent substrate release from Hrd1p
Next we tested whether Cdc48p can release substrate bound to Hrd1p. We first formed a complex between bead-immobilized SBP-tagged Hrd1p and fluorescently labeled CPY*, and then incubated the beads with the ubiquitination machinery. After removal of the ubiquitination machinery, the beads were incubated with Cdc48p in the presence Ufd1p/Npl4p and ATP. About 50% of poly-ubiquitinated CPY* was released from the beads (Figure 4A, lane 6; quantification in Figure 4B). No release above background was seen if ATP was depleted with hexokinase/glucose (Hk/G) (Figure 4A, lane 7), or if Cdc48p or Ufd1/Npl4p were omitted (lanes 4 and 5). Similar results were obtained with sCPY* (Figures S4A,B).
Figure 4. Substrate Release from Hrd1p by Cdc48p ATPase.
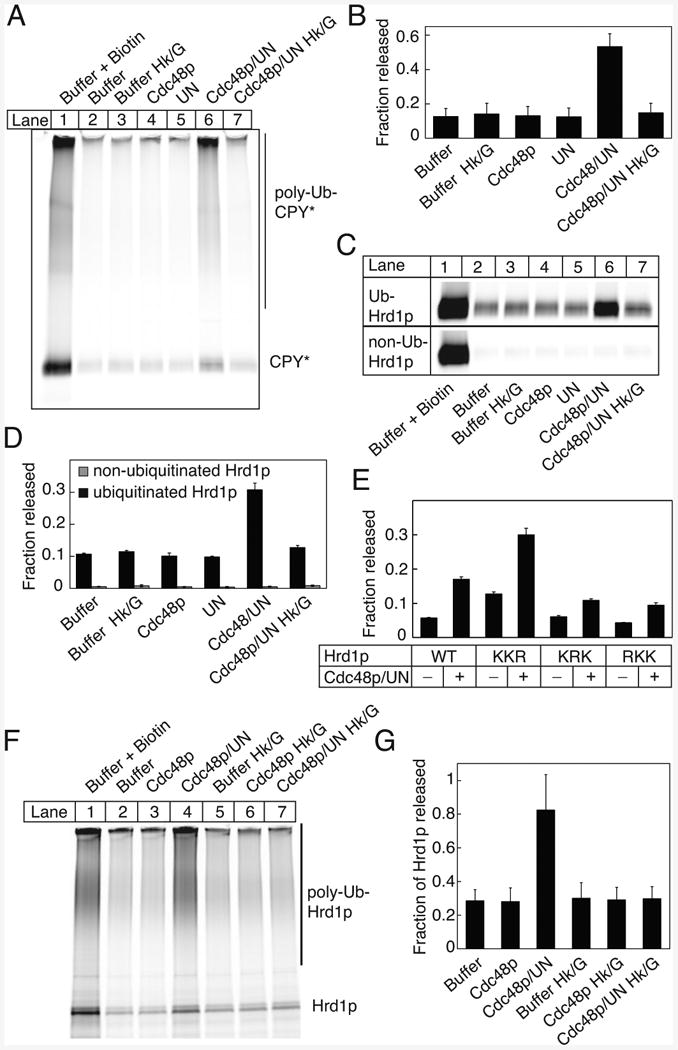
(A) A bead-immobilized complex of 500 nM SBP-tagged Hrd1p and 100 nM fluorescently labeled CPY* was incubated with the ubiquitination machinery. The beads were washed and incubated with 100 nM Cdc48p and ATP in the absence or presence of 100 nM Ufd1/Npl4p (UN) complex, or with UN alone. Where indicated, ATP was depleted with hexokinase/glucose (Hk/G). The released material was analyzed by SDS-PAGE and fluorescence scanning. The total releasable amount of CPY* was determined by incubating the beads with biotin.
(B) Quantification (means and standard deviations) of released poly-ubiquitinated CPY* determined from four experiments as shown in (A). The released fraction is expressed relative to the total releasable material.
(C) Bead-immobilized SBP-tagged Hrd1p was ubiquitinated (Ub-Hrd1p) for 1 h. The beads were washed, incubated with fluorescently labeled CPY*, and treated as in (A). The lower panel shows the same experiment with non-ubiquitinated Hrd1p (non-Ub-Hrd1p).
(D) Quantification (means and standard deviations) of released non-ubiquitinated CPY* determined from four experiments as shown in (C) (released from Ub-Hrd1p, black columns; released from non-Ub-Hrd1p, grey columns).
(E) Immobilized Hrd1p mutants with Lys to Arg mutations in three different regions were ubiquitinated for 30 min, and Cdc48p-dependent release of unmodified CPY* was tested as in (C). Shown are means and standard deviation of three experiments.
(F) Bead-immobilized Hrd1p was incubated with untagged, fluorescently labeled Hrd1p. After ubiquitination and washing, the beads were incubated with the indicated components. The material released from the beads was analyzed by SDS-PAGE and fluorescence scanning.
(G) Quantification of three experiments performed as in (E) (means and standard deviations).
See also Figure S4.
We noticed that non-ubiquitinated CPY* and sCPY* were also released from Hrd1p by Cdc48p activity (Figures 4A and S4A). This was confirmed in experiments in which CPY* was added after the ubiquitination reaction, so that only Hrd1p was modified (Figure 4C; quantification in Figure 4D). Some CPY* was released from the beads without Cdc48p action, likely because ubiquitinated Hrd1p is partially dissociating from the beads. No release was seen for substrate bound to non-ubiquitinated Hrd1p (Figures 4C,D). Similar results were obtained with sCPY* and sCPY*-DHFR (Figures S4C,D), although more spontaneous release was seen with sCPY*, consistent with its reduced binding affinity for Hrd1p. Together, these data indicate that the Cdc48p ATPase complex is first recruited to poly-ubiquitinated Hrd1p and then uses ATP hydrolysis to release both poly-ubiquitinated and non-ubiquitinated substrate from Hrd1p.
To test whether auto-ubiquitination at a specific site is required for the release of non-ubiquitinated substrate, we employed Lys-mutants of Hrd1p (Figure 4E). The release was significantly reduced with the RKK and KRK mutants, but was even stimulated with the KKR mutant. Thus, ubiquitin chains attached to either one of the N-terminal regions, but not the C-terminal tail, are required for efficient release of non-modified substrate from Hrd1p.
We next tested whether substrate release from Hrd1p is mediated by the dissociation of Hrd1p oligomers. Streptavidin beads were incubated with a mixture of Hrd1p-SBP and fluorescently labeled Hrd1p, so that labeled Hrd1p was bound to the beads through Hrd1p-SBP. After poly-ubiquitination, the addition of Cdc48p, Ufd1p/Npl4p, and ATP led to the dissociation of ∼80% of labeled poly-ubiquitinated Hrd1p (Figure 4F, lane 4; Figure 4G). Significantly less Hrd1p was released when ATP was depleted with Hk/G, or in the absence of Ufd1/Npl4p (Figures 4F, lanes 7 and 3; Figure 4G). These results indicate that the Cdc48p ATPase causes the dissociation of poly-ubiquitinated Hrd1p oligomers. As monomers bind substrate more weakly (Figure 1G), this may explain the release of substrate from Hrd1p, particularly the release of non-ubiquitinated substrate, which itself cannot interact with the Cdc48p complex. Cdc48p complex added directly to the ubiquitination reaction reduced substrate modification (Figure S4E), consistent with increased substrate release. Auto-ubiquitination was not affected (Figure S4E), as expected from an intra-molecular reaction (Figure 2D).
Involvement of the DUB enzyme Otu1p in ERAD
Since the DUB enzyme Yod1p had been implicated in ERAD-L (Ernst et al., 2009; Rumpf and Jentsch, 2006), we next investigated the role of its yeast homolog Otu1p. Expression of an inactive Otu1p mutant (Otu1p C120S), but not wild type Otu1p, strongly inhibited the degradation of the ERAD-L substrate sCPY*-DHFR in S. cerevisiae cells (Figures 5A,B). The Hrd1p levels were not greatly affected (Figure S5A). The degradation of the ERAD-C substrate Erg1p was also inhibited (Figure 5C,D). Although Otu1p C120S expression inhibited cell growth (Figure S5B), the effect on ERAD occurred in viable cells, as demonstrated by the degradation of the cytosolic proteasome substrate Deg1-LacZ (Figures 5C,D; and data not shown). As Deg1-LacZ degradation does not depend on Cdc48p (Ravid et al., 2006), the Otu1p mutant seems to affect only Cdc48p-dependent substrates. Indeed, deletion of the Cdc48p-interacting Ubx domain from Otu1p C120S greatly reduced the inhibition (Figures 5A,B). Otu1p does not seem to be the only DUB involved in ERAD, as a yeast strain lacking Otu1p did not show ERAD-L defects (Figures 5E,F).
Figure 5. Cdc48p-dependent function of Otu1p in vivo.
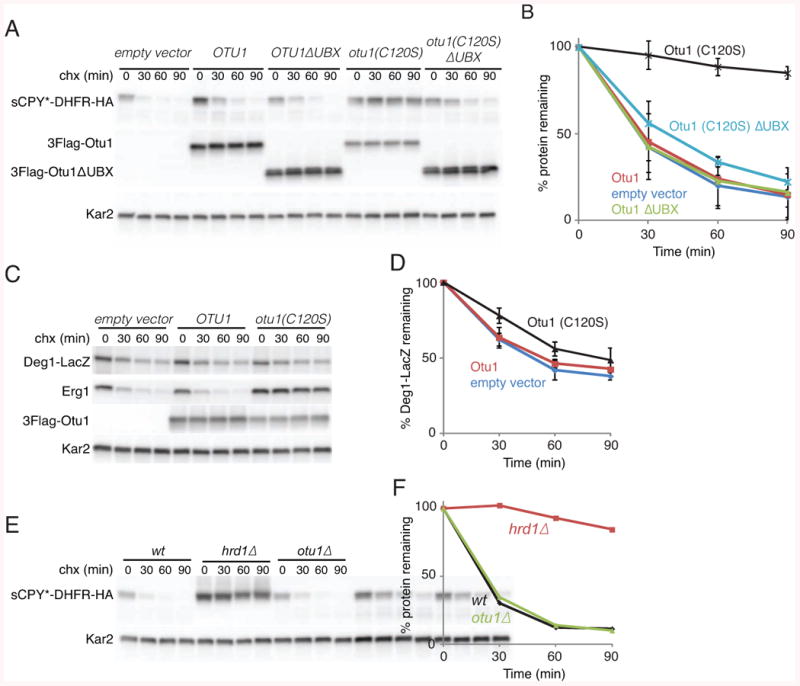
(A) The degradation of a fusion of sCPY* with DHFR and a hemagglutinin (HA) tag (sCPY*-DHFR-HA) was tested in S. cerevisiae. The cells were transformed with an empty vector or plasmids expressing FLAG-tagged wild type or mutant Otu1p (Otu1p (C120S)) from a Gal1 promoter. Where indicated, Otu1p variants lacking their Ubx domains were expressed instead. The samples were analyzed at different time points after addition of cycloheximide (chx) by SDS-PAGE and immunoblotting with anti-HA and anti-FLAG antibodies. Loading controls were performed with Kar2p antibodies.
(B) Quantification of two experiments as in (A) (means and standard deviations).
(C) As in (A), but following simultaneously the degradation of Erg1p and Deg1-LacZ with antibodies to the endogenous protein and to LacZ, respectively.
(D) Quantification of three experiments as in (C) (means and standard deviations).
(E) The degradation of sCPY*-DHFR-HA was analyzed in cells lacking Otu1p and wild type (wt) cells. Cells lacking Hrd1p were analyzed in parallel.
(F) Quantification of the experiment in (E).
See also Figure S5.
To analyze Otu1p in vitro, we expressed a His-tagged version and purified it by Ni-affinity chromatography and gel filtration (Figure S1C). Otu1p de-ubiquitinated modified fluorescently labeled Hrd1p or CPY* efficiently only when Cdc48p and Ufd1/Npl4p were present (Figure 6A-C). Much less de-ubiquitination was observed with Otu1p and Cdc48p alone, even though they interact with one another, consistent with the slow reaction previously observed (Rumpf and Jentsch, 2006). Addition of Ufd1p/Npl4p alone significantly accelerated Otu1p action (Figures 6A,B), in contrast to another ubiquitin-binding protein (Rad23p) (data not shown). As expected, the Otu1p C120S mutant was inactive (Figure 6B). ATP hydrolysis by Cdc48p stimulated de-ubiquitination by Otu1p, as shown with an ATPase-deficient Cdc48p mutant (E588Q mutation) and by ATP-depletion with Hk/G (Figure 6B). Otu1p needs to bind to the Cdc48p complex, as deletion of the Ubx domain drastically reduced DUB activity (Figure 6B). Similar results were obtained with poly-ubiquitinated Hrd1p containing fluorescently labeled ubiquitin, instead of labeled Hrd1p (Figures S6A,B). Otu1p removes short ubiquitin chains, rather than individual ubiquitin molecules, and it does not completely de-ubiquitinate Hrd1p.
Figure 6. Cdc48p-dependent in vitro de-ubiquitination by Otu1p.
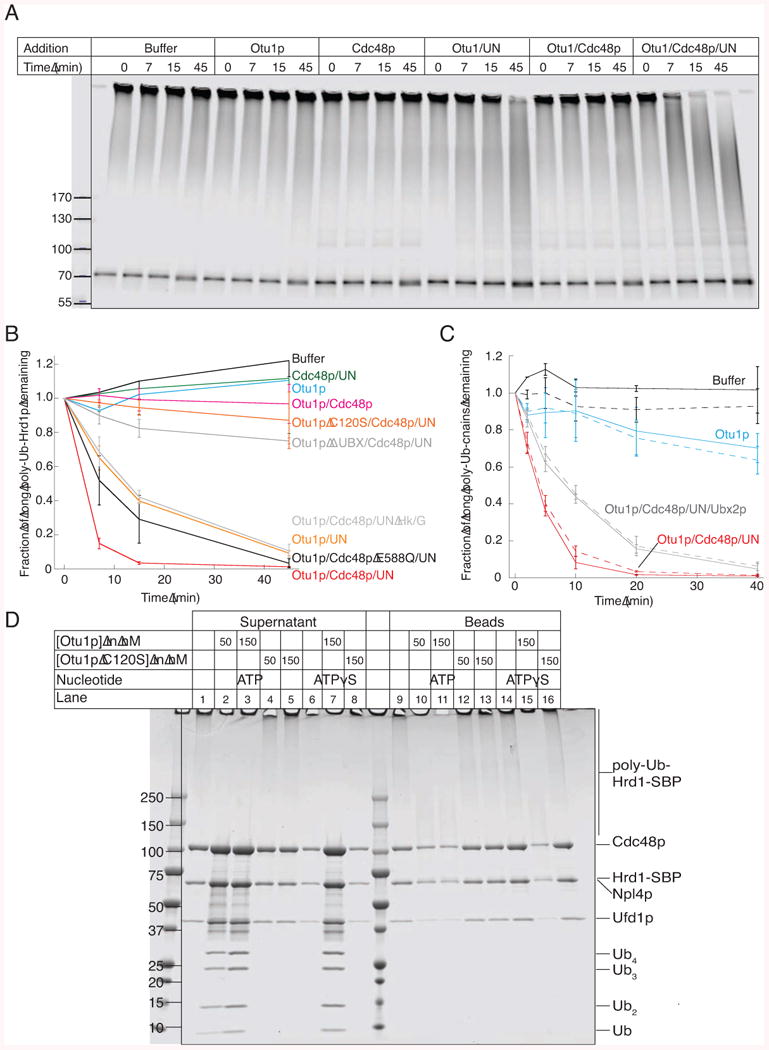
(A) Bead-immobilized fluorescently labeled Hrd1p-SBP was incubated with the ubiquitination machinery. After washing, Hrd1p was eluted from the beads with biotin and incubated with the indicated components (Ufd1/Npl4p; UN) for different time periods in the presence of ATP. Hrd1p was in a 30-fold excess over Otu1p, whereas all other components were about equimolar to Hrd1p. The samples were analyzed by SDS-PAGE and fluorescence-scanning.
(B) Quantification of experiments performed as in (A). The disappearance of the longest ubiquitin chains was quantified under different conditions (means and standard deviation of three experiments). ATP was depleted with hexokinase/glucose (Hk/G). Where indicated, an ATPase-defective Cdc48p mutant (Cdc48p E588Q) or an Otu1p mutant lacking the Ubx domain were used.
(C) Bead-immobilized complexes of Hrd1p-SBP and CPY*, labeled with different fluorescent dyes, were treated as in (A). The de-ubiquitination of modified Hrd1p and CPY* was followed in parallel (solid and broken lines, respectively). Shown are the means and standard deviations of three experiments.
(D) Bead-immobilized ubiquitinated Hrd1-SBP was incubated with Cdc48p complex in the presence of ATPγS. After washing, the beads were incubated for 1 h with the indicated components. Supernatants and beads were analyzed by SDS-PAGE and Coomassie staining.
See also Figure S6.
We found that Otu1p inhibited Cdc48p-dependent substrate-release by only ∼50%, even at high concentrations (data not shown). Thus, Cdc48p often functions before Otu1p had a chance to make the ubiquitin chains too short. On the other hand, experiments with bead-immobilized poly-ubiquitinated Hrd1p showed that extended incubation with Otu1p causes most of the Cdc48p complex to eventually dissociate, both in the absence or presence of ATP hydrolysis (Figure 6D; see supernatants in lanes 7 and 3 versus 6 and 1). The inactive Otu1p C120S mutant had no effect. These data suggest that Cdc48p function generally precedes Otu1p-mediated trimming of the ubiquitin chains. Interestingly, Ubx2p inhibited Cdc48p-dependent de-ubiquitination (Figure 6C), suggesting that in vivo it could help to prevent the premature function of Otu1p at the membrane.
Cdc48p-dependent protein extraction from reconstituted proteoliposomes
Finally, we tested whether the ERAD-L reactions would also occur with Hrd1p reconstituted into proteoliposomes. Proteoliposomes were generated by detergent removal from a mixture of purified Hrd1p and synthetic phospholipids, both in DMNG. We also added a fluorescently labeled lipid (Texas-Red phosphatidyl ethanolamine; TR-PE) prior to detergent removal. The reconstituted vesicles, including labeled Hrd1p and lipid, floated in a Nycodenz-step gradient (Figure 7A). The majority of Hrd1p was found in the second fraction from the top, while much of the lipid floated all the way to the top, indicating that some vesicles contain no protein and others Hrd1p oligomers. Most Hrd1p molecules have their cytoplasmic domain exposed to the outside of the vesicles, as demonstrated by the accessibility of a C-terminal TEV cleavage site to the TEV protease (Figure 7B). When labeled Hrd1p-containing proteoliposomes or protein-free liposomes were mixed with fluorescently labeled CPY*, only ∼10% of substrate floated with the proteoliposomes (Figure 7C; blue and black columns). Thus, substrate binds only weakly to the cytoplasmic side of membrane-incorporated Hrd1p. Next we co-reconstituted labeled CPY* and Hrd1p into vesicles. Flotation experiments showed that essentially all substrate co-migrated with the reconstituted vesicles (Figure 7C; red columns). The efficiency of co-flotation correlated with the binding affinity of substrate for Hrd1p in detergent; ∼40% were found for sCPY* (not shown), whereas wild type CPY did not float at all (Figure 7C). Addition of trypsin to the proteoliposomes showed that labeled CPY* was not protected by the lipid bilayer (Figure 7D), indicating that the substrate is bound to the transmembrane domain of Hrd1p with segment(s) exposed to the outside of the vesicles.
Figure 7. Membrane Extraction of Poly-ubiquitinated Proteins by Cdc48p.
(A) Proteoliposomes containing fluorescently labeled Hrd1p-SBP and Texas-red labeled phosphatidyl ethanolamine (TR-PE) were subjected to flotation in a Nycodenz gradient. Fractions were collected from the top, and analyzed by SDS-PAGE and fluorescence scanning of the gel. The lipid content of the fractions was determined by absorbance at 590 nm.
(B) Proteoliposomes containing Hrd1p-SBP with a TEV-cleavage site at the C-terminus were treated with TEV protease in the absence or presence of DMNG. The samples were analyzed by SDS-PAGE and staining with Coomassie blue.
(C) Fluorescently labeled CPY* was incubated with either protein-free liposomes or proteoliposomes containing Hrd1p, or it was co-reconstituted with Hrd1p into vesicles (black, light blue and red bars, respectively). The samples were subjected to flotation in a Nycodenz gradient and fractions were analyzed by SDS-PAGE and fluorescence scanning. Wild type CPY was used as a control.
(D) Proteoliposomes containing Hrd1p and labeled CPY* were incubated with trypsin in the absence or presence of DMNG. The samples were analyzed by SDS-PAGE and fluorescence scanning.
(E) Proteoliposomes were generated by co-reconstitution of Hrd1p with either CPY* or wild type CPY. Hrd1p and substrate were labeled with different fluorophores. The vesicles were incubated with the ubiquitination machinery for different time periods, and samples were analyzed by SDS-PAGE and fluorescence scanning. For Hrd1p, the gel was cropped to only show the disappearance of unmodified protein.
(F) The disappearance of unmodified protein in (D) was quantitated. Solid and broken lines show the modification of substrate and Hrd1p, respectively.
(G) Fluorescently labeled Hrd1p and CPY* were co-reconstituted into proteoliposomes. The vesicles were incubated with the ubiquitination machinery, followed by incubation in the absence or presence of the Cdc48p complex (Cdc48p/UN). The vesicles were floated in a Nycodenz gradient and fractions analyzed by SDS-PAGE and fluorescence scanning.
(H) Experiments as in (G) were quantified by determining the total fluorescence in the bottom two fractions (material released from the vesicles) as a fraction of the total fluorescence in the gradient (mean and standard deviation of at least three experiments). Where indicated, ATP was depleted with hexokinase/glucose (Hk/G) or an ATPase-deficient Cdc48p mutant (Cdc48p E588Q) was used.
(I) As in (G), but with Hrd1p mutants carrying Lys to Arg mutations in three different regions. Quantification of three experiments was done as in (H).
See also Figure S7.
Next we incubated proteoliposomes containing labeled Hrd1p and substrate with the ubiquitination machinery. About 80% of both Hrd1p and CPY* were poly-ubiquitinated (Figure 7E,F). Wild type CPY remained largely unmodifed. About 90% of poly-ubquitinated Hrd1p and CPY* floated with the vesicles in a Nycodenz gradient (Figure 7G; lanes 1-6), indicating that poly-ubiquitination alone does not extract proteins from the membrane. Ubiquitination increased the density of the vesicles. Unmodified Hrd1p and CPY* also shifted their position in the gradient, consistent with the observation that the vesicles contain multiple Hrd1p molecules, some of which become poly-ubiquitinated.
Finally, we tested whether poly-ubiquitinated Hrd1p- and CPY*-molecules are extracted from the membrane by the Cdc48p complex. Proteoliposomes containing fluorescently labeled Hrd1p and CPY* were incubated with the ubiquitination machinery, followed by the addition of Cdc48p and Ufd1p/Npl4p. The sample was then subjected to flotation in a Nycodenz gradient. Approximately 20-25% of CPY* and up to 35% of Hrd1p were found in the bottom fractions (Figures 7G, lanes 7-12; quantification in Figure 7H), indicating that they were no longer associated with the liposomes. Longer incubation times or higher Cdc48p complex concentrations did not increase the efficiency of membrane extraction (Figure S7A,B). No membrane extraction was observed in the absence of ubiquitination, when Cdc48p or Ufd1p/Npl4p were omitted, when an ATPase-defective Cdc48p mutant was used, or when ATP was depleted with Hk/G (Figure 7H). The extracted Hrd1p and CPY* proteins carried relatively short ubiquitin chains (Figure 7G); most of the longer chains remained in the floated fractions, suggesting that they are poor substrates for the Cdc48p complex.
Otu1p addition inhibited the membrane extraction of poly-ubiquitinated proteins to a maximum of ∼50% (Figures S7C,D), similar to the effect of Otu1p on substrate release from Hrd1p in detergent (data not shown). The inactive Otu1p C120S mutant had no effect on membrane extraction, even when added in a 10-fold excess over Cdc48p (Figure S7D), suggesting that its effect in vivo (Figures 5A,B) is caused at a step following substrate release into the cytosol.
All three Hrd1p mutants with Lys mutations showed reduced extraction of poly-ubiquitinated Hrd1p from the membrane (Figure 7I), consistent with their lowered level of modification (Figure S2D). The site of modification does not seem to be important, in contrast to the release of unmodified substrate from poly-ubiquitinated Hrd1 in detergent (Figure 4E). Although less pronounced, the site of modification also affected the extraction of poly-ubiquitinated substrate from Hrd1p-containing proteoliposomes, as the RKK mutant had a significantly stronger effect on substrate- than Hrd1p- extraction (Figure 7I). The KKR mutant was not affected in membrane extraction of the substrate (Figure 7I), indicating that modification of the C-terminal tail is not required for substrate release from Hrd1p, and suggesting that substrate is not obligatorily extracted as a complex with Hrd1p.
Discussion
We have reproduced key steps of ERAD-L with purified protein components, both in detergent and in reconstituted proteoliposomes. Our minimal in vitro system mimics essential aspects of ERAD-L in vivo when Hrd1p is overexpressed (basic ERAD-L). In both systems, glycosylated and non-glycosylated misfolded proteins serve as substrates for Hrd1p, and Hrd1p itself is poly-ubiquitinated and extracted from the membrane. Since Hrd1p appears to be the only membrane component required for a basic ERAD-L reaction in vivo, and because Hrd1p is sufficient for the binding and membrane extraction of misfolded proteins in vitro, it is likely to form a channel for the transport of misfolded proteins through the ER membrane.
Our in vitro system allowed us to break down the basic ERAD-L process into individual steps. First, Hrd1p binds unfolded polypeptides through its membrane-spanning domain. Next, both Hrd1p and substrate are poly-ubiquitinated, resulting in the recruitment of the Cdc48p ATPase complex, a process mediated by the cofactor Ufd1p/Npl4p and facilitated by the adaptor protein Ubx2p. Then, the poly-ubiquitinated proteins are extracted from the membrane by the Cdc48p complex in an ATP hydrolysis-dependent reaction. Finally, Otu1p trims the poly-ubiquitin chains in a Cdc48p complex-dependent manner, resulting in the dissociation of the Cdc48p complex from substrate.
In basic ERAD-L, substrates are exclusively selected by Hrd1p. We show that Hrd1p in detergent discriminates folded from unfolded polypeptides. Hrd1p uses its hydrophobic TM segments to bind unfolded polypeptide segments. Oligomeric Hrd1p may provide more TMs for substrate interaction than monomeric Hrd1p, explaining why it has a higher affinity. Hrd1p does not interact with all hydrophobic polypeptide segments, since it does not bind Ubx2p (Figure S3C) or a fragment of Usa1p lacking the N-terminal cytoplasmic interaction domain (data not shown). Perhaps, a loosely folded polypeptide structure is also required for substrate recognition. It seems likely that several TMs in Hrd1p interact with an unfolded polypeptide chain because mutations scattered throughout the membrane-embedded domain of Hrd1p affect different substrates to varying degrees (Sato et al., 2009). Although some substrate interaction may be caused simply by the hydrophobicity of the TMs, the membrane-embedded domain of Hrd1p must have unique properties, as it needs to be folded, and only little substrate interaction is seen with purified Usa1p containing two TM segments (data not shown).
Insertion of a polypeptide loop into the Hrd1p channel could be the first step in the actual retro-translocation process. Such a model would be analogous to loop insertion of a signal sequence-containing polypeptide into the Sec61/SecY channel during forward translocation (Park and Rapoport, 2012). In both cases, a substrate segment would reach the other side of the membrane and the binding of a hydrophobic region to the channel would provide the driving force for polypeptide chain insertion. Our experiments show that a misfolded substrate co-reconstituted with Hrd1p indeed exposes a segment to the outside of vesicles. As expected, substrate does not bind to Hrd1p from the cytoplasmic side when added after reconstitution. However, it remains to be shown that reconstituted Hrd1p can bind substrate on the luminal side of the membrane, as suggested by experiments in intact yeast cells overexpressing Hrd1p (Carvalho et al., 2010). A system in which the starting proteoliposomes contain only Hrd1p or substrate and are subsequently fused will also address whether actual retro-translocation can be reproduced with the purified components.
Similarly to forward translocation, a polypeptide chain may be able to slide back and forth in a Hrd1p channel, but there has to be energy input to achieve net movement into the cytosol. One possibility is that the attachment of poly-ubiquitin chains to the substrate would bias polypeptide sliding, providing a ratcheting mechanism for translocation. However, poly-ubiquitination alone is insufficient to completely move a polypeptide chain from the ER membrane into the cytosol, as shown both in vivo (Flierman et al., 2003) and by our in vitro experiments; this requires the function of the Cdc48p complex as well. In fact, our data indicate that the Cdc48p complex is sufficient to move poly-ubiquitinated proteins into the cytosol, although other cytosolic factors could be stimulatory. Current models assume that the Cdc48p-binding poly-ubiquitin chains need to be attached to the substrate, but our data raise the possibility that the crucial modification is on Hrd1p itself. We show that Cdc48p releases unmodified substrate from poly-ubiquitinated Hrd1p in detergent, probably by disassembling Hrd1p oligomers into smaller assemblies that have a lower affinity for substrate. Indeed, for Cdc48p's segregase activity, it is not necessary that the ubiquitin chains are attached to the protein extracted from the membrane, as shown for the generation of the transcription factors Mga2p and Spt23p. Here, Cdc48p action releases an unmodified polypeptide (the p90 fragment) into the cytosol by acting on poly-ubiquitin chains attached to an associated membrane-anchored protein (the p120 precursor) (Shcherbik and Haines, 2007). Auto-ubiquitination of Hrd1p followed by Cdc48p function could provide the driving force for retro-translocation by allowing substrate segments to move through the membrane by multiple rounds of binding to and release from Hrd1p. Such a model does not exclude a role for substrate ubiquitination, which could determine directionality of polypeptide movement by a ratcheting mechanism or by interaction with the Cdc48p complex.
A model in which auto-ubiquitination of Hrd1p is a crucial modification event would be consistent with studies that showed that substrates, in which all Lys residues are removed, continue to be degraded; in these cases, the ubiquitination machinery was still required, suggesting that a protein other than the substrate is ubiquitinated (Hassink et al., 2006; Wang et al., 2013; Yu and Kopito, 1999). There are also ERAD-L substrates, such as prepro-a-factor and cholera toxin, which are never ubiquitinated and whose retro-translocation can be blocked with a dominant-negative DUB (Bernardi et al., 2013), again indicating ubiquitination of another component. Protease-protection studies showed that ubiquitination is required to expose a substrate segment to the cytosol (Jarosch et al., 2002), and photo-crosslinking demonstrated that the ubiquitination activity of Hrd1p and Cdc48p action are needed for an early Hrd1p-substrate interaction on the luminal side of the membrane (Carvalho et al., 2010). These results suggest that ubiquitination by Hrd1 p of a component other than substrate is crucial for ERAD-L.
Although a role for auto-ubiquitination of Hrd1p is attractive, it is difficult to exclude that at least some Hrd1 p modification is the result of a non-specific side reaction that has been observed with other ligases. Indeed, we found that ubiquitination of the C-terminal tail of Hrd1p has no effect on substrate modification and release. There is also little evidence that Hrd1p is poly-ubiquitinated in wild type yeast cells. It is therefore possible that Hrd1p modification and Cdc48p-dependent extraction serve to regulate Hrd1p levels in the membrane. However, because auto-ubiquitination occurs in an intramolecular reaction, our results argue that Hrd1p molecules do not recognize each other as unfolded ERAD-M substrates. Regardless of whether auto-ubiquitination is required for ERAD or as a regulatory mechanism, it is minimized in wild type cells.
Our in vitro results faithfully recapitulate what is observed in vivo in Hrd1p-overexpressing yeast cells. However, they do not recapitulate substrate selection as seen in wild type cells, where the additional ERAD components Yos9p, Hrd3p, and Der1p ensure that only genuine substrates are degraded (Denic et al., 2006; Gauss et al., 2006; Mehnert et al., 2013; Xie and Ng, 2010). Our results indicate that recognition of an unfolded polypeptide by Hrd1p is the last checkpoint before substrate is committed to retro-translocation and degradation. This may explain why bypassing all upstream steps in Hrd1p-overexpressing cells slows, but does not prevent, cell growth. In addition, Hrd1p may provide the main checkpoint for non-glycosylated ERAD substrates. The additional ERAD components may also minimize excessive auto-ubiquitination and membrane extraction of Hrd1p. Hrd3p has a particularly important role, as in its absence, Hrd1p is poly-ubiquitinated and degraded (Gardner et al., 2000; Plemper et al., 1999). Perhaps, Hrd3p is regulating auto-ubiquitination of Hrd1p in response to substrate binding.
Our results support the idea that DUBs, specifically Otu1p, play a role in ERAD. We show that Otu1p is only activated after recruitment by the Cdc48p complex. This ensures that poly-ubiquitination, Cdc48p recruitment, and de-ubiquitination occur in a sequential manner. Our data suggest that Cdc48p-mediated membrane extraction precedes Otu1p function, so that most substrate de-ubiquitination occurs in the cytosol. Ubx2p could help to prevent premature de-ubiquitination at the membrane by competing with Otu1p for Cdc48p binding. The kinetic delay would guarantee that Otu1p does not interfere with Cdc48p's function as a segregase. The sequential action of Cdc48p and Otu1p would be further enhanced by the ubiquitin ligase Hrd1p counteracting de-ubiquitination at the membrane, but not in the cytosol. A function of Otu1p downstream of Cdc48p's segregase activity explains why overexpression of wild type Otu1p does not affect ERAD in vivo. The inactive Otu1p mutant would inhibit by interacting with poly-ubiquitin chains bound to the Cdc48p complex, preventing the dissociation of Ufd1p/Npl4p, and blocking the access of other DUBs. Indeed, expression of the equivalent Yod1p mutant in mammalian cells leads to the accumulation of ubiquitinated substrate bound to the ATPase complex (Ernst et al., 2009). The inactive Otu1p mutant does not inhibit membrane extraction in vitro, because there is probably no need for recycling of the Cdc48p complex. Otu1p/Yod1p probably function not only in ERAD, but also in other processes involving the Cdc48p/Ufd1p/Npl4p complex.
Finally, our results suggest that ERAD-L proceeds through an intermediate in which the substrate is bound to the membrane-embedded domain of Hrd1p, a situation that resembles the recognition of an ERAD-M substrate. Thus, ERAD-M substrates may enter the same process at a later stage, explaining why the same ubiquitin ligase (Hrd1p) is involved, and why ERAD-M requires only a subset of the ERAD-L components. Many other ubiquitin ligases, such as Doa10p involved in ERAD-C, are also multi-spanning membrane proteins. As proposed previously for Doa10p (Swanson et al., 2001), the membrane-spanning domains may serve as conduits for polypeptides through the membrane.
Experimental Procedures
Details of protein purifications and experimental procedures are described in Extended Experimental Procedures.
Protein Expression and Purification
All proteins are from S. cerevisiae. Uba1p, Ubx2p, and the substrates CPY*, sCPY*, sCPY*-DHFR, and CPY, as well as Hrd1p and its variants were expressed in S. cerevisiae under the Gal1 promoter. All other proteins were expressed in E. coli strain BL21 DE3 RIPL. The membrane proteins Ubx2p and Hrd1p and its variants were solubilized in DMNG. ERAD substrates were extracted from a crude membrane fraction with urea. Hrd1p and its variants were first purified by streptavidin affinity chromatography utilizing a C-terminal streptavidin-binding peptide (SBP) tag, followed by gel filtration. All other proteins were purified as His-tagged variants by Ni-affinity chromatography, followed by ion exchange chromatography and/or gel filtration. Unless noted otherwise, the tags were proteolytically removed.
Labeling with Fluorescent Dyes
The substrates sCPY*, sCPY*-DHFR, sCPY*-GFP, DHFR, and GFP were labeled at a C-terminally attached Cys residue with DyLight 800 maleimide. CPY* and CPY were labeled with a N-hydroxy-succinimidyl ester of DyLight 800. Hrd1p was labeled using the sortase technique.
Ubiquitination Assays
All ubiquitination assays were performed at 30°C. Unless indicated otherwise, the concentrations of the components of the ubiquitination machinery were: 100 nM Uba1p, 1 μM Ubc7p, 1μM Cue1p-c, 100 nM Hrd1p, 100 nM substrate, 100 μM ubiquitin, 2.5 mM ATP.
Binding and Release Experiments
All binding experiments were performed at room temperature with Hrd1p immobilized on magnetic streptavidin beads via its C-terminal SBP-tag. Unbound and bound fluorescent material was analyzed by SDS-PAGE and fluorescence scanning of the gel in an Odyssey scanner (Li-COR). Binding of substrates or Cdc48p to ubiquitinated Hrd1p was tested by first immobilizing Hrd1p-SBP on beads at room temperature for 1h and then adding the ubiquitination machinery.
Reconstitution into Proteoliposomes and Density Gradients
For reconstitution of Hrd1p, protein-free liposomes (final lipid concentration 10 mM, containing 0.5 mol% Texas Red labeled phosphatidyl ethanolamine) were mixed with DMNG (15 mM), Hrd1p (2 μM) and, optionally, with substrate (0.5 μM), and incubated for 1h at room temperature. This mixture was applied to detergent-removal spin columns (Pierce).
In vivo ERAD-Substrate Degradation Experiments
Cycloheximide shutoff experiments were performed essentially as described (Carvalho et al., 2010).
Supplementary Material
Figure S1. Characterization of the Purified ERAD Components and Substrates (Related to Figure 1)
Figure S2. Ubiquitination of Substrate and Hrd1p (Related to Figure 2)
Figure S3. Recruitment of Cdc48p to Ubiquitinated Hrd1p (Related to Figure 3)
Figure S4. Release of Substrate from Hrd1p by Cdc48p (Related to Figure 4)
Figure S5. Otu1p function in vivo (Related to Figure 5).
Figure S6. Cdc48p-dependent in vitro de-ubiquitination by Otu1p (Related to Figure 6).
Figure S7. Membrane Extraction of Poly-ubiquitinated Proteins by Cdc48p (Related to Figure 7).
Highlights.
- The ubiquitin ligase Hrd1p discriminates between folded and unfolded proteins
- Hrd1p and the Cdc48p ATPase complex cooperate to extract proteins from the membrane
- The de-ubiquitinating enzyme Otu1p is recruited and activated by the Cdc48p complex
- The results suggest a model for protein retro-translocation through the ER membrane
Acknowledgments
We thank Robert Oliete for preliminary experiments, Ryan Baldridge, Alexandra Boye-Doe, Angelyn Larkin, Nicholas Bodnar, and Stefan Schoebel for help with some in vitro experiments, and Randy King, Ryan Baldridge, and Adrian Salic for critical reading of the manuscript. A.S. is supported by an Otto Hahn Fellowship of the Max Planck Society, A.R. by a “La Caixa” graduate fellowship, and T.A.R. by an NIH grant (GM052586). T.A.R. is a Howard Hughes Medical Institute Investigator.
Footnotes
Supplemental Information: Supplemental information includes Extended Experimental Procedures and seven figures, and can be found with this article online at…
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Alexander Stein, Email: alexander_stein@hms.harvard.edu.
Tom A. Rapoport, Email: tom_rapoport@hms.harvard.edu.
References
- Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001a;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001b;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Inoue T, Schultz A, Tsai B. A deubiquitinase negatively regulates retro-translocation of nonubiquitinated substrates. Mol Biol Cell. 2013;24:3545–3556. doi: 10.1091/mbc.E13-06-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo J, Wolf DH. A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae. FEBS Lett. 1999;448:244–248. doi: 10.1016/s0014-5793(99)00362-2. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger A, Knop M, Wolf DH. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur J Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- Flierman D, Ye Y, Dai M, Chau V, Rapoport TA. Polyubiquitin serves as a recognition signal, rather than a ratcheting molecule, during retrotranslocation of proteins across the endoplasmic reticulum membrane. J Biol Chem. 2003;278:34774–34782. doi: 10.1074/jbc.M303360200. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann P, Buchberger A, Schindelin H. Hierarchical binding of cofactors to the AAA ATPase p97. Structure. 2011;19:833–843. doi: 10.1016/j.str.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Hassink GC, Barel MT, Van Voorden SB, Kikkert M, Wiertz EJ. Ubiquitination of MHC class I heavy chains is essential for dislocation by human cytomegalovirus-encoded US2 but not US11. J Biol Chem. 2006;281:30063–30071. doi: 10.1074/jbc.M602248200. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ye Y. Roles of p97-associated deubiquitinases in protein quality control at the endoplasmic reticulum. Curr Protein Pept Sci. 2012;13:436–446. doi: 10.2174/138920312802430608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M, Sommer T, Jarosch E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat Cell Biol. 2013 doi: 10.1038/ncb2882. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Wang Y, Warren G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002;21:5645–5652. doi: 10.1093/emboj/cdf579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annual review of biophysics. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci. 1999;112(Pt 22):4123–4134. doi: 10.1242/jcs.112.22.4123. [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- Shcherbik N, Haines DS. Cdc48p(Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol Cell. 2007;25:385–397. doi: 10.1016/j.molcel.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yu YY, Myers N, Hansen TH. Decoupling the role of ubiquitination for the dislocation versus degradation of major histocompatibility complex (MHC) class I proteins during endoplasmic reticulum-associated degradation (ERAD) J Biol Chem. 2013;288:23295–23306. doi: 10.1074/jbc.M113.482018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Ng DT. ERAD substrate recognition in budding yeast. Semin Cell Dev Biol. 2010;21:533–539. doi: 10.1016/j.semcdb.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Yu H, Kopito RR. The role of multiubiquitination in dislocation and degradation of the alpha subunit of the T cell antigen receptor. J Biol Chem. 1999;274:36852–36858. doi: 10.1074/jbc.274.52.36852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of the Purified ERAD Components and Substrates (Related to Figure 1)
Figure S2. Ubiquitination of Substrate and Hrd1p (Related to Figure 2)
Figure S3. Recruitment of Cdc48p to Ubiquitinated Hrd1p (Related to Figure 3)
Figure S4. Release of Substrate from Hrd1p by Cdc48p (Related to Figure 4)
Figure S5. Otu1p function in vivo (Related to Figure 5).
Figure S6. Cdc48p-dependent in vitro de-ubiquitination by Otu1p (Related to Figure 6).
Figure S7. Membrane Extraction of Poly-ubiquitinated Proteins by Cdc48p (Related to Figure 7).