

Abstract
Complex Cu-GGHYrFK-amide (1-Cu) was previously reported as a novel metallotherapeutic that catalytically inactivates stem loop IIb of the Hepatitis C Virus (HCV) Internal Ribosomal Entry Site (IRES) RNA and demonstrates significant antiviral activity in a cellular HCV replicon assay. Herein are described additional studies focused on understanding the cleavage mechanism, as well as the relationship of catalyst configuration to structural recognition and site-selective cleavage of the structured RNA motif. These are advanced by use of a combination of MALDI-TOF mass spectrometry, melting temperature determination, and computational analysis to develop a structural model for binding and reactivity toward SLIIb of the IRES RNA. In addition, the binding, reactivity, and structural chemistry of the all d-amino acid form of this metallopeptide, complex 2-Cu, is reported and compared to complex 1-Cu. In vitro RNA binding and cleavage assays for complex 2-Cu show a KD of 76 ± 3 nM, and Michaelis-Menten parameters of kcat of 0.14 ± 0.01 min−1 and KM of 7.9 ± 1.2 µM, with a turnover number exceeding 40. In a luciferase-based cellular replicon assay Cu-GGhyrfk-amide shows activity similar to the parent peptide, complex 1-Cu, with IC50 of 1.9 ± 0.4 µM and cytotoxicity exceeding 100 µM. RT-PCR experiments confirm a significant reduction in HCV RNA levels in replicon assays for up to nine days when treated with complex 1-Cu in three day dosing increments. This study shows the influence that the α-carbon stereocenter has for this the new class of compounds, while detailed mass spectrometry and computational analysis provide new insights into the mechanisms of recognition, binding, and reactivity.
Keywords: HCV IRES RNA, copper ATCUN motif, RNA cleavage, stem-loop, catalytic, metallodrug
INTRODUCTION
Catalytic metallodrugs represent a new class of compounds with the potential to achieve high therapeutic activity with low toxicity.[1–2] Incorporation of a reactive metal-binding domain allows for the irreversible inactivation of a target protein or RNA and the potential for multiple turnover to irreversibly modify and inactivate many copies of a target, resulting in a therapeutic effect at lower dosage.[3] Previous work has shown that the tetrapeptide sequence YrFK-amide (where the arginine is in the d-conformation) binds to stem loop IIb (SLIIb) of the Internal Ribosomal Entry Site (IRES) of Hepatitis C Virus (HCV) RNA[4] and that incorporation of a metal-binding ATCUN[5] (amino terminal copper and nickel binding, XXH, where X ≠ P) motif results in catalytic degradation of the target RNA under oxidative conditions.[4] The SLIIb domain of IRES RNA has been shown to be important for the initiation of translation for HCV-derived proteins and represents a promising target for therapeutic intervention.[6–7] In fact, the activity of the copper-peptide complex Cu-GGHYrFK-amide (1-Cu) was demonstrated in a cellular replicon assay and exhibited additive to slightly synergic activity when given in combination with recombinant interferon alpha-2b,[4] one of the current treatments for HCV infection.
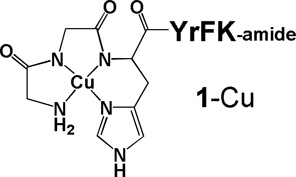
The incorporation of d-amino acids into a peptide sequence is a common approach to improve the stability of a peptide toward digestion by proteases, a recurrent problem for the use of peptides in vivo.[8–9] It also addresses a fundamentally interesting question regarding the impact of stereochemical inversion on binding and reactivity for the all d-form stereoisomer (Gly excluded), relative to the parent peptide, and provides additional insights into the mechanism of action. Stereochemical change at the α-carbon alters the conformation of the peptide and therefore potentially affects its ability to interact with its target. A complex set of factors, including binding affinity, off-rate for metallopeptide release from the inactivated target, orientation of the metal relative to the site of reaction, reduction potential of the metal cofactor, and cellular uptake and stability, will determine the efficacy of the metallopeptide complex. If conversion of the amino acids to the d-form negatively impacts any one of these factors it could be detrimental to its effectiveness as a therapeutic. However, there is extensive precedent for retention of bioactivity for proteins and peptides following conversion to the d-amino acid form. Examples include HIV-1 protease where the d-form of the enzyme retained activity but with inverted specificity,[10] a peptide derived from the N-terminus of HIV-1 gp41 retained activity in the opposite configuration,[11] while the all-d form of the Rev peptide showed enhanced binding to its RRE RNA target[12], in addition to the d-configuration form of channel-forming peptides that are used as antibiotics.[13] In the case of an amyloid beta assembly inhibitor peptide, the observed activity also increased.[14] The binding of the YrFK-amide targeting peptide to the SLIIb of the HCV IRES was only recently reported and there is no structural information available on the mode of binding.[4] This current report investigates the impact, following conversion of the peptide sequence GGHYrFK-amide to the corresponding sequence containing all d-amino acids, GGhyrfk-amide (2), on binding as well as solution and cellular reactivity. Computational analysis provides a model for how this class of peptides interacts with and promotes cleavage of its target RNA, which is supported by mass spectrometric analysis of product fragments and RNA melting profiles. Mass spectrometric experiments also support specific primary mechanisms for RNA cleavage by this class of metallodrug.
RESULTS
Metallopeptide Binding and Thermal Melts
It has been shown that the isolated stem loop IIb domain of the HCV IRES maintains the structure adopted in the full length RNA and is a useful probe for in vitro assays.[15] Binding and cleavage assays were performed using 5'-fluorescein labeled SLIIb. A dissociation constant (KD) of 76 nM for GGhyrfk-amide binding to SLIIb RNA was determined by monitoring the shift in the tyrosine emission of the peptide as a function of RNA concentration under the conditions shown in Figure SM1. Next, the melting temperature of SLIIb was determined in the presence and absence of peptides and the results are shown in Figure 1. Melting profiles were fit to a three state consecutive model (with an initial melt, TM1, of the lower stem; and a subsequent melt of the upper stem, TM2 as illustrated in Figure 1 (right)). Figure 1 (left) also shows fits for conditions of RNA alone, and in the presence of 50 µM GGHYrFK-amide or 50 µM GGhyrfk-amide. To further compare the binding profile for the two forms of the peptide, the salt dependence of thermal melts with and without the addition of 50 µM peptide was studied (Figure 2). At high to intermediate salt concentrations, no noticeable shift in the melting temperature was observed. At lower salt concentrations, however, both of the peptides show an increase in TM1 on the order of 2 to 4 K, but only the d-amino acid form experienced a significant shift in TM2 of up to 18 K. Error limits are typically greater for TM1, relative to TM2 because the fitting is more variable for the first phase.
Figure 1.

(Left) Variation of fluorescein emission as a function of temperature in 20 mM HEPES, pH 7.4, with 1 mM NaCl. SLIIb alone (open triangles, TM1 = 304.6 ± 0.8, TM2 = 329.3 ± 0.3), SLIIb plus 50 µM GGHYrFK-amide (1) (black squares, TM1 = 325.3 ± 3.8, TM2 = 330.8 ± 0.4), SLIIb plus 50 µM GGhyrfk-amide (2) (open circles, TM1 = 317.8 ± 5.9, TM2 = 332.3 ± 0.9). Fits are shown to a three state consecutive melt model, with the proposed melts corresponding to TM1 and then TM2 (model shown on the right).
Figure 2.

Variation of shifts in IRES SLIIb melting temperature (ΔTM1 and ΔTM2) in the presence of peptides, relative to the SLIIb RNA alone (indicated by open triangles (top) or the black line (bottom)), SLIIb RNA and 50 µM GGHYrFK-amide (1) (black squares), and SLIIb RNA and 50 µM GGhyrfk-amide (2) (open circles). [RNA] = 1 µM; [peptide] = 50 µM, [HEPES] = 20 mM, pH = 7.4.
RNA Cleavage Reactivity
The reactivity of the metal complexes was determined by following the degradation of 5’-fluorescein labeled SLIIb by time-dependent fluorescence spectrophotometry. Initial velocities were plotted as a function of catalyst concentration to generate a pseudo-Michaelis-Menten plot to generate KM and kcat values (Figure 3). A turnover number was estimated based on the limiting amount of peptide catalyst consumed by a specific amount of RNA. The turnover number for the all d-amino acid metallopeptide, complex 2-Cu, could not be directly determined because it exceeded the ratio of catalyst:RNA that was practical to use, but the turnover number was greater than 40 compared to the previously determined value of ~ 32 determined for complex 1-Cu.
Figure 3.

Pseudo Michaelis-Menten profile for reactivity of Cu-GGhyrfk-amide with fluorescein-labeled SLIIb RNA. [RNA] = 1 µM, [H2O2] = 1 mM, [ascorbic acid] = 1 mM, [HEPES] = 20 mM, [NaCl] = 100 mM, pH = 7.4. KM = 7.9 µM, Vmax = 0.14 µM RNA/min, kcat = 0.14 min−1.
MALDI-TOF Mass Spectrometry
MALDI-TOF analysis of RNA cleavage products is complicated by the presence of a large number of fragmentation peaks that are present even in the absence of catalyst (Figure 4, left), and has been reported for other systems.[16] The identification of cleavage products was determined by considering only those peaks that exhibited time dependence and were not observed in controls of either RNA alone, RNA in the presence of the coreagents (ascorbate and H2O2), or in the absence of catalyst. By use of these criteria it can be seen in Figure 4 (right) that the major product peaks are centered on m/z = 5100 and m/z = 6400. The new peaks and their assignments are shown in Tables SM1 and SM2. Figure 5 shows the sites of cleavage observed based on the time dependence of a position in the RNA for complexes 1-Cu and 2-Cu and how they map to the structure (based upon the data in Figures SM2 and SM3). This experiment reports the evaluation of initial velocities from the time-dependent change of mass spectrometric signal intensities and is intrinsically more error prone. However, the general trends are clear.
Figure 4.

Mass spectrometric analysis of RNA cleavage by copper-peptide complexes. RNA alone (dash lines), and after reaction with 1-Cu (left, black) or 2-Cu (right, black). New product peaks are centered on m/z = 5100 and m/z = 6400.
Figure 5.

Summary of the rates of appearance of cleavage products at sites on SLIIb after reaction with 1-Cu (black squares) and 2-Cu (open circles).
In Silico - Dynamics and Docking
The Cu-ATCUN sequence containing the targeting domain was simulated to investigate the site of localization of the peptide/complex on the RNA. In all cases, the complex was made flexible except for aromatic carbons, peptide bonds, and the metal binding domain. The geometry around the metal was based on the X-ray crystal structure of Cu-GGH and was restricted to a square planar configuration.[17] As a result of the high number of flexible bonds already in the complex, the RNA was restricted to its initial state.
Figure 6 provides a summary of the results for docking of each of the complexes to the RNA. Complexes 1-Cu and 2-Cu do not bind at the same location. Cluster analysis was performed with an RMSD = 8 Å for the complex (Figure 6). Further analysis with a particular cluster was guided by positioning of the metal binding domain based on the experimental reaction data obtained from mass spectrometric (Table SM3, complex 1-Cu; Table SM4, complex 2-Cu).
Figure 6.

The metal ion, shown as a gray sphere, for each bound metallopeptide conformer following docking to the top conformation of the NMR structure of SLIIb in Autodock. Positions of the docked metal ion are shown for each conformation of the RNA-complexed metallopeptide 1-Cu (left) and the RNA-complexed metallopeptide 2-Cu (right). Included in the supplementary materials (Figure SM9) is a colored image which also includes the assignment of each of the metal ions to its nearest residue.
HCV Cellular Replicon Assay
The activity of metallodrug 2-Cu was evaluated in cell culture by use of a cellular HCV replicon assay that mimics the native HCV replication system as described previously. The results are shown in Table 1 and are compared to the data for the parent metallodrug, complex 1-Cu. The diastereomer of complex 1-Cu showed activity similar to the parent metallopeptide (IC50 = 1.9 µM for complex 2-Cu versus an IC50 = 0.6 µM for complex 1-Cu). As in the case of complex 1-Cu, no activity was observed for controls of the RNA binding domain alone, Cu-GGH alone, free copper, or the peptide without copper. Recombinant IFNα-2b was used as a positive control. The Cu-GGh complex was not tested, but is not expected to differ from the Cu-GGH response, because both GGH and GGh lack targeting domains and inversion of configuration of one stereocenter would not reasonably be expected to promote additional binding contacts or significantly change the intrinsic reactivity of the copper center.
Table 1.
HCV cellular replicon data for all d-amino acid analog of complex 1-Cu.
| Compound | Antiviral IC50 (µM) |
Cytotoxicity TC50 (µM) |
Selectivity Index (TI) |
|---|---|---|---|
| [GGHYrFK-amide]-Cu2+, 1-Cu | 0.6 ± 0.3 | > 100 | > 172 |
| [GGhyrfk-amide]-Cu2+, 2-Cu | 1.9 ± 0.4 | > 100 | >52 |
| YrFK-amide | > 100 | > 100 | na |
| yrfk-amide | > 100 | > 100 | na |
| [GGH]-Cu2+ | > 100 | > 100 | na |
| Cu2+(aq) | > 100 | > 100 | na |
na = not applicable
RT-PCR
To confirm the proposed mode of action (RNA degradation) for the metallopeptide in a cellular environment the disappearance of target RNA in cellular replicon assays by RT-PCR experiments was demonstrated. These were performed to look at RNA levels for both HCV RNA and rRNA (as a control) in the replicon assays. It can be seen in Figure 7 that there is a preference for HCV RNA as the normalized copies of HCV RNA approach zero after 9 days at the highest concentration used. A small reduction in levels of rRNA is also observed.
Figure 7.

RT-PCR results showing the preferential cleavage of HCV RNA (left) over ribosomal RNA (right) at increasing dosages of 1-Cu, where medium and complex are replenished at 3 day intervals. 0 µM (black square), 2.5 µM (white circle), 5 µM (black triangle), 10 µM (white diamond), 20 µM (black star). The graph on the left is normalized to the RNA levels in the absence of metallopeptide. A colored image is included in supplementary material (Figure SM10).
DISCUSSION
Binding and Thermal Melts
Sample melting profiles are shown in Figure 1 and melting temperatures were determined by measuring the change in fluorescence emission of fluorescein-labeled RNA as a function of temperature. The resulting melting profiles were fit to a three state consecutive model for melting of the SLIIb secondary structure, consistent with initial melting of the double stranded region at the 3’- and 5’-ends prior to melting of the double-stranded region near the upper loop (Figure 1, left). This model also provided the best and most consistent fit to the data. Other fits that were tried were a two state model and a three state nonconsecutive model. Figure 2 shows that addition of peptide to the RNA caused the melting temperature to increase, consistent with the peptide-RNA complex being more stable than free RNA, and is consistent with the observation that these assays had to be performed at lower salt concentrations in order to see a noticeable shift in melting temperature where higher salt concentrations compete with the peptide for binding to the negatively charged RNA. This suggests a mode of binding for 2, in particular, that is dominated by electrostatic interactions.
Clearly the relative change in melting temperatures does not directly correlate with relative KD‘s. In particular, larger changes in TM’s accrue from the weaker binding complex 2-Cu. However, this reflects the distinct binding modes for complexes complex 1-Cu and complex 2-Cu. It is generally accepted that interactions with the phosphate backbone stabilize base-paired RNA and in turn increase the TM, while interactions with bases tend to destabilize and cause a decrease. The trends observed for change in the TM are consistent with the binding locations supported by reactivity and modeling experiments. Complex 2-Cu is expected to bind in the central TM1 domain, whereas complex 1-Cu appears centered more in the TM2 region, which is a smaller region within the RNA with fewer base pairs and consequently does not require as much energy to promote unfolding. Also note the TM reflects base pair melting, and so metallopeptide binding could also stabilize base paired domains by reducing the degrees of freedom of the flanking domain.
The original peptide GGHYrFK-amide exhibits surprisingly tight binding (KD = 44 nM) for such a small peptide. Therefore, it might be expected that all of the amino acid residues are important for binding. While there is no current structure of the peptide bound to the IRES SLIIb, it would be expected that the lysine and the arginine make electrostatic and hydrogen bond contacts with the negatively-charged RNA backbone. The tyrosine, phenylalanine, and/or arginine could also be involved in stacking interactions with the bases. The peptide backbone would be flexible but tighter binding could also be achieved by preordering the side chains, for example by π-stacking between the tyrosine and phenylalanine of YrFK-amide (and/or arginine) or by hydrogen bonding between functional groups.
Following inversion of stereochemistry at the α-carbons, the relative positioning of the amino acid side chains changes and intramolecular interactions would still be possible but with differences in orientation, as noted in the case of d-Rev peptide binding to stem-loop IIb RRE RNA.[12] Only a modest change in KD is observed following conversion to the all d-amino acid peptide (44 ± 2 nM for 1 versus 76 ± 3 nM for 2), consistent with flexibility in the longer side chains of lysine and arginine that can more readily adjust their positions. This is also consistent with the effect of increasing [NaCl] on shifts in melting temperature for the IRES SLIIb in the presence and absence of the two peptides as shown in Figure 2. As expected, both TM1 and TM2 for RNA alone increase with increasing [NaCl] due to stabilization of the negatively charged phosphate backbone by sodium binding. The original peptide GGHYrFK-amide does not show a significant effect on TM2, and ΔTM2 ~ 1 K, presumably due to a balance of stabilizing electrostatic interactions and destabilizing interactions with the bases. If the binding of GGhyrfk-amide is more dominated by electrostatics, little or no shift would be observed at high [NaCl] due to the loss of electrostatic interactions with the target RNA and at lower [NaCl] there would be a stabilizing effect, consistent with the data shown in Figure 2. For both 1 and 2, there is an increase in TM1 at low salt concentrations (for 1 ΔTM1 ~ 2 to 4 K and for 2 ΔTM1 ~ 18 K). These differences are a reflection of differences in targeting of 1 versus 2, and is consistent with the dominance of electrostatic contacts of 2 compared to 1 or a reflection of binding differences with the RNA as discussed in the sites of reactivity section.
Reactivity
Reaction kinetics were monitored by following the change in fluorescein emission over the course of the reaction as previously described for complex 1-Cu. Initial velocities were plotted as a function of Cu-GGhyrfk-amide concentration to generate a pseudo Michaelis-Menten plot (Figure 3). A kcat value of 0.14 min−1 and a Km of 7.9 µM were determined (Table 2). The parent peptide 1 demonstrated a KD (44 nM), much lower than its KM (850 nM), which is consistent with a high affinity binding peptide where release of the RNA from the peptide catalyst is most likely rate determining. The previously reported data for complex 1-Cu was obtained under conditions of fixed catalyst concentration with varying [RNA], whereas complex 2-Cu showed inconsistent behavior at high [RNA] and so was studied under pseudo Michaelis-Menten conditions although the values are directly comparable. The kcat value for complex 1-Cu is slightly greater than the value obtained for complex 2-Cu (0.53 min−1 versus 0.14 min−1) and the KM is about an order of magnitude higher (0.85 ± 0.09 µM versus 7.9 ± 1.2 µM), consistent with different binding modes that affect the chemistry without significantly changing the overall affinity. For complex 1-Cu, it was estimated by comparing the KM and KD that k2 ~ 18 x k−1. In the case of complex 2-Cu, the k2 ~ 9 × k−1 and therefore k2 is also significant relative to k−1 and chemistry is fast relative to dissociation from the RNA. This is consistent with a system where catalyst release, not chemistry or initial binding, is determining the catalytic efficiency, kcat/KM, which is still significantly better for complex 1-Cu. However, kcat/KM is not the only parameter that is important for determining the activity of a catalytic metallodrug. A comparison of turnover numbers is also worthy of consideration. An exact turnover number could not be determined for complex 2-Cu except that it is greater than 40. This is higher than the turnover number of 32 previously reported for complex 1-Cu, and is consistent with the opportunity for more effective turnover with weaker binding. This reflects the complexity of factors that ultimately determine the efficacy of a catalytic metallodrug. A classical reversible inhibitor would show reduced efficacy with reduced binding, such as in the case of a mutation of the drug target that promotes resistance by the virus. The catalytic metallodrug approach, however, provides the possibility for the retention, or enhancement, of activity upon mutation of the target binding site. This emphasizes the fact that standard predictors for the efficacy of a drug, such as binding affinity, are not necessarily the best predictors for the efficacy of a catalytic metallodrug. As long as specificity can be achieved, the catalyst can still be effective. Along with the targeting domain, specificity is also achieved by a double filter mechanism where both binding and positioning of the metal binding domain must be optimized for a given target.
Table 2.
Michaelis-Menten parameters for degradation of SLIIb.
| Compound | kcat (min−1) |
KM (µM) |
kcat/KM (µM−1 min−1) |
KD* (nM) |
Turnover Number |
ref |
|---|---|---|---|---|---|---|
| 1-Cu | 0.53 ± 0.02 | 0.85 ± 0.09 | 0.62 | 44 ± 2 | 32 | [4] |
| 2-Cu | 0.14 ± 0.01 | 7.9 ± 1.2 | 0.018 | 76 ± 3 | > 40 | This work |
KD values listed are for the free peptide.
Sites of Reactivity
The mechanism and site of cleavage for complex 1-Cu was probed by MALDI-TOF mass spectrometry to map the sites of cleavage (Figures 5, SM2, SM3). RNA alone, in the absence of reaction with catalytic metallodrug, showed fragmentation during the MALDI process (Figure 4). This has been shown previously for other RNA systems. Cleavage products from reaction with complex 1-Cu were analyzed by considering those peaks that met strict requirements of having a signal to noise ratio greater than five and time dependence. These peaks were then compared to the predicted masses for expected products based on hydrolysis and known pathways for the oxidation of DNA. It should be noted that pathways for the oxidative cleavage of DNA are well characterized, but those for RNA oxidation are not.[18] Correlations to the pathways for DNA can be made, however, even if hydrogen abstraction is expected to be more difficult for RNA than for DNA. Oxidation of RNA is likely facilitated by the high oxidation state Cu3+ and formation of intermediate ROS, a characteristic of Cu-ATCUN complexes.[2, 19–24]
Reaction of complex 1-Cu with isolated SLIIb showed reaction products primarily centered on m/z = 5100 and m/z = 6400 (Figure 4, right). The assignments for major new peaks, along with errors in ppm, are shown in Table SM1. Overall trends show common residues for reaction (U14, A15, C18, A19, and G21) and these sites are mapped to the three dimensional structure of SLIIb and shown in Figure 8 (left). A recent paper performed selective 2’ hydroxyl acylation analyzed by primer extension (SHAPE) on the full length HCV IRES and showed high accessibility at U14 and A15, consistent with the reactivity observed.[25] Therefore, the observed preference for reaction in this area reflects both the placement and orientation of the metal ion, as well as the intrinsic reactivity and accessibility of these residues as shown in Figure SM5. Previous work by Kalliampakou et al. examined mutations of the apical region of SLIIb and showed that single substitutions at the proposed residues (U14, A15, C18, and A19)[26] cause dramatic reductions in IRES mediated translation, with a ~ 30–60% reduction for a single residue. This demonstrates the importance of these residues, but also suggests that activity for complex 1-Cu would be expected even if dissociation of the RNA strands does not occur after cleavage, and particularly in the context of the full length IRES and under physiological conditions where most of the hydrogen bonding interactions would remain intact. Clearly structural perturbations at the positions of those bases have a pronounced effect on translation, and cleavage at those positions would be expected to have a similar impact.
Figure 8.

Sites of reactivity based on MALDI assignments of fragments that show a time dependence in the presence of 1-Cu with SLIIb (left) and 2-Cu with SLIIb (right). The scale reflects the relative initial velocity for reaction at each position with time (per minute) as described in the experimental section. The darker a residue is the faster (black indicated the fastest) the observed rate was. A colored representation of the figure with values can be found in the supplementary materials (Figure SM11)
The sites where cleavage occurs for complex 2-Cu were further probed by MALDI-TOF mass spectrometry (Table SM2) as described for complex 1-Cu. Reactions of complex 2-Cu with SLIIb showed few similar reactive residues to those of complex 1-Cu and instead exhibited a wider distribution of sites of reactivity located away from the apical residues (Figure 8). As mentioned for complex 1-Cu, the proposed sites of cleavage at the top loop have been shown to be important for translation. This is consistent with a system that occasionally binds to one of these apical residues similar to complex 1-Cu, at A15, A19, and G21, but as a result of the different stereochemistry around the α-carbons complex 2-Cu prefers the residues between A6 and G9.
Reactions were also performed both in the presence and absence of coreagents. It can be seen from Figures SM6 and SM7 that the overall reactivity in the absence of coreagents is negligible compared to reactions with coreagents (Figure SM2 and SM3), consistent with a requirement for the coreagents in promoting the reactivity of the catalyst. Previous work has suggested that the ATCUN motif mediates cleavage through a metal-associated reactive oxygen species,[21] and the ability of the ATCUN motif to produce this reactive intermediate is enhanced in the presence of coreagents. An unexpected product from the 3’-overhang that is observed for the reaction of both complexes 1-Cu and 2-Cu is the 2’,3’-cyclic phosphate, in addition to the 3’-phosphate, and for the 5’-overhang the major product is the 5’OH. Given the absence of such a reaction product with complex 1-Cu alone, the former cyclic phosphate product is consistent with a hydrolytic mechanism (Figure 9) that proceeds through a high oxidation state copper center serving as a Lewis acid to promote deprotonation of the 2’-OH, as shown in Figure 9A. The rate of cyclic phosphate formation (Figure 10) is greater for complex 1-Cu versus complex 2-Cu (kobs 0.063 ± 0.017 min−1 for complex 1-Cu and kobs = 0.021 ± 0.003 min−1 for complex 2-Cu). These rates directly correlate with the observed differences in kcat values (0.53 ± 0.02 min−1 for complex 1-Cu vs. 0.14 ± 0.01 min−1 for complex 2-Cu) for each metallopeptide. Variability between kobs and kcat most likely reflect solution and methodology differences between the solution fluorescence assay and mass spectrometric detection.
Figure 9.

Under oxidative conditions, catalysts 1-Cu and 2-Cu can promote RNA cleavage chemistry either through “hydrolytic” (A) or “oxidative” (B) mechanisms promoted through the transient formation of higher valent Cu3+. The illustration shows initial intermediates proposed for RNA cleavage mechanisms. The requirement for redox coreagents to effect cleavage supports the requirement for Cu3+ formation and transient intermediates are shown that invoke either a role as Lewis acid in a classical hydrolysis mechanism involving a cyclic phosphate ester (A), where the Cu3+ promotes formation of an outer or inner sphere hydroxide that deprotonates the 2’-hydroxyl and/or stabilizes the build-up of negative charge density in the transition state of cyclic phosphate ester formation; or (B), by an oxidative pathway involving a copper-associated reactive oxygen species that is positioned to mediate hydrogen abstraction from the C4’ or C5’ hydrogens. For the analogous reactions for oxidative cleavage of DNA, both C4’-H or C5’-H hydrogen abstraction are most common although the oligonucleotide 3’-phosphate product can be observed for oxidative C–H scission at any ribose carbon.[29] For RNA, the oxidative reaction (B) is suggested to most likely occur by C4’-H or C5’-H hydrogen abstraction because these hydrogens are typically the most accessible and the pathways are less compromised by the presence of the C2’-OH.[28]
Figure 10.

(Left) The time dependent formation of 2’,3’-cyclic phosphates in the presence of 1-Cu (black squares) and 2-Cu (empty circles) as determined by MALDI-MS. (Right) The time-dependent formation of 3’-phosphates in the presence of 1-Cu (black squares) and 2-Cu (empty circles).
For complex 1-Cu the primary cleavage reaction appears to proceed via formation of the 2’,3’-cyclic phosphate product and the accompanying 5’-OH fragment (Figure SM8). Minor production of 3’-phosphate products can arise either from hydrolysis of the 2’, 3’-cyclic phosphates,[27] or by an alternative hydrogen abstraction pathway analogous to those for DNA. While 3’-phosphate formation can in principal occur as a result of hydrogen abstraction at any position on the ribose, for RNA hydrogen abstraction is expected to be most likely at C4’ or C5’ [28] and, when the observed product types are taken into account, the most likely hydrogen abstraction pathway involves C5’-H abstraction (Figure 9B) which generates a 3’-phosphate and 5’-aldehyde (the mass difference of the 5’aldehyde product compared to the 5’-OH product is only 2 amu and, at the high molecular weights under consideration, are essentially indistinguishable by MALDI-MS; consequently these products are not differentiated in Figure SM8). Because the C5’ hydrogens are located next to the phosphate ester, these hydrogens are more accessible to solvent and would also be more accessible to the copper ion of the complex. It should again be noted, however, that this hydrogen abstraction mechanism does not account for the production of 2’, 3’-cyclic phosphates, and so the “hydrolysis”-type mechanism is required and is a significant, most likely dominant component of the overall RNA cleavage reaction.
As an alternative to a direct hydrogen abstraction pathway, one can invoke 3’-phosphate formation entirely through hydrolysis of the 2’,3’-cyclic phosphate, especially in the presence of a powerful Lewis acid.[27] However, the absence of significant 3’-phosphate formation for reactions catalyzed by complex 1-Cu, relative to one third of the products derived from reaction mediated by complex 2-Cu, suggest independent reaction routes and varying rates of reaction for “hydrolysis” versus “oxidative” paths (Figure 9). Based upon the kobs values noted earlier, it would be expected that complex 1-Cu would generate 3’-phosphates at a higher rate than complex 2-Cu if this product were formed by rapid hydrolysis of cyclic phosphates - but this is not observed (Figure 10, right). Such variability in reactivity is expected because each catalyst will likely have a distinct orientation for the catalytic metal relative to scissile bonds on the RNA, and the intrinsic reactivities toward each available pathway (in this case hydrolysis versus hydrogen abstraction) will be locally determined by the stereoelectronic constraints and in particular solvent accessibility imposed by each reaction site.
Representative Binding Model
In order to further evaluate the proposed binding model shown in Figure 8, and derived from experimental RNA cleavage profiles, computational studies were performed to generate energy optimized structures for complexes 1-Cu and 2-Cu using a B3LYP DFT calculation in Gaussian 09 (Figure SM12). Involvement of the tyrosine and phenylalanine aromatic rings in contributions to RNA binding would be expected to involve interactions with nucleic acid bases, whereas contributions from arginine should involve either electrostatic, H-bonding and/or pi-stacking interactions, while lysine would be expected to engage primarily in electrostatic and H-bonding interactions with the phosphate backbone. Two hundred structures were simulated and the localization of the second lowest energy cluster is shown in the left box of Figure 6 for complex 1-Cu, and the lowest energy cluster is shown in the right box of Figure 6 for complex 2-Cu. These clusters, and the clusters of metals that they contain, are chosen as the working model for the proposed sites of reactivity because they are in the best agreement with the experimentally-determined cleavage sites determined by mass spectrometry (Figure 11), where reactivity is centered on the copper ion. In the case of 1-Cu the results demonstrate that the thermodynamically more stable conformation is not necessarily the most reactive, an important point that is often under-appreciated in studies of catalysts. It is also noteworthy that the common docking sites based on clustering are different for each complex, again emphasizing the effect that conversion to d-amino acids has on the conformation of the isolated peptide in addition to how it interacts with the target RNA.
Figure 11.

Comparison of the reactive sites based upon the MALDI-TOF MS (shaded bases on the RNA) versus the simulated binding proposed by Autodock (gray spheres represent copper atoms) for 1-Cu (left), and 2-Cu (right). A color representative figure can be found in the supplemental material (Figure SM13).
HCV Replicon Assays
Complex 2-Cu was also tested in the HCV cellular replicon assay previously described to assess activity in vivo. This cellular replicon assay is a widely accepted measure of drug efficacy for HCV treatment. The copper complex of 2 was found to have an IC50 of 1.9 µM, which is comparable to the previously reported data for complex 1-Cu (IC50 = 0.6 µM), and it exhibited no cytotoxicity up to 100 µM (Table 1). Despite a ~10 fold higher KM and ~34 fold worse kcat/KM, the observed cellular activity was approximately the same. This is a reflection of the complex combination of factors that determine the activity of these metallodrugs (Figure SM4), in vivo stability, and the possible limiting step of cellular uptake, which is not addressed in this work. Importantly, the all d-amino acid analog also maintains the ability to be taken up by cells and the lack of toxicity shows the specificity for the virus.
To further confirm that the mechanism of action in the cellular replicon assays is consistent with that proposed for catalytic metallodrugs, real-time polymerase chain reaction (RT-PCR) was performed to measure the RNA levels for both HCV RNA and ribosomal RNA (rRNA). The results can be seen in Figure 7 (left) and show a clear preference for HCV RNA over rRNA. Significantly, complex 1-Cu still showed activity up to final day tested, 9 days later, and reduced HCV RNA levels to close to zero (Figure 7, left, data is normalized). In contrast, the rRNA levels decreased much less although there was still a small decrease relative to the control. This data is complicated by the fact that it will reflect both the amount of RNA being made as well as the amount of RNA being consumed. Also, the rate of signal amplification can be different for the two RNAs which further complicates any quantitative comparison. The data for copies of rRNA (Figure 7, right, not normalized) is shown and it is seen that the amount of rRNA being made over time increases both with and without complex 1-Cu but that the rRNA levels are slightly higher in the absence of catalyst. This could be due to small levels of background cleavage of the rRNA but is consistent with the idea that higher levels of rRNA are being made to compensate for the presence of the IRES, thereby reducing the effective concentration of rRNA available for use by the cell. In the presence of catalytic metallodrug, however, the reduction in the concentration of IRES means that more rRNA is available for the cell to use and, therefore, it does not need to make as much. Regardless, there is a clear preference for the HCV RNA and no significant overall cellular toxicity was observed as evidenced by the TC50 values (greater than 100 µM). All of these are consistent with the metallodrug behaving in the cellular replicon assays in the similar way that is proposed for catalytic metallodrugs and shown for the in vitro experiments.
CONCLUSIONS
As determined by observation of predominately 2’, 3’ – cyclic phosphates and 3’-phosphates products a mechanism associated with a Cu(III) intermediate acting as a strong Lewis acid is proposed to facilitate the irreversible cleavage of the HCV IRES RNA for complexes 1-Cu and 2-Cu. In addition, the diasteromeric analog of complex 1-Cu, which incorporates d-configuration amino acids, complex 2-Cu, was investigated and shows potential for use in vivo. The assays described show that the chiral analog maintains activity but changes selectivity in the recognition of the stem loop. This illustrates the importance of the stereochemical environment of the α-carbons. Cu-GGhyrfk binds to the target RNA with a KD of 76 nM, and, despite differences in catalytic efficiency (kcat/Km = 0.0018 µM−1min−1 for complex 2-Cu versus 0.62 µM−1min−1, for complex 1-Cu), both show good activity in an FDA approved HCV replicon assay. This similar efficacy, combined with potentially higher in vivo stability, should lead to a more effective catalytic metallodrug in more complex systems such as animal models or humans.
EXPERIMENTAL SECTION
Materials
RNA was purchased from Dharmacon, part of Thermo Fisher Scientific (Lafayette, CO). The sequence for the IRES SLIIb RNA was 5’-fluorescein-GGCAGAAAGCGUCUAGCCAUGGCGUUAGUA UGCC-3’, for the IRES SLIV RNA was 5’-fluorescein-GGACCGUGCACCAUGAGCACGAAUCC-3’, and for HIV Rev response element (RRE) RNA was 5’-fluorescein-UUGGUCUGGGCGCAGCGC AAGCUGACGGUAC AGGCC-3’. The sequence of the 5-mer RNA used for calibration in the MALDI-TOF experiments was 5’-fluorescein(T)-UGUG-3’. All RNA was annealed by heating to 95 °C and then cooled slowly to room temperature before use. Sodium chloride, sodium hydroxide, and acetonitrile were purchased from Fisher and HEPES, ammonium citrate, and 3-hydroxypicolinic acid were purchased from Sigma. C18 ZipTips were obtained from Millipore. All experiments were performed using diethyl pyrocarbonate (DEPC) treated water and autoclaved pipette tips and eppendorfs. Copper complexes form quickly and were made by mixing in a 1.1:1 peptide:CuCl2 ratio and waiting at least 5 min for complex to form. Structures were rendered in the Visualization Applet for RNA (VARNA)[30] for RNA secondary structures and the PyMOL Molecular Graphics System, Version 1.5.0.4 (Schrödinger, LLC) or AutoDockTools, for three dimensional structures.
Synthesis and Characterization
Peptides were purchased from Genemed Synthesis, Inc. (South San Francisco, CA) following synthesis by standard automated amino acid coupling methods using the appropriate l-or d-configuration amino acids. Peptide purity was established by both HPLC and mass spectrometric characterization, and concentrations were established both by monitoring the tyrosine absorbance at 274 nm (ε = 1400 M−1cm−1), and by titrating copper ion from stock copper(II) chloride solutions into a fixed amount of peptide at 37 °C every 5 min and fitting the absorbance at 240 nm and 525 nm.
Binding Constant Determination
For GGhyrfk-amide, RNA binding experiments were performed in the presence of 84 nM GGhyrfk-amide (2) in 20 mM HEPES (pH 7.4), 100 mM NaCl. Aliquots of unlabeled IRES SLIIb RNA were added and tyrosine emission was monitored (λex = 280 nm, λem = 313 nm). Data were then fit to a one-site binding model using Origin 7.0 software.
Melting Temperatures
Melting temperatures were measured by monitoring fluorescein emission (ex = 485 nm, em = 518 nm) with increasing temperature (15 to 95 degrees Celsius heated at a rate of 3 degrees Celsius per minute) in the presence or absence of metal-free peptide and/or varying [NaCl]. Melting temperatures were determined under conditions of 20 mM HEPES, pH 7.4, and 1 µM fluorescein-labeled SLIIb IRES. Melting profiles were fit to a three-state consecutive melting model using Origin 7.0 software to obtain TM1 and TM2. The values shown are an average of at least 3 trials.
In Silico Docking
The peptide structures were optimized with Gaussian 09 vA01* using an Amber MM/MD force field in an aqueous solvent, followed by a more thorough refinement with a DFT/B3LYP/3-21G force field.[31] Solvent interactions, water in this case, were considered by using the recent universal solvation model, SMD, by Truhlar and co-workers.[32] The structures of the metal complexes were optimized by attaching the crystal structure for CuGGH (inverting the stereocenter for the d-His in complex 2-Cu) to the peptide and submitting the complexes using the above-mentioned force field and solvation model. The charge for the complex structures was set to +3 with a multiplicity of 1, because this represents overall charge of the complex (Arg and Lys and the N-terminus each contribute +1, the copper contributes +2 and the two deprotonated backbone amides each have a −1 charge).
The Cu-GGH atoms in the complexes were held fixed during the geometry optimization of the complexes by freezing the atoms in Gaussian 09. The optimized peptides and complexes were used for docking simulations. The solution state NMR structure of stem loop IIb (SLIIb) of the HCV IRES RNA is readily available from the protein data bank (PDB: 1P5N). The top solution state NMR structure (out of the 20 available) was used for docking simulations using AutoDock 4.2. The entire SLIIb domain was considered for docking. In all cases, the complex was made flexible except for aromatic carbons, peptide bonds, and the metal binding domain. The Lamarckian Genetic Algorithm allows a large degree of flexibility in the selection of the initial starting position: essentially searching a local area, identifying the thermodynamically most favorable orientation, and then “mutating” to a different spot on the RNA. Comparison of the docking in this manner provides a measure of how readily the complex is able to be position in a manner to perform effective chemistry.
Autodock does not have parameters for copper so an iron atom was substituted to mimic the geometry of the copper with a charge of +0.8. Iron and copper should have similar ionic radii and the iron simulates the charge on the copper ion. A value of +0.8 is used to simulate the +2 charge in order to compensate for the tendency of Autodock to overestimate electrostatic interactions.[33] The geometry around the metal was based on the X-ray crystal structure of Cu-GGH and was restricted to a square planar configuration.[17] a Lamarckian Genetic Algorithm and the following parameters were used for docking: population size of 150, a random starting position and conformation, a maximal mutation of 2Å in translation and 50° in rotations (elitism of 5), iterations of Solis and Wets local search of 300, torsional degrees of freedom of 23 for the peptide complex, an external grid energy of 1000 kcal/mol, a mutation rate of 0.02 and a crossover rate of 0.8, and local search rate of 0.06. Simulations were performed with a maximum of 107 energy evaluations and a maximum of 27000 generations. The total number of hybrid GA-LS runs was set to 200, yielding an output of 200 clusters. Docking analyses were performed on the Unix system at the Ohio Supercomputer Center using the OAKLEY cluster platform with 12 CPU processors running on 1 computing node and a total wall time of 120 hours
Reaction Kinetics via Fluorescence
HCV IRES RNA cleavage was monitored in vitro by fluorescence using 5’ fluorescein end-labeled RNA with excitation and emission wavelengths of 485 nm and 518 nm, respectively. Reactions were carried out at 25 °C in reaction volumes of 100 µL in the presence of 1 mM ascorbic acid and 1 mM H2O2 in HEPES buffer (pH = 7.4, 100 mM NaCl) with 1 µM fluorescein labeled IRES SLIIb and analyzed according to the change in fluorescence observed as the reaction occurred. Both a time-dependence and a concentration-dependence of catalyst activity were observed. The initial velocity of the time dependence plot was used to generate the pseudo Michaelis-Menten plots which were then fit to the Michaelis Menten equation. All fits were performed using Origin 7.0 software. The values shown are an average of at least three trials. A turnover number was estimated based on the limiting amount of peptide catalyst consumed by a specific amount of RNA.
Mass Spectrometry
Reactions for MALDI-TOF analysis were run as described above, but using 10 µM fluorescein labeled IRES SLIIb and 10 µM copper-peptide incubated for up to two hours. Reactions were then quenched by being placed on ice and ZipTipped. ZipTipping was performed using C18 ZipTips from Millipore Co. in order to desalt the reaction mixtures prior to mass spectrometric analysis. ZipTips were wetted with a 50:50 mixture of acetonitrile:water and equilibrated with 2 M triethylammonium acetate (TEAA), pH 7.0. The reaction mixture was then bound to the ZipTip, washed with nanopure water, and eluted with 50:50 acetonitrile:water. These samples were spotted onto a Bruker ground steel 96 target microScout plate by first spotting with 1 µL of a matrix solution containing 0.3 M 4-hydroxypicolinic acid (HPA) and 30 mM ammonium citrate in 30% acetonitrile, drying, spotting with 1 µL of a 2:1 RNA:matrix mixture, and allowed to dry. A calibration mixture containing 3 RNAs covering a range of molecular weights (Fl-5mer, Fl-IRES SLIV, and Fl-RRE, with molecular weights of 2057.5, 8861.5, and 12172.5 amu, respectively) was used to calibrate the instrument. All MALDI-TOF MS analysis was performed on a Bruker MicroFlex LRF instrument equipped with a gridless reflectron, using negative ion mode and reflectron mode. The pulsed ion extraction time was 1200 ns. Typically, at least 1000 shots were summed per spectrum to acquire an accurate representation of the reaction. Data analysis was performed using Bruker flexAnalysis software. Assignment of peaks was performed by comparison of each peak list with the expected masses for possible cleavage products using the MassDaddy Perl script analysis program.[2, 20] Only m/z values > 1500 amu and those with a signal to noise ratio greater than five were considered, since excessive spectral crowding occurred at lower m/z ranges. Peaks which showed time dependence were then compared to controls containing SLIIb RNA in the presence of copper-peptide. Reactions for the time dependent assay were collected at: 2 min, 10 min, 20 min, 30 min, 45 min, 60 min, 90, and 120 min. A comparison to controls of two hour reactions containing SLIIb RNA with 1 mM ascorbic acid and/or 1 mM H2O2 as well as SLIIb RNA alone was used to identify new products of cleavage by the metallopeptide. Heat maps and initial rates were generated by first summing the total change at a position within the RNA sequence and determining the fraction of associated change at each time point. An apparent initial rate was then determined from the linear region of the data.
HCV Cellular Replicon Assay
A stable cell line ET (luc-ubi-neo/ET) was employed in the assay. The ET is a Huh7 human hepatoma cell line that contains an HCV RNA replicon with a stable luciferase (Luc) reporter and three cell culture-adaptive mutations. The HCV RNA replicon antiviral evaluation assay examined the effects of compounds at six half-log concentrations each. Human interferon alpha-2b was included in each run as a positive control compound. Sub-confluent cultures of the ET line were plated out into 96-well plates that were dedicated for the analysis of cell numbers (cytotoxicity) or antiviral activity, and various concentrations of metallodrugs and controls were added to the appropriate wells the following day. Cells were processed 72 hours later when the cells were still sub-confluent. Six half-log serial dilutions of the compound were performed, and values derived for IC50 (the concentration that inhibited virus replication by 50%), TC50 (the concentration that lowered cell viability by 50%) and TI (the selectivity index: TC50/IC50). HCV RNA replicon levels were assessed as the replicon-derived Luc activity. The toxic concentration of drug that reduced cell numbers (cytotoxicity) was assessed by the CytoTox-1 cell proliferation colorimetric assay (Promega).
RT-PCR
From the above replicon assays, medium was replenished and compound was added every 3 days for a total of 9 days. The cells were collected at 0, 3, 6 and 9 days for RNA extraction and measurement of HCV RNA copies by qRT-PCR (TaqMan).[34] Ribosomal RNA determined simultaneously using qRT-PCR was used for calibration of HCV RNA reduction.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health [HL093446 and AA016712]. This work was also supported in part by an allocation of computing time from the Ohio Supercomputer Center and we thank Jason Brown (OSU Chemistry) for help with the computational studies. The Bruker Microflex instrument used for all MALDI-TOF analysis was provided by a grant from the Ohio BioProducts Innovation Center. We would like to thank Dr. Jeff Joyner for his help with the MALDI-TOF experiments. We thank Dr. Zhuhui Huang and staff at Southern Research Institute for carrying out the cellular replicon and RT-PCR studies.
Footnotes
Supporting Information Available. Tabulation of peak masses from mass spectrometric studies of cleavage reactions and structural details from modeling of RNA-metallopeptide complexes, plots of reactivity profiles for each base site on the RNA target, fluorescence plots of metallopeptide binding, and information on NMR structures used for modeling.
REFERENCES
- 1.Cowan JA. Pure Appl. Chem. 2008;80:1799–1810. [Google Scholar]
- 2.Joyner JC, Cowan JA. J. Am. Chem. Soc. 2011;133:9912–9922. doi: 10.1021/ja203057z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hocharoen L, Cowan JA. Chem. Eur. J. 2009;15:8670–8676. doi: 10.1002/chem.200900821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford S, Cowan JA. Chem. Commun. 2012;48:3118–3120. doi: 10.1039/c2cc17377h. [DOI] [PubMed] [Google Scholar]
- 5.Lau S-J, Kruck TPA, Sarkar B. J. Biol. Chem. 1974;249:5878–5884. [PubMed] [Google Scholar]
- 6.Honda M, Beard MR, Ping L-H, Lemon SM. J. Virol. 1999;73:1165–1174. doi: 10.1128/jvi.73.2.1165-1174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Nat. Struct. Biol. 2003;10:1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 8.Erspamer V, Melchiorri P, Broccardo M, Erspamer GF, Falaschi P, Improota G, Negri L, Renda T. Peptides. 1981;2(Suppl 2):7–16. doi: 10.1016/0196-9781(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 9.Montecucchi PC, De Castiglione R, Piani S, Gozzini L, Erspamer V. Int. J. Pept. Protein Res. 1981;17:275–283. doi: 10.1111/j.1399-3011.1981.tb01993.x. [DOI] [PubMed] [Google Scholar]
- 10.Milton RCL, Milton SCF, Kent SBH. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 11.Pritsker M, Jones P, Blumenthal R, Shai Y. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7287–7292. doi: 10.1073/pnas.95.13.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Litovchick A, Rando RR. RNA. 2003;9:937–948. doi: 10.1261/rna.2172103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wade D, Boman A, Waahlin B, Drain CM, Andreu D, Boman HG, Merrifield RB. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4761–4765. doi: 10.1073/pnas.87.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boule M, Sarazin P, Stea D, Lacombe D, Tremblay P, Gervais F. J. Biol. Chem. 2003;278:34874–34881. doi: 10.1074/jbc.M212694200. [DOI] [PubMed] [Google Scholar]
- 15.Kim I, Lukavsky PJ, Puglisi JD. J. Am. Chem. Soc. 2002;124:9338–9339. doi: 10.1021/ja026647w. [DOI] [PubMed] [Google Scholar]
- 16.Andersen TE, Kirpekar F, Haselmann KF. J. Am. Soc. Mass Spectrom. 2006;17:1353–1368. doi: 10.1016/j.jasms.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Camerman N, Camerman A, Sarkar B. Can. J. Chem. 1976;54:1309–1316. [Google Scholar]
- 18.Thorp HH. Chem. Biol. 2000;7:R33–R36. doi: 10.1016/s1074-5521(00)00080-6. [DOI] [PubMed] [Google Scholar]
- 19.Joyner JC, Hocharoen L, Cowan JA. J. Am. Chem. Soc. 2012;134:3396–3410. doi: 10.1021/ja208791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joyner JC, Keuper K, Cowan JA. Nucl. Acids Res. 2013;41:e2. doi: 10.1093/nar/gks811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joyner JC, Reichfield J, Cowan JA. J. Am. Chem. Soc. 2011;133:15613–15626. doi: 10.1021/ja2052599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bossu FP, Chellappa KL, Margerum DW. J. Am. Chem. Soc. 1977;99:2195–2203. doi: 10.1021/ja00449a028. [DOI] [PubMed] [Google Scholar]
- 23.Margerum DW, Owens GD. Metal Ions Biol. Syst. 1981;12:75–132. [Google Scholar]
- 24.McDonald MR, Fredericks FC, Margerum DW. Inorg. Chem. 1997;36:3119–3124. doi: 10.1021/ic9608713. [DOI] [PubMed] [Google Scholar]
- 25.Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Structure. 2011;19:1456–1466. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalliampakou KI, Psaridi-Linardaki L, Mavromara P. FEBS Letts. 2002;511:79–84. doi: 10.1016/s0014-5793(01)03300-2. [DOI] [PubMed] [Google Scholar]
- 27.Markham RS, D J. J. Biochemistry. 1952;52:552–557. [Google Scholar]
- 28.Balasubramanian W BP, Tullius TD. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9738–9743. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 30.Darty K, Denise A, Ponty Y. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian, Inc., Wallingford CT. 2009. [Google Scholar]
- 32.Saielli G. J. Phys. Chem. A. 2010;114:7261–7265. doi: 10.1021/jp103783j. [DOI] [PubMed] [Google Scholar]
- 33.Park H, Kim S, Kim YE, Lim S-J. Chem Med Chem. 2010;5:591–597. doi: 10.1002/cmdc.200900500. [DOI] [PubMed] [Google Scholar]
- 34.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Nucl. Acids Res. 2005;33:e179/171–e179/179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.