

Summary
LIGHT initiates intracellular signaling via engagement of the two TNF receptors, HVEM and LTβR. In humans, LIGHT is neutralized by DcR3, a unique soluble member of the TNFR superfamily, which tightly binds LIGHT and inhibits its interactions with HVEM and LTβR. DcR3 also neutralizes two other TNF ligands, FasL and TL1A. Due to its ability to neutralize three distinct different ligands, DcR3 contributes to a wide range of biological and pathological processes, including cancer and autoimmune diseases. However, the mechanisms which support the broad specificity of DcR3 remain to be fully defined. We report the structures of LIGHT and the LIGHT:DcR3 complex, which reveal the structural basis for the DcR3-mediated neutralization of LIGHT and afford new insights into DcR3 function and binding promiscuity. Based on these structures, we designed novel LIGHT mutants with altered affinities for DcR3 and HVEM, which may represent new mechanistically informative probe reagents.
Introduction
Myriad interactions involving secreted and cell surface proteins provide the stimulatory and inhibitory signals that determine the course, strength and duration of mammalian immune responses. Members of the tumor necrosis factor/tumor necrosis factor receptor (TNF/TNFR) superfamilies make important contribution to these processes (Cai and Freeman, 2009), and these same molecules are associated with numerous autoimmune diseases. Furthermore, the associated signaling pathways are co-opted by infectious agents and malignancies for evasion of the host immune response. These properties make the members of the TNF/TNFR superfamilies prime targets for a wide range of immunotherapies involving function modulating monoclonal antibodies (mAbs) and multimeric soluble versions of these molecules themselves. For example, Remicade, a mAb against TNF, which results in blockade of the TNFR signaling pathway by sequestration of TNF, is used to treat autoimmune disorders such as rheumatoid arthritis, ulcerative colitis and Crohn’s disease. Enbrel is a soluble TNFR-Ig fusion protein, formed from the extracellular domain of TNFR and the Fc region of immunoglobulin (Ig), which binds with high affinity to TNF. This interaction prevents TNF from engaging and activating cell surface associated TNFR, resulting in clinically significant reductions in inflammatory responses in rheumatoid arthritis, juvenile rheumatoid arthritis, psoriasis, psoriatic arthritis and ankylosing spondylitis (Bendtzen, 2012).
Due to their important roles in immunity, pathogenesis and clinical applications, the structures of these molecules and complexes have been extensively studied. The TNF ligands are type-II integral membrane proteins that typically form compact trimeric assemblies, in which each protomer adopts a beta sandwich “jelly-roll” structure (Eck and Sprang, 1989). TNF receptors are type-I membrane proteins containing between one to six cysteine-rich domains (CRDs), which bind to the interprotomer interface formed between adjacent TNF ligand monomers (Tansey and Szymkowski, 2009). These interactions typically result in hexameric assemblies, with 3:3 (or sometimes 2:3) receptor:ligand stoichiometries, which span interacting cells. The ligand-mediated clustering of TNFRs triggers the recruitment of adaptor proteins, such as death domain adaptor proteins and TNF receptor associated factors (TRAFs), which control diverse downstream signaling pathways (Ashkenazi, 2002).
LIGHT (homologous to Lymphotoxin, exhibits Inducible expression and competes with HSV Glycoprotein D for Herpesvirus entry mediator, a receptor expressed on T cells) is a member of the TNF superfamily that is transiently induced on activated T cells (Mauri et al., 1998). LIGHT is recognized by two differentially expressed TNFR superfamily members, herpes simplex virus entry mediator (HVEM) and lymphotoxin beta receptor (LTβR) (Mauri et al., 1998; Zhai et al., 1998). LIGHT functions as part of a costimulatory circuit to boost T cell proliferation and cytokine production through its interaction with HVEM (Tamada et al., 2000; Zhai et al., 1998). Blockade of LIGHT signaling by HVEM-Ig and LTβR-Ig fusion proteins dramatically reduces T cell proliferation and cytotoxic T cell activity (Tamada et al., 2000; Wang et al., 2001). Furthermore, constitutive expression of LIGHT on T cells in transgenic mouse models results in abnormal T cell activation, leading to diverse autoimmune responses and severe inflammation in the gut (Shaikh et al., 2001; Wang et al., 2001).
In contrast to the expression of HVEM on lymphocytes, LTβR is widely expressed on epithelial, stromal, and myeloid cells but not lymphocytes, consistent with a role in mediating signals between lymphocytes and nearby epithelial and stromal cells (Ware, 2005). Engagement of LIGHT by LTβR initiates multiple signaling pathways, including JNK/c-Jun and NF-κB activation (Chang et al., 2002; Kim et al., 2005). LTβR signaling in intestinal epithelial cells elicits a protective response against mucosal bacterial C. rodentium infection (Wang et al., 2010). Signaling through LTβR initiated by soluble LIGHT induces cell death of certain tumor cell lines such as HT-29 and MDA-MB-231 (Browning et al., 1996; Rooney et al., 2000; Yu et al., 1999; Zhai et al., 1998).
Human LIGHT also binds Decoy Receptor 3 (DcR3; also referred to as TNFRSF6B), which contains 4 cysteine rich domains (CRDs) at its N-terminus and an approximately 100 residue C-terminal segment that binds heparin sulfate proteoglycans (HSPG) (Zhan et al., 2011). Unlike other members of the TNFR superfamily, which are cellular receptors capable of transducing signals, DcR3 lacks transmembrane and cytoplasmic segments, resulting in a soluble secreted molecule capable of interacting with multiple cell types (Connor et al., 2012; You et al., 2008; Zhan et al., 2011). DcR3 exhibits broad specificity, recognizing three TNF ligands, LIGHT, FasL and TL1A, through canonical interactions involving the CRDs (Chang et al., 2006; Hsu et al., 2004; Zhan et al., 2011). These interactions result in blockade of the associated signaling pathways by virtue of interfering with recognition between the TNF ligands and their cognate receptors.
Defining the precise contributions of DcR3 to adaptive and innate immunity is challenging due to the diverse functions of the three TNF ligands that it neutralizes (Figure 1). DcR3 disrupts the activation signal initiated by LIGHT, resulting in reduced T cell activation (Zhang et al., 2001). Inhibition of the TL1A signaling pathway also down-regulates the T cell immune response and is a potential strategy for the treatment of autoimmune diseases (Young and Tovey, 2006). In contrast, blockade of the Fas: FasL interaction, which induces T cell apoptosis and restrains the immune response, may contribute to the development of autoimmune disorders (Funke et al., 2009; Hayashi et al., 2007). Systemic expression of human DcR3 in mice (DcR3 is not present in mice, while LIGHT, TL1A and FasL orthologues are present in mice) attenuates the T cell immune response, suggesting an overall negative role of DcR3 in the regulation of the immune system (Hsu et al., 2005). Consistent with an attenuator role for DcR3, it has been demonstrated that DcR3 is present at a low level in a wide range of tissues from healthy humans, while it is highly elevated in patients with cancer (Wu et al., 2003). Tumor-secreted DcR3 functions as a decoy receptor for FasL and LIGHT on lymphocytes to prevent transmission of apoptotic signals via tumor-expressed Fas and LTβR, thus providing a mechanism for immune evasion by the malignant cells (Ashkenazi, 2002). The role of DcR3 is further complicated by contributions of the C-terminal region which was recently reported to bind heparin sulfate proteoglycans (HSPG) and trigger apoptosis of antigen-presenting cells (APC) (Chang et al., 2006; Hsu et al., 2004; You et al., 2008).
Figure 1.

The interaction network of LIGHT and DcR3 within the TNF/TNFR superfamilies. TNF ligands FasL (orange), LIGHT (blue) and TL1A (magenta) bind to Fas (light orange), DR3 (dark magenta), and HVEM (green) and LTβR (dark blue), respectively, transducing proliferative (+) or apoptotic (−) signals to different cell types. Engagement of TL1A with DR3 expressed on T cells promotes proliferation whereas interaction of TL1A with DR3 expressed on osteoblasts induces apoptosis. The signals transduced by FasL, LIGHT and TL1A can all be blocked by DcR3 (red), which competes for binding with the cognate signaling receptors. See also Figure S7.
Despite the roles of LIGHT and DcR3 in regulating the immune response (Figure 1), the structural basis of LIGHT and DcR3 engagement and function remains to be elucidated. Herein we report the biochemical and structural properties of LIGHT and the LIGHT:DcR3 complex. These structures define the physical determinants contributing to ligand-receptor recognition and specificity, and provide the basis for designing novel LIGHT mutants with altered selectivities towards its multiple ligands. This work has revealed the structural basis of DcR3-mediated neutralization of LIGHT and enabled the design of novel molecules to antagonize and probe DcR3 function.
RESULTS
Overall Structure of the LIGHT:DcR3 Complex
We determined the crystal structures of LIGHT and LIGHT in complex with DcR3 (Table 1). Soluble LIGHT (residues L83-V240, numbering from the initiation codon) crystallized with one canonical TNF-like trimer in the asymmetric unit. The complex between LIGHT and DcR3 (residues V30-S195) revealed two 3:3 hetero-hexameric assemblies typical of the TNF and TNFR superfamilies. We define the conventional binding interface between LIGHT and DcR3 in the hexameric assembly as the “cis” interaction interface and the interface between two hexameric assemblies as the “trans” interaction interface. Based on the structure of the LIGHT:DcR3 complex, we designed two mutants of LIGHT that retain approximately wild type affinity for DcR3 but have significantly reduced affinity for HVEM. Consideration of all local and crystallographic symmetry operators results in an unusual dodecameric structure formed by the intimate packing of the two conventional hetero-hexameric assemblies in the crystal (Figure S1).
Table 1.
Data Collection and Refinement Statistics
| LIGHT | LIGHT:DcR3 | LIGHT Mutant2 | Mutant1:DcR3 | Mutant2:DcR3 | |
|---|---|---|---|---|---|
|
Data Collection
| |||||
| Wavelength used (Å) | 0.9791 | 1.075 | 1.075 | 1.075 | 1.075 |
| Resolution range (Å) | 2.59–50.00 | 2.40–50.00 | 2.25–50.00 | 2.27–50.00 | 2.78–50.00 |
| Space group | I212121 | P213 | P21 | P213 | P213 |
| Unit cell (Å) | a=94.7, b=100.0, c=124.4 | a=b=c= 149.1 | a=59.6, b=46.9, c=70.4 | a=b=c= 149.3 | a=b=c= 148.8 |
| Unit cell (°) | β=98.02 | ||||
| Unique reflections (N) | 18664 | 43288 | 18578 | 51516 | 27912 |
| Redundancy | 7.8 (8.1) | 11.2 (11.2) | 7.3 (7.1) | 11.6 (11.3) | 12.5 (12.8) |
| Completeness | 100(100) | 100(100) | 100(99.5) | 100(100) | 99.9(100) |
| I/sigma | 7.3 (2.0) | 27.2 (3.5) | 24.7 (4.7) | 23.1 (4.4) | 20.1 (4.1) |
| Rmerge | 0.136 (0.950) | 0.102 (0.810) | 0.079 (0.448) | 0.104 (0.650) | 0.140 (0.691) |
|
| |||||
| Refinement
| |||||
| Resolution range (Å) | 2.59–46.12 | 2.40–19.93 | 2.25–20.00 | 2.27–37.35 | 2.78–49.63 |
| Rwork | 0.234 (0.350) | 0.194 (0.277) | 0.211 (0.244) | 0.195 (0.237) | 0.178 (0.266) |
| Rfree | 0.286 (0.410) | 0.232 (0.292) | 0.286 (0.339) | 0.238 (0.292) | 0.235 (0.356) |
| Average B factor (Å2) | 74 | 41 | 41 | 46 | 45 |
| Rms bond (Å) | 0.004 | 0.018 | 0.015 | 0.023 | 0.017 |
| Rms angles (°) | 1.274 | 1.961 | 1.885 | 2.099 | 1.983 |
|
| |||||
| PDB code | 4EN0 | 4J6G | 4KG8 | 4KGQ | 4KGG |
Rmerge=ΣhklΣi|Ii(hkl)−<I(hkl)>|/ΣhklΣiIi(hkl).
Rwork=Σ|Fc−Fo|/ΣFo.
Parentheses indicate statistics for the highest resolution bin.
The conventional binding interface between LIGHT and DcR3
The asymmetric unit of the LIGHT:DcR3 crystal structure consists of two independent chains of LIGHT and two independent chains of DcR3 which, upon application of crystallographic three-fold rotational operators, form two canonical three-fold symmetric TNF:TNFR hetero-hexameric assemblies. The two hetero-hexamers are related to each other by a non-crystallographic two-fold rotational operator perpendicular to the crystallographic three-fold axis. LIGHT adopts the typical jelly-roll fold of the TNF family (Eck and Sprang, 1989), with the inner and outer beta-sheets composed of strands A′AHCF and B′BGDE, respectively (Figure S2). Each DcR3 molecule contacts the surface formed by two adjacent LIGHT protomers, resulting in an assembly with overall 3:3 receptor:ligand stoichiometry in which no direct contacts occur between individual receptor molecules (Figure 2). The two independent copies of LIGHT and DcR3 are highly similar with RMSDs of 0.2Å (for 138 aligned LIGHT Cα atoms) and 0.9Å (for 158 aligned DcR3 Cα atoms), respectively. The two independent hetero-hexamers exhibit nearly identical binding interfaces with buried solvent accessible areas of 1060Å2 for chains A (LIGHT) and C (DcR3) and 1020 Å2 for chains B (LIGHT) and D (DcR3) (these values refer to the buried surface area associated with a single DcR3 chain interacting with the LIGHT trimer). All subsequent discussions are based on the structure of the complex formed by chain A (LIGHT) and chain C (DcR3).
Figure 2.

Overall structure and determinants of LIGHT: DcR3 recognition. (A) Side view (top panel) and bottom view (bottom panel) of the overall structure of LIGHT:DcR3 complex. LIGHT is shown as a cartoon (blue and red) and DcR3 is shown as surface (grey). One LIGHT homo-trimer binds three DcR3 molecules at interprotomer interfaces. (B) The GH loop (magenta) makes polar contacts with DcR3 (grey and green). (C) The DE loop (magenta) makes polar contacts with DcR3 (grey and green). See also Figure S3.
The LIGHT:DcR3 interface is largely formed by residues contributed by the AA′, CD, DE, EF and GH loops of LIGHT and CRD2 and CRD3 of DcR3 (Figure 2; Figure S2). The binding surface of LIGHT can be divided into two parts: the lower region proximal to the ligand-associated plasma membrane and the upper region distal to the plasma membrane (Zhan et al., 2011). Loops of AA′, DE and GH in the lower region of LIGHT contribute to the interaction with CRD2 of DcR3, while the CD and EF loops in the upper region of LIGHT contact CRD3 of DcR3.
A number of ionic and hydrogen bonding interactions support the contact between the lower region of LIGHT and DcR3, with DE loop residues T170-E178 interacting with Y78-E86 and R89 at the beginning and N92 in the middle of the DcR3 CRD2 (Figure 2). Polar interactions are formed by LIGHT DE loop residues R172, Y173, E175, E176 and E178 contacting Q80, Y84, R89 and N92 of DcR3. Among these, Y173 is conserved in all three DcR3 ligands, LIGHT, TL1A and FasL (Zhan et al., 2009). In the LIGHT:DcR3 structure, the side chain of Y173 extends into a geometrically complementary cavity of DcR3 and forms a hydrogen bond with the main chain amide nitrogen of DcR3 Q80 (Figure S3). The Y173F mutant of LIGHT exhibited significantly decreased binding to its receptors HVEM and LTβR, suggesting the general importance of this residue in receptor recognition(Rooney et al., 2000). The main chain oxygen and a side chain guanidinium nitrogen of LIGHT R172 form polar contacts with the main chain amide nitrogen and oxygen of DcR3 Y84, respectively. The main chain oxygen of LIGHT E175 forms a hydrogen bond with the side chain of DcR3 R89.
An additional patch of polar contacts is formed between the GH loop of LIGHT and CRD2 of DcR3, with residues R226-T231 of the GH loop interacting with residues N92-E99 in DcR3 CRD2 (Figure 2). The main chain carbonyl oxygen of LIGHT R228 forms a hydrogen bond with the amide nitrogen of DcR3 L94 (Figure 2). In addition, the side chain of LIGHT R228 forms a salt bridge with the side chain of DcR3 E99. The main chain carbonyl oxygen of LIGHT D229 also forms a hydrogen bond with the side chain nitrogen of N92 (Figure 2). Notably, the LIGHT R228E mutation resulted in a significant decrease in affinity for HVEM, but did not significantly affect the interaction with LTβR (Chen et al., 2003). Although the GH loop appears important for the interaction with DcR3, the residues in this region are not conserved among LIGHT, TL1A and FasL, suggesting that the diversity of this loop may provide some of the specificity determinants required for each ligand to recognize its own signaling receptors while maintaining binding with DcR3.
Also of note are G100-N102 and E115-G119 of the AA′ loop of LIGHT, which contact CRD2 of DcR3 at the binding interface. The mutation of LIGHT residues E115-L120 significantly reduced the affinity for DcR3, suggesting an important role of this loop in recognition (Morishige et al., 2010).
The upper region of the interface accounts for approximately 40% of the total buried solvent accessible area of LIGHT in the conventional LIGHT:DcR3 binding interface, with the LIGHT CD (G150-V152 and T161) and EF (R195-W198) loops interacting with DcR3 CRD3 residues H122-L127. Hydrophobic interactions play important roles, with F125 of DcR3 occupying a hydrophobic pocket formed by LIGHT residues V152, V196 and W198. The side chain of DcR3 L127 is also close to the same hydrophobic pocket on LIGHT. The only observed polar contact in the upper region is the potential hydrogen bond between the side chain indole nitrogen of LIGHT W198 and the main chain oxygen of DcR3 A123.
Comparison of LIGHT:DcR3 and TL1A:DcR3 complexes
The structures of the LIGHT:DcR3 and TL1A:DcR3 complexes revealed generally similar interaction patterns between the two ligands and DcR3 in the lower region, with analogous loops of LIGHT and TL1A contacting the same surfaces on DcR3. The interfaces in the upper region of the LIGHT:DcR3 and TL1A:DcR3 complexes are less similar due to detailed conformational differences in DcR3, although comparable areas on DcR3 contact analogous loops in the ligands.
The AA′, DE and GH loops of TL1A in the lower region interact predominately with CRD2 of DcR3 in a manner similar to LIGHT, with the DE loop also being one of the major determinants contacting DcR3 (Figure 3). Multiple residues are involved in polar contacts, including the side chains and/or main chains of DcR3 Y78-E86. We previously hypothesized that the broad specificity of DcR3 arises from the recognition of invariant backbone determinants and conserved side chains in the DE loops of the three ligands (Zhan et al., 2011). Comparison of LIGHT:DcR3 and TL1A:DcR3 structures supports this notion. For example, the side chain hydroxyls of TL1A Y188/LIGHT Y173 make polar contacts with the main chain nitrogen of DcR3 Q80 (Figure S3; N.B.: the numbering for TL1A follows that found in UNIPROT entry O95407 including the signal peptide sequence). Mutation of TL1A Y188 severely compromised DcR3 binding highlighting the importance of this interaction (Zhan et al., 2009). The carbonyl oxygen of TL1A S187 (analogous to LIGHT R172) forms a hydrogen bond with the main chain amide of DcR3 Y84. The main chain carbonyl oxygens of TL1A E200 and P201 (analogous to LIGHT E175 and E176) form potential hydrogen bonds with a guanidinium nitrogen on the side chain of DcR3 R89.
Figure 3.

Comparison of the structures of the LIGHT:DcR3 and TL1A:DcR3 complexes reveals similar interaction determinants on both ligand and receptor. The upper and lower regions are separated by a blue dashed line. The upper region of the interface is colored blue. The DE loop of the lower region is colored yellow and the other parts of the lower region are colored red. (A) “Open book” view of LIGHT:DcR3 binding surfaces. The DcR3 molecule was rotated around one axis by 180° to expose the binding interface. (B) “Open book” view of TL1A:DcR3 binding surfaces. (C) Alignment of the three DcR3 ligands LIGHT, TL1A and FasL shows less than 35% sequence identity. The binding residues shaded red, yellow and blue in A and B are correspondingly colored. See also Figure S4.
As observed in the LIGHT:DcR3 complex, additional interaction determinants in the lower region of TL1A are contributed by the AA′ and GH loops (Figure 3). Two separate segments of TL1A in the AA′ loop (V101-R103 and E120-A126) and the tip of the GH loop (Y238-E241) are buried at the interface with the end of DcR3 CRD2 (Figure 3). Among these interactions, the main chain carbonyl oxygen of TL1A T239 (analogous to LIGHT R228) makes a hydrogen bond with the amide nitrogen of DcR3 L94, while the side chain carbonyl oxygen of TL1A E241 forms a polar contact with the side chain amide nitrogen of DcR3 N92 (Figure S4).
Although the interfaces involving the upper regions of TL1A and LIGHT utilize similar surfaces, the detailed spatial organization of DcR3 in this region is distinct in the two complexes. Structural superposition of the LIGHT:DcR3 and TL1A:DcR3 complexes, based on alignment of the LIGHT and TL1A ligands, showed that most of DcR3 CRD2 aligns well (Figure S5) (Hasegawa and Holm, 2009). However, relative to the TL1A:DcR3 complex, when bound to LIGHT, the C-terminal portion of DcR3 is shifted (~15°), resulting in a more angular DcR3 architecture and a different spatial organization of the upper binding regions (Figure S5). This variation results from distinct interactions in the upper region; LIGHT binds to DcR3 mainly through hydrophobic interactions, while TL1A utilizes polar contacts. For example, G150-V152 of LIGHT, which contributes to this interface (see above), has no polar contacts with DcR3. In contrast, residue TL1A R156 (analogous to LIGHT G150) contacts the main chain oxygen of G124 on DcR3 through a side chain guanidinium nitrogen.
DcR3-responsiveness of LIGHT
The asymmetric unit of the LIGHT crystals contains a tightly packed trimeric assembly typical of TNF ligands. Each protomer buries 2130 Å2 of solvent accessible surface area in the trimer, similar to that observed in TL1A (1960 Å2, PDB entry 2QE3) and other TNF ligands (TNF alpha, 2370 Å2, PDB entry 1TNF; lymphotoxin alpha, 1950 Å2, PDB entry 1TNR; TRAIL, 2180 Å2, PDB entry 1D2Q). Light scattering analysis revealed an apparent molecular weight of 55 KDa, consistent with the calculated molecular weight (52 KDa) of trimeric LIGHT (Figure S6). Superposition of the receptor-free LIGHT and TL1A (PDB entry 2QE3) monomers shows a similar overall scaffold with RMSD of 1.4 Å for 124 structurally equivalent Cα atoms and RMSD of 1.5 Å for superposition of the intact trimers (Figure S2). DcR3-bound and - unbound LIGHT are similar with a RMSD of 1.0 Å for 138 aligned Cα atoms (Figure 4). The largest deviations are located in loops involved in contacting DcR3, suggesting that these segments of LIGHT are conformationally responsive to receptor engagement. The most significant structural alterations involve T170-E178 in the DE loop, G100-N102 in the AA′ loop and R226-T231 in the GH loop (Figure 4). Significant DcR3-dependent structural changes were also observed in the analogous loops in TL1A (Zhan et al., 2011).
Figure 4.
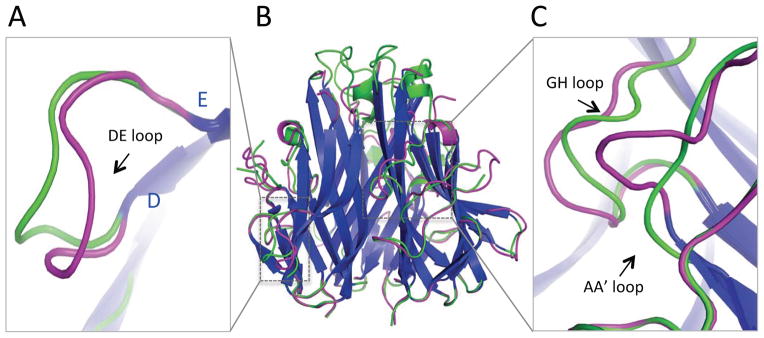
DcR3-dependent conformational changes of LIGHT. (A) Zoom in view of the DE loop shows significant DcR3-dependent structural alterations. (B) Superposition of LIGHT unbound (green) or bound (magenta) with DcR3 shows slight changes in the two parallel beta-sheets (blue) but significant conformational changes in the loops. (C) Zoom in view of the AA′ and GH loops shows significant DcR3-dependent structural alterations. See also Figure S8.
Loop transplant from TL1A to LIGHT
Comparison of the TL1A:DcR3 and LIGHT:DcR3 complexes indicates that TL1A and LIGHT employ similar strategies for recognition of the same surfaces on DcR3; however, the DcR3 recognition loops in the ligands share low sequence identity (Figure 3). These same segments in LIGHT and TL1A confer binding specificities for their cognate signaling receptors (i.e., DR3 for TL1A; HVEM and LTβR for LIGHT). Based on these observation, we hypothesized that transplantation of loops from TL1A to the corresponding region of LIGHT might result in LIGHT mutants possessing wild type affinity for DcR3 and reduced affinity for the signaling receptors of LIGHT (Figure 5).
Figure 5.
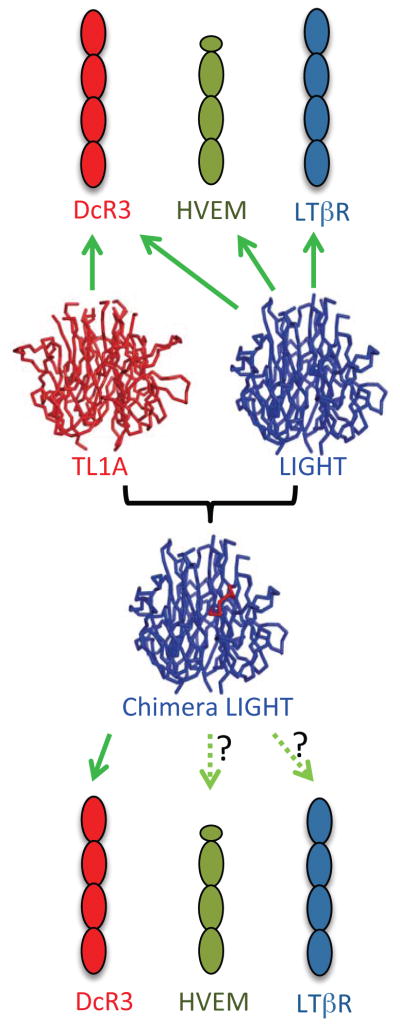
Rationale for designing LIGHT mutants. TL1A (red ribbon) and LIGHT (blue ribbon) both bind to DcR3; LIGHT also binds HVEM and LTβR, while TL1A does not. Loop transplantation of the binding sites from TL1A to LIGHT result in chimeric proteins which possess binding to DcR3 but reduced interactions with HVEM and LTβR. See also Figure S2.
Loops from both the lower (GH loops, residues R226-T231) and upper (EF loops, residues R195-W198) regions of TL1A were selected for transplantation into LIGHT and designated as mutant 1 and mutant 2, respectively (Figure S5; Table 2). The chimeric proteins were expressed in E. coli and exhibited apparent molecular weights similar to wild type LIGHT, as determined by size exclusion chromatography (SEC). Wild type LIGHT and LIGHT mutants were qualitatively evaluated for interactions with HVEM and LTβR by SEC. As with wild type LIGHT, significant peak shifts were observed upon mixing each mutant with DcR3, consistent with the mutants retaining binding to DcR3 (Figure S7). However, when mixed with HVEM, both mutants exhibited SEC traces differing from the wild type protein, consistent with impaired binding to HVEM (Figure S7).
Table 2.
Binding affinities of LIGHT with Receptors
| Mutation residues | KD (nM) | ||||
|---|---|---|---|---|---|
| 195–198 | 226–231 | DcR3 | HVEM | LTβR | |
| Wild Type | RVWW | RLRDGT | 16.7±0.3 | 18.7±0.2 | 11.2±0.6 |
| Mutant 1 | RVWW | DYTKED | 36.0±1.8 | 2500±2200 | 21.0±3.3 |
| Mutant 2 | SNWF | RLRDGT | 153±54 | 271±37 | 17.2±14 |
To more accurately quantify the interactions of the mutants with DcR3-Ig, HVEM-Ig and LTβR-Ig, surface plasmon resonance (SPR) was employed to measure equilibrium dissociation constants (KDs). Mutant 1, which has the loop transplanted from the lower interaction region, largely retains nearly wild type binding affinity to DcR3 (i.e., approximately 2-fold decrease in the affinity comparing to wild type LIGHT) (Table 2). However, mutant 2, which has the loop transplanted from the upper region, exhibits approximately 10-fold loss in affinity for DcR3 (Table 2). Mutant 1 and mutant 2 exhibited approximately 100-fold and 10-fold reductions in affinity for HVEM, respectively, suggesting that the GH loop may contribute more to the binding of HVEM than the EF loop. Both mutants bound LTβR with affinities close to those exhibited by the wild type protein.
Crystal structures of LIGHT mutant 1 and mutant 2 bound to DcR3 show that the interfaces and overall structural features present in the wild type structure are preserved in these complexes (Table 1; Figure S1). In both LIGHT mutants, residue Y173 occupies the hydrophobic groove of DcR3 and forms a hydrogen bond with main chain of Q80 in a fashion indistinguishable from the wild type LIGHT:DcR3 complex (Figure S3). In the mutant 1 LIGHT:DcR3 complex, the binding pattern of the transplanted loop is very similar to that observed in the TL1A:DcR3 structure (PDB entry 3K51), in which the TL1A T239 (analogous to LIGHT R228) carbonyl oxygen makes a main chain contact with the DcR3 L94 amide nitrogen, and TL1A E241 (analogous to LIGHT G230) and DcR3 N92 side chains interact via hydrogen bonding (Figure S4).
Like wild type LIGHT and TL1A, DcR3-dependent conformational alterations were present in the analogous loops on LIGHT mutant 2 (Figure S8). Notably, unlike mutant 1:DcR3 complex, the transplanted loop in mutant 2 does not fully mimic the conformation observed in the TL1A:DcR3 complex. Although the backbone of the transplanted loop in LIGHT mutant 2 superimposes well with the same loop in TL1A, the side chains adopt different conformations compared to TL1A (Figure S4).
Effects on Signaling
It has been reported that LIGHT-associated pathways inhibit proliferation and trigger the apoptosis of HT-29 tumor cells, possibly through engagement of HVEM and LTβR. To assess if the altered binding affinities of the LIGHT mutants resulted in new functional properties, we examined their effects over a range of concentrations (0, 50ng/mL, 100ng/mL or 200ng/mL) on HT-29 tumor cells. Our results show that wild type LIGHT induced apoptosis and reduced the viability of HT-29 cells in a dose-dependent manner, reaching saturation at a concentration of ~100ng/mL (Figure 6). Under the conditions employed, ~10% of untreated HT-29 cells undergo apoptosis, while treatment with wild type LIGHT results in a three-fold increase in apoptosis at the concentration of 100ng/mL. The two LIGHT mutants also elicited dose-dependent effects; however, each mutant exhibited reduced activity, eliciting only a two-fold increase in apoptosis relative to the untreated cells at 100ng/mL. Wild type LIGHT (100ng/mL) decreases the viable cells of HT-29 to about 70% of the control group. The mutant proteins exhibit reduced effects on HT-29 cell viability (~80% of control group) (Figure 6).
Figure 6.
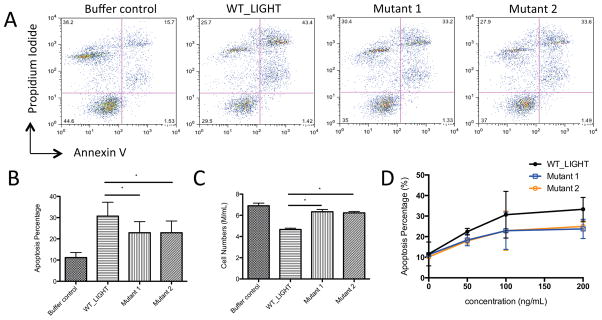
Biological activities of LIGHT on HT-29 tumor cells. (A) FACS analysis of the apoptosis of HT-29 cells. The population of late apoptotic cells is indicated as propidium iodide and annexin-V double positive events in the upper right quadrant. (B) The apoptosis percentages of HT-29 from different treatments (wild type and mutants) at protein concentrations of 100ng/mL. Stars indicate P values below 0.05% by paired t-test; error bars are calculated from experiments performed in triplicate. (C) The effect of different treatments on inhibition of HT-29 growth at protein concentrations of 100ng/mL. (D) The apoptosis percentage of HT-29 from treatments at different protein concentrations (0, 50, 100, 200 ng/mL). Stars indicate P values below 0.05% by paired t-test; error bars are calculated from experiments performed in triplicate. See also Figure S1.
Since both mutants show little change in affinity for LTβR, but significant reductions in affinities for HVEM (~100 fold reduction for mutant 1 and 10 fold reduction for mutant 2), the reduced induction of apoptosis rate and cell viability are likely the result of impaired LIGHT:HVEM signaling. Although the two mutants compromise the biological activities of LIGHT, at saturation the effect is incomplete, suggesting the involvement of LIGHT:LTβR or other unknown LIGHT-related pathways.
Unusual Higher Order Assembly of the LIGHT:DcR3 Complex
While the LIGHT:DcR3 complex described above is typical of the TNF/TNFR superfamilies, crystal packing results in an unusual dimer of hetero-hexamers. Two canonical LIGHT:DcR3 hexamers form an interlocking assembly in which one hexamer is rotated by 180 degrees perpendicular to the threefold axis and rotated by 40 degrees about the threefold axis to form the dodecamer. Contacts between LIGHT and DcR3 chains within the canonical hexamer are referred to as “cis” interactions, while contacts between the canonical hexamers are termed “trans” interactions (Figure 7). The dodecamer is stabilized by trans interactions between DcR3 CRD4 and the AA′ and DE loops of LIGHT, and by anti-parallel trans contacts between two DcR3 molecules (Figure S9). No trans contacts occur between the LIGHT trimers. The solvent accessible surface area buried between each trans interacting LIGHT and DcR3 is 560 Å2 and involves hydrophobic interactions and hydrogen bonds. The buried surface area associated with each DcR3-DcR3 interaction is 460 Å2 and includes many hydrogen bonds. The N-linked glycan on N173 of DcR3 contacts the adjacent anti-parallel DcR3 molecule, further stabilizing the assembly (Figure S9). The other potential N-linked glycan site is LIGHT N102, which is not near the cis and trans interaction surfaces, and is thus unlikely to impact the interactions between LIGHT and DcR3. The total buried solvent accessible surface area for the trans interaction is ~5300 Å2, compared to ~3100 Å2 for the total buried surface area associated with the canonical 3:3 LIGHT-DcR3 cis interaction. Interestingly, the structures of the LIGHT mutants in complex with DcR3 show crystal packing (i.e., the unusual dodecamer) similar to the wild type LIGHT:DcR3 complex, despite different crystallization conditions (different pH, salt and precipitants).
Figure 7.
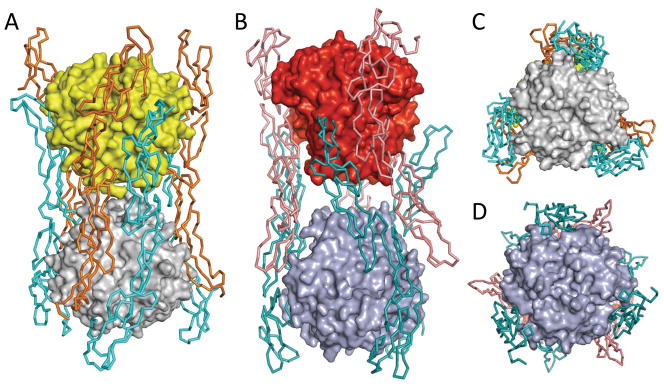
LIGHT:DcR3 forms interlocking hexamers similar to those observed in TNF:TNFR2 (PDB entry 3ALQ). The ligands LIGHT and TNF are both shown as surface representations. The receptors DcR3 and TNFR2 are shown as ribbons. (A) Side view of the LIGHT:DcR3 complex. For clarity, one canonical LIGHT:DcR3 hexamer is colored grey (LIGHT) and cyan (DcR3) and the other is colored yellow (LIGHT) and orange (DcR3). (B) Side view of the TNF:TNFR2 complex. For clarity, one canonical TNF:TNFR2 hexamer is colored light blue (TNF) and teal (TNFR2) and the other is colored red (TNF) and salmon (TNFR2). (C) Bottom view of the interlocking hexamers of LIGHT:DcR3 complex. (D) Boom view of the interlocking hexamers of the TNF:TNFR2 complex. See also Figure S6.
Although the structure of LIGHT:DcR3 suggests a model in which the trans interlocking hexamer plays some biological role, light scattering analysis shows that the LIGHT:DcR3 complex exists as a single hetero-hexamer in solution (Figure S6). Furthermore, we could not detect interactions between cell surface expressed LIGHT and soluble DcR3. Drosophila S2 cells were separately transfected with plasmids encoding full length LIGHT:red fluorophore (mCherry) fusions or full length LIGHT: green fluorophore (eGFP) fusions. Addition of the soluble DcR3 failed to direct the formation of red:green cell conjugates (data not shown). This behavior indicates that two interlocking hexamers are only weakly associated and that the observed dodecamer may be the consequence of crystal contacts.
DISCUSSION
We determined the crystal structures of LIGHT and the LIGHT:DcR3 complex, as well as the structures of LIGHT mutants in complex with DcR3. All of these structures exhibited typical TNF/TNFR superfamily organization, with the receptor binding along the length of the interprotomer interfaces of the compact LIGHT trimer and a 3:3 receptor:ligand stoichiometry. These structures revealed the chemical and physical determinants specific for the LIGHT:DcR3 interaction and the mechanisms contributing to the broad specificity of DcR3. Consistent with the TL1A:DcR3 structure, CRD2 of DcR3 contributes the major determinants for recognition of the lower region of LIGHT. Despite the modest sequence identity between LIGHT and TL1A (<35%), and the even greater divergence in the loop regions, DcR3 utilizes the same surface for recognition of both ligands. This broad specificity appears, at least in part, to be the due to the recognition of invariant main chain residues and conserved side chains present in LIGHT, TL1A and FasL.
As observed in the TL1A:DcR3 complex, the two parallel beta sheets, which form the core of LIGHT, are insensitive to DcR3 binding, while the loops involved in DcR3 recognition exhibit receptor-dependent structural rearrangements. Determinants in these loops are also involved in recognition of the cognate signaling receptors for TL1A, LIGHT and FasL. The segregation of these determinants in relatively flexible and readily deformable loop segments affords a facile mechanism for adoption of local conformations required for each ligand to optimally engage DcR3, as well as their cognate signaling receptors. In the upper region of the interaction interface, flexibility between CRD2 and CRD3 also contributes to the ability of DcR3 to recognize LIGHT, TL1A and FasL.
Based on these structures, two mutants of LIGHT were generated by transplanting loops from TL1A. Mutant 1 and mutant 2 exhibited 2-fold and 10-fold reductions in affinities for DcR3 relative to wild type LIGHT. Both mutants show compromised binding (mutant 1 has 100 fold reduction and mutant 2 has 10 fold reduction) to HVEM, but little difference in binding to LTβR relative to wild type LIGHT. The two loop transplant mutants of LIGHT provide an opportunity to dissect the roles of HVEM and LTβR in HT-29 biology. Our results show that both mutants have compromised activities for inducing apoptosis compared to wild type LIGHT. We conclude the reduced apoptosis effect is most likely the result of affinity reduction to HVEM, indicating that interaction of LIGHT and HVEM plays a role in HT-29 apoptosis.
Application of all crystallographic and non-crystallographic symmetry operators results in the formation of an interlocking dodecameric assembly, which was observed under a number of distinct crystallization conditions (Figure S1). Interestingly, the TNF:TNFR2 structure reported by (Mukai et al., 2010) exhibits an arrangement of interlocking hexamers similar to that observed in the LIGHT:DcR3 crystal structure (Figure 7). The overall structure of the LIGHT:DcR3 dodecamer is more compact than the TNF:TNFR2 complex, as the distance between the centers of mass of the two LIGHT trimers is ~59Å compared to ~68Å for the two TNF trimers (Figure 7). In addition, the receptors embracing the ligands are more constrained in the LIGHT:DcR3 complex than in the TNF:TNFR2 complex, with C-terminus of TNFR2 extending away from the ligands (Figure 7). It was suggested that since TNFR2 is a membrane-anchored protein the interlocking dodecamer observed in the crystalline state would be inaccessible for plasma membrane associated TNF and TNFR2; however, as the TNF receptors can also exist as soluble species, these steric constraints may be relaxed in certain circumstances (Mukai et al., 2010; Spoettl et al., 2007). DcR3 exists solely as a soluble molecule, obviating steric conflicts that might preclude formation of the interlocking assembly in vivo. Interestingly, a natural variant of LIGHT, E214K, which is close to the trans interaction interface (Figure S9), has been implicated in inflammatory diseases (Cheung et al., 2010). However, solution and cell-based studies failed to detect this higher order assembly and at present no direct experimental evidence exists supporting a physiological role for the interlocking dodecamer.
In conclusion, we have determined the crystal structures of LIGHT and the LIGHT:DcR3 complex that defined the determinants responsible for the broad ligand recognition properties of DcR3. Based on these structures, chimeric LIGHT molecules were constructed which allowed for the biochemical and functional dissection of LIGHT and provided insights into the roles of the LIGHT:HVEM and LIGHT:LTβR interactions.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of human LIGHT and DcR3
LIGHT cDNA was synthesized commercially (Genscript). The extracellular domain (L83-V240) was cloned into the pMT/BiP/V5-His A vector (Invitrogen) and cotransfected into Drosophila S2 cells with the pCoBlast (Invitrogen) plasmid at a 20:1 ratio. A stable cell line was selected with Blasticidin following the manufacture’s protocol (Invitrogen). LIGHT expression was induced with copper sulfate (500 μM final concentration). LIGHT protein from filtered culture supernatant was purified by Ni-NTA column (QIAGEN) and size exclusion chromatography (HiLoad Superdex 75; Amersham). The cloning, expression and purification of DcR3 has been described previously (Zhan et al., 2011).
LIGHT mutants were generated by cDNA synthesis and the corresponding cDNAs cloned into pET3a. All LIGHT mutants were expressed and refolded using published methods (Zhang et al., 2002). Briefly, LIGHT mutants were expressed in E. coli and refolded from inclusion bodies and purified by size exclusion chromatography using Superdex 200 gel-filtration columns.
Crystallization, Data Collection, and Structure Determination
LIGHT protein (10 mg/mL in HEPES, pH 7.0) was crystallized by sitting drop vapor diffusion using 0.5uL of protein and 0.5uL of precipitant composed of 1.26M monobasic sodium phosphate monohydrate, 0.14M dibasic potassium phosphate and 10% (V/V) 0.2M NDSB-211, pH 5.6, at 17°C. LIGHT:DcR3 complex (5 mg/mL in HEPES, pH 7.0) was crystallized by sitting drop vapor diffusion by combining 0.5 uL of protein and 0.5 uL of precipitant composed of 0.2M sodium chloride, 0.1M sodium citrate/citric acid buffer and 1.0M dibasic ammonium phosphate, pH 5.5, at 17°C. All crystals were flash-cooled in mother liquor supplemented with 20% glycerol. LIGHT and LIGHT:DcR3 crystals exhibited diffraction consistent with the space group I212121 (94.69, 99.98, 124.38) and P213 (a=b=c=149.06), respectively.
All mutant protein samples were crystallized by sitting drop vapor diffusion by combining 0.5uL of protein (5 mg/mL in HEPES, pH 7.0) and 0.5uL of precipitant at 17°C. The final crystallization conditions were: LIGHT mutant 2, 0.2M dibasic ammonium citrate, and 20% (W/V) PEG 3350, pH 5.0; mutant 1 LIGHT:DcR3 complex, 0.1M imidazole:HCl, 2.5M sodium chloride, pH 8.0; mutant 2 LIGHT:DcR3, 0.2M magnesium chloride, 0.1M tris:HCl, 2.5M sodium chloride, pH 7.0. LIGHT mutant 2 crystals were flashed-cooled and mounted directly. Crystals of mutant 1 LIGHT:DcR3 and mutant 2 LIGHT:DcR3 complexes were mounted in mother liquor supplemented with 20% glycerol. LIGHT mutant 2 exhibited diffraction consistent with space group P21 (a=59.64, b=46.91, c=70.40, β=98.02) and the two mutant LIGHT:DcR3 complexes both exhibited diffraction consistent with space group P213 (Mutant 1, a=b=c=149.27; Mutant 2, a=b=c=148.75).
All diffraction data were collected at beam line X29 of the National Synchrotron Light Source, Brookhaven National Laboratory. Data were integrated and scaled with HKL2000 and further processed with the programs within the CCP4 software package (Otwinowski and Minor, 1997; Winn et al., 2011). The models were further built using Coot and refined by PHENIX or REFMAC5 (Adams et al., 2010; Emsley et al., 2010; Murshudov et al., 1997) without NCS restraints. The figures in the paper were generated using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrodinger, LLC). The solvent accessible surface areas were calculated by online server PDBePISA with probe radius of 1.4 Å (Krissinel and Henrick, 2007).
Size-Exclusion Chromatography – Multi-Angle Light Scattering (SEC-MALS)
A solution of LIGHT and DCR3 (1 mg/mL) in buffer containing 20 mM HEPES, 150 mM NaCl, 1mM EDTA, pH 7.5 or pH 5.5, was subjected to size exclusion chromatography using a WTC030N5 (Wyatt Technology Corporation) column coupled to a Shimadzu HPLC system. LIGHT and DCR3 were also mixed in molar ratios of 1:1, 2:1 and 1:2 and subjected to SEC-MALS. Light scattering measurements were performed downstream, using a miniDawn TREOS instrument connected to the column output, followed by Optilab rEX refractive index analysis (Wyatt Technology Corporation). Control experiments were carried out with BSA diluted in the same buffer as the sample. Data from these experiments were collected and interpreted using ASTRA software (version 6.0.3.16).
Surface Plasmon Resonance Binding Assay
The recombinant DcR3-Ig, HVEM-Ig and LTβR-Ig were purchased from R&D Systems and immobilized on a CM5 sensor chip (GE Life Sciences). The binding of soluble wild type and mutant LIGHTs to these receptors was examined at 25°C using a BIAcore 3000 optical biosensor. The wild type and mutants of LIGHT were injected and flowed over the chip at concentrations ranging from 3nM to 0.3μM at a flow rate of 20μL/min. The signals derived from different concentration were corrected with the response of the blank channel (no protein immobilized in the channel). The resulting data were plotted and analyzed with Prism 5 (Graphpad Software) using the one site-total model [equation: ; Bmax: maximum specific binding; Kd: equilibrium dissociation constant].
Biological activity Assay
0.5 × 106 HT-29 cells were cultured in 6-well plates with 10% fetal bovine serum, DMEM medium. Cells were cultured overnight and treated with (0, 50ng/mL, 100ng/mL or 200ng/mL) purified LIGHT or LIGHT mutants for 72h. The endotoxin level in all protein samples was less than 0.25EU/mL as determined by LAL method (Genescript endotoxin assay Kit). The numbers of live cells in each treatment were determined by the trypan blue exclusion method (Zhai et al., 1998). The cells were also analyzed by Annexin-V staining (BD Pharmingen) to measure apoptosis (Zhai et al., 1998). Briefly, HT-29 cells were washed twice with cold PBS and resuspended in binding buffer to a concentration of approximately 1 × 106 cells/mL. 100μL of resuspended cells were mixed with 5μL of Annexin V and 5μL of Propidium Iodide. After gentle mixing and incubation for 15 min at room temperature in the dark, 400μL of binding buffer was added and the samples subjected to flow cytometry analysis. The experiments are performed in triplicate and the significance of the resulting data was evaluated by the paired t-test.
Supplementary Material
Highlights.
LIGHT binds to DcR3 with 3:3 stoichiometry
DcR3 interacts with invariant atoms in the DE loop of LIGHT
Loop transplantation from TL1A to LIGHT generates novel mutants
Acknowledgments
We thank the staff of X29A beam lines at the National Synchrotron Light Source for the help. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. Data for this study were measured at beamline X29A of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources (P41RR012408) and the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health. We also thank Dr. Steven M. Larson for providing the HT-29 cell line, Dr. Teresa DiLorenzo for critical reading of the paper. This work was supported by National Institute of Health Grant GM094662 (to S.C.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nature reviews Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- Bendtzen K. Anti-TNF-alpha biotherapies: perspectives for evidence-based personalized medicine. Immunotherapy. 2012;4:1167–1179. doi: 10.2217/imt.12.114. [DOI] [PubMed] [Google Scholar]
- Browning JL, Miatkowski K, Sizing I, Griffiths D, Zafari M, Benjamin CD, Meier W, Mackay F. Signaling through the lymphotoxin beta receptor induces the death of some adenocarcinoma tumor lines. The Journal of experimental medicine. 1996;183:867–878. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunological reviews. 2009;229:244–258. doi: 10.1111/j.1600-065X.2009.00783.x. [DOI] [PubMed] [Google Scholar]
- Chang YC, Chan YH, Jackson DG, Hsieh SL. The glycosaminoglycan-binding domain of decoy receptor 3 is essential for induction of monocyte adhesion. J Immunol. 2006;176:173–180. doi: 10.4049/jimmunol.176.1.173. [DOI] [PubMed] [Google Scholar]
- Chang YH, Hsieh SL, Chen MC, Lin WW. Lymphotoxin beta receptor induces interleukin 8 gene expression via NF-kappaB and AP-1 activation. Experimental cell research. 2002;278:166–174. doi: 10.1006/excr.2002.5573. [DOI] [PubMed] [Google Scholar]
- Chen MC, Hwang MJ, Chou YC, Chen WH, Cheng G, Nakano H, Luh TY, Mai SC, Hsieh SL. The role of apoptosis signal-regulating kinase 1 in lymphotoxin-beta receptor-mediated cell death. The Journal of biological chemistry. 2003;278:16073–16081. doi: 10.1074/jbc.M208661200. [DOI] [PubMed] [Google Scholar]
- Cheung TC, Coppieters K, Sanjo H, Oborne LM, Norris PS, Coddington A, Granger SW, Elewaut D, Ware CF. Polymorphic variants of LIGHT (TNF superfamily-14) alter receptor avidity and bioavailability. J Immunol. 2010;185:1949–1958. doi: 10.4049/jimmunol.1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JP, Felder M, Kapur A, Onujiogu N. DcR3 binds to ovarian cancer via heparan sulfate proteoglycans and modulates tumor cells response to platinum with corresponding alteration in the expression of BRCA1. BMC cancer. 2012;12:176. doi: 10.1186/1471-2407-12-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck MJ, Sprang SR. The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. The Journal of biological chemistry. 1989;264:17595–17605. doi: 10.2210/pdb1tnf/pdb. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke B, Autschbach F, Kim S, Lasitschka F, Strauch U, Rogler G, Gdynia G, Li L, Gretz N, Macher-Goeppinger S, et al. Functional characterisation of decoy receptor 3 in Crohn’s disease. Gut. 2009;58:483–491. doi: 10.1136/gut.2008.148908. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Holm L. Advances and pitfalls of protein structural alignment. Current opinion in structural biology. 2009;19:341–348. doi: 10.1016/j.sbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Miura Y, Nishiyama T, Mitani M, Tateishi K, Sakai Y, Hashiramoto A, Kurosaka M, Shiozawa S, Doita M. Decoy receptor 3 expressed in rheumatoid synovial fibroblasts protects the cells against Fas-induced apoptosis. Arthritis and rheumatism. 2007;56:1067–1075. doi: 10.1002/art.22494. [DOI] [PubMed] [Google Scholar]
- Hsu MJ, Lin WW, Tsao WC, Chang YC, Hsu TL, Chiu AW, Chio CC, Hsieh SL. Enhanced adhesion of monocytes via reverse signaling triggered by decoy receptor 3. Experimental cell research. 2004;292:241–251. doi: 10.1016/j.yexcr.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Hsu TL, Wu YY, Chang YC, Yang CY, Lai MZ, Su WB, Hsieh SL. Attenuation of Th1 response in decoy receptor 3 transgenic mice. J Immunol. 2005;175:5135–5145. doi: 10.4049/jimmunol.175.8.5135. [DOI] [PubMed] [Google Scholar]
- Kim YS, Nedospasov SA, Liu ZG. TRAF2 plays a key, nonredundant role in LIGHT-lymphotoxin beta receptor signaling. Molecular and cellular biology. 2005;25:2130–2137. doi: 10.1128/MCB.25.6.2130-2137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- Morishige T, Yoshioka Y, Inakura H, Tanabe A, Yao X, Tsunoda S, Tsutsumi Y, Mukai Y, Okada N, Nakagawa S. Creation of a LIGHT mutant with the capacity to evade the decoy receptor for cancer therapy. Biomaterials. 2010;31:3357–3363. doi: 10.1016/j.biomaterials.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, Yamagata Y, Tsutsumi Y. Solution of the structure of the TNF-TNFR2 complex. Science signaling. 2010;3:ra83. doi: 10.1126/scisignal.2000954. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Rooney IA, Butrovich KD, Glass AA, Borboroglu S, Benedict CA, Whitbeck JC, Cohen GH, Eisenberg RJ, Ware CF. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. The Journal of biological chemistry. 2000;275:14307–14315. doi: 10.1074/jbc.275.19.14307. [DOI] [PubMed] [Google Scholar]
- Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, Cheroutre H, Ware CF. Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol. 2001;167:6330–6337. doi: 10.4049/jimmunol.167.11.6330. [DOI] [PubMed] [Google Scholar]
- Spoettl T, Hausmann M, Klebl F, Dirmeier A, Klump B, Hoffmann J, Herfarth H, Timmer A, Rogler G. Serum soluble TNF receptor I and II levels correlate with disease activity in IBD patients. Inflammatory bowel diseases. 2007;13:727–732. doi: 10.1002/ibd.20107. [DOI] [PubMed] [Google Scholar]
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, Chen L. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. doi: 10.4049/jimmunol.164.8.4105. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Szymkowski DE. The TNF superfamily in 2009: new pathways, new indications, and new drugs. Drug discovery today. 2009;14:1082–1088. doi: 10.1016/j.drudis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, Tamada K, Chen L, Fu YX. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. The Journal of clinical investigation. 2001;108:1771–1780. doi: 10.1172/JCI13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, Tumanov AV. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. 2010;32:403–413. doi: 10.1016/j.immuni.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annual review of immunology. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Han B, Sheng H, Lin M, Moore PA, Zhang J, Wu J. Clinical significance of detecting elevated serum DcR3/TR6/M68 in malignant tumor patients. International journal of cancer Journal international du cancer. 2003;105:724–732. doi: 10.1002/ijc.11138. [DOI] [PubMed] [Google Scholar]
- You RI, Chang YC, Chen PM, Wang WS, Hsu TL, Yang CY, Lee CT, Hsieh SL. Apoptosis of dendritic cells induced by decoy receptor 3 (DcR3) Blood. 2008;111:1480–1488. doi: 10.1182/blood-2007-09-114850. [DOI] [PubMed] [Google Scholar]
- Young HA, Tovey MG. TL1A: a mediator of gut inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8303–8304. doi: 10.1073/pnas.0602655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. The Journal of biological chemistry. 1999;274:13733–13736. doi: 10.1074/jbc.274.20.13733. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, Jiang GW, Lu J, Tan J, Ugustus M, et al. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. The Journal of clinical investigation. 1998;102:1142–1151. doi: 10.1172/JCI3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C, Patskovsky Y, Yan Q, Li Z, Ramagopal U, Cheng H, Brenowitz M, Hui X, Nathenson SG, Almo SC. Decoy strategies: the structure of TL1A:DcR3 complex. Structure. 2011;19:162–171. doi: 10.1016/j.str.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C, Yan Q, Patskovsky Y, Li Z, Toro R, Meyer A, Cheng H, Brenowitz M, Nathenson SG, Almo SC. Biochemical and structural characterization of the human TL1A ectodomain. Biochemistry. 2009;48:7636–7645. doi: 10.1021/bi900031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Salcedo TW, Wan X, Ullrich S, Hu B, Gregorio T, Feng P, Qi S, Chen H, Cho YH, et al. Modulation of T-cell responses to alloantigens by TR6/DcR3. The Journal of clinical investigation. 2001;107:1459–1468. doi: 10.1172/JCI12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Schwartz JC, Almo SC, Nathenson SG. Expression, refolding, purification, molecular characterization, crystallization, and preliminary X-ray analysis of the receptor binding domain of human B7-2. Protein expression and purification. 2002;25:105–113. doi: 10.1006/prep.2002.1616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.