

Abstract
Background and Objective
Dipyrone is an analgesic and antipyretic drug usually prescribed for patients with inflammatory conditions. We recently identified dipyrone as an anti-apoptotic agent by screening a library of 1040 compounds for their ability to inhibit cytochrome c release from isolated mitochondria. We investigated the potential neuroprotective properties of dipyrone in cerebral ischemia.
Methods
We evaluated the protective effects of dipyrone in experimental models of neuronal hypoxia/ischemia, including an oxygen/glucose deprivation model in primary cerebrocortical neurons and a focal cerebral ischemia model in mice.
Results
Dipyrone reduced hypoxia/ischemia injury in both cellular and animal models. Dipyrone inhibited the release of cytochrome c and other mitochondrial apoptogenic factors from mitochondria into the cytoplasm, and attenuated subsequent caspase-9 and caspase-3 activation both in vitro and in vivo. Moreover, dipyrone prevented ischemia-induced changes in Bcl-2 and tBid, and ameliorated OGD-mediated loss of mitochondrial membrane potential. Dipyrone also inhibited ischemia-induced reactive microgliosis. In the cellular models evaluated, dipyrone did not inhibit OGD-induced COX-2 activation.
Conclusion
This study demonstrates that dipyrone is remarkably neuroprotective in cerebral ischemia, and its COX-independent protective properties are, at least in part, due to the inhibition of mitochondrial cell death cascades.
Keywords: Cerebral ischemia, Cytochrome c release, Dipyrone, Drug screen, Mitochondria, Neuroprotection
INTRODUCTION
Stroke is the third leading cause of death in industrialized countries and a major cause of long-term disability. More than 80% of all strokes are a consequence of a permanent or prolonged arterial occlusion, resulting in energy failure with hypoxic-ischemic neuronal death 1–4.
Molecular changes in the mitochondria are central to many cell death pathways, particularly those caused by ischemia/hypoxia 5–7. A critical cell death triggering event is the release of cytochrome c and other mitochondrial proteins (e.g., cytochrome c, Smac/Diablo, apoptosis inducing factor [AIF], and endonucease G) into the cytoplasm in response to a variety of apoptotic stimuli, which then trigger an orchestrated set of events leading to swift cellular demise. The balance of Bcl-2 family proteins, concentration of cytosolic calcium, and reactive oxygen species regulate the key steps that result in the release of these factors into the cytoplasm 2, 8–12. Therefore, blocking release of these mitochondrial factors should inhibit cell death.
Release of cytochrome c, which is considered to be a commitment step in this mitochondrial cell death cascade, induces assembly of the apoptosome (i.e. cytochrome c, Apaf-1, and procaspase-9), resulting in caspase-9 activation. Caspase-9 activates caspase-3, which is a terminal effector caspase, catalyzing the final steps in the cell death cascade 4, 12–14. Preventing this molecular process blocks cells from entering a late, irreversible phase in the cell death pathway 10, 12. Consequently, pathways which modulate cytochrome c release are targets for potential therapies for neurodegenerative disease 15–18.
As described in our recent report 19, we screened a library of 1040 FDA-approved drugs and other bioactive compounds for their ability to inhibit cytochrome c release from purified mitochondria. We identified 21 compounds which strongly inhibited Ca2+-induced mitochondrial cytochrome c release. Of these 21 compounds, 16 indeed inhibited neuronal cell death, some exhibiting an IC50 in the nanomolar range. The most potent drug from the cellular model, dipyrone, was evaluated in this report to determine its neuroprotective activity in primary cerebrocortical neurons (PCNs) exposed to oxygen/glucose deprivation (OGD) and in mice subjected to middle cerebral artery occlusion (MCAO).
Dipyrone (a.k.a. metamizol) is an analgesic and antipyretic drug without apparent anti-inflammatory properties 20–22. Rare, but severe agranulocytosis has been linked to dipyrone, and therefore its clinical use is prohibited in the United States. Due to its effectiveness, low cost, and the low incidence of agranulocytosis, it is still commonly used in many countries 20, 23. Similar to other non-steroidal anti-inflammatory drugs (NSAIDs), the antipyretic action of dipyrone is associated with its inhibition of cyclooxygenase (COX) and the consequent decrease in prostaglandin synthesis in the peripheral and central nervous systems at high concentrations24, 25. Subcutaneous (s.c.) or intracerebroventricular (i.c.v.) administration of dipyrone at high doses (500 mg/kg and 10mg, respectively) decreases basal core temperature in normal mice and prevents fever induced by interleukin-1 beta (IL-1β), lipoploysaccharide (LPS), and tumor necrosis factor-alpha (TNF-α) 24, 26. Intraperitoneal (i.p.) treatment with a single high dose of dipyrone (100mg/kg) prevents cerebral ischemia-reperfusion induced hyperthermia in rats, and thereby reduces neuronal damage 27. Having found that dipyrone inhibits cytochrome c release in a cell-free system and that it has strong antiapoptotic activity in a striatal cell line, we evaluated the drug’s protective effects in experimental models of in vitro hypoxia and in vivo ischemia. We evaluated PCNs subjected to OGD. The cellular model was intended to evaluate the direct anti-apoptotic property of dipyrone where temperature related protection would not play a role as it may occur in vivo. Experiments on cultured cells were complemented by in vivo trials using mice subjected to MCAO. In this focal cerebral ischemia model, a low concentration of dipyrone administered either systemically or via direct intra-cerebro-ventricular (i.c.v.) delivery was used to exclude the possibility of antipyretic-related protection. We evaluated whether dipyrone can ameliorate hypoxic-ischemic injury, and whether this neuroprotective property is associated with inhibition of mitochondrial cell death pathways.
MATERIALS AND METHODS
Animals, induction of focal cerebral ischemia and drug treatment
Animal experiments were performed in accordance with the guidelines for the care and use of laboratory animals and were approved by the Animal Care Committee of Harvard Medical School.C57BL/6 male mice, weighing 22–25 g, were assigned randomly to treatment groups. Animals were allowed free access to food and water in a temperature-controlled environment at 25°C before and after surgery.
Mice were initially anesthetized with 2% (v/v) isoflurane in 70% N2O and 30% O2. Isoflurane concentration was thereafter titrated to 1.5% during the surgery. During all surgical procedures, rectal temperature was maintained between 37.0 and 37.5°C with a heating pad (Harvard Apparatus, Cambridge, MA). Focal cerebral ischemia was induced by MCAO as described previously 28. Briefly, under the operating microscope, the right common carotid artery (CCA) was exposed, and the right external carotid artery (ECA) and the right internal carotid artery (ICA) were isolated. The ECA was ligated distally, and a silicon-coated 7-0 nylon suture (180–220 μm diameter) was introduced into the ECA thorough a small incision on the stump of the ECA and pushed up the ICA until resistance was felt and the filament was inserted 9 to 10 mm from the carotid bifurcation, effectively blocking the middle cerebral artery (MCA). A laser-Doppler probe (Probe 407, Perimed, Jarfalla, Sweden) was attached to the skull at the right temporal fossa (6mm lateral and 1mm posterior of bregma) to continually monitor cerebral blood flow (CBF). The surgical procedure was considered adequate if >70% reduction in regional CBF occurred immediately after MCA occlusion; otherwise mice were excluded. Data were expressed as a mean percentage of the pre-ischemic baseline values. In order to control for experimental variability caused by the operation, control animals were subjected to the same procedures without occlusion of blood flow. Blood pressure was not different among mice of the various groups.
Dipyrone (Sigma Co., St. Louis, MO) was dissolved in 0.9% saline and made fresh daily. Dipyrone (100ng) or saline, which served as vehicle-control, was injected intracerebroventricularly (i.c.v.; 1.0 mm lateral, 0.5 mm posterior, 2.3 mm deep of bregma) 30 minutes before ischemia. The volume of the i.c.v. injections was 2 μl, delivered over 30 seconds. For systemic treatment, dipyrone (10mg/kg) or saline was administered by intraperitoneal (i.p.) injection 30 minutes before ischemia in a volume of 10 ml/kg body weight.
Neurobehavioral test
Each animal was assessed for neurological deficits 2 and 24 hours after onset of focal ischemia. The following scoring was used: 0, no observable deficits; 1, forelimb flexion when lifted by the tail; 2, forelimb flexion and consistently reduced resistance to lateral push; 3, forelimb flexion, reduced resistance to lateral push, and unilateral circling toward the paretic side; 4, forelimb flexion and ambulation inability or difficulty 29–32.
Infarct assessment by TTC staining
After 24 hours of ischemia, animals were anesthetized again and decapitated. Brains were rapidly removed and chilled in ice-cold saline for 10 minutes. Coronal sections (1-mm-thick, n=7) were cut by use of a mouse brain matrix (RBM 2000C, ASI Instruments), then stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma Co., St. Louis, MO) at 37°C for 15 minutes. After scanning, the infarct (i.e., unstained) area on each slice was evaluated by digital imaging with a computerized image analyzer (SPOT 3.5 Biometrics software, Silicon Graphics Inc., Mountain View, CA), and the infarct volume was calculated by summing up the infarct area in the seven slices. Infarct volumes were expressed as a percentage of the contralateral hemisphere volume by using an indirect method to compensate for edema formation in the ipsilateral hemisphere 31, 33.
Vc = volume of intact contralateral [left] hemisphere
Vi = volume of intact regions of the ipsilateral [right] hemisphere
Mitochondrial Physiology
Mitochondrial respiration was measured using the oxygen sensitive fluorescent dye A65-N1 (Luxcel) with isolated rat liver mitochondria in a buffer containing 250 mM sucrose, 10 mM Hepes/KOH, 2 mM K2HPO4, pH 7.4. ADP (300 μM) or FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone, 100 μM) was added to assess state 3 respiration and uncoupled respiration, respectively. Time-resolved fluorescence was measured at specified excitation/emission wavelength (λex/em) of 380/640 nm as described elsewhere 34.
The resistance of isolated mitochondria to calcium overload was assessed in a buffer containing 250 mM sucrose, 10 mM HEPES, 5 mM K-succinate, 1 mM KH2PO4, 2.5 μM EDTA, pH 7.4. Isolated rat liver mitochondria (0.5 mg/ml) were challenged with 10 μM CaCl2. Ca2+ fluxes and mitochondrial membrane potential (ΔΨm) was measured using fluorescent dye Ca-Green-5N (0.3 μM, λex/em=488/535 nm) and TMRM (Tetramethylrhodamine methyl ester, 0.1 μM, λex/em=546/590 nm), respectively. Mitochondrial swelling was simultaneously measured as changes of light scattering at λ =540 nm.
Primary cerebrocortical neurons (PCNs) culture and oxygen/glucose deprivation (OGD) treatment
Culture of PCNs and OGD treatment were performed as previously described 28. Cerebral cortex of mouse embryos at day 15 (E15) were freed from meninges and separated from the olfactory bulb and hippocampus. Trypsinized cells were suspended in medium [neurobasal medium (NBM) with 2% (vol/vol) B27 supplement, 2 mM glutamine, 100 units/ml penicillin and streptomycin (GIBCO)] and seeded at a density of 2×104 per cm2 on polylysine-coated dishes. Cells were used for experiments on day 7 of culture. PCNs were preincubated with or without dipyrone (1μM) for 2 hours, and then challenged with OGD. The culture medium was replaced by glucose-free Earle’s balanced salt solution, and cells were placed in an anaerobic chamber with BBL GasPak Plus (BD Pharmingen), which was used to delete oxygen to <100 ppm within 90 min. Control cells were incubated in Earle’s balanced salt solution with glucose in a normoxic incubator for the same period. After 3 hours, OGD was terminated by a return to normal culture conditions.
Lactate dehydrogenase (LDH) assay
The extent of cell death was determined by the LDH assay performed according to the manufacturer’s instructions (Roche Products). Briefly, each reaction mixture (100 μl) was added to conditioned media (100 μl) removed from dishes after centrifugation at 250 ×g for 10 min. Absorbance of samples at 490 nm was measured in an ELISA reader after 15 min of incubation at room temperature. The same volume of blank medium was used as the background control.
Rhodamine 123 staining
After OGD treatment, the cultured (living) PCNs were directly incubated with 2 μmol/L rhodamine 123 (Molecular Probes) for five minutes at room temperature followed by rinsing three times with phosphate-buffered saline (five minutes per rinse) 17, 35. Digital images were taken with a Nikon ECLIPSE TE200 fluorescence microscope and processed with IP LAB software. Reduced green rhodamine 123 fluorescence indicates dissipated ΔΨm.
Tissue and cell fractionation
Samples of total lysate and cytosolic/mitochondrial fraction from brain tissues (ipsilateral hemisphere minus 2 mm from the frontal and occipital poles) or cultured cells were prepared as described previously 36, 37. Briefly, for total protein lysate samples, brain tissues or cells were homogenized on ice in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 142.5 mM KCl, 5 mM MgCl2, 10 mM Hepes, pH 7.4) with protease inhibitor cocktail (Sigma, St. Louis, MO) and PMSF (0.1 mg/ml; Fluka, Switzerland). Lysates were centrifuged twice at 10,000 ×g for 20 min at 4°C. The supernatants were obtained for western blot analysis of caspase-3, caspase-9, Bcl-2, and Bid. For cytosolic fractionation, tissue or cells were gently homogenized in cold buffer (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 2 mM EDTA, 1 mM DTT, 10 mM Hepes, pH 7.4, plus protease inhibitor cocktail and 0.1 mg/ml PMSF), followed by a 700 × g centrifugation for five minutes at 4 °C, the supernatant was centrifuged again at 15,000 × g for 25 minutes at 4°C and used as the cytosolic component for the determination of released cytochrome c, Smac/Diablo, and AIF by western blot. The final pellets were lysed completely by RIPA buffer for 15 min on ice and obtained as mitochondrial fraction.
Western blot
Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA). Samples of total protein lysates and cytosolic/mitochondrial fractions (50–100 μg protein) were run on 12% or 15% SDS-PAGE gels and probed with the requisite antibodies. Antibodies were purchased from the following suppliers: antibody to pro- or active caspase-3, caspase-9, Cell Signaling Technology (Beverly, MA); Bcl-2 antibody, Santa Cruz Biotechnology (Santa Cruz, CA); Bid antibody, R&D systems (Minneapolis, MN); Cytochrome c antibody, BD PharMingen (San Diego, CA); Smac/Diablo antibody, Novus Biologicals (Littleton, CO); AIF antibody, Sigma. (St. Louis, MO). β-actin (Sigma) or COX IV (Abcam) was used as a loading control.
Cyclooxygenase-2 (COX-2) fluorescent assay
COX-2 activity was determined by the COX fluorescent assay (Cayman chemical, Ann Arbor, MI). Briefly, cultured cells were collected by centrifugation at 1,000 × g for 10 min at 4°C, and sonicated in cold buffer (100 mM Tris-Hcl, pH 7.5, plus protease inhibitor cocktail and 0.1 mg/ml PMSF), then centrifuged at 10,000 × g for 15 min at 4°C to get supernatant as the cell lysate sample. Lysates (10μl) were assayed and fluorescence was recorded using an excitation wavelength of 544 nm and emission wavelength of 595 nm.
IL-1β immunoassay
Generation of mature IL-1β was measured using an ELISA kit (R&D systems, Minneapolis, MN). Brains were removed eight hours after the beginning of permanent ischemia. The ipsilateral hemisphere minus 2 mm from the frontal and occipital poles was dissected out, weighed, and homogenized in lysis buffer (0.1 M PBS with 0.1% TritonX-100, 4 mM EDTA, plus protease-inhibitor cocktail and 0.1 mg/ml PMSF). Homogenates were centrifuged at 50,000 × g for 30 min at 4°C, and 50μl supernatant was used for each measurement 30. Supernatants from PCN cultures following OGD treatment were also assayed for mature IL-1β.
Immunohistochemistry
After 24 hours of ischemia, mice were anesthetized and transcardially perfused with cold PBS and then with 4% paraformaldehyde/PBS. Brains were dissected out, post-fixed for 24 hours, cryoprotected with 30% sucrose/PBS, then frozen in the presence of OCT medium (Sakura Finetek, Torrance, CA), and stored at −80°C until used. 20-μm-thick coronal cryosections were cut and stained with CD11b antibody (1:100, Chemicon, Temecula, CA) with the ABC Elite kit (Vector Laboratories, Burlingame, CA) and visualized with diaminobenzidine. Samples were counterstained with hematoxylin (Sigma). Brain sections were also stained with Iba1 antibody (1:750, Wako, Richmond, VA) and Alexa Fluor 488 secondary antibody, or AIF antibody (1:100, Cell Signaling) and Alexa Fluor 546 secondary antibody, and these fluorescent slices were counterstained with DAPI (Sigma).
RESULTS
Dipyrone inhibited cytochrome c release from isolated mitochondria, but did not affect mitochondrial respiration or permeability transition in isolated mitochondria
As described above, dipyrone was initially identified from a library of 1040 compounds by screening for their ability to inhibit cytochrome c release in a cell-free assay with isolated mitochondria. In a dose-dependent manner, dipyrone inhibited Ca2+ induced cytochrome c release in isolated mitochondria by 22.5% or 46.4% (P< 0.01) at 10 or 20 μM, respectively 19.
In addition to playing a vital role in apoptotic cell death, the chief function of the mitochondria is to generate energy for cellular activity. Energy generation involves the processes of aerobic respiration coupled with the tricarboxylic acid cycle and the mitochondrial electron transport chain. To evaluate the effects of dipyrone on mitochondrial metabolism, we monitored the rate of oxidative phosphorylation (assessed as state 3 respiration) and maximal respiratory capacity of the isolated mitochondria using ADP (Fig. 1A) and FCCP, respectively. Dipyrone did not significantly affect mitochondrial state 3 respiration at the concentrations lower than 5 mM (range tested from 0.1 to 10000 μM). Greater concentrations apparently were toxic, with estimated IC50 for the respiration inhibition was found to be 10 ± 2 mM. The inhibitory effect appeared the same for ADP and FCCP (data not shown). These results suggested that primary mitochondrial functions were maintained with dipyrone present up to millimolar concentrations.
Figure 1.

Dipyrone does not affect normal mitochondrial functions in isolated mitochondria. A, Dipyrone did not significantly alter mitochondrial respiration in the isolated mitochondria over a range of concentrations from 0.1 to 1000 μM. ADP stimulated mitochondrial state 3 respiration rate was determined using the oxygen sensitive fluorescent dye A65-N1 with isolated rat liver mitochondria (0.5mg/ml). Values are presented as mean ± S.E.M. (n=5). B, Dipyrone did not affect Ca2+-induced mPT in the isolated mitochondria over a range of concentrations from 0.1 to 1000 μM. Mitochondrial mPT was assessed by swelling amplitude with the determination of light scattering changes. Values are presented as mean ± S.E.M. (n=5). C, Typical multi-parameter measurements of the Ca2+-induced mPT in the presence of dipyrone at 0 to 100 μM. “Ca-fluxes”, “ΔΨm”, and “Swelling” represent changes in extramitochondrial Ca2+ concentration, mitochondrial membrane potential, and light scattering, respectively. To induce mPT, after preincubation with dipyrone, 10 μM Ca2+ ions was added to aliquot of rat liver mitochondria (0.5mg/ml) at time point “0”. Fluorescent dyes Ca-Green-5N (0.3 μM) and TMRM (0.1 μM) were used to measure Ca-fluxes and ΔΨm, respectively. Swelling was simultaneously measured as changes of light scattering.
The putative mitochondrial permeability transition pore consists of a multimeric complex of proteins spanning the inner and outer membranes. Its opening is a component of mitochondrial dysfunction, resulting in the release of proapoptotic cytosolic proteins38. Having observed that dipyrone inhibited cytochrome c release from purified mitochondria, we next evaluated the effect of dipyrone on mitochondrial permeability transition (mPT) by simultaneously assessing Ca2+-efflux, mitochondrial membrane potential (ΔΨm) and mitochondrial swelling with the stimulation of exogenous Ca2+. As shown in Figure 1B and 1C, dipyrone did not have any significant effects on mPT induction at concentrations lower than 5 mM (range tested, 0.1 μM to 10 mM). The slight acceleration of mPT induction at 10 mM assessed by swelling (Fig. 1B) correlated with toxic effect of the dipyrone on the respiration. Typical kinetic changes of Ca-efflux, ΔΨm and swelling in the presence of dipyrone at 0 to 100 μM were presented on Figure 1C. These data indicated that dipyrone was not a direct mPT inhibitor, and its inhibitory effects on cytochrome c release were not a result of interfering with mitochondrial physiology, including Ca2+ transport or membrane potential change in isolated mitochondria.
Dipyrone inhibited OGD-induced PCNs cell death
We previously reported using a striatal cell line expressing mutant huntingtin, a cellular model of Huntington’s disease, that dipyrone inhibited cell death with an IC50 in the low nanomolar range 19. We therefore evaluated whether dipyrone inhibited neuronal cell death in an in vitro model of hypoxic injury. As reported, PCNs exposed to three hours of OGD die in a time-dependent fashion28. We therefore evaluated whether dipyrone could inhibit cell death in this primary neuronal model system, in particular if it did in such low concentrations. As measured by the LDH assay, dipyrone inhibited OGD-mediated cell death over a range of 0.1 nM to 1 μM (Fig. 2A, 2B). Dipyrone rescued OGD-challenged PCNs with IC50 of 0.2nM and maximum protection of 81%.
Figure 2.
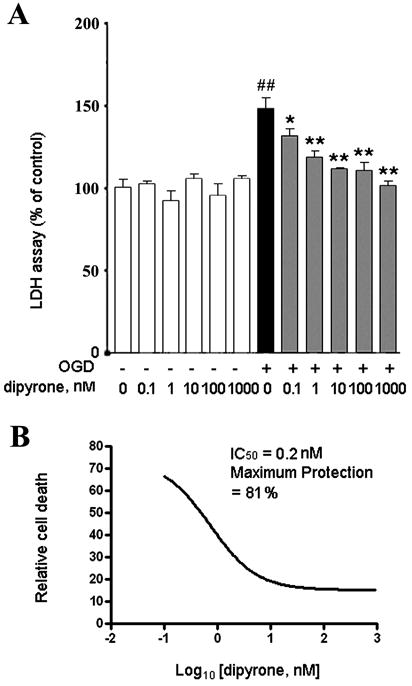
Dipyrone inhibits neuronal cell death of PCNs challenged by OGD. PCNs were pretreated for 2 hours with dipyrone over a wide range of concentrations (0.1 to 1000 nM), and then subjected to 3 hours of OGD. Cell death was evaluated by the LDH assay 16 hours after completion of OGD. Data from three independent experiments are presented as mean ± S.E.M. A, The extent of cell death is graphed against dipyrone concentration. Values are normalized with respect to the release of LDH in a null control (i.e., a cell culture that was not drug-treated and was never challenged by OGD). The black bar corresponds to the extent of cell death upon challenge with OGD in the absence of drug. Each grey bars corresponds to the extent of cell death upon OGD-treatment in the presence of the indicated concentration of dipyrone. ##, P < 0.01 versus the null control (i.e., cells not subject to OGD and not treated with dipyrone). *, P < 0.05; **, P < 0.01 versus the OGD-treated culture without dipyrone. B, The sigmoidal dose-response fitting curve is generated by the Graphpad Prism 4 software (Graphpad Software Inc., San Diego, CA). The data in figure A are plotted as a semi-logarithmic graph of the extent of cell death as a function of dipyrone concentration. The IC50 for dipyrone is 0.2 nM and the maximum protection is 81%.
Dipyrone inhibited OGD-mediated morphologic changes and release of mitochondrial apoptogenic factors in PCNs but did not affect COX-2 activation
Observation of PCNs by phase-contrast and fluorescent microscopy demonstrated that OGD caused nuclear shrinkage as well as loss of axo-dendritic arborization. Dipyrone partially restored normal neuronal morphology (Fig. 3A). Different from the results in isolated mitochondria, another manifestation of dipyrone’s rescue of PCNs was the preservation of ΔΨm. Proper ΔΨm was critical for appropriate cellular bioenergetic homeostasis, and its loss was an important event associated with progression of mitochondrial dysfunction, leading to cell death. In healthy cells, rhodamine 123 staining demonstrated a high intensity/punctuate pattern of fluorescence due to preferential uptake by negatively charged mitochondria (Fig. 3B). Following OGD, cellular fluorescence became diffuse due to mitochondrial depolarization. Dipyrone countered OGD-associated dissipation of rhodamine 123 fluorescence, indicating that ΔΨm was maintained. These data provide evidence that dipyrone not only inhibited cell death, but based on cellular morphology and ΔΨ m, protected cellular structure and function.
Figure 3.

Dipyrone inhibits the release of apoptogenic factors from mitochondria, thereby rescuing cells from cell death. PCNs were pretreated for 2 hours with 1 μM dipyrone and challenged by 3-hour OGD. 16 hours after completion of OGD, cells were used for examinations. A, Cell morphology was studied by phase-contrast (200× magnification) and immunofluorescence microscopy (200× and 600× magnification). Apoptosis is evident from the change in the cells’ shape upon OGD. Pretreatment with dipyrone decreases the extent of cell death. Immunostaining with antibodies against MAP2 (red) and DAPI (blue) reveals the cells’ dendrites and nuclei, respectively. Scale bars, 40 μm. B, Dipyrone retains mitochondrial membrane potential (ΔΨm) in PCNs after OGD. The living cells were stained with 2 μmol/L rhodamine 123 to determine ΔΨm. Green fluorescence resulted from the accumulation of rhodamine 123 within negatively charged mitochondria, and diminished fluorescence, as was observed with apoptotic cells, indicated mitochondrial depolarization and the dissipation of ΔΨm. C, D, The release of mitochondrial factors and concomitant activation of caspase in response to OGD was analyzed by western blot. Cells were fractionated to obtain either cytosolic components or total cell lysates, and then used respectively for determination of cytochrome c and AIF (cytosolic fractions, C), or caspase-3 (total lysates, D). β-actin staining was used as an internal loading control. The blots are representative of three independent experiments. E, COX-2 activity was detected by the COX fluorescent assay in cell lysates. OGD caused a statistically significant increase in COX fluorescence in PCNs, and this increase there was not diminished by preincubation with dipyrone. Data are presented as mean ± S.E.M. of four independent experiments. #, P < 0.05 versus null-control group.
OGD induces the translocation of cytochrome c and AIF from the mitochondria to the cytoplasm 11, 28. Moreover, these two molecules play important roles in the triggering of terminal caspase-dependent and caspase-independent cell death pathways. Given that dipyrone inhibited cytochrome c release from purified mitochondria, and cytochrome c was released in OGD treated PCNs, we next evaluated whether dipyrone-mediated neuroprotection was associated with inhibition of OGD-induced cytochrome c and AIF release. Indeed, dipyrone blocked release of these two mitochondrial factors into the cytoplasm. As expected for an event downstream of mitochondrial changes, activation of the “executioner” caspase-3 was also decreased in the presence of dipyrone (Fig. 3C, 3D).
Dipyrone is a typical NSAID exhibiting non-selective inhibition of COX-2, which is believed to be the target of its pharmacological activity 25. COX is the key rate-limiting enzyme in the synthesis of prostaglandins from arachidonic acid. Three isoforms of the enzyme have been identified in mammalian cells. COX-2 is an inducible enzyme expressed in association with inflammation, including in the central nervous system. To determine whether dipyrone-mediated neuroprotection correlated with its ability to inhibit COX-2, we analyzed COX-2 activities in cell lysates from PCNs subjected to OGD. We detected a significant increase in COX-2 activity following OGD. Dipyrone (1 μM) did not inhibit OGD-induced COX-2 activation (Fig. 3E). This result suggested that the mechanism of dipyrone-mediated protection in this model was not related to its ability to inhibit COX-2.
Neuroprotection of systemic Dipyrone administration in focal cerebral ischemia-induced injury
As demonstrated above, dipyrone inhibited OGD-induced PCN cell death. Neuroprotection observed in cell culture was independent of any effect on temperature. We therefore speculated that dipyrone has neuroprotective properties in vivo beyond what can be attributed to the drug’s antipyretic effect. In particular, we were interested in whether the drug’s neuroprotective properties could be dissociated from its action as an antipyretic. Dipyrone was administered intraperitoneally in the mouse permanent focal cerebral ischemia model induced by intraluminal suture occlusion of the middle cerebral artery (MCA). Regional cerebral blood flow and body temperature were monitored before and during ischemia (Fig. 4C). Dipyrone, at the dose of 10 mg/kg, had no antipyretic effect on core temperature at 2 or 24 hours following MCA occlusion. However, significant improvements in neurobehavioral score were observed in the dipyrone-treated group at both 2 hours and 24 hours of ischemia compared with the vehicle-treated group (n=8, P<0.01) (Fig. 4A). Brain lesion size was determined 24 hours after the onset of ischemia by staining with 2% TTC. Correlating with improved behavioral score, we found that dipyrone-treatment reduced cerebral infarct volume by 42.8% compared with that of vehicle-treated mice (27.0% ± 4.0% vs 47.2% ± 2.9%; n=8, P<0.01) (Fig. 4B).
Figure 4.
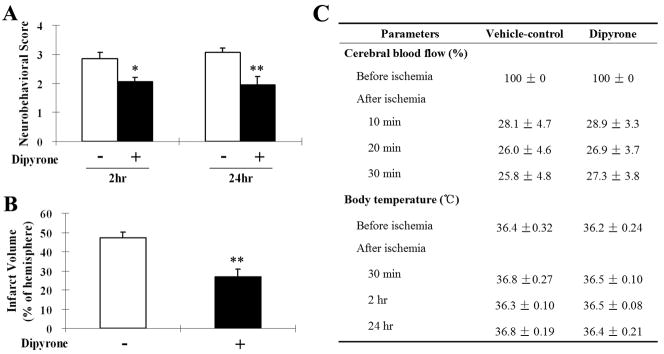
Improvements in neurological behavior (A) and infarct volume (B) of systemic administration of dipyrone (i.p.) in the mouse model of focal cerebral ischemia. Cerebral ischemia was induced by intraluminal suture occlusion of middle cerebral artery. Dipyrone (10 mg/kg) was intraperitoneally administered 30 minutes before the onset of MCAO. A, Treatment with dipyrone decreased the neurological score of mice tested both 2 and 24 hours after the onset of MCAO. (Using the scale described in the Methods section, a higher neurological score corresponds to a greater behavioral deficit.) B, Dipyrone also decreased the volume of infracts in the brain resulting from MCAO. Infarct volume was determined by 2% TTC staining of coronal brain sections after 24 hours of ischemia. C, Physiological parameters were monitored before and after the onset of ischemia. No statistical difference was found in body (rectal) temperature between the vehicle and the dipyrone-treated group. Data are presented as mean ± S.E.M. (n=9). *, P < 0.05; **, P < 0.01 versus vehicle-control group.
Intracerebroventricular injection with dipyrone diminished neurological deficit and reduced brain lesion size from focal cerebral ischemia
To minimize the known peripheral side effects following systemic administration, we evaluated the effect of dipyrone when delivered directly into the brain by intracerebroventricular (i.c.v.) injection. Mice injected with dipyrone or saline vehicle control were subject to permanent focal cerebral ischemia by MCAO. During the surgical procedure and subsequent recovery from anesthesia, the rectal temperature of all animals was maintained at a level between 37.0 and 37.5°C with a temperature-control system. After 24 hours of ischemia (but not after only 2 hours), compared with sham-operated group, the core temperature in vehicle group, as well as in dipyrone-treated group, both slightly increased (the difference was not statistically significant); and the temperature in these two groups was almost same at all time points (Fig. 5D). Therefore, i.c.v. injection of 100ng of dipyrone had no antipyretic effect following MCAO. As shown in Figure 5A, i.c.v. injection with dipyrone significantly improved the animals’ neurobehavioral score both at 2 and 24 hours of ischemia relative to vehicle-treated controls (n=10, P<0.05). Lesion size was measured 24 hours following the onset of ischemia as described for the systemic drug administration (Fig. 5B). Dipyrone also reduced MCAO-induced infarct volume from a control value of 48.5% ± 5.6% to 27.3% ± 5.8% (n=10, P<0.05) (Fig. 5C).
Figure 5.

Improvements in neurological behavior (A) and infarct volume (B, C) of intracerebroventricularly (i.c.v.) injection of dipyrone in the mouse model of focal cerebral ischemia. Cerebral ischemia was induced by intraluminal suture occlusion of middle cerebral artery. Dipyrone (100 ng) was i.c.v. delivered 30 min before the onset of MCAO. A, Neurobehavioral scores were assessed 2 and 24 hours after ischemia as scale described in the Methods section. B, The TTC-stained coronal brain sections are from representative animals of sham-operated group, vehicle-control group and dipyrone-treated group, respectively. Brains were removed after 24 hours of MCAO, and coronal sections thereof were stained with 2% TTC. The infarcted tissue is white, whereas live tissue is darkly stained by TTC. C, Brain infarct volume was determined by serial reconstruction from coronal sections prepared by staining with 2% TTC 24 hours after ischemia as in B. D, Physiological parameters were monitored before and after the onset of ischemia. No statistical difference was found in body (rectal) temperature between the vehicle and the dipyrone-treated group. Data are presented as mean ± S.E.M. (n=10). *, P < 0.05 versus vehicle-control group.
The paraffin sections were dewaxed and rehydrated for morphological staining with hematoxylin-eosin (H&E). The morphologically stained sections were examined using light microscopy. The results of H&E staining indicated that a large number of degenerative cells (pyknosis of nuclei, shrinkage or disruption of cytoplasm and the formation of numerous intra- and intercellular vacuoles) appeared in the cortex and striatum at 24 hour of ischemia. As shown in Fig. 6, these morphological changes observed by H&E staining were partially prevented upon the treatment with dipyrone.
Figure 6.

Improvements in morphology with the treatment of dipyrone (i.c.v.) in the mouse model of focal cerebral ischemia. Cerebral ischemia was induced by intraluminal suture occlusion of middle cerebral artery. Dipyrone (100 ng) was i.c.v. delivered 30 min before the onset of MCAO. Morphological H&E staining was performed in paraffin sections from brain of control and treated mice at 24 h after ischemia. Low- and high-power images are from the cortical (A–F) and striatal (G–K) brain sections of sham (A, D, G, J), vehicle-control (B, E, H, K) and dipyrone treated mice (C, F, I, L), respectively. Note that some dying cells showed features of degeneration: nuclear pyknosis, cell shrinkage and vacuolated cytoplasm. Scale bar, 100 μm.
Intracerebroventricular injection of dipyrone inhibited the release of mitochondrial apoptogenic factors and activation of caspases in ischemic brain
The first indication that dipyrone has anti-apoptotic activity was its potent inhibition of cytochrome c release from isolated mitochondria. Moreover, when administered to PCNs challenged with OGD, dipyrone inhibited the release of mitochondrial apoptogenic factors and caspase-3 activation (Fig. 3C, 3D). We next determined whether i.c.v. dipyrone inhibited the release of apoptogenic factors from mitochondria in ischemic brain territory. Following 12 hours of MCAO, the cytosolic and mitochondrial fractions of ischemic tissue were separated, and the respective levels of mitochondrial apoptogenic factors were measured by western blot. Western blot analysis revealed an increase of cytochrome c, smac/Diablo, and AIF in the cytosolic fractions and a corresponding decrease in the mitochondrial fractions after cerebral ischemia, and this documented translocation from mitochondria to cytosol of apoptogenic factors 17, 18 decreased substantially upon i.c.v. injection with dipyrone (Fig. 7A–C). After the release from the mitochondrial intermembrane space, AIF tends to redistribute to the nucleus and induce apoptotic phenotypes, including chromatin condensation as well as DNA fragmentation. As shown in Fig. 7D, revealed by immunofluorescent staining of brain sections, the release and subcellular redistribution of released AIF into the nucleus induced by ischemia was partially rescued by the treatment of dipyrone.
Figure 7.


Dipyrone delivered by intracerebroventricular (i.c.v.) injection inhibits molecular events associated with cell death in ischemic brain tissue. Cerebral ischemia was induced by intraluminal suture, thereby occluding the middle cerebral artery. 100 ng of dipyrone was administered by i.c.v. injection 30 minutes before the onset of MCAO. After 12 hours of ischemia, brains were removed and ischemic territories were isolated. Cytosolic fractions, mitochondrial fractions or total lysates were prepared by homogenization and centrifugation as described in the Methods section. A–C, Samples of the cytosolic (n=4) and mitochondrial (n=3) fractions were analyzed by western blot using antibodies to cytochrome c (A), Smac (B), and AIF(C). D, The release and nuclear redistribution of AIF in the brain after ischemia. Scale bar, 10 μm. E, Samples of total lysate were analyzed by western blot with antibodies to pro- and active caspase-3, caspase-9, Bcl-2, and Bid (n=3). β-actin or COX IV staining was used as an internal loading control to the cytosolic fractions/total lysates or mitochondrial fractions, respectively. Each lane in the blots represents a separate mouse. The bar graphs were generated by densitometry, and the values are presented as mean ± S.E.M. #, P < 0.05; ##, P < 0.01 versus sham-operated group. *, P < 0.05; **, P < 0.01 versus vehicle-control group.
Once in the cytosol, cytochrome c binds to Apaf-1, procaspase-9, and dATP to form the “apoptosome”. This molecular assembly drives the activation of caspase-9 via autocatalytic proteolysis. Mature caspase-9 activates “executioner” caspase-3 from its inert precursor 13, 14. The process is regulated, in part, by the antagonistic action of pro-apoptotic tBid and anti-apoptotic Bcl-2. These proteins bind to the membrane of mitochondria and play an important role in regulating the release of apoptogenic factors.
Having established that dipyrone inhibited the release of mitochondrial apoptogenic factors and diminished the extent of cell death, we investigated its effects upon molecular processes linking this cause with its effect. To this end, we measured the activities of caspases-3 and -9, the degree of Bcl-2 expression, and the extent of Bid cleavage in ischemic and non-ischemic territory. Dipyrone inhibited ischemia-induced activation of caspase-3 and caspase-9. Moreover, dipyrone inhibited ischemia-induced reduction of Bcl-2 and generation of tBid (Fig. 7E). These findings may explain dipyrone’s inhibitory activities of cytochrome c release and thereafter caspase-3 activation, and provide a possible explanation as to why dipyrone prevented ΔΨm loss in cells, but not in isolated mitochondria.
Dipyrone inhibited the generation of mature IL-1β in hypoxic PCNs and ischemic brain
Caspase-1 is unique in its ability to generate mature IL-1β from its protein precursor 39, 40. Consequently, measuring the level of mature IL-1β is a sensitive caliper of caspase-1 activity. In fact, the first indication that caspase-1 was activated by acute insult to the CNS was the elevation of mature IL-1β levels observed following ischemic and traumatic brain injury 29, 30. As measured by an ELISA assay, in both the in vivo and the in vitro models of cerebral ischemia, we observed a dramatic increase in the concentration of mature IL-1β. Whereas brain tissue from sham-operated mice contained 23.0 ± 3.9 pg/g IL-1β, there was 48.0 ± 6.7 pg/g in tissue that had been rendered ischemic by eight hours of MCAO (Fig. 8A). In mice given an i.c.v. injection of 100 ng of dipyrone, however, the concentration of IL-1β in ischemic tissue (21.9 ± 1.9 pg/g) resembled that for brains from sham-operated mice. Administering dipyrone had a comparable effect in the cellular model of ischemia: PCNs exhibited dramatically higher levels of IL-1β levels following three hours of OGD, but this increase was essentially eliminated by drug treatment (Fig. 8B).
Figure 8.
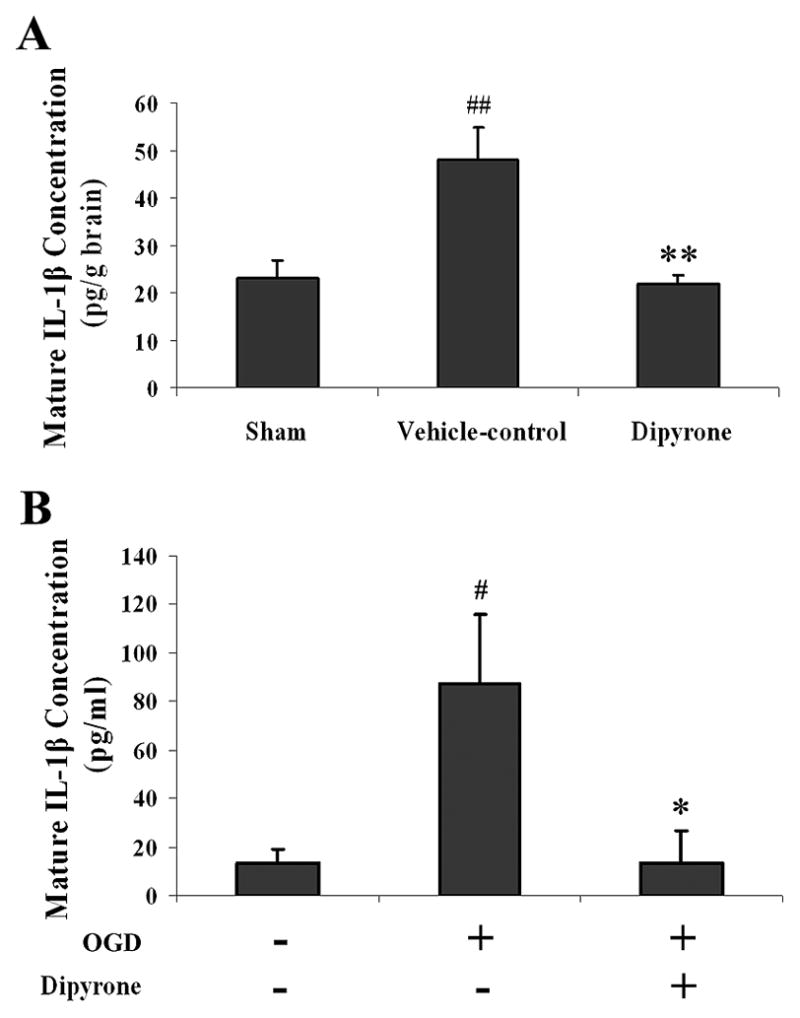
Treatment with dipyrone eliminates the surge of IL-1β generated in brain tissue upon focal ischemia and in PCNs upon OGD. Mature IL-1β levels were determined specifically with an ELISA kit in both in vivo and in vitro models. Values are presented as mean ± S.E.M. A, Mature IL-1β concentrations in ischemic brain territories. Dipyrone (100 ng) was administered by i.c.v. 30 minutes before the onset of MCAO. Ischemic brain tissues were isolated after 8 hours of ischemia. Subsequently, supernatants were prepared for IL-1β measurement (n=5~6). ##, P < 0.01 versus sham-operated group. **, P < 0.01 versus vehicle-control group. B, Mature IL-1β concentrations in PCNs. PCNs were pretreated with 1 μM dipyrone for 2 hours and then challenged with 3-hour OGD. 16 hours after the termination of OGD, cells were harvested, and lysates thereof were tested for IL-1β by ELISA (n=3). #, P < 0.05 versus null-control group (i.e., cells not subject to OGD and not treated with dipyrone). *, P < 0.05 versus naïve cells challenged with OGD.
Dipyrone attenuated microglial activation induced by focal cerebral ischemia
Microglial activation has been implicated in the pathogenesis of ischemic stroke 41, 42. Although activated microglia scavenge dead cells from the CNS, it is believed that this activation causes autoimmune responses which although debated, may lead to exacerbation of brain injury. Since microglia also derive from myeloid precursor cells, accordingly at the site of pathology, it is difficult to distinguish activated microglia from infiltrating macrophages that were derived from blood monocytes. Therefore, two microglial phenotypic markers, CD11b and Iba1, were both used in this study. CD11b has been widely used as a surface marker to detect activated microglia. As shown in Fig. 9A, relatively few microglial cells exhibited weak immunoreactivity to CD11b in the brain of sham-operated mice; whereas following cerebral ischemia, there was a large increase in CD11b immunoreactivity. I.C.V. injection of dipyrone dramatically reduced CD11b-immunoreactivity. Immunofluorescent staining with another marker-Iba1 (Fig. 9B), also confirmed that dipyrone attenuated microglial activation induced by focal cerebral ischemia in mice. These results indicate that dipyrone-mediated neuroprotection was associated with significant inhibition of ischemia-mediated reactive microgliosis.
Figure 9.

Dipyrone attenuates microglia activation induced by focal cerebral ischemia in mouse. Dipyrone (100 ng) was administered by i.c.v. 30 minutes before the onset of MCAO. Mice were sacrificed 24 hours after MCAO, and brains were removed and fixed. 20-μm-thick coronal cryosections were prepared and immunostained with antibody to CD11b (dark stained, A) or Iba1 (green, B). Hematoxylin or DAPI (blue) staining was used as the counterstain, respectively. As phenotypic marker of microglia, the increased expression of CD11b or Iba1 corresponds to severity of microglial activation. Scale bars, 150 μm.
DISCUSSION
Neuronal loss from aberrant apoptosis increases damage from ischemic or traumatic brain injury and drives neurodegenerative diseases 13, 14, 43. Consequently, proteins mediating pathways of apoptosis - caspases, Bcl-2 family proteins, and mitochondrial factors - are attractive targets for pharmacotherapy. Targeting these molecular processes, we developed a multi-step screen for experimental drugs starting with a cell-free assay, and our hypothesis is that inhibition of cytochrome c release is a potential therapeutic target for neuroprotection in neurodegenerative diseases. To test this hypothesis and identify novel neuroprotective agents, a library of 1040 compounds was screened for inhibitors of Ca2+-induced cytochrome c release from isolated mitochondria. A secondary screen was performed using a mutant huntingtin-induced death of a striatal cell line 19. Based on the screen results, dipyrone was identified as the most promising drug with potent inhibition of cytochrome c release and the most significant protection in striatal cells. Therefore in the present study, we evaluated the ability of dipyrone to mediate protection in complementary in vivo and in vitro models of neuronal hypoxic/ischemic injury. Our results demonstrated that dipyrone significantly decreased cytochrome c release both in vivo and in vitro, and thereby diminished ischemia/hypoxia-induced caspase activation
We found that nanomolar concentrations of dipyrone protected both a striatal cell line expressing mutant huntingtin (IC50=14.6 nM by MTS assay), as well as PCNs exposed to OGD (0.2 nM by LDH assay). By contrast, >30 μM dipyrone was necessary for significant toxicity in striatal cells 19; >300 μM for leukocytes 44. Moreover, the toxicity of dipyrone at high concentrations in leukemia cell lines was considered to be due to its apoptogenic effect, and this bipartite effect was implicated in the induction of agranulocytosis, the main significant side effect of dipyrone. Direct drug delivery into the CNS should minimize the risk of any systemic side effects. Therefore, in our in vivo experiments, we delivered a relatively low dose (100ng) of dipyrone by direct i.c.v. injection. Consequently, given the data we generated, we concluded that at the doses evaluated dipyrone was neuroprotective due to its antiapoptotic and not its antipyretic effect. These results support our contention that dipyrone’s anti-apoptotic effect underlies the drug’s benefit following cerebral ischemia.
To our knowledge, this is the first report to demonstrate that dipyrone’s antiapoptotic function underlies the neuroprotection which it exerts during cerebral ischemia. This property was different from the drug’s ability to inhibit Cox-2 and subsequent prostaglandin synthesis, the mechanism that usually accounts for its antipyretic/analgesic actions 21, 24. By contrast, as demonstrated in this study, dipyrone had alternative effects relevant to its protective effects: it prevented the drop in Bcl-2 expression and the increase in tBid levels associated with cerebral ischemia. As described above, Bcl-2 and tBid bound to the mitochondria regulating the release of mitochondrial apoptogenic factors. Therefore, we found that dipyrone could slow OGD associated dissipation of ΔΨm in PCNs, but did not affect Ca2+-induced mPT in isolated mitochondria. Dipyrone inhibited the production of mature IL-1β, a process that confirms that the drug inhibits caspase-1 activation. Caspase-1 plays a role in the cleavage of Bid and generation of tBid in cerebral ischemia 28, suggesting a potential feedback mechanism between caspase-1 activation and downstream mitochondrial pathways.
Dipyrone is widely administered as an antipyretic drug in Europe, Asia, Africa, and South America. Its clinical usage is prohibited in the US, however, due to the association with agranulocytosis. The frequency of the latter syndrome, a deficiency of innate immunity, is 3–19 per million among patients given dipyrone 45, 46. The potential benefit of dipyrone in the treatment of cerebral ischemia and stroke among serious and critical patients certainly outweighs the danger from this possible side effect. Moreover, the induction of agranulocytosis results from apoptosis that mostly follows treatment with high doses of dipyrone. Consequently, this side effect can be avoided by controlling drug levels and administering in a targeted manner.
In conclusion, we have demonstrated that dipyrone is significantly neuroprotective in both cellular and animal models of cerebral ischemia. Although the precise target for its mechanism of action has not been identified, dipyrone clearly has beneficial effects at the levels of the mitochondria, and key molecular targets critical for the modulation of hypoxic/ischemia injury. Therefore, dipyrone may be developed for use as a neuroprotective agent.
Acknowledgments
Sources of Funding and Acknowledgements
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant RO1 NS051756 (R.M.F.), RO1 NS039324 (R.M.F.), KO1 NS055072 (X.W.) and Hereditary Disease Foundation Grant (X.W.). We thank Ethan Shimony for editorial assistance.
References
- 1.Mohr JP, Caplan LR, Melski JW, et al. The Harvard Cooperative Stroke Registry: a prospective registry. Neurology. 1978;28:754–762. doi: 10.1212/wnl.28.8.754. [DOI] [PubMed] [Google Scholar]
- 2.Scorrano L, Oakes SA, Opferman JT, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 3.Silvestrelli G, Corea F, Paciaroni M, et al. The Perugia hospital-based Stroke Registry: report of the 2nd year. Clin Exp Hypertens. 2002;24:485–491. doi: 10.1081/ceh-120015324. [DOI] [PubMed] [Google Scholar]
- 4.Benchoua A, Guegan C, Couriaud C, et al. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001;21:7127–7134. doi: 10.1523/JNEUROSCI.21-18-07127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenner C, Kroemer G. Apoptosis. Mitochondria--the death signal integrators. Science. 2000;289:1150–1151. doi: 10.1126/science.289.5482.1150. [DOI] [PubMed] [Google Scholar]
- 6.Kristal BS, Stavrovskaya IG, Narayanan MV, et al. The mitochondrial permeability transition as a target for neuroprotection. J Bioenerg Biomembr. 2004;36:309–312. doi: 10.1023/B:JOBB.0000041759.35731.70. [DOI] [PubMed] [Google Scholar]
- 7.Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic Biol Med. 2005;38:687–697. doi: 10.1016/j.freeradbiomed.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 8.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 10.Martinou I, Desagher S, Eskes R, et al. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plesnila N, Zhu C, Culmsee C, et al. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:458–466. doi: 10.1097/00004647-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome C release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid Redox Signal. 2005;7:1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- 13.Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- 14.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 15.Narayanan MV, Zhang W, Stavrovskaya IG, et al. Promethazine: a novel application as a neuroprotectant that reduces ischemia-mediated injury by inhibiting mitochondrial dysfunction. Clin Neurosurg. 2004;51:102–107. [PubMed] [Google Scholar]
- 16.Teng YD, Choi H, Onario RC, et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101:3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang WH, Wang H, Wang X, et al. Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke. 2008;39:455–462. doi: 10.1161/STROKEAHA.107.496810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Zhu S, Pei Z, et al. Inhibitors of cytochrome c release with therapeutic potential for Huntington’s disease. J Neurosci. 2008;28:9473–9485. doi: 10.1523/JNEUROSCI.1867-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brogden RN. Pyrazolone derivatives. Drugs. 1986;32 (Suppl 4):60–70. doi: 10.2165/00003495-198600324-00006. [DOI] [PubMed] [Google Scholar]
- 21.Brune K. Comparative pharmacology of ‘non-opioid’ analgesics. Med Toxicol. 1986;1 (Suppl 1):1–9. [PubMed] [Google Scholar]
- 22.Schug SA, Manopas A. Update on the role of non-opioids for postoperative pain treatment. Best Pract Res Clin Anaesthesiol. 2007;21:15–30. doi: 10.1016/j.bpa.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Arellano F, Sacristan JA. Metamizole: reassessment of its therapeutic role. Eur J Clin Pharmacol. 1990;38:617–619. doi: 10.1007/BF00278592. [DOI] [PubMed] [Google Scholar]
- 24.De Souza GE, Cardoso RA, Melo MC, et al. A comparative study of the antipyretic effects of indomethacin and dipyrone in rats. Inflamm Res. 2002;51:24–32. doi: 10.1007/pl00000278. [DOI] [PubMed] [Google Scholar]
- 25.Pierre SC, Schmidt R, Brenneis C, et al. Inhibition of cyclooxygenases by dipyrone. Br J Pharmacol. 2007;151:494–503. doi: 10.1038/sj.bjp.0707239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada SG, Otterness IG, Stitt JT. A study of the mechanism of action of the mild analgesic dipyrone. Agents Actions. 1994;41:188–192. doi: 10.1007/BF02001915. [DOI] [PubMed] [Google Scholar]
- 27.Coimbra C, Drake M, Boris-Moller F, Wieloch T. Long-lasting neuroprotective effect of postischemic hypothermia and treatment with an anti-inflammatory/antipyretic drug. Evidence for chronic encephalopathic processes following ischemia. Stroke. 1996;27:1578–1585. doi: 10.1161/01.str.27.9.1578. [DOI] [PubMed] [Google Scholar]
- 28.Zhang WH, Wang X, Narayanan M, et al. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci U S A. 2003;100:16012–16017. doi: 10.1073/pnas.2534856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedlander RM, Gagliardini V, Hara H, et al. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hara H, Friedlander RM, Gagliardini V, et al. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Wang L, Li J, Wang XL. 2-(1-Hydroxypentyl)-benzoate increases cerebral blood flow and reduces infarct volume in rats model of transient focal cerebral ischemia. J Pharmacol Exp Ther. 2006;317:973–979. doi: 10.1124/jpet.105.098517. [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Chan PH, Chen J, et al. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 33.Swanson RA, Morton MT, Tsao-Wu G, et al. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 34.Will Y, Hynes J, Ogurtsov VI, Papkovsky DB. Analysis of mitochondrial function using phosphorescent oxygen-sensitive probes. Nat Protoc. 2006;1:2563–2572. doi: 10.1038/nprot.2006.351. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Figueroa BE, Stavrovskaya IG, et al. Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guegan C, Vila M, Teismann P, et al. Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol Cell Neurosci. 2002;20:553–562. doi: 10.1006/mcne.2002.1136. [DOI] [PubMed] [Google Scholar]
- 37.Zhu S, Li M, Figueroa BE, et al. Prophylactic creatine administration mediates neuroprotection in cerebral ischemia in mice. J Neurosci. 2004;24:5909–5912. doi: 10.1523/JNEUROSCI.1278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stavrovskaya IG, Narayanan MV, Zhang W, et al. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200:211–222. doi: 10.1084/jem.20032053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuida K, Lippke JA, Ku G, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 40.Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 41.Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- 42.Yenari MA, Xu L, Tang XN, et al. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 43.Dhib-Jalbut S, Arnold DL, Cleveland DW, et al. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. J Neuroimmunol. 2006;176:198–215. doi: 10.1016/j.jneuroim.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 44.Pompeia C, Boaventura MF, Curi R. Antiapoptotic effect of dipyrone on HL-60, Jurkat and Raji cell lines submitted to UV irradiation, arachidonic acid and cycloheximide treatments. Int Immunopharmacol. 2001;1:2173–2182. doi: 10.1016/s1567-5769(01)00144-8. [DOI] [PubMed] [Google Scholar]
- 45.Laporte JR, Carne X. Blood dyscrasias and the relative safety of non-narcotic analgesics. Lancet. 1987;1:809. doi: 10.1016/s0140-6736(87)92836-4. [DOI] [PubMed] [Google Scholar]
- 46.Risks of agranulocytosis and aplastic anemia. A first report of their relation to drug use with special reference to analgesics. The International Agranulocytosis and Aplastic Anemia Study. Jama. 1986;256:1749–1757. [PubMed] [Google Scholar]