

Abstract
A radio frequency-free electromagnetostatic (EMS) cell devised for electron-capture dissociation (ECD) of ions has been retrofitted into the collision-induced dissociation (CID) section of a triple quadrupole mass spectrometer to enable recording of ECD product-ion mass spectra and simultaneous recording of ECD-CID product-ion mass spectra. This modified instrument can be used to produce easily interpretable ECD and ECD-CID product-ion mass spectra of tyrosine-phosphorylated peptides that cover over 50% of their respective amino-acid sequences and readily identify their respective sites of phosphorylation. ECD fragmentation of doubly protonated, tyrosine-phosphorylated peptides, which was difficult to observe with FT-ICR instruments, occurs efficiently in the EMS cell.
INTRODUCTION
In modern proteomic applications of tandem mass spectrometry (MS/MS), proteins and peptides can be induced to fragment by a number of physicochemical processes, the most common in use being collision-induced dissociation (CID) [1], electron-transfer dissociation (ETD) [2], and electron-capture dissociation (ECD) [3–4]. Each of these latter three forms of dissociation has its advantages and disadvantages. CID is broadly applicable and operationally robust, but it frequently leaves gaps in the sequence-data and generally causes peptides to fragment excessively, destroying information about modifications in the process. ETD and ECD by contrast exhibit little selectivity for any particular amino acids (proline and amino acids associated with disulfide bonds are the outstanding exceptions) and generally preserve labile post-translational modifications, e.g., phosphorylation, O-glycosylation, and N-glycosylation, thereby making it possible to reliably determine the sites and nature of such modifications [3–4]. ETD and ECD are not as broadly applicable as CID, but both have been commercialized and are used as complements to CID. The only commercial access to ECD, however, remains on FT-ICR mass spectrometers, and in this form, its use in proteomics studies has to date been relatively limited.
Barely less than a decade ago, three groups of investigators independently succeeded in observing ECD in a linear ion trap [5–6], a three dimensional (3D) ion trap [7], and a digital 3D ion trap [8] respectively. In all three approaches, it was necessary to use He as a moderating gas either to convert some of the translational energy of the electrons into rotational energy about the magnetic field lines, to compensate for the unavoidable transfer of energy from the radiofrequency (RF) field to the electrons, or both. In the two 3D ion-trap demonstrations [7–8], ECD occurred in the analyzer itself, whereas, in the linear ion-trap demonstration [5–6], it took place in a custom-designed cell.
In 2008, the present authors described an electromagnetostatic (EMS), electron-inducing dissociation cell that can be retrofitted into virtually any existing type of mass spectrometer [9]. When installed in a triple quadrupole (QqQ) [9–11] or a hybrid quadrupole/time-of-flight (QqTOF) [12] mass spectrometer, this device makes it possible to record ECD product-ion spectra of authentic, unmodified peptides without the aid of cooling gas or phase-specific electron injection into the cell. Additionally, ECD can be induced in these modified instruments simultaneously with CID to produce ECD-CID product-ion spectra exhibiting golden complementary pairs [13] of peptide fragments [10,12]. The authors have also shown that electron ionization dissociation (EID) [14–15] can be induced with an EMS cell retrofitted into a QqTOF [12]. Disregarding the obvious loss in resolution, mass spectra recorded in all of the preceding experiments exhibit fragmentation patterns that are practically identical to those exhibited in corresponding mass spectra produced with Fourier transform ion cyclotron resonance (FT-ICR) instruments. When ECD-based sequencing of phosphopeptides is performed in an FT-ICR mass spectrometer, no loss of phosphoric acid, phosphate, or water from the precursor or fragment ions is observed, thus, allowing direct assignment of phosphorylation sites [16]. In this report, the authors demonstrate that, as in its FT-ICR counterpart, phosphorylation is preserved when tyrosine-phosphorylated peptides undergo ECD in the EMS cell, a capability that had not been previously evaluated.
EXPERIMENTAL DETAILS
Materials and Sample Preparation
Peptide standards tyrosine kinase peptide 3 [Arg-Arg-Leu-Ile-Glu-Asp-Ala-Glu-Tyr-Ala-Ala-Arg-Gly-NH2], MW = 1518; phosphorylated tyrosine kinase peptide 3 [Arg-Arg-Leu-Ile-Glu-Asp-Ala-Glu-Tyr(PO3H2)-Ala-Ala-Arg-Gly-NH2], MW = 1598; p60 c-src (521–533) [Thr-Ser-Glu-Pro-Gln-Tyr-Gln-Pro-Gly-Glu-Asn-Leu], MW = 1463; and Pp60 c-src (521–533) [Thr-Ser-Glu-Pro-Gln-Tyr(PO3H2)-Gln-Pro-Gly-Glu-Asn-Leu], MW = 1543 were purchased from American Peptide Co. (Sunnyvale, CA, USA). The peptide standards were each dissolved in 50% MeOH containing 0.1% formic acid to a final concentration of 20–50 μg/mL.
Mass spectrometry
Mass spectra were recorded using a commercial, electrospray ionization (ESI), QqQ mass spectrometer system (Agilent G6460: Agilent Technologies, Santa Clara, CA, U.S.A.). The QqQ was modified by shortening the original RF hexapole CID cell by 40 mm and inserting the radio-frequency-free (RFF) EMS ECD cell described in the succeeding subsection into the space created between the first quadrupole and the shortened CID cell (Figure 1A). A solution of each of the peptide standards was electrosprayed at a flow rate of 10 μL/min. Precursor ions selected by the first quadrupole mass analyzer were transferred through the EMS ECD cell wherein they merged with low energy electrons. Ions exiting the EMS cell passed through the shortened CID cell, which in this study was operated in both transmission only and CID modes; Ar instead of N2 was used as the collision gas in the CID cell as a conservative precaution against burning out the EMS cell’s electron emitter. By minimally heating, the temperature of the filament was raised just enough during the experiments reported here to provide recordable ECD spectra. Consequently, the original filament has not burned out despite hundreds of hours of operation, and its lifetime expectancy remains to be determined. The second quadrupole mass analyzer was tuned to operate with a resolving power of ~2,500 FWHM, which allowed good ion transmission and unit-resolution mass spectra of essentially all of the ECD fragments and nondissociated precursors to be recorded.
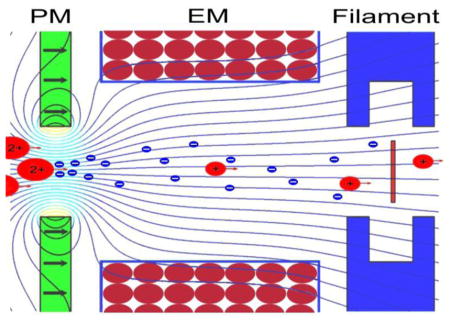
Figure 1.

(A) Schematic of the EMS ECD cell retrofitted into the Agilent G6460 QqQ mass spectrometer used in this study; (B) enlarged schematic of the EMS ECD cell.
FT-ICR ECD product-ion mass spectra shown in this report were produced from two aliquots of the phosphorylated tyrosine kinase peptide 3 taken from the supply purchased for this study. One aliquot was sent to the University of California – Los Angeles (UCLA) Molecular Instrumentation Center to be analyzed using its Bruker 15T SolariX FT mass spectrometer system, and the other was given to the Oregon State University Organic Mass Spectrometry Facility to be analyzed using its Thermo-Finnigan 7T LTQ FT Ultra linear ion trap mass spectrometer system.
EMS ECD Cell
The EMS cell used in this study comprised in the following order from entrance to exit: an electrostatic lens, a permanent magnet followed by an electromagnet, and a heated filament within a holder that also served as an electrostatic lens (Figure 1B). The electromagnet was spooled on a bobbin with an outer diameter of 70 mm, an inner diameter of 6.0 mm, and width of 15 mm. The bobbin and lenses were machined from titanium to avoid perturbing the magnetic field used to control and confine the electrons emitted from the heated filament. The permanent magnet was an axially polarized, Sm2Co17 disc-magnet (25.4 mm, 1.0 mm thick, 3.0 mm aperture on center) with an operating temperature up to 350°C (grade SM2435, Chino Magnetism Co., Fairfield, NJ, USA). The filament, which served as the ECD cell’s source of electrons, was a loop (~1.0 mm diameter) of tungsten-rhenium wire (Figure 1B, insert) similar to filaments described in previous reports [9–10]. The emitter and its titanium holder were located concentric with the axis of the cell at the ions’ exit. For this study, the filament and permanent magnet potentials were set at +15 V, the potential on the first electrostatic lens at −15 V, the potential on the electromagnet at +21 V, and the potential on the last electrostatic lens at +10 V.
RESULTS AND DISCUSSION
While traversing an EMS cell, ions must pass through a hot, transmissive electron emitter (~3,000 °C) located either at the cell’s entrance [10–11], center [12] or exit (Figure 1). Hence in passing through an EMS cell, the ions are briefly exposed to thermal radiation similar to that sometimes used to induce or at least facilitate peptide dissociation in FT-ICR cells. Although authentic, unmodified peptides clearly survive this in-flight passage through the an EMS cell to produce analytically useful ECD product-ion spectra [9–12], there has been no demonstration that tyrosine phosphorylation or other peptide modifications survive passage through an EMS ECD cell intact.
ECD products of the [M+2H]2+ precursors of tyrosine kinase peptide 3 (p1518) and phosphorylated tyrosine kinase peptide 3 (Pp1598) were easily produced in the EMS ECD cell retrofitted into the Agilent G6460 (Figures 2A and 2B); the product-ion spectra exhibit signals corresponding to the reduced precursor ion [M+2H]+ and almost exclusively to a series of c-fragments comprising doublets of cx and cx ions [17] (Figure 2B, unit resolution insert). The [M+2H]2+ precursor of Pp1598 proved refractory to ECD in the Bruker and Thermo-Finnigan ICR cells, and therefore, extra effort was required to acquire the ECD product-ion spectra of this particular peptide (Figures 2C and S1 in Supplemental Materials) with these two instruments. Assignments of the fragment ions was based on the G6460’s mass accuracy at unit resolution; subsequently, they were confirmed by comparison to highly mass-accurate spectra recorded respectively on the Bruker 15T SolariX FT and Thermo-Finnigan 7T LTQ FT systems (Figure 2C plus Figure S1 and Table S1 in Supplemental Materials). For clarity, only b, c, y, and z fragment ions and reduced precursor ions are labeled in the spectra and tables.
Figure 2.

ECD product-ion mass spectra of the [M+2H]2+ precursors of (A) nonphosphorylated p1518 and (B) phosphorylated Pp1598 produced with the EMS cell retrofitted into the Agilent G6460 QqQ mass spectrometer. (C) ECD product-ion mass spectrum of the [M+2H]2+ precursor of phosphorylated Pp1598 produced with the Bruker 15T SolariX FT mass spectrometer. (D) CID product-ion spectrum of the [M+2H]2+ precursor of phosphorylated Pp1598 produced with the shortened CID cell retrofitted into the Agilent G6460 QqQ.
Signals corresponding to z11 and z12 are present in the p1518 spectrum (Figure 2A), and a signal for z12 is present in the Pp1598 spectrum (Figure 2B). The additional 80 u in nominal mass evident between the c8 and c9 signals in the Pp1598 spectra over the 163 u between these two signals in the p1518 spectrum unambiguously points to phosphorylation on the tyrosine at position 9. As is usually the case for ECD, these product-ion spectra exhibit easily interpretable series of c and z fragment peaks and almost no extraneous background signals. The scaling factors in Figures 2A–B indicate that the ECD efficiencies for the EMS ECD and the FT ICR cells are comparable for this peptide and are both on the order of 1%. The relative signal intensities of the c2 and c7 through c11 fragments in the QqQ ECD product-ion mass spectrum of Pp1598 (Figure 2B) are similar to those observed in the ECD product-ion spectrum of this same peptide produced in the ICR cell of the Bruker 15T SolariX FT mass spectrometer (Figure 2C). The two mass spectra do, however, display noticeable differences in relative signal intensities for the c3 through c5, c12, and z12 fragments, and moreover, there are peaks in the FT ICR spectrum (Figure 2C and Table S1 in Supplemental materials) that do not appear in the QqQ spectrum (Figure 2B). The intense peak that appears nominally at m/z 399 in the FT ICR spectrum (labeled ω2 in Figure 2C) is not an ECD fragment but, rather, a second harmonic of the doubly protonated precursor generated in the FT ICR cell. The relative intensities of the assigned fragments in the ECD product-ion mass spectrum of the doubly protonated Pp1598 produced by the Thermo-Finnigan 7T LTQ FT (Figure S1 in Supplemental Materials) differ from those produced by the Bruker 15T SolariX FT (Figure 2C) and, thus, even more from those produced by the modified Agilent QqQ (Figure 2B). The interested reader can refer to Supplemental materials to assign structures to signals not assigned in Figures 2C.
The CID product-ion spectrum of the [M+2H]2+ precursor of Pp1598 recorded with the Agilent G6460 (Figure 2D) displays, as is typical of CID product-ion spectra of peptides, a relatively large number of fragment signals (≳100) with intensities well above background; the majority of these signals are not readily interpretable and as such compose the chemical background that characterizes CID peptide-product-ion spectra in general and can confound extraction of amino-acid sequence information. In this particular instance, the CID spectrum accounts for less coverage of the peptide’s amino-acid sequence than do the ECD spectra but, because it exhibits y4 and y5, still reveals the peptide’s site of phosphorylation.
ECD product-ion mass spectra of the [M+3H]3+ precursor of Pp1598 were easily produced with the EMS ECD cell mounted in the Agilent G6460 (Figure 3A) as well as with both the Bruker 15T SolariX FT and Thermo-Finnigan 7T LTQ FT (Figures 3B and S2 in Supplemental Materials). These product-ion spectra are more similar in appearance than are the product-ion mass spectra produced by these two instrument types from the doubly protonated precursor of Pp1598 (Figures 2B, 2C, and S1). For this particular peptide, the ECD spectra of [M+3H]3+ from both types of instrument display a series of z-ions that ranges from z4 to z12 as well as the series of c-ions seen in the doubly protonated spectra that, save for one residue, spans Pp1598’s entire sequence. Consequently, the presence of the phosphorylated tyrosine can be readily discerned not only by the m/z-separation between the c8 and c9 peaks but also by that between the z4 and z5 peaks. Aside from the obvious differences in resolution and mass accuracy, the principal feature that distinguishes the triply protonated spectrum recorded by the Bruker FT ICR mass spectrometer from that recorded by the Q(EMS)qQ instrument is the much larger abundance of C11 in the FT ICR spectrum (Figure 3B) relative to that in the Q(EMS)qQ spectrum (Figure 3A). The differences in relative intensities are somewhat greater between the Q(EMS)qQ and the Thermo-Finnigan FT ICR product-ion spectra of the triply protonated precursor of Pp1598 (Figures 3A and S2).
Figure 3.

ECD product-ion mass spectra of the [M+3H]3+ precursor of phosphorylated Pp1598 produced with (A) the EMS cell retrofitted into the Agilent G6460 QqQ mass spectrometer and (B) the Bruker 15T SolariX FT mass spectrometer.
The FT ICR spectra contains more unidentified peaks than spectra from the Q(EMS)qQ, while the relative intensities of the c- and z-ions in the FT ICR product-ion spectra differ for the corresponding ions in the Q(EMS)qQ ECD product-ion spectra. Evidence in the literature supports these differences could arise from the distinct experimental conditions producing the two sets of data. In the FT-ICR cell, electrons, precursor ions, and many ECD product-ions circulate together for periods of at least a millisecond or more, during which time some ECD fragments can be formed through a radical cascade mechanism that renders their signal intensities very dependent on experimental time scale [18]. By contrast, ions pass through a small volume of high electron density for a fraction of a microsecond in the EMS ECD device [11]. Following their brief exposure (at least three orders of magnitude less than in an FT ICR cell) to electrons in the EMS cell, precursors decay much as do metastable ions, and the ECD fragment abundances depend strongly on flight time to the detector [9]. In addition, precursor and fragment ions are exposed to thermal radiation as they pass through the hot zone (2500–3000 °C) created by the electron emitter located at the exit of the EMS cell (Figure 1B). Despite these substantial differences in experimental conditions, the ECD product-ion spectra for the expected c and z ions produced by the two types of instruments are remarkably similar and both preserve labile tyrosine phosphorylation.
As mentioned in the second paragraph of this section, the ECD product-ion spectrum of Pp1598’s [M+2H]2+ precursor (Figure 2B) was produced without difficulty on the Q(EMS)qQ instrument whereas the corresponding spectra of this same precursor (Figures 2C and S1) were produced with difficulty on the Bruker and Thermo-Finnigan FT ICR instruments. This contrasting behavior of doubly protonated Pp1598 in the two types of ECD cell might be a consequence of salt-bridges [19], which could form in Pp1598 between the phosphogroup and a protonated arginine, hindering dissociation of the c/z-complexes produced by the N–Cα cleavages of this peptide. It has long been known that prior to unimolecular decomposition of certain isolated organic ions, the incipient ion and neutral fragments can remain associated for an extensive length of time [20]. The occurrence of this phenomenon in the particular case of ECD of doubly protonated phosphorylated peptides in ICR cells reduces efficiency of dissociation; the latter can be improved by performing ECD on triply charged precursor ions or by an input of external energy [19]. The Bruker and Thermo-Finnigan ECD product-ion spectra of the doubly and triply protonated Pp1598 seem to be an example of this phenomenon. The higher dissociation efficiency of the Q(EMS)qQ ECD product-ion spectra of the doubly protonated Pp1598 suggests that the complementary c/z-fragments are acquiring extra energy before they exit the EMS cell, very likely from the thermal radiation given off by the electron emitter at the cell’s exit (Figure 1B). This latter possibility is supported by the fact that, upon turning the EMS cell around 180° so that the ECD products exited the cell without passing through the electron emitter, efficiency of dissociation decreased. If systematic investigation bears this effect out, ECD analyses carried out in an EMS cell would gain an additional benefit.
The product-ion spectra of the [M+2H]2+ precursors of the nonphosphorylated p60 c-src (Figure 4A) and its phosphorylated analogue Pp60 c-src (Figure 4B) display peaks corresponding to the reduced precursor ion [M+2H]+ and partial series of c and z fragments; the site of phosphorylation on the tyrosine at position 7 is clearly indicated by the 80-u increase in mass between c6 and c7 in the spectrum of Pp60 c-src (Figure 4B). Between them, the c- and z-fragments in these two product-ion spectra cover nearly all of the two peptides’ sequences. The cyclic structure of the prolines at positions 5 and 9 block N–Cα cleavage of the glutamic acid–proline and glutamine–proline bonds respectively [18,21–22]. Consequently, peaks corresponding to c8 and z9 are absent in the spectra of both of the c-src peptides, and the peak corresponding to c9 has relatively low intensity. Weak signals for b7, b8, y5, y6, and y9 are present in the ECD spectra of both peptides (Figures 4A and 4B). These peaks may be evidence of the proline effect [23–25] wherein protonated, proline-containing peptides are labile to fragmentation N-terminal to the proline residues. During operation of the Q(EMS)qQ in the ECD-only mode, a minimal amount of gas (Ar in this instance) is fed into the CID cell to boost transmission; in order to avoid CID under this condition, the energy of the ions is lowered below the normal threshold for onset of CID. Nevertheless, appearance of the minor b and y ions in the ECD product-ion spectra of the proline containing p60 and Pp60 c-src ions would seem to indicate that the ions still had sufficient energy in this particular case to induce CID of the labile bonds N-terminal to the proline residues in these peptides.
Figure 4.

ECD product-ion mass spectra of (A) nonphosphorylated p60 c-src and (B) phosphorylated Pp60 c-src; (C) combined ECD/CID spectrum of phosphorylated Pp60 c-src.
Retrofitting an EMS cell into a mass spectrometer preserves its original CID capability while enabling combined ECD/CID analyses either by using the EMS cell for ECD and high-energy CID [10,12] or by using the EMS cell for ECD and the shortened collision/cooling cell for CID. The latter combination was used in this study to record an ECD/CID spectrum of Pp60 c-src (Figure 4C). Increasing the precursors’ kinetic energy through the CID cell while maintaining ECD conditions in the EMS cell, which is upstream of the CID cell (Figure 1A), amplified the signals corresponding to the two b and three y ions seen in ECD-only spectra (Figures 4A and 4B) and produced a new signal corresponding to b11. These CID fragments complement the ECD fragments whose signals remain essentially unaltered in the simultaneously recorded spectrum (Figure 4C). The strong y9 peak and even more intense b8 and y5 peaks result respectively from cleavage of the peptide’s glutamic acid-proline and glutamine-proline bonds. These bonds, which are known to be refractory to ECD [18, 21–22], cleave readily under CID due to the proline effect [23–25]. The combined ECD/CID spectrum (Figure 4C) thus accounts for nearly the entire sequence (10 out of a total of 12 possible cleavages) of Pp60 c-src. Incidentally, the b7/c7 and b11/c11 fragment-ion pairs present in this spectrum are simultaneously recorded CID/ECD golden complementary pairs [13]. This combined spectrum demonstrates, as have previously reported ECD/CID product-ion spectra [10,12], that ECD produced by an EMS cell retrofitted into a QqQ or QTOF retains the CID functionality of the original instrument, thereby making it possible to produce complementary sequence information that can enhance the specificity of peptide identifications.
CONCLUSION
The data presented in this report clearly demonstrate that tyrosine phosphorylation remains intact during the passage of the peptide’s doubly and triply protonated precursors through an EMS ECD cell retrofitted into a commercial QqQ despite brief exposure to the radiation emitted by the hot (~3,000 °C) filament used as the source of electrons. In fact, ECD fragmentation of a doubly protonated, tyrosine-phosphorylated peptide was more efficient in the EMS cell than in the two FT-ICR cells used in this study suggesting that the electron emitter’s radiation might assist dissociation of the c/z-complexes produced by N–Cα cleavages. Furthermore except for mass resolution and mass accuracy, the ECD product-ion spectra recorded with the modified QqQ exhibit as much qualitative information as contained in those produced from the same peptides by commercial FT ICR mass spectrometers. Those qualitative differences that where observed as well as the differences in relative signal intensities evident in the ECD spectra produced by the EMS and ICR cells and additional ion signals evident in the ICR ECD spectra are attributed to substantial differences in the experimental conditions that prevail in the two types of cells.
As is widely acknowledged and demonstrated directly in this report by the example of Pp1598, the lower fragmentation efficiency of ECD is generally compensated in its product-ion spectra by higher or more readily retrievable informational content relative to that in CID product-ion spectra, stressing therefore that the two fragmentation processes should be viewed as complementary. The fact that ECD and CID can be performed simultaneously in a Q(EMS)qQ, such as the one used in this study, adds emphasis to this latter observation. In the particular example presented in this report, the ECD produces information that confirms the presence of tyrosine phosphorylation but provides only partial amino-acid sequence information due to suppressed bond cleavages at the proline residues, whereas the CID, which yields scant information about the phosphorylation, supplies the proline-sequence information missed by the ECD – widespread availability of a capability such as this would increase the specificity of tandem mass analyses of peptides.
The experimental results presented in this report also raise the possibility of performing targeted multiple reaction monitoring (MRM) of peptides with triple quadrupole mass spectrometers based on ECD transitions or combined ECD/CID transitions. This capability could have widespread applicability in pharmaceutical and clinical analyses. Recent reports indicate that MRM-based mass-spectrometric techniques have the sensitivity, specificity, and capability for rapid, reproducible, multiplexed quantification of proteins in clinical assays [26–27]. Combing the capability of ECD-based sequencing with existing CID-based sequencing in MRM analyses could significantly increase the specificity and thereby increase the utility of targeted tandem mass spectrometry in proteomics.
Supplementary Material
Acknowledgments
Various aspects of this research were supported by grants from the NSF (CHE-0924027), the Oregon Nanoscience and Microtechnologies Institute (#09-31 #3.5), NIH NCRR (R01RR026275), NIH NIEHS (ES00210–Environmental Health Sciences Center), and Agilent Technologies (#2443). The authors thank Peter D. Hoffman (Department of Environmental & Molecular Toxicology, Oregon State University, Corvallis, Oregon, USA) for his assistance in collecting some of the data presented in this manuscript, Elsworth T. Hinke and Larry M. Nelson (Department of Chemistry, Oregon State University, Corvallis, Oregon, USA) for their assistance in fabricating components of the EMS ECD cell used in this study, Piriya Wongkongkathep and Dr. Joseph Loo (Department of Chemistry and Biochemistry, University of California-Los Angeles, USA) for graciously acquiring and labeling the ECD FT-ICR product-ion mass spectra displayed in this report, and George Stafford (Agilent Technologies, Santa Clara, California, USA) for generously reading the manuscript and offering several useful suggestions for improving its content.
References
- 1.Mayer PM, Poon C. The mechanisms of collisional activation of ions in mass spectrometry. Mass Spectrom Rev. 2009;28:608–639. doi: 10.1002/mas.20225. [DOI] [PubMed] [Google Scholar]
- 2.Kim MS, Pandey A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics. 2012;12:530–542. doi: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakhtiar R, Guan Z. Electron capture dissociation mass spectrometry in characterization of peptides and proteins. Biotechnol Lett. 2006;28:1047–1059. doi: 10.1007/s10529-006-9065-z. [DOI] [PubMed] [Google Scholar]
- 4.Zubarev R. Electron capture dissociation and other ion-electron fragmentation reactions. In: Laskin J, Lifshitz C, editors. Principles of Mass Spectrometry Applied to Biomolecules. John Wiley & Sons, Inc; Hoboken, NJ: 2006. p. 475. [Google Scholar]
- 5.Baba T, Hashimoto Y, Hasegawa H, Hirabayashi A, Waki I. Electron capture dissociation in a radio frequency ion trap. Anal Chem. 2004;76:4263–4266. doi: 10.1021/ac049309h. [DOI] [PubMed] [Google Scholar]
- 6.Satake H, Hasegawa H, Hirabayashi A, Hashimoto M, Baba T. Fast multiple electron capture dissociation in a linear radio frequency quadrupole ion trap. Anal Chem. 2007;79:8755–8761. doi: 10.1021/ac071462z. [DOI] [PubMed] [Google Scholar]
- 7.Silivra OA, Kjeldsen F, Ivonin IA, Zubarev R. Electron capture dissociation of polypeptides in a three-dimensional quadrupole ion trap: implementation and first results. J Am Soc Mass Spectrom. 2005;16:22–27. doi: 10.1016/j.jasms.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Ding L, Brancia FL. Electron capture dissociation in a digital ion trap mass spectrometer. Anal Chem. 2006;78:1995–2000. doi: 10.1021/ac0519007. [DOI] [PubMed] [Google Scholar]
- 9.Voinov VG, Deinzer ML, Barofsky DF. Electron capture dissociation in a linear radiofrequency-free magnetic cell. Rapid Commun Mass Spectrom. 2008;22:3087–3088. doi: 10.1002/rcm.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voinov VG, Beckman JS, Deinzer ML, Barofsky DF. Electron-capture dissociation (ECD), collision-induced dissociation (CID) and ECD/CID in a linear radio-frequency-free magnetic cell. Rapid Commun Mass Spectrom. 2009;23:3028–3030. doi: 10.1002/rcm.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voinov VG, Deinzer ML, Barofsky DF. A radio-frequency-free cell for electron capture dissociation in tandem mass spectrometry. Anal Chem. 2009;81:1238–1243. doi: 10.1021/ac802084w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voinov VG, Deinzer ML, Beckman JS, Barofsky DF. Electron capture, collision-induced, and electron capture-collision induced dissociation in Q-TOF. J Am Soc Mass Spectrom. 2011;22:607–611. doi: 10.1007/s13361-010-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horn DM, Zubarev RA, McLafferty FW. Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budnik BA, Zubarev RA. MH2+ ion production from protonated polypeptides by electron impact: observation and determination of ionization energies and a cross-section. Chem Phys Lett. 2000;316:19–23. [Google Scholar]
- 15.Fung YM, Adams CM, Zubarev RA. Electron ionization dissociation of singly and multiply charged peptides. J Am Chem Soc. 2009;131:9977–9985. doi: 10.1021/ja8087407. [DOI] [PubMed] [Google Scholar]
- 16.Mann M, Ong SE, Grønborg M, Steen H, ONJ, Pandey A. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 17.Kjeldsen F, Haselmann KF, Budnik BA, Jensen F, Zubarev RA. Dissociative capture of hot (3–13 eV) electrons by polypeptide polycations: an efficient process accompanied by secondary fragmentation. Chem Phys Lett. 2002;356:201–206. [Google Scholar]
- 18.Leymarie N, Berg EA, McComb ME, O’Connor PB, Grogan J, Oppenheim FG, Costello CE. Tandem mass spectrometry for structural characterization of proline-rich proteins: application to salivary PRP-3. Anal Chem. 2002;74:4124–4132. doi: 10.1021/ac0255835. [DOI] [PubMed] [Google Scholar]
- 19.Creese AJ, Cooper HJ. The effect of phosphorylation on the electron capture dissociation of peptide ions. J Am Soc Mass Spectrom. 2008;19:1263–1274. doi: 10.1016/j.jasms.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longevialle P, Lefèvre O, Mollova N, Bouchoux G. Further arguments concerning a ‘rotational effect’ in the unimolecular fragmentations of organic ions in the gas phase. Rapid Commun Mass Spectrom. 1998;12:57–60. [Google Scholar]
- 21.Stensballe A, Jensen ON, Olsen JV, Haselmann KF, Zubarev RA. Electron capture dissociation of singly and multiply phosphorylated peptides. Rapid Commun Mass Spectrom. 2000;14:1793–1800. doi: 10.1002/1097-0231(20001015)14:19<1793::AID-RCM95>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 22.Nishikaze T, Takayama M. Influence of charge state and amino acid composition on hydrogen transfer in electron capture dissociation of peptides. J Am Soc Mass Spectrom. 2010;21:1979–1988. doi: 10.1016/j.jasms.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz BL, Bursey MM. Some proline substituent effects in the tandem mass spectrum of protonated pentaalanine. Biol Mass Spectrom. 1992;21:92–96. doi: 10.1002/bms.1200210206. [DOI] [PubMed] [Google Scholar]
- 24.Vaisar T, Urban J. Probing the proline effect in CID of protonated peptides. J Mass Spectrom. 1996;31:1185–1187. doi: 10.1002/(SICI)1096-9888(199610)31:10<1185::AID-JMS396>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 25.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 26.Kitteringham NR, Jenkins RE, Lane CS, Elliott VL, Park BK. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J Chromatogr B: Anal Technol Biomed Life Sci. 2009;877:1229–1239. doi: 10.1016/j.jchromb.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Chambers AG, Percy AJ, Simon R, Borchers CH. MRM for the verification of cancer biomarker proteins: recent applications to human plasma and serum. Expert Rev Proteomics. 2014;11:137–148. doi: 10.1586/14789450.2014.877346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.