

Abstract
Background
Aggression is a major public health issue and is integral to several mental health disorders. Antiepileptic drugs may reduce aggression by acting on the central nervous system to reduce neuronal hyper‐excitability associated with aggression.
Objectives
To evaluate the efficacy of antiepileptic drugs in reducing aggression and associated impulsivity.
Search methods
We searched CENTRAL, MEDLINE, EMBASE, CINAHL, PsycINFO, metaRegister of Controlled Trials (mRCT) and ClinicalTrials.gov to April 2009. We also searched Cochrane Schizophrenia Group's register of trials on aggression, National Research Record and handsearched for studies.
Selection criteria
Prospective, placebo‐controlled trials of antiepileptic drugs taken regularly by individuals with recurrent aggression to reduce the frequency or intensity of aggressive outbursts.
Data collection and analysis
Three authors independently selected studies and two authors independently extracted data. We calculated standardised mean differences (SMDs), with odds ratios (ORs) for dichotomous data.
Main results
Fourteen studies with data from 672 participants met the inclusion criteria. Five different antiepileptic drugs were examined. Sodium valproate/divalproex was superior to placebo for outpatient men with recurrent impulsive aggression, for impulsively aggressive adults with cluster B personality disorders, and for youths with conduct disorder, but not for children and adolescents with pervasive developmental disorder. Carbamazepine was superior to placebo in reducing acts of self‐directed aggression in women with borderline personality disorder, but not in children with conduct disorder. Oxcarbazepine was superior to placebo for verbal aggression and aggression against objects in adult outpatients. Phenytoin was superior to placebo on the frequency of aggressive acts in male prisoners and in outpatient men including those with personality disorder, but not on the frequency of 'behavioral incidents' in delinquent boys.
Authors' conclusions
The authors consider that the body of evidence summarised in this review is insufficient to allow any firm conclusion to be drawn about the use of antiepileptic medication in the treatment of aggression and associated impulsivity. Four antiepileptics (valproate/divalproex, carbamazepine, oxcarbazepine and phenytoin) were effective, compared to placebo, in reducing aggression in at least one study, although for three drugs (valproate, carbamazepine and phenytoin) at least one other study showed no statistically significant difference between treatment and control conditions. Side effects were more commonly noted for the intervention group although adverse effects were not well reported. Absence of information does not necessarily mean that the treatment is safe, nor that the potential gains from the medication necessarily balance the risk of an adverse event occurring. Further research is needed.
Plain language summary
Antiepileptic drugs for treating recurrent aggression
Various medicines, which are collectively termed 'antiepileptic drugs', have been used to treat persistent aggression. This review systematically examines the evidence supporting this practice. From the evidence available, we were unable to draw any firm conclusion about using these medicines to treat aggression. Four antiepileptic drugs (valproate/divalproex, carbamazepine, oxcarbazepine and phenytoin) helped to reduce aggression in at least one study. However, for three of these drugs (valproate, carbamazepine and phenytoin) we found at least one other study where there was no significant improvement. Further research is needed to clarify which antiepileptic drugs are effective for whom. Such research is best carried out using carefully designed clinical trials. Such trials need to take account of the type of aggression displayed, the severity of the aggression, and any other disorders experienced by the participants.
Summary of findings
for the main comparison.
| Antiepileptic medication compared with placebo for individuals with recurrent aggression | ||||||
|
Patient or population: Any individual experiencing recurrent aggressive outbursts or episodes Settings: Any (including hospital inpatient, outpatient, community and custodial settings) Intervention: Any antiepileptic drug1 Comparison: Placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With antiepileptic medication | |||||
|
Aggression OAS, OAS‐M, MOAS, self‐reported incidents, CPRS subscale (endpoints fall between 2 weeks and 6 months) |
Unclear2 | See comment | Not estimable | 606 (12 studies; 14 comparisons) |
+++O moderate | Quantitative synthesis not possible since 11 of the 14 comparisons met criteria for skewed data Descriptively, 8 of the 14 comparisons demonstrated significantly lower aggression scores in the intervention condition |
|
Impulsivity Q‐sort, Porteus Maze Test (endpoints fall between 2 weeks and 6 months) |
Unclear2 | See comment | Not estimable | 43 (2 studies; 2 comparisons) |
+OOO very low | Quantitative synthesis not possible since both comparisons met criteria for skewed data Descriptively, neither of the 2 comparisons demonstrated significantly lower impulsivity scores in the intervention or control conditions |
|
Hostility BPRS hostility sub scale, CPRS hostility subscale, Q‐sort, speech content analysis (endpoints fall between 1 and 6 months) |
Unclear2 | See comment | Not estimable | 143 (4 studies; 4 comparisons) |
++OO low | Quantitative synthesis not possible since all 4 comparisons met criteria for skewed data Descriptively, one of the 4 comparisons demonstrated significantly lower hostility scores in the intervention condition |
|
Anger Self‐reported outbursts, Rosenweig Picture Frustration Test (endpoints fall between 2 weeks and 6 months) |
Unclear2 | See comment | Not estimable | 46 (2 studies; 2 comparisons) |
++OO low | Quantitative synthesis not possible since 1 of the 2 comparisons met criteria for skewed data Descriptively, neither of the 2 comparisons demonstrated significantly lower impulsivity scores in the intervention or control conditions |
|
Anger‐Hostility POMS anger‐hostility subscale (endpoints fall between 1 and 6 months) |
Unclear2 | See comment | Not estimable | 125 (3 studies; 3 comparisons) |
++OO low | Quantitative synthesis not possible since all 3 comparisons met criteria for skewed data Descriptively, 1 of the 3 comparisons demonstrated significantly lower hostility scores in the intervention condition |
|
Non‐compliance leaving the study early (endpoints fall between 1 and 6 months) |
Low‐risk population | Not estimable | 411 (6 studies; 8 comparisons) |
+++O moderate | None of the 8 comparisons demonstrated any significant difference between experimental and control conditions Meta‐analysis of all 8 comparisons: OR 1.14; 95%CI 0.77 to 1.70, P = 0.52, Analysis 6.1 |
|
| See comment | See comment | |||||
| Medium‐risk population | ||||||
| See comment | See comment | |||||
| High‐risk population | ||||||
| See comment | See comment | |||||
|
Adverse events, any (endpoints fall between 1 6 six months) |
Low‐risk population | Not estimable | 300 (3 studies; 3 comparisons) |
+++O moderate | Two of the 3 comparisons found a significantly higher proportion of participants having any adverse effect in the intervention compared to the control condition Meta‐analysis of 3 comparisons: OR 3.48; 95% CI 1.68 to 7.21, P < 0.001, Analysis 6.2 |
|
| See comment | See comment | |||||
| Medium‐risk population | ||||||
| See comment | See comment | |||||
| High‐risk population | ||||||
| See comment | See comment | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; CPRS: Children's Psychiatric Rating Scale; BPRS: British Psychiatric Rating Scale; OAS: Overt Aggression Scale; MOAS: Modified Overt Aggression Scale; OAS‐M: Overt Aggression Scale ‐ Modified; POMS: Profile of Moods Scale | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Table summarises findings for antiepileptic drugs as a class.
2Unclear, since (a) this outcome is measured as a continuous (scale) variable using several instruments that are not directly equivalent, and (b) we have been unable to find normative data for individuals with recurrent aggression in the general population. We have not used baseline or control group data from the included studies to estimate risk or cut‐off scores on scale measures because of the diversity of the sample populations.
Background
Description of the condition
The term aggression has been used to define a broad range of behaviours which generally involve non‐accidental harm. This includes violence, defined by the World Health Organization as "the intentional use of physical force or power, threatened or actual, against oneself, another person, or against a group or community, that either results in or has a high likelihood of resulting in injury, death or psychological harm, maldevelopment or deprivation". Worldwide, violence is considered to be a major public health issue (World Health Organization 2002), and survey data on criminal violence in the UK has revealed that in the year 2006/7 there were just under 2½ million violent crimes in private households. Aggression is also used to describe other disturbed behaviour, such as intentional damage to property or hostile agitation.
Two broad subtypes of aggression have been identified – instrumental and reactive (Barratt 1999; Berkowitz 1993; Vitiello 1997). Instrumental aggression is more controlled and consciously goal‐directed. Reactive aggression, which is considered to be far more prevalent (Nelson 2007), involves an emotionally‐driven reaction to frustration or perceived threat. A key feature of reactive aggression is impulsivity. Impulsivity describes a tendency to act on impulse, i.e. a sudden or involuntary inclination or tendency to act without premeditation or reflection.
There is a well‐established link between mental disorder and aggression. Aggressive behaviour is an integral part of some disorders. The conventional diagnostic systems, ICD‐10 and DSM‐IV, have been shaped by a medical model that relies on the identification of symptoms to determine pathology. However, mental disorder diagnoses, especially those associated with aggression, give undue prominence to behaviours rather than underlying process disturbances. Consequently there is the tautological problem that the diagnostic criteria are used both to identify the disorder and to characterise the disorder. However many mental disorders, including antisocial personality disorder and conduct disorder, appear to be aetiologically heterogeneous rather than distinct unitary phenomena. In the case of ‘intermittent explosive disorder’, extreme aggression is the defining feature. In this condition there is failure to resist aggressive impulses with a disproportionate response to stress (DSM‐IV‐TR; American Psychiatric Association 2000). A study of psychiatric outpatients found the life‐time prevalence of intermittent explosive disorder to be 7.3% (Kessler 2006).
A history of aggression can contribute to the diagnosis of certain personality disorders. A "very low tolerance to frustration and a low threshold for discharge of aggression"’ is a diagnostic feature of dissocial personality disorder under ICD‐10 (World Health Organization 1993). The broader category of antisocial personality disorder within DSM‐IV includes the criterion ‘irritability and aggressiveness as indicated by repeated physical fights or assaults’. Key features of borderline personality disorder under DSM‐IV are emotional dysregulation and impulsivity, which may be manifest in "inappropriate, intense anger or difficulty controlling anger (e.g. frequent displays of temper, constant anger, recurrent physical fights)". The ICD‐10 equivalent, ‘emotionally unstable personality disorder’, refers to "liability to outbursts of anger or violence". Impulsivity is also a diagnostic criterion in these two personality disorders.
Certain developmental disorders may also present with aggressive behaviour. Two of the four conduct disorder symptom clusters involve aggression (DSM‐IV‐TR). The ‘aggression to people and animals’ cluster include items such as "often initiates fights", "has used a weapon" and "has been physically cruel to people". There is also a ‘destruction of property’ cluster which includes "has deliberately destroyed others’ property". Oppositional defiant disorder is characterised by "a pattern of negativistic, hostile and defiant behaviour" (DSM‐IV‐TR).
There are other mental disorders which do not have aggression as a specific diagnostic criterion, but which have been found to be associated with an increased risk of aggression. Substance abuse disorders are strongly associated with aggression (Nestor 2002). Epidemiological studies have established a modest but significant relationship between psychosis and violence (Arseneault 2000; Brennan 2000; Fazel 2006). In the developmental period, attention‐deficit hyperactivity disorder, which involves a loss of impulse control, is associated with aggression (Staller 2007). Across mental disorders, key traits that are associated with aggression are impulse control and affect regulation (Nestor 2002).
It has long been recognised that aggression can be a feature of organic brain conditions (Haller 2006). Aggressiveness may be specifically associated with frontal and temporal lobe damage (Anderson 1999; Hawkins 2000). A diagnostic criterion of organic personality disorder is ‘irritability and/or outbursts of anger and aggression’ (World Health Organization 1993). A specific relationship between epilepsy and aggression has also been found in adults (Kanemoto 1999; van Elst 2000) and children (Connor 1996).
Description of the intervention
The use of antiepileptic drugs in the treatment of aggression has a relatively long track‐record (for example Stein 1992; Tyrer 1988) and there are some supporting data (Goedhard 2006). Using a double‐blind cross‐over design, phenytoin has been reported to be associated with a reduction in the frequency of acts of aggression by individuals in a community sample (Stanford 2001) and in a prison sample (Barratt 1997). A similar effect has been found with carbamazepine and sodium valproate/divalproex (Gardner 1986; Hollander 2003; Hollander 2005; Kavoussi 1989; Lindenmayer 2000). The effect may be specific to impulsive aggression as opposed to instrumental aggression (Barratt 1997; Stanford 2001). There is some empirical support for the anti‐aggression effect of other antiepileptics, such as lamotrigine (Tritt 2005) and topiramate (Gobbi 2006).
An antiepileptic drug is defined here as one normally used prophylactically to reduce the frequency and/or severity of epileptic seizures. We consider pharmacological interventions where an antiepileptic drug is given not only as monotherapy but also as an adjunctive intervention. The review focuses on studies of persistent/serious aggression. We include, but have not confined ourselves to, studies of individuals resistant to other interventions.
How the intervention might work
Impulsive reactive aggression reflects a high arousal state (Haller 2006; Nelson 2007). It has been proposed that repeated acts of aggression, specifically of a reactive sort, may be underpinned by neuronal hyperexcitability, and there is some empirical support for this (Keele 2005). Therefore drugs which reduce neuronal excitability, and specifically those classed as antiepileptics or anticonvulsants, could theoretically be expected to lower the likelihood of reactive aggressive outburst.
The precise mechanisms by which this occurs are not fully understood. At a neurochemical level, an imbalance of gabaminergic systems is thought to be associated with aggression (Nelson 2007; Siever 2008), and gamma‐aminobutyric acid (GABA) has been found to suppress aggression in animal models (Miczek 2002). Therefore it follows that pharmacological antiepileptic agents that act on gabaminergic neurons, such as carbamazepine, phenytoin and valproate, may have a beneficial effect on aggressive behaviour.
Why it is important to do this review
Poorly controlled aggression is prevalent in society. It results in emotional and physical damage to victims, and is associated with considerable costs and distress. Medication has potential to provide one treatment option, and may be particularly relevant to individuals who seek treatment but who find difficulty engaging in psychological interventions. There is some evidence that antiepileptic drugs may be effective, but there are currently no clinical guidelines. A systematic review of the current evidence base is therefore timely.
Objectives
This review aims to evaluate the efficacy of antiepileptic drugs in reducing aggression and associated impulsivity.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials in which participants have been randomly allocated to an experimental group and a control group.
Types of participants
Individuals in any setting who have displayed recurrent aggression as defined by the authors of the study. We included studies with participants diagnosed with a mental illness, personality disorder, impulse disorder, or who have received a brain injury, but excluded studies of people with dementia. We would have included studies where antiepileptics were used to treat people with epilepsy, but only if the participants had also displayed recurrent aggression; however no studies of this type were identified.
Types of interventions
Any antiepileptic drug (including all antiepileptic drugs listed in Section 4.8 of the British National Formulary) in any dosage, used for any length of time, and measured against a placebo.
For the purpose if this review, an antiepileptic drug is defined as one used prophylactically to reduce the frequency and/or severity of epileptic seizures. Drugs used in the acute treatment of status epilepticus but which are not generally prescribed for the prophylaxis of epileptic seizures (such as chlormethiazole, diazepam, fosphenytoin, lorazepam, midazolam and paraldehyde) were excluded. We did not include studies where antiepileptic drugs were used to treat acute presentations as in rapid tranquillisation procedures.
We included studies where the antiepileptic drug being evaluated was given as an adjunct to another drug not classed as an antiepileptic or as an adjunct to a psychological intervention. Comparisons might therefore include:
antiepileptic versus placebo;
antiepileptic plus other drug treatment versus placebo plus (the same) other drug treatment; and
antiepileptic plus psychological treatment versus placebo plus (the same) psychological treatment.
Types of outcome measures
A number of validated instruments have been developed for the measurement of aggression. To date, however, no single measure has been identified as superior and we therefore anticipated finding a range of outcome measures in studies identified for inclusion in this review.
Primary outcomes
Aggression (observer‐reported): reduction in aggressive behaviour; continuous outcome, measured through improvement in scores on the Modified Overt Aggression Scale (MOAS; Malone 1994) or the Overt Aggression Scale‐Modified (OAS‐M, Coccaro 1991) or similar validated instrument; or as number of observed incidents per participant per specified time period.
Aggression (self‐reported): reduction in aggressive behaviour or aggressive feelings; continuous outcome, measured through improvement in scores on the Aggression Questionnaire (AQ; Buss 1992), the Positive and Negative Syndrome Scale (PANSS; Kay 1989) or similar validated instrument.
Secondary outcomes
Impulsivity: self‐reported improvement in impulsivity; continuous outcome, measured through reduction in scores on the Barratt Impulsivity Scale (BIS: Patton 1995) or similar validated instrument.
Hostility: improvement in hostility; continuous outcome, measured through reduction in scores on the hostility subscale of the observer‐reported Brief Psychiatric Rating Scale, on the hostility subscale of the self‐reported SCL‐90 (Derogartis 1973) or using a similar validated instrument.
Anger: self‐reported improvement in anger expression and control; continuous outcome, measured through reduction in scores on the State‐Trait Anger Expression Inventory‐2 (STAXI‐II; Spielberger 1999) or similar validated instrument.
Quality of life: self‐reported improvement in overall quality of life; continuous outcome, measured through improvement in scores on the European quality of life instrument (EuroQol; EuroQoL group 1990) or similar validated instrument.
Satisfaction with care: continuous outcome; measured through improvement in scores on the Client Satisfaction Questionnaire (CSQ‐8; Attkisson 1982) or similar validated instrument.
Non‐compliance: measured as proportion of participants discontinuing treatment; dichotomous.
Adverse events: measured as incidence of overall adverse events and of the three most common adverse events; dichotomous outcome, measured as numbers reporting, or dichotomised as number reporting at least one event.
We had intended to divide outcomes into short‐term (within one month), medium‐term (between one month and six months) and long‐term (between six and 12 months) for comparison using, where possible, meta‐analysis. On this occasion, however, the data extracted on the primary outcome measure (aggression) did not lend themselves to meta‐analysis and so such a comparison was not attempted.
Search methods for identification of studies
Electronic searches
We searched the following electronic databases:
Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2009; Issue 1);
MEDLINE (searched 1966 to April 2009);
EMBASE (searched 1980 to April 2009);
CINAHL (searched 1982 to April 2009);
PsycINFO (searched 1872 to April 2009);
Cochrane Schizophrenia Group's register of trials on aggression (searched 22/11/07);
National Research Record, (NRR), (searched 2007 (Issue 4));
metaRegister of Controlled Trials, (mRCT), including Clinical Trials.gov, (searched 8/4/09)
Details of search terms are given in Appendix 1, Appendix 2, Appendix 3, Appendix 4, Appendix 5, Appendix 6 and Appendix 7. No language or date restrictions were applied.The CSG's register of trials on aggression and the NRR were not searched after 2007 as no new records had been added past that date.
Searching other resources
We handsearched the reference lists of included and excluded studies for additional relevant trials. We also examined bibliographies of systematic review articles published in the last five years to identify relevant studies. We contacted authors of relevant studies to enquire about other sources of information and the first author of each included study for information regarding unpublished data. We contacted all pharmaceutical companies listed in the current British National Formulary as manufacturing medication licensed for treating epilepsy requesting information about any published or unpublished trials using antiepileptic drugs in the treatment of people with recurrent aggression.
Data collection and analysis
Selection of studies
Three authors (NH, MF & HJ) independently identified, read and reviewed titles and abstracts against the inclusion criteria. Two authors (NH, MF) independently assessed full copies of studies which appeared to meet the inclusion criteria. Uncertainty concerning the appropriateness of one study for inclusion in the review was resolved through consultation with a third author (RN). Authors were not blinded to the name(s) of the study author(s), their institution(s) or publication sources at any stage of the review.
Data extraction and management
Two authors (NH & MF) extracted data independently using a data extraction form and entered data into RevMan 5 (RevMan 2008). Where data were not available in the published trial reports, we attempted to contact the trial investigators for the missing information.
Assessment of risk of bias in included studies
For each included study, two authors (NH & MF) independently completed the Cochrane Collaboration’s tool for assessing risk of bias (Higgins 2008, section 8.5.1). We assessed the degree to which:
the allocation sequence was adequately generated (‘sequence generation’);
the allocation was adequately concealed (‘allocation concealment’);
knowledge of the allocated interventions was adequately prevented during the study (‘blinding’);
incomplete outcome data were adequately addressed;
reports of the study were free of suggestion of selective outcome reporting; and
the study was apparently free of other problems that could put it at high risk of bias.
Each domain was allocated one of three possible categories for each of the included studies: ‘Yes’ for low risk of bias, ‘No’ for high risk of bias, and ‘Unclear’ where the risk of bias was uncertain or unknown.
Measures of treatment effect
For dichotomous (binary) data, we used the odds ratio with a 95% confidence interval to summarise results within each study. The odds ratio was chosen because it has statistical advantages relating to its sampling distribution and its suitability for modelling, and is a relative measure and so can be used to combine studies. For continuous data, such as the measurement of impulsiveness on a scale, we had intended to compare the mean score for each outcome as determined by a standardised tool between the two groups to give a mean difference (MD), again with a 95% confidence interval. We had intended to use the standardised mean difference (SMD) where different outcome measures of the same construct were reported. On this occasion, however, all continuous data from the 14 trials in this review met our criteria for skewness. We define skewness as occurring when, for a scale or measure with positive values and a minimum value of zero, the mean is less than twice the standard deviation (Altman 1996). All skewed continuous data are reported in separate tables, together with results of any statistical analyses conducted by the trial investigators; we did not attempt to calculate treatment effect sizes for this data to minimise the risk of applying parametric statistics to data that departs significantly from a normal distribution.
Where possible, endpoint data are presented. Where both endpoint and change data were available for the same outcomes, then only the former are reported.
We had intended that the statistical comparisons on primary and secondary measures would be categorised into one of three specific follow‐up periods (within the first month; between one and six months; between six and 12 months) and meta‐analysed within these categories. For 12 of the 14 studies included in this review, however, the endpoint results fell within the one‐to‐six‐month period and the four meta‐analyses we report all fall within this category.
Unit of analysis issues
a) Cluster‐randomised trials
Had we found trials that used clustered randomisation, we anticipated that study investigators would have presented their results after appropriately controlling for clustering effects (using, for example, robust standard errors or hierarchical linear models). If a cluster‐randomised trial had been found where it was unclear whether appropriate controls had been used for clustering, we planned to contact the study investigators for further information. If appropriate controls had not been used, individual participant data would have been requested and re‐analysed using multilevel models which control for clustering. Following this, we planned that effect sizes and standard errors would be meta‐analysed in RevMan using the generic inverse method (Higgins 2008). If appropriate controls had not been used and individual participant data had not been available, statistical guidance would have been sought from the Cochrane Methods Group and external experts as to which method to apply to the published results in attempt to control for clustering. If there had been insufficient information to control for clustering, outcome data would have been entered into RevMan using individuals as the units of analysis, and sensitivity analysis then used to assess the potential biasing effects of inadequately controlled clustered trials (Donner 2001). In this review, however, no cluster‐randomised trials were identified.
b) Cross‐over trials
Had we been able to conduct meta‐analysis combining the results of cross‐over trials, we planned to use the inverse variance methods recommended by Elbourne (Elbourne 2002). Had the data presented from a cross‐over trial been restricted (and more information was not available from the original investigators), we planned to use the presented data within the first phase only, up to the point of cross‐over.
c) Multi‐arm trials
Multi‐arm trials were considered for inclusion in the review if (a) at least one arm constituted a relevant intervention with an antiepileptic drug, and (b) there was one arm which constituted a placebo condition. Separate data extractions were made for each pair‐wise comparison between a relevant intervention and the placebo condition.
Dealing with missing data
We attempted to contact the original investigators to request any missing data and information on whether or not it could be assumed to be ‘missing at random’. For dichotomous data, we report missing data and dropouts for each included study. We report the number of participants included in the final analysis as a proportion of all participants in each study. We provide reasons for missing data in the narrative summary within the 'Risk of bias' tables. For missing continuous data, we provide a qualitative summary. We attempted to contact the investigators if the standard deviations of the outcome measures for each group were not given. Had meta‐analyses been attempted where these standard deviations were unavailable, we planned to impute standard deviations using relevant data (for example, standard deviations or correlation coefficients) from other similar studies (Follmann 1992) but only if, after seeking statistical advice, to do so was deemed practical and appropriate.
We considered people leaving early to have had the negative outcome, except for adverse effects such as death. We had planned to report separately all data from studies where more than 50% of participants in any group were lost to follow up, and to exclude these from any meta‐analysis. However, on this occasion we identified only one study (Mattes 2005) where the proportion of participants lost to follow up compared to the number randomised was 52.5%. Since this was the only study on oxcarbazepine, the results are already presented separately.
We had planned to subject the impact of including studies with high attrition rates (25% to 50%) to sensitivity analysis in comparison with those with low attrition rates (0% to 49%). If inclusion of data from this group had resulted in a substantive change in the estimate of effect of the primary outcomes, we would not have added data from these studies to trials with less attrition, but would have presented them separately, and we had planned to use this approach for continuous outcomes where the proportion missing can be taken as an indication of potential bias. For dichotomous (binary) outcomes, however, the higher the ratio of participants with missing data to participants with events, the greater potential there is for bias. We had therefore planned to report this ratio for each binary outcome, and to allow for it when interpreting results of any sensitivity analysis. On this occasion, however, the data extracted were insufficient for this procedure to be executed.
Assessment of heterogeneity
We assessed the extent of between‐trial differences and the consistency of results of any meta‐analysis in three ways: by visual inspection of the forest plots, by performing the Chi2 test of heterogeneity (where a significance level less than 0.10 was interpreted as evidence of heterogeneity), and by examining the I2 statistic (Higgins 2008; section 9.5.2). The I2 statistic describes approximately the proportion of variation in point estimates due to heterogeneity rather than sampling error. We considered I2 values less than 30% as indicating low heterogeneity, values in the range 31% to 69% as indicating moderate heterogeneity, and values greater than 70% as indicating high heterogeneity. We would have attempted to identify any significant determinants of heterogeneity categorised at moderate or high.
Assessment of reporting biases
We had planned to draw funnel plots (effect size versus standard error) to assess publication bias if sufficient studies had been found. Asymmetry of the plots may indicate publication bias, although they may also represent a true relationship between trial size and effect size. If such a relationship had been identified, the clinical diversity of the studies would have been further examined as a possible explanation (Egger 1997). On this occasion, however, we were able to perform only four meta‐analyses (none of which were on primary outcomes) which were insufficient for this procedure to be executed.
Data synthesis
We planned to perform meta‐analysis using a fixed‐effect model where we considered studies to have sufficiently similar participants, interventions, comparators and outcome measures. In carrying out such meta‐analysis, the weight given to each study is such that the more precise estimates (from larger studies with more events) are given more weight.
For meta‐analysis of dichotomous data, we used Mantel‐Haenszel methods because they have been shown to have better statistical properties that other fixed‐effect methods (such as Peto and inverse variance) when the study size is small or when the odds ratios are not close to unity.
The weighted average of the results of all the available studies would have been used, if appropriate, to provide an estimate of the effect of antiepileptic drugs for aggression and impulsiveness. Where appropriate and if a sufficient number of studies had been found, we planned to use regression techniques to investigate the effects of differences in the study characteristics on the estimate of the treatment effects. Statistical advice would have been sought before attempting meta‐regression and if performed would have been executed using a random‐effects model. On this occasion, however, the data extracted were insufficient for this procedure to be executed.
Subgroup analysis and investigation of heterogeneity
If sufficient studies had been found, we planned to undertake subgroup analysis to examine the effect on primary outcomes of:
participants’ age;
participants’ principal diagnosis (e.g. personality disorder, learning disability, ADHD);
setting (inpatient, custodial, outpatient/community);
class of antiepileptic drug.
On finding a number of studies with participants aged less than 18 years, we planned to perform sensitivity analysis to explore the effect of including/excluding this younger sample. On this occasion, however, the data extracted were insufficient for this procedure to be executed.
Sensitivity analysis
If there had been sufficient data, we planned to undertake sensitivity analyses to investigate the robustness of the overall findings in relation to certain study characteristics with a priori sensitivity analyses planned for:
concealment of allocation;
blinding of outcome assessors; and
extent of dropouts.
On this occasion, however, the data extracted were insufficient for this procedure to be executed.
Results
Description of studies
Results of the search
Electronic searches to April 2009 identified 2258 records (767 through MEDLINE, 506 through EMBASE, 332 through CENTRAL, 545 through PsycINFO, 57 through CINAHL, 44 through the Cochrane Schizophrenia Group's register of trials on aggression, five through ClinicalTrials.gov, and two through the National Research Records/metaRegister of Controlled Trials). Removing obvious duplicates resulted in 1916 records. Detailed inspection of abstracts resulted in removal of 1820 that were clearly irrelevant, resulting in 96 records for which a full copy was obtained. After inspecting these 96 papers we removed 48 because of the clear lack of random allocation or absence of a placebo control group; 48 studies remained.
Included studies
Of the 48 studies, we identified 14 that met fully the inclusion criteria (Barratt 1991; Barratt 1997; Conners 1971; Cueva 1996; Donovan 2000; Gardner 1986; Gottschalk 1973; Hellings 2005; Hollander 2003; Mattes 2005; Mattes 2008; Rosenblatt 1976; Stanford 2001; Stanford 2005). These 14 studies involved a total of 16 comparisons of an antiepileptic drug against placebo. There were some important differences between the studies. We summarise these differences and the main study characteristics below. Further details are provided in the Characteristics of included studies table.
Design
Of these 14 placebo‐controlled studies, eight were parallel trials and six were cross‐over trials. Of the eight parallel trials, six were two‐condition comparisons of an antiepileptic drug against placebo (Cueva 1996; Gottschalk 1973; Hellings 2005; Hollander 2003; Mattes 2005; Mattes 2008), one study was a three‐condition comparison of two drugs against placebo, one of which was not an antiepileptic (Conners 1971), and one was a four‐condition comparison involving three antiepileptic drugs against placebo (Stanford 2005). When evaluating the six cross‐over trials for inclusion in the review, we first considered whether the cross‐over design was suitable for the condition being studied. Cross‐over trials are suitable for evaluating interventions with a temporary effect in the treatment of stable conditions, and where long‐term follow up is not required (Higgins 2008, p.500). On this basis, the cross‐over design was considered suitable since recurrent aggression is a reasonably stable condition and long‐term follow up, though desirable, is not essential for evaluating the effects of antiepileptic medication. Of the six studies with cross‐over designs, five were two‐condition studies of an antiepileptic drug against placebo (Barratt 1997; Donovan 2000; Gardner 1986; Rosenblatt 1976; Stanford 2001), and one was a three‐condition study of an antiepileptic drug at two dose regimes against placebo (Barratt 1991).
Sample sizes
There was a considerable variation in sample size between studies. Overall, the number of participants initially randomised per study ranged from 13 to 246 (mean 54.4; median 34), and the number of participants completing per study ranged from 8 to 145 (mean 33.3; median 23.5). The distribution of sample size across the studies was uneven. Whereas one large multi‐centre trial (Hollander 2003) randomised 246 participants and the other multi‐centre trial (Barratt 1997) reported on a subgroup of 60 randomised participants, the remaining 13 studies involved less than 50 participants, with sample sizes ranging from 13 to 48.
Setting
All 14 studies were carried out in North America. Two were multi‐centre trials (Barratt 1997; Hollander 2003), 10 were single‐centre trials, and two did not give sufficient information to allow classification. The trials took place in a number of very different settings encompassing custodial, hospital and community environments. Three studies involved participants who were prisoners (Barratt 1991; Barratt 1997; Gottschalk 1973), one involved boys at a residential centre for delinquent juveniles (Conners 1971) and one involved hospitalised children (Cueva 1996). The remaining nine studies involved participants in outpatient settings.
Participants
An inclusion criterion for this review was that participants must have been recruited on the basis of having displayed recurrent aggression. Investigators defined recurrent aggression in various ways (see Characteristics of included studies table), but all required either a chronic pattern of aggressive behaviour or some minimum frequency of observed or reported acts of aggression over a recent time interval. Several studies required additional indicators of aggression or hostility: a diagnosis of intermittent explosive disorder was an inclusion criterion for Mattes 2005 and Mattes 2008; a score of 8 or higher on the irritability subscale of the Buss‐Durkee Hostility Inventory was an inclusion criterion for Stanford 2001 and Stanford 2005; and a minimum score of 15 on the Overt Aggression Scale (OAS) was required by Hollander 2003. Rosenblatt 1976 recruited participants who had complained of difficulty in controlling physically punitive impulses towards (or were suspected of physical assault on) their own children.
In 10 of the 14 included studies, the aggression was described specifically as directed against others, against property or both. Donovan 2000 had inclusion criteria that included either oppositional defiant disorder or conduct disorder (both diagnosed under DSM‐IV) with, additionally, an explosive temper defined as four or more outbursts of rage, property destruction or fighting per month on minimal provocation. Where the trial investigators appeared to have included violence towards the self (i.e. self‐harm) in their definition of aggression, we excluded the study only if participants had been recruited solely on the basis of having self‐harmed. We therefore included Hellings 2005 with an inclusion criterion of significant aggression to self, others, or property at least three times a week, and Gardner 1986 who recruited on the basis of an extensive history of ‘behavioural dyscontrol’ which included aggression towards the self as well as towards others and property.
In eight of the 14 included studies, participants were recruited on the basis of meeting criteria for a formal clinical diagnosis in addition to having displayed recurrent aggression: pervasive developmental disorder (Hellings 2005), conduct disorder (Cueva 1996), conduct disorder or oppositional defiant disorder (Donovan 2000), borderline personality disorder (Gardner 1986), personality disorder (Stanford 2001), intermittent explosive disorder (Mattes 2005; Mattes 2008), and either intermittent explosive disorder, post‐traumatic stress disorder or cluster B personality disorder (Hollander 2003).
There were considerable differences between the studies in terms of sex and age of the participants recruited. Participants were restricted to males in six studies (Barratt 1991; Barratt 1997; Conners 1971; Gottschalk 1973; Stanford 2001; Stanford 2005). One study was female‐only (Gardner 1986). The remaining seven studies had a mix of male and female participants. Ten of the 14 studies involved adult participants. One was restricted to children, aged 5 to 12 years (Cueva 1996). Three involved young people described by the authors at ‘youths’ or ‘juveniles’ in which age ranges were 9 to 14 years (Conners 1971), 10 to 12 years (Hellings 2005) and 10 to 18 years (Donovan 2000).
Interventions
Five antiepileptic drugs were compared to placebo in the 14 included studies. In each case, the route of administration was oral (by tablets, capsules or liquid). Studies varied in the way they reported the dose administered to the treatment group: a fixed daily dose (mg/day), a fixed daily dose in relation to body weight (mg/kg/day), or a dose adjusted in attempt to achieve a target blood serum concentration (μg/ml). Full details are provided in the Characteristics of included studies table but can be summarised as follows.
Four studies involved valproate (full name: sodium valproate) (Hellings 2005 target 20 mg/kg/day; Stanford 2005 750 mg/day) or divalproex (full name: divalproex sodium) (Donovan 2000 target 90 μg/ml, range 750 to 1500 mg/day; Hollander 2003 target 80 to 120 μg/ml, max 30 mg/kg/day).
Three studies involved carbamazepine (Cueva 1996 mean 683 mg/day, range 400 to 800 mg/day; Gardner 1986 mean 820 mg/day, range 200 to 1200 mg/day; Stanford 2005 450 mg/day).
Seven studies involved phenytoin/diphenylhydantoin (Barratt 1991 100 mg/day and 300 mg/day, Barratt 1997 300 mg/day; Stanford 2001 300 mg/day; Stanford 2005 300 mg/day; Conners 1971 200 mg/day; Gottschalk 1973 300 mg/day; Rosenblatt 1976 400 mg/day).
One study involved levetiracetam (Mattes 2008 mean 1738 mg/day).
One study involved oxcarbazepine (Mattes 2005 mean 1500 mg/day).
In identifying these five antiepileptic drugs, we took account of the following.
Phenytoin is described as diphenylhydantoin in some early studies (the name was subsequently shortened), but both terms refer to the same drug (Marson 2009); we therefore consider them together in this review.
Divalproex sodium is an equimolar compound of sodium valproate and valproic acid; because the two drugs are regarded as equivalent in efficacy and have similar side effect profiles, we consider them together in this review.
Oxcarbazepine is a recently developed analogue of carbamazepine which is claimed to have a lower potential for drug interactions and to induce liver enzymes to a lesser extent than carbamazepine than carbamazepine; because the two drugs are essentially different agents and are likely to have different efficacies and side effect profiles, we consider them separately in this review.
The duration of the interventions ranged between two and 24 weeks (mean 8.1 weeks; median 6.0 weeks). None of the 14 studies followed up participants beyond the end of the intervention period. The duration of the trials ranged between two and 24 weeks (mean 12.2 weeks; median 12.5 weeks).
Outcomes
Primary outcomes
For the primary outcome of observer‐reported aggression, one study reported the frequency of aggressive incidents as observed by a third party (Barratt 1991). Five studies reported measurements made using the Overt Aggression Scale (OAS) in its original form (Barratt 1997; Cueva 1996; Hellings 2005; Stanford 2001; Stanford 2005). One study (Donovan 2000) reported aggression using the Modified Overt Aggression Scale (MOAS) in conjunction with scores from six 'irritability' items from the anger‐hostility subscale of the SCL‐90, with the combined scores dichotomised to improved/not improved. One study (Cueva 1996) reported additional aggression data using the aggression subscale of the Children’s Psychiatric Rating Scale (CPRS).
For the primary outcome of self‐reported aggression, one study (Hollander 2003) used the Overt Aggression Scale ‐ Modified (OAS‐M) which takes the form of a clinician‐rated semi‐structured interview. Two studies (Mattes 2005; Mattes 2008) measured aggression using the OAS‐M with slight revision by the trial investigators. One study (Gardner 1986) reported the frequency of aggressive incidents as reported by the participants in response to questioning by the trial investigators.
Three studies (Conners 1971; Gottschalk 1973; Rosenblatt 1976), all carried out more than 30 years ago, did not include any formal measure of primary outcome (aggression) as defined in the protocol of this review.
Secondary outcomes
Studies varied widely in terms of choice of secondary outcomes. Two studies included a measure of impulsivity: Conners 1971 reported Porteus Maze scores, and Rosenblatt 1976 reported data from the impulsiveness subscale of a Q‐sort scale developed by the investigators. Four studies included a measure of hostility: Cueva 1996 reported scores from the hostility subscale of the Children’s Psychiatric Rating Scale (CPRS), Mattes 2005 and Mattes 2008 reported scores from the hostility subscale of the British Psychiatric Rating scale (BPRS), Gottschalk 1973 reported using three hostility subscales from a speech content analysis, and Rosenblatt 1976 reported a hostility measure from a Q‐sort scale developed by the investigators. Two studies included a measure of anger: Conners 1971 reported using the Rosenweig Picture Frustration Test categories, and Gardner 1986 provided data on the self‐reported presence or absence of angry outbursts. No studies included any measure of quality of life. No studies included any measure of satisfaction with treatment. Eight studies included information on non‐compliance as the proportion of participants discontinuing treatment (Donovan 2000; Gardner 1986; Hellings 2005; Hollander 2003; Mattes 2005; Mattes 2008; Rosenblatt 1976; Stanford 2005). Four studies provided data on adverse events (Cueva 1996; Hellings 2005; Hollander 2003; Mattes 2008).
Other relevant outcomes
Three studies reported using a combined measure of anger and hostility as a subscale of the Profile of Moods Scale (POMS) (Barratt 1991; Barratt 1997; Stanford 2001). The review authors were unable to decide whether this should be classified as a measure of anger or of hostility, and so considered anger‐hostility as a unique secondary outcome measure (and additional to those defined in the original protocol).
Three studies (Cueva 1996; Hellings 2005; Hollander 2003) reported on weight change in the participants. Because change in weight can be viewed both positively and negatively, we classify both weight gain and weight loss as ‘adverse events’ in this review and summarise all data on weight change where this is supplied by the investigators.
Excluded studies
The remaining 34 studies that did not meet all the inclusion criteria were categorised as excluded studies. Twenty‐four were excluded because, on close inspection and following translation into English and contact with the investigators where necessary, it became clear that participants had not been selected on the basis of having displayed recurrent aggression. Two were excluded because participants had been allocated alternately and not at random ('quasi‐randomisation'), four because of lack of a placebo arm, two because they were subgroup analyses of studies already included that provided no additional data, one because it was a brief summary of another excluded study and one because it was an overview of several non‐randomised studies. Reasons for exclusion of each of these 34 studies are given in the Characteristics of excluded studies table.
Risk of bias in included studies
There was considerable variation in how quality aspects were reported. We attempted to contact the investigators wherever the available trial reports provided insufficient information for decisions to be made about the likely risk of bias, and were successful in respect of six studies. The older studies tended to report quality poorly, and in three that were more than 30 years old (Conners 1971; Gottschalk 1973; Rosenblatt 1976) we were unsuccessful in our attempts to contact the trial investigators for clarification. Full details of our assessment of the risk of bias for each included study are provided as tables within the Characteristics of included studies section.
Allocation
The generation of allocation sequence was considered adequate in six studies; two where allocation was by throwing a dice (Mattes 2005; Mattes 2008) and four where allocation was based on random numbers which were computer‐generated or derived from a table (Cueva 1996; Donovan 2000; Stanford 2001; Stanford 2005). Adequacy of sequence generation was classified as ‘unclear’ in the remaining studies where the investigators reported that participants had been allocated at random but provided no further information on how this had been achieved.
Concealment of the allocation sequence was considered adequate in six studies (Cueva 1996; Donovan 2000; Mattes 2005; Mattes 2008; Stanford 2001; Stanford 2005) where the authors considered that there was sufficient evidence that the person enrolling participants could not have foreseen assignment. Adequacy of sequence concealment was classified as ‘unclear’ in the remaining studies.
Blinding
Review authors judged that adequate blinding had been assured and was adequate for participants and personnel in 12 studies, and for outcome assessors in 11 studies, and that it was unlikely that this blinding could have been broken. The remaining studies (Barratt 1991; Barratt 1997; Rosenblatt 1976) were each described by the trial investigators as being double‐blind, but were classified as 'unclear' on the basis that there was insufficient information to reach a decision on the adequacy of the blinding.
Incomplete outcome data
Most studies appeared to have adequately addressed incomplete outcome data. Four studies were classified as 'unclear' (Barratt 1991; Barratt 1997; Cueva 1996; Gardner 1986) because reasons for attrition were not clearly stated or because information was lacking on how missing data were distributed between the treatment groups. Three studies reported exclusions following randomisation on account of additional medication or a serious adverse effect (Gardner 1986), because a measure was not completed post‐baseline (Hollander 2003), or because the investigators were reporting a subgroup analysis (Barratt 1997).
The rate of attrition varied significantly between studies. Two studies reported no attrition (Conners 1971; Gottschalk 1973). Attrition rates, calculated as number completing in comparison with number randomised initially, ranged from zero to 60.0% (mean 30.6%; SD 19.4%; median 31.4%). Attrition rates, calculated as number completing in comparison with number randomised less those excluded by the trial investigators, ranged from zero to 52.5% (mean 26.2%; SD 17.6%; median 24.3%). Mean attrition rates by antiepileptic drug, calculated as number completing in comparison with number randomised initially, were as follows: valproate 20.2%, two studies; divalproex 33.1%, two studies; carbamazepine 21.1%, three studies; phenytoin 32.7%, seven studies; levetiracetam 52.5%, one study; oxcarbazepine 50.0%, one study.
Of the 12 studies with attrition, five (Cueva 1996; Hellings 2005; Hollander 2003; Mattes 2005; Mattes 2008) reported undertaking an intention‐to‐treat analysis for at least one measurement of the primary outcome (aggression). Donovan 2000 provided an intention‐to‐treat analysis only for the initial phase of their cross‐over trial. The remaining studies provided analysis only for those participants classed by the investigators as 'completers'. Attrition at follow up was not relevant as none of the 14 studies followed up participants beyond the end of the intervention period.
Selective reporting
Most studies appear to have reported on all the measures they set out to use in as far as can be discerned from the published reports without access to the original protocols, and at all time scales. In the only exception (Cueva 1996) the investigators stated that Cognitive Battery Ratings would be reported elsewhere but gave no further details. In this case, the risk of bias was classified as ‘unclear’ since although these results appear unavailable, neither the primary nor the secondary outcomes specified in this review rely on cognitive testing.
Other potential sources of bias
Five studies appeared to be free of any other sources of bias. Nine studies were classified as ‘unclear’ for reasons which included the possibility of bias in the selection of two subgroups for analysis (Barratt 1997), from potential carry‐over effects in the absence of a washout period between phases of a cross‐over trial (Donovan 2000), from recruitment procedures that may have included participants who had self‐harmed without displaying any other form of aggression (Gardner 1986), from the use of speech content analysis as a measure of hostility (Gottschalk 1973), from use of the OAS‐M instrument with small but apparently non‐validated revisions (Mattes 2005; Mattes 2008), and from presenting results from the first week only of cross‐over trial (Rosenblatt 1976).
Effects of interventions
See: Table 1
1. Comparison 1: valproate/divalproex versus placebo
Four studies were included in this comparison: Hellings 2005 (outpatient children and adolescents with pervasive developmental disorder; dose 20 mg/kg/day; n = 30), Stanford 2005 (outpatient men; dose 750 mg/day; n = 20), Donovan 2000 (outpatient youths with conduct disorder or oppositional defiant disorder; dose 750 to 1500 mg/day; n = 20), and Hollander 2003 (adult outpatients with cluster B personality disorder, intermittent explosive disorder or post‐traumatic stress disorder; dose max 30 mg/kg/day; n = 246).
1.1 Aggression (observer‐reported)
Two studies (Hellings 2005; Stanford 2005) examined the impact of valproate on observer‐reported aggression using the Overt Aggression Scale (OAS). In both cases, the data provided are skewed and it is not possible to include them in a meta‐analysis.
Hellings 2005 reports skewed summary data (see Table 8) indicating no statistically significant difference between valproate and placebo conditions for OAS total scores averaged over weeks six, seven and eight of this eight‐week intervention (P = 0.96; two‐sided Wilcoxon rank sum test; intention‐to‐treat analysis by the trial investigators).
1. Comparison 1: valproate versus placebo: aggression, observer‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Hellings 2005 20 mg/kg/day |
OAS total score, at 8 wks (mean of wks 6, 7 & 8) | 16 | 5.86 | 3.84 | 14 | 5.72 | 4.62 | P = 0.96 (2‐sided Wilcoxon rank sum test) | Favours neither condition |
|
Stanford 2005 750 mg/day |
OAS aggression score, at 2 wks | 7 | 2.02 | 1.95 | 8 | 4.38 | 1.86 |
F1,13 = 16.92 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.57; power = 0.97); treatment OAS aggression score significantly lower compared with the placebo group. Significant main effect by time (3 interventions of which valproate was one; baseline, 2, 4, 6 wks); F3,75 = 3.41;P = 0.02; partial eta2 = 0.12; power = 0.78). |
Favours valproate Completer analysis (see note 1) |
|
Stanford 2005 750 mg/day |
OAS aggression score, at 4 wks | 7 | 2.80 | 2.30 | 8 | 4.49 | 2.14 |
F1,13 = 16.92 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.57; power = 0.97); treatment OAS aggression score significantly lower compared with the placebo group Significant main effect by time (3 interventions of which valproate was one; baseline, 2, 4, 6 wks); F3,75 = 3.41;P = 0.02; partial eta2 = 0.12; power = 0.78) |
Favours valproate Completer analysis (see note 1) |
|
Stanford 2005 750 mg/day |
OAS aggression score, at 6 wks | 7 | 0.62 | 1.99 | 8 | 5.40 | 1.86 |
F1,13 = 16.92 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.57; power = 0.97); treatment OAS aggression score significantly lower compared with the placebo group Significant main effect by time (three interventions of which valproate was one; baseline, 2, 4, 6 wks); F3,75 = 3.41; P = 0.02; partial eta2 = 0.12; power = 0.78). |
Favours valproate Completer analysis (see note 1) |
1. Data extracted from graph provided in study paper and confirmed by inspection of original Excel file supplied by lead author (email to J. Dennis 22 January 2009)
OAS = Overt Aggression Scale; wks = weeks
Stanford 2005 reports skewed summary data (see Table 8) for OAS aggression scores at three time points (two weeks; four weeks; endpoint at six weeks) indicating a statistically significant lower aggression score overall for the valproate condition compared with the placebo condition (P = 0.001; repeated measures ANOVA; analysis of completers conducted by the trial investigators). The investigators also report a statistically significant main effect by time for the three interventions in their study, of which valproate was one (P = 0.02; completer analysis by the trial investigators).
A third study (Donovan 2000) reports data indicating a statistically significant difference between divalproex and placebo conditions on number improved (calculated as > 69% reduction in MOAS + SCL‐90 'irritability' scores at endpoint (six weeks) (OR 18.00; 95% CI 1.27 to 255.74, P = 0.03, Analysis 1.8), favouring divalproex.
1.8. Analysis.
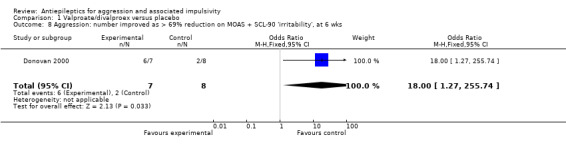
Comparison 1 Valproate/divalproex versus placebo, Outcome 8 Aggression: number improved as > 69% reduction on MOAS + SCL‐90 'irritability', at 6 wks.
1.2 Aggression (self‐reported)
Hollander 2003 reports skewed summary data (see Table 9) for OAS‐M aggression scores for the sample overall, and for three subgroups of participants classified by diagnosis as having intermittent explosive disorder (IED), post‐traumatic stress disorder (PTSD) and Cluster B personality disorder respectively. There was no statistically significant difference between conditions on OAS‐M aggression scores over the last four weeks of the intervention for the sample overall (P = 0.989), for the IED subgroup (P = 0.108), or for the PTSD subgroup (P = 0.679) (all analyses Wilcoxon rank‐sum test, van Elteren analysis; intention‐to‐treat analysis conducted by the trial investigators). However, Hollander 2003 reports a statistically significant difference between divalproex and placebo conditions on OAS‐M aggression scores over the last four weeks of the intervention for the cluster B personality disorder subgroup (P = 0.047), favouring divalproex.
2. Comparison 1: divalproex versus placebo: aggression, self‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Hollander 2003 max 30 mg/kg/day |
OAS‐M aggression score, all participants, mean over last 4 wks | 116 | 34.5 median 10.6 |
71.3 | 117 | 32.1 median 12.3 |
57.2 | Z = 0.000 (Wilcoxon rank‐sum test, van Elteren analyses; P = 0.989) | Favours neither condition |
|
Hollander 2003 max 30 mg/kg/day |
OAS‐M aggression score, IED subgroup, mean over last 4 wks | 55 | 28.9 median 13.0 |
39.1 | 54 | 28.9 median 9.0 |
59.9 | Z = 2.580 (Wilcoxon rank‐sum test, van Elteren analyses; P = 0.108) | Favours neither condition |
|
Hollander 2003 max 30 mg/kg/day |
OAS‐M aggression score, PTSD subgroup, mean over last 4 wks | 18 | 64.2 median 14.9) |
13.6 | 15 | 22.9 median 14.3 |
27.1 | Z = 0.173 (Wilcoxon rank‐sum test, van Elteren analyses; P = 0.679) | Favours neither condition |
|
Hollander 2003 max 30 mg/kg/day |
OAS‐M aggression score, Cluster B PD subgroup, mean over last 4 wks | 43 | 29.2 median 8.3 |
66.1 | 48 | 38.6 median 16.3 |
61.1 | Z = 3.952 (Wilcoxon rank‐sum test, van Elteren analyses; P = 0.047) | Favours divalproex |
IED = intermittent explosive disorder; OAS = Overt Aggression Scale; PTSD = post‐traumatic stress disorder; wks = weeks; PD = personality disorder
1.3 Impulsivity
No study set out to examine this as an outcome.
1.4 Hostility
No study set out to examine this as an outcome.
1.5 Anger
No study set out to examine this as an outcome.
1.5a Anger‐hostility
No study set out to examine this as an outcome.
1.6 Quality of life
No study set out to examine this as an outcome.
1.7 Satisfaction with care
No study set out to examine this as an outcome.
1.8 Non‐compliance
Donovan 2000, Hellings 2005, Hollander 2003 and Stanford 2005 reported on the outcome of leaving the study early. Results of meta‐analysis of data from these four studies indicate no statistically significant difference between valproate/divalproex and placebo conditions (OR 1.23; 95% CI 0.77 to 1.96, P = 0.38, I2 = 0%; P value for heterogeneity 0.96, Analysis 1.7). Hollander 2003 additionally provides data on leaving the study early for the subgroup of participants with cluster B personality disorder, again indicating no statistically significant difference between divalproex and placebo conditions (OR 1.08; 95% CI 0.48 to 2.41, P = 0.85, Analysis 1.11).
1.7. Analysis.

Comparison 1 Valproate/divalproex versus placebo, Outcome 7 Non‐compliance: leaving the study early, any reason.
1.11. Analysis.
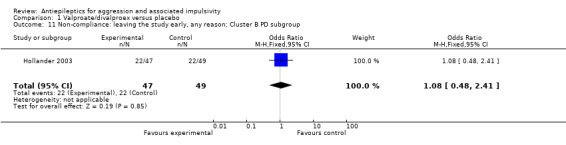
Comparison 1 Valproate/divalproex versus placebo, Outcome 11 Non‐compliance: leaving the study early, any reason; Cluster B PD subgroup.
1.9 Adverse events
Neither Donovan 2000 nor Stanford 2005 provide data on adverse events. Meta‐analysis of data from Hellings 2005 and Hollander 2003 on numbers with any adverse event indicates a statistically significant difference between valproate/divalproex and placebo conditions, favouring placebo (OR 3.07; 95% CI 1.42 to 6.65, P = 0.004, I2 = 0%; P value for heterogeneity 0.80, Analysis 1.2). The three most frequently occurring adverse events reported by Hellings 2005 were headache, rash and increased appetite; those reported by Hollander 2003 were somnolence, headache and nausea.
1.2. Analysis.
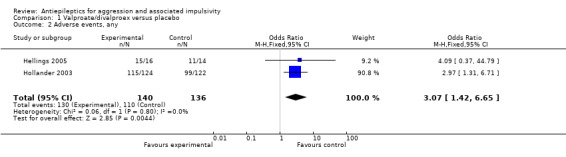
Comparison 1 Valproate/divalproex versus placebo, Outcome 2 Adverse events, any.
For the adverse event of headache, a meta‐analysis of data from these two studies indicates no statistically significant difference between treatment and placebo conditions (OR 0.76; 95% CI 0.44 to 1.32, P = 0.33, I2 = 0%; P value for heterogeneity 0.32, Analysis 1.4). For Hellings 2005, no statistically significant difference between conditions is indicated for the adverse event of rash (OR 7.80; 95% CI 0.80 to 75.64, P = 0.08, Analysis 1.3), but a statistically significant difference emerged for increased appetite (OR 7.71; 95% CI 1.28 to 46.36, P = 0.03, Analysis 1.6), favouring the placebo condition. For Hollander 2003, a statistically significant difference between conditions is indicated for the presence of nausea (OR 2.81; 95% CI 1.44 to 5.47, P = 0.002, Analysis 1.9), again favouring placebo. There was no statistically significant difference between treatment and placebo conditions for presence of somnolence (OR 1.64; 95% CI 0.94 to 2.87, P = 0.08, Analysis 1.10).
1.4. Analysis.

Comparison 1 Valproate/divalproex versus placebo, Outcome 4 Adverse events, headache.
1.3. Analysis.
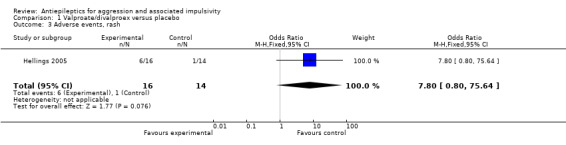
Comparison 1 Valproate/divalproex versus placebo, Outcome 3 Adverse events, rash.
1.6. Analysis.
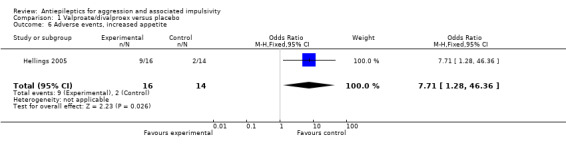
Comparison 1 Valproate/divalproex versus placebo, Outcome 6 Adverse events, increased appetite.
1.9. Analysis.
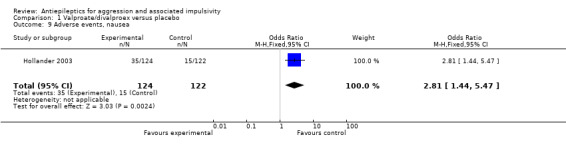
Comparison 1 Valproate/divalproex versus placebo, Outcome 9 Adverse events, nausea.
1.10. Analysis.
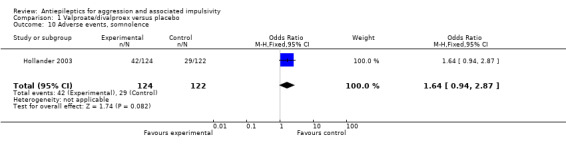
Comparison 1 Valproate/divalproex versus placebo, Outcome 10 Adverse events, somnolence.
In terms of change in weight, neither Donovan 2000 nor Stanford 2005 provide data. Meta‐analysis of data from Hellings 2005 and Hollander 2003 indicates a statistically significant difference between valproate/divalproex and placebo conditions, with weight gain significantly more likely in the divalproex group (OR 2.42; 95% CI 1.10 to 5.31, P = 0.03, I2 = 0%; P value for heterogeneity 0.75, Analysis 1.5). Hellings 2005 also provides summarised continuous data on weight gain over the course of the study in the valproate group (mean 1.98 kg, SD 1.88, n = 16) and the placebo group (mean 1.10 kg, SD 1.10, n = 14). Interpretation of such data is not attempted here, but should take account of the age of the participants (range 10 to 12 years) and the possibility of significant variability between individuals in growth rates, and hence rate of weight increase over time, at this stage of development.
1.5. Analysis.

Comparison 1 Valproate/divalproex versus placebo, Outcome 5 Adverse events, weight gain.
1.10 Other outcomes
Hellings 2005 additionally provides data on the number of overall clinical responders (as assessed by the principal investigator) indicating no statistically significant difference between valproate and placebo conditions (OR 2.25; 95% CI 0.48 to 10.60, P = 0.31; Analysis 1.1). We report this as a post‐hoc analysis since overall clinical response is not a prospectively stated outcome for this review.
1.1. Analysis.
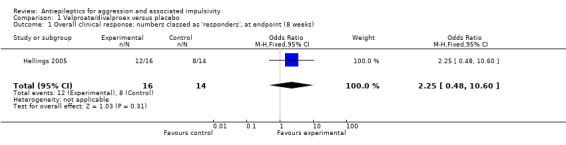
Comparison 1 Valproate/divalproex versus placebo, Outcome 1 Overall clinical response: numbers classed as 'responders', at endpoint (8 weeks).
2. Comparison 2: carbamazepine versus placebo
Three studies were included in this comparison: Cueva 1996 (inpatient children with conduct disorder; dose mean 683 mg/day; n = 24), Gardner 1986 (outpatient women with borderline personality disorder; dose mean 820 mg/day; n = 11) and Stanford 2005 (outpatient men; dose 450 mg/day; n = 20).
2.1 Aggression (observer‐reported)
Cueva 1996 reports skewed summary data (see Table 10) for OAS total scores at two time points (four weeks; endpoint at six weeks) indicating no statistically significantly difference overall between carbamazepine and placebo conditions at either time point (fitted slopes analysis via regression model; completer analysis by the trial investigators). Cueva 1996 also reports skewed summary data (see Table 10) for CPRS aggression subscale scores at two time points (four weeks; endpoint at six weeks) indicating no statistically significantly difference overall between conditions at either time point (ANCOVA; completer analysis by the trial investigators). Stanford 2005 reports skewed summary data (see Table 10) for OAS aggression scores at three time points (two weeks; four weeks; endpoint at six weeks) indicating no statistically significantly difference overall between carbamazepine and placebo conditions (P = 0.083; repeated measures ANOVA; completer analysis by the trial investigators).
3. Comparison 2: carbamazepine versus placebo: aggression, observer‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Cueva 1996 Mean 683 mg/day |
CPRS aggression subscale, at 4 wks | See note 1 | 3.42 | 1.73 | See note 1 | 3.20 | 1.46 | No significant difference between groups (ANCOVA) | Favours neither condition Completer analysis (see note 1) |
|
Cueva 1996 Mean 683 mg/day |
CPRS aggression subscale, at 6 wks | See note 1 | 3.08 | 1.60 | See note 1 | 3.18 | 1.34 | No significant difference between groups (ANCOVA) | Favours neither condition Completer analysis (see note 2) |
|
Cueva 1996 Mean 683 mg/day |
OAS total score; via regression model, at 4 wks | 13 | No data | 11 | No data | No significant difference between groups (fitted slopes analysis) | Favours neither condition | ||
|
Cueva 1996 Mean 683 mg/day |
OAS total score; via regression model, at 6 wks | 13 | No data | 11 | No data | No significant difference between groups (fitted slopes analysis) | Favours neither condition | ||
|
Stanford 2005 450 mg/day |
OAS aggression score, at 2 wks | 7 | 3.62 | 1.95 | 8 | 4.38 | 1.86 | Treatment mean OAS aggression scores not significantly lower compared with the placebo group (repeated measures ANOVA; P = 0.083). | Favours neither condition Completer analysis (see note 2) |
|
Stanford 2005 450 mg/day |
OAS aggression score, at 4 wks | 7 | 4.21 | 2.30 | 8 | 4.49 | 2.14 | Treatment mean OAS aggression scores not significantly lower compared with the placebo group (repeated measures ANOVA; P = 0.083). | Favours neither condition Completer analysis (see note 2) |
|
Stanford 2005 450 mg/day |
OAS aggression score, at 6 wks | 7 | 1.86 | 1.99 | 8 | 5.40 | 1.86 | Treatment mean OAS aggression scores not significantly lower compared with the placebo group (repeated measures ANOVA; P = 0.083). | Favours neither condition Completer analysis (see note 2) |
1. 22 of 24 completed, but trial investigators do not report the distribution of dropouts between conditions
2. Data extracted from graph provided in study paper and confirmed by inspection of original Excel file supplied by lead author (email to J. Dennis 22 January 2009)
CPRS = Children's Psychiatric Rating Scale; OAS = Overt Aggression Scale; wks = weeks
2.2 Aggression (self‐reported)
Gardner 1986 reports data indicating a statistically significant difference between carbamazepine and placebo conditions in number of self‐reported aggressive acts towards others, objects or self over the last three weeks of the intervention (OR 0.06; 95% CI 0.01 to 0.63, P = 0.02, Analysis 2.10), favouring carbamazepine. However, Gardner 1986 also reports data indicating no statistically significant difference between conditions for number of self‐reported aggressive acts towards others and objects (and not self) over the last three weeks of the intervention (OR 0.12; 95% CI 0.01 to 1.29, P = 0.08, Analysis 2.9). For the other two studies, the data provided on aggression are skewed and it is not possible to include them in a meta‐analysis.
2.10. Analysis.
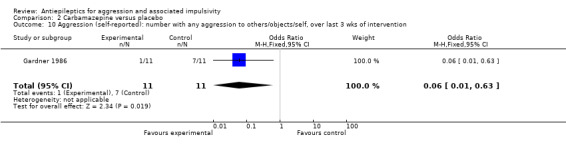
Comparison 2 Carbamazepine versus placebo, Outcome 10 Aggression (self‐reported): number with any aggression to others/objects/self, over last 3 wks of intervention.
2.9. Analysis.
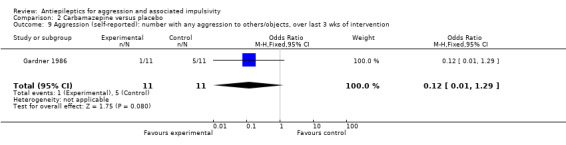
Comparison 2 Carbamazepine versus placebo, Outcome 9 Aggression (self‐reported): number with any aggression to others/objects, over last 3 wks of intervention.
2.3 Impulsivity
No study set out to examine this as an outcome.
2.4 Hostility
Cueva 1996 reports skewed summary data (see Table 11) indicating no statistically significant difference between carbamazepine and placebo conditions for mean CPRS hostility subscale scores at two time points (four weeks; endpoint at six weeks) (ANCOVA; completer analysis by the trial investigators).
4. Comparison 2: carbamazepine versus placebo: hostility (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Cueva 1996 Mean 683 mg/day |
CPRS hostility subscale, at 4 wks | see note 1 | 1.58 | 0.74 | see note 1 | 1.78 | 1.05 | No significant difference between groups (ANCOVA) | Favours neither condition Completer analysis (see note 1) |
|
Cueva 1996 Mean 683 mg/day |
CPRS hostility subscale, at 6 wks | see note 1 | 1.81 | 1.20 | see note 1 | 1.73 | 0.68 | No significant difference between groups (ANCOVA) | Favours neither condition Completer analysis (see note 1) |
1. 22 of 24 completed, but trial investigators do not report the distribution of dropouts between conditions.
CPRS = Children's Psychiatric Rating Scale; wks = weeks
2.5 Anger
Gardner 1986 reports data indicating no statistically significant difference between conditions for number displaying more than one angry outburst over six weeks (OR 0.12; 95% CI 0.01 to 1.29, P = 0.08, Analysis 2.12) and for numbers displaying any angry outbursts over six weeks (OR 0.18; 95% CI 0.02 to 1.92, P = 0.15, Analysis 2.13).
2.12. Analysis.
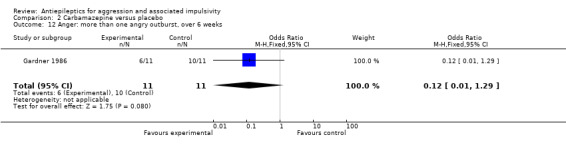
Comparison 2 Carbamazepine versus placebo, Outcome 12 Anger: more than one angry outburst, over 6 weeks.
2.13. Analysis.

Comparison 2 Carbamazepine versus placebo, Outcome 13 Anger: any angry outburst, over 6 weeks.
2.5a Anger‐hostility
No study set out to examine this as an outcome.
2.6 Quality of life
No study set out to examine this as an outcome.
2.7 Satisfaction with care
No study set out to examine this as an outcome.
2.8 Non‐compliance
Only Stanford 2005 provides data on non‐compliance; these indicate no statistically significant difference between carbamazepine and placebo conditions for the outcome of leaving the study early (OR 0.76; 95% CI 0.10 to 5.96, P = 0.80, Analysis 2.8). Gardner 1986 provides no data, but notes that compliance with medication appeared to be 'excellent' and compliance in completing the daily and weekly forms was extremely high (90% completed).
2.8. Analysis.
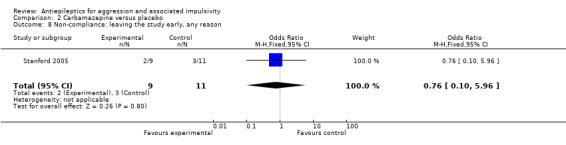
Comparison 2 Carbamazepine versus placebo, Outcome 8 Non‐compliance: leaving the study early, any reason.
2.9 Adverse events
Gardner 1986 and Stanford 2005 do not provide data on adverse events. Cueva 1996 reports the most frequently occurring adverse events as headache, rash/dermatitis, dizziness and stomach ache. Cueva 1996 reports data indicating no statistically significant difference between conditions for presence of any adverse event (OR 10.00; 95% CI 0.94 to 105.92, P = 0.06, Analysis 2.1), for rash/dermatitis (OR 8.57; 95% CI 0.84 to 87.83, P = 0.07, Analysis 2.2), or for headache (OR 19.93; 95% CI 0.97 to 408.44, P = 0.05, Analysis 2.3), but found a statistically significant difference for dizziness (OR 26.54; 95% CI 1.30 to 543.78, P = 0.03, Analysis 2.4), favouring the placebo group. There was no statistically significant difference between the carbamazepine and placebo conditions for presence of stomach ache (OR 0.80; 95% CI 0.13 to 5.09, P = 0.81, Analysis 2.5). Cueva 1996 additionally provides data on leucopenia which we include here on account of the potential seriousness of this adverse event. These data indicate no statistically significant difference between carbamazepine and placebo conditions (OR 8.57; 95% CI 0.84 to 87.83, P = 0.07, Analysis 2.11).
2.1. Analysis.
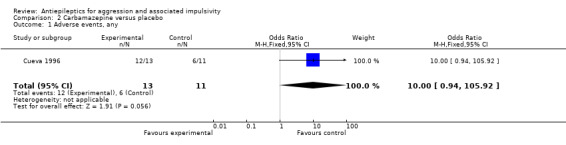
Comparison 2 Carbamazepine versus placebo, Outcome 1 Adverse events, any.
2.2. Analysis.
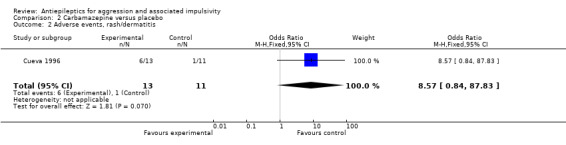
Comparison 2 Carbamazepine versus placebo, Outcome 2 Adverse events, rash/dermatitis.
2.3. Analysis.

Comparison 2 Carbamazepine versus placebo, Outcome 3 Adverse events, headache.
2.4. Analysis.
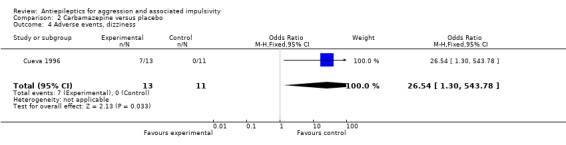
Comparison 2 Carbamazepine versus placebo, Outcome 4 Adverse events, dizziness.
2.5. Analysis.
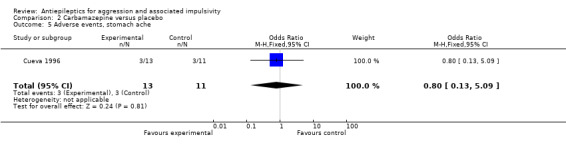
Comparison 2 Carbamazepine versus placebo, Outcome 5 Adverse events, stomach ache.
2.11. Analysis.
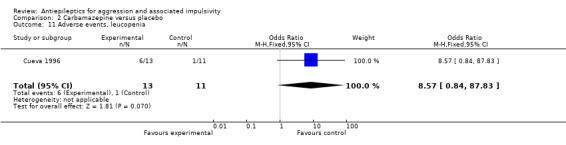
Comparison 2 Carbamazepine versus placebo, Outcome 11 Adverse events, leucopenia.
In terms of change in weight, neither Gardner 1986 nor Stanford 2005 provide data. Cueva 1996 reports data indicating no statistically significant difference between carbamazepine and placebo conditions for weight loss (OR 0.40; 95% CI 0.05 to 3.12, P = 0.38, Analysis 2.6) or weight gain (OR 2.50; 95% CI 0.32 to 19.53, P = 0.38, Analysis 2.7).
2.6. Analysis.

Comparison 2 Carbamazepine versus placebo, Outcome 6 Adverse events, weight loss.
2.7. Analysis.
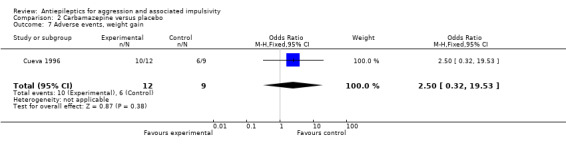
Comparison 2 Carbamazepine versus placebo, Outcome 7 Adverse events, weight gain.
3. Comparison 3: phenytoin/diphenylhydantoin versus placebo
Seven studies were included in this comparison: Barratt 1991 (incarcerated men; dose 100 mg/day and 300 mg/day; n = 19), Barratt 1997 (incarcerated men; dose 300 mg/day; n = 126 with analysis of 60), Gottschalk 1973 (incarcerated men; 300 mg/day; n = 42), Rosenblatt 1976 (outpatients; dose 400 mg/day; n = 13), Stanford 2001 (outpatient men with personality disorder; dose 300 mg/day; n = 46), Stanford 2005 (outpatient men; dose 300 mg/day; n = 20) and Conners 1971 (boys at a residential training facility, dose 200 mg/day; n = 30).
3.1 Aggression (observer‐reported)
Gottschalk 1973 and Rosenblatt 1976 provide no data on this outcome. Stanford 2005 reports skewed summary data (see Table 12) for OAS aggression scores at three time points (two weeks; four weeks; endpoint at six weeks) indicating a statistically significant difference between conditions (P = 0.001, repeated measures ANOVA; completer analysis by the trial investigators), favouring phenytoin. Barratt 1991 reports skewed summary data (see Table 12) for phenytoin versus placebo at two dose regimes (100 mg/day and 300 mg/day). These data indicate no statistically significant difference between conditions at the 100 mg/day dose for change from baseline in frequency of aggressive acts at endpoint (four weeks) (Wilcoxon's signed ranks test; completer analysis by the trial investigators). However, frequency of aggressive acts appeared significantly reduced from baseline at the 300 mg/day dose during the phenytoin but not the placebo condition (P < 0.001, Wilcoxon's signed ranks test; completer analysis by the trial investigators). Barratt 1997 reports skewed summary data (see Table 12) for both impulsive and non‐impulsive subgroups. For the impulsive subgroup, these data indicate a statistically significant difference between conditions at endpoint (six weeks) for mean frequency of aggressive acts (P < 0.01) and for mean intensity of aggressive acts (P < 0.01), favouring phenytoin in both cases. For the non‐impulsive subgroup, the data indicate no statistically significant difference between conditions at endpoint (six weeks) for either mean frequency or mean intensity of aggressive acts. All analyses were Geissner‐Greenhouse adjusted ANOVAs, conducted by the trial investigators). Stanford 2001 reports skewed summary data (see Table 12) indicating a statistically significant difference between conditions for OAS frequency of impulsive‐aggressive outbursts per week at endpoint (six weeks) (P = 0.008, repeated measures ANOVA; completer analysis by the trial investigators), favouring phenytoin. Conners 1971 reports data on frequency of 'behavioral incidents' together with a statistical analysis that indicate no statistically significant difference between conditions, but not all such incidents were necessarily aggressive and no further details are provided. Conners 1971 also reports summary data on both home ('cottage') staff and teacher ratings of overall symptoms with statistical analyses that again indicate no statistically significant difference between conditions, but these rating were not solely of aggression and no further details are provided.
5. Comparison 3: phenytoin/diphenylhydantoin versus placebo: aggression, observer‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Stanford 2005 300 mg/day |
OAS aggression score, at 2 wks | 7 | 2.17 | 1.95 | 8 | 4.38 | 1.86 |
F1,13 = 19.84 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.60; power = 0.98); treatment OAS aggression score significantly lower compared with the placebo group Significant main effect by time (3 interventions of which phenytoin was one; baseline, 2, 4, 6 wks); F3,75 = 3.41; P = 0.02; partial eta2 = 0.12; power = 0.78) |
Favours phenytoin Completer analysis (see note 1) |
|
Stanford 2005 300 mg/day |
OAS aggression score, at 4 wks | 7 | 1.28 | 2.30 | 8 | 4.49 | 2.14 |
F1,13 = 19.84 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.60; power = 0.98); treatment OAS aggression score significantly lower compared with the placebo group Significant main effect by time (3 interventions of which phenytoin was one; baseline, 2, 4, 6 wks); F3,75 = 3.41; P = 0.02; partial eta2 = 0.12; power = 0.78). |
Favours phenytoin Completer analysis (see note 1) |
|
Stanford 2005 300 mg/day |
OAS aggression score, at 6 wks | 7 | 2.38 | 1.99 | 8 | 5.40 | 1.86 |
F1,13 = 19.84 (repeated measures ANOVA; P = 0.001; partial eta2 = 0.60; power = 0.98); treatment OAS aggression score significantly lower compared with the placebo group Significant main effect by time (3 interventions of which phenytoin was one; baseline, 2, 4, 6 wks); F3,75 = 3.41; P = 0.02; partial eta2 = 0.12; power = 0.78) |
Favours phenytoin Completer analysis (see note 1) |
|
Barratt 1991 100 mg/day |
Frequency of aggressive acts, change from baseline, at 4 wks | 13 |
No data | 13 |
No data | Frequency of aggressive acts not significantly reduced from baseline during either treatment or placebo conditions (Wilcoxon’s signed ranks test) | Favours neither condition Completer analysis (no data available) |
||
|
Barratt 1991 300 mg/day |
Frequency of aggressive acts, change from baseline, at 4 wks | 13 |
No data | 13 |
No data | Frequency of aggressive acts significantly reduced from baseline (Wilcoxon’s signed ranks test; P < 0.001) during treatment but not placebo condition |
Favours phenytoin Completer analysis (no data available) |
||
|
Barratt 1997 300 mg/day |
Frequency of aggressive acts per wk, mean, at 6 wks | 60 | 0.33 | No data | 60 | 0.51 | No data | F1,58 = 9.64 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.001) |
Favours phenytoin (see below for subgroup analysis) |
|
Barratt 1997 300 mg/day |
Intensity of aggressive acts, mean, at 6 wks | 60 | 2.61 | No data | 60 | 3.96 | No data | F1,58 = 8.23 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) |
Favours phenytoin (see below for subgroup analysis) |
|
Barratt 1997 300 mg/day |
Frequency of aggressive acts per wk, mean, impulsive subgroup, at 6 wks | 30 | 0.20 | 0.19 | 30 | 0.52 | 0.46 | Subgroup effect (impulsive vs. non‐impulsive) F1,58 = 9.21 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) Subgroup by drug‐placebo effect F1,58 = 9.50 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) |
Favours phenytoin (impulsive aggression subgroup) |
|
Barratt 1997 300 mg/day |
Intensity of aggressive acts, mean, impulsive subgroup, at 6 wks | 30 | 2.11 | 1.20 | 30 | 4.16 | 1.92 | Subgroup effect (impulsive vs. non‐impulsive) F1,58 = 4.78 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.05) Subgroup by drug‐placebo effect F1,58 = 9.74 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; p<0.01). |
Favours phenytoin (impulsive aggression subgroup) |
|
Barratt 1997 300 mg/day |
Frequency of aggressive acts per week, mean, non‐impulsive subgroup, at 6 wks | 30 | 0.42 | 0.24 | 30 | 0.51 | 0.48 | Subgroup effect (impulsive vs. non‐impulsive) F1,58 = 9.21 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) Subgroup by drug‐placebo effect F1,58 = 9.50 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) |
Favours neither condition (non‐impulsive aggression subgroup) |
|
Barratt 1997 300 mg/day |
Intensity of aggressive acts, mean, non‐impulsive subgroup, at 6 wks | 30 | 3.40 | 1.29 | 30 | 3.76 | 1.59 | Subgroup effect (impulsive vs. non‐impulsive) F1,58 = 4.78 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.05) Subgroup by drug‐placebo effect F1,58 = 9.74 (repeated measure ANOVA, Geissner‐Greenhouse adjusted; P < 0.01) |
Favours neither condition (non‐impulsive aggression subgroup) |
|
Stanford 2001 300 mg/day |
Frequency of impulsive‐aggressive outbursts, from OAS, mean no outbursts/week, over 6 wks | 23 | 0.60 | 0.41 | 23 | 0.97 | 0.68 |
F1,21 = 8.44 (repeated measures ANOVA; P = 0.008) Frequency of impulsive‐aggressive outbursts significantly reduced from baseline for phenytoin (F1,21 = 9.37; P = 0.006) |
Favours phenytoin Completer analysis (see note 2) |
1. Data presented here extracted from graph provided as figure 1 in study paper and confirmed by inspection of original Excel file supplied by lead author (email to J. Dennis 22 January 2009); statistics from study paper.
2. Data presented here extracted from graph provided as figure 1 in study paper; statistics from study paper.
OAS = Overt Aggression Scale; wks = weeks
3.2 Aggression (self‐reported)
No study set out to examine this as an outcome.
3.3 Impulsivity
Rosenblatt 1976 reports skewed summary data (see Table 13) indicating no statistically significant difference between diphenylhydantoin and placebo conditions for change from baseline in aggressive impulsiveness subscale scores on their Q‐sort 'A' measure (Mann Whitney U test; analysis conducted by the trial investigators).
6. Comparison 3: phenytoin/diphenylhydantoin versus placebo: impulsivity (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Conners 1971 200 mg/day |
Porteus Maze test quotient, at 2 wks | 15 | 112.90 | 22.60 | 15 | 101.60 | 26.20 | No significant differences between conditions (one‐way ANOVA) | Favours neither condition |
|
Conners 1971 200 mg/day |
Porteus Maze qualitative score, at 2 wks | 15 | 36.90 | 26.40 | 15 | 28.10 | 14.18 | No significant differences between conditions (one‐way ANOVA) | Favours neither condition |
|
Rosenblatt 1976 400 mg/day |
Q‐sort 'A' aggressive impulsiveness subscale, mean % change from baseline, at end of trial | 5 | ‐31% | No data | 3 | ‐31% | No data | No significant differences between conditions (Mann Whitney U test) | Favours neither condition |
wks = weeks
Conners 1971 reports skewed summary data (see Table 13) indicating no statistically significant difference between conditions for Porteus Maze test quotient scores (one‐way ANOVA; analysis conducted by the trial investigators), or for Porteus Maze qualitative scores (one‐way ANOVA; analysis conducted by the trial investigators) at endpoint (two weeks).
3.4 Hostility
Rosenblatt 1976 reports skewed summary data (see Table 14) indicating no statistically significant difference between diphenylhydantoin and placebo conditions for change from baseline in hostility subscale scores on their Q‐sort 'A' and 'B' measures (Mann Whitney U test; analysis conducted by the trial investigators). Gottschalk 1973 reports skewed summary data (see Table 14) indicating no statistically significant difference between conditions at endpoint (24 weeks) on speech content analysis hostility‐out scores (t = 1.21), on hostility‐in scores (t = ‐0.76) and on ambivalent hostility scores (t = ‐1.77). All were fitted slopes analyses conducted by the trial investigators.
7. Comparison 3: phenytoin/diphenylhydantoin versus placebo: hostility (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Gottschalk 1973 300 mg/day |
Speech content analysis, hostility‐out, fitted slope at 24 wks | 24 | 0.093 | 0.433 | 18 | ‐0.038 | 0.162 | t = 1.21 (no significant differences between conditions; fitted slopes analysis) | Favours neither condition |
|
Gottschalk 1973 300 mg/day |
Speech content analysis, hostility‐in, fitted slope at 24 wks | 24 | ‐0.037 | 0.128 | 18 | ‐0.007 | 0.124 | t = ‐0.76 (no significant differences between conditions; fitted slopes analysis) | Favours neither condition |
|
Gottschalk 1973 300 mg/day |
Speech content analysis, amb. hostility, fitted slope at 24 wks | 24 | ‐0.045 | 0.203 | 18 | ‐0.038 | 0.162 | t = ‐1.77 (no significant differences between conditions; fitted slopes analysis) | Favours neither condition |
|
Rosenblatt 1976 400 mg/day |
Q‐sort 'A' hostility subscales, mean % change from baseline, at end of trial | 5 | ‐37% | No data | 3 | ‐6% | No data | No significant differences between conditions (Mann Whitney U test) | Favours neither condition |
|
Rosenblatt 1976 400 mg/day |
Q‐sort 'B' hostility subscales, mean % change from baseline, at end of trial | 5 | ‐5% | No data | 3 | ‐53% | No data | No significant differences between conditions (Mann Whitney U test) | Favours neither condition |
wks = weeks
3.5 Anger
Conners 1971 reports skewed summary data (see Table 15) indicating no statistically significant difference between conditions for Rosenweig Picture Frustration Test categories at endpoint (two weeks) (one‐way Kurskal‐Wallis ANOVA; analysis conducted by the trial investigators).
8. Comparison 3: phenytoin/diphenylhydantoin versus placebo: anger (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Conners 1971 200 mg/day |
Rosenzweig Picture Frustration Test Categories, at 2 wks | 15 | Positive 4.4 Neutral 4.8 Negative 4.2 |
No data | 15 | Positive 4.4 Neutral 4.5 Negative 4.2 |
No data | No significant differences between conditions for the 3 categories (one‐way Kurskal‐Wallis ANOVA) | Favours neither condition |
3.5a Anger‐hostility
Barratt 1991 reports skewed summary data (see Table 16) for phenytoin versus placebo at two dose regimes (100 mg/day and 300 mg/day). These data indicate no statistically significant difference between conditions for change from baseline on the POMS anger‐hostility subscale at endpoint (four weeks) at 100 mg/day dose and also at 300 mg/day dose (ANOVA, Geissner‐Greenhouse adjusted; completer analysis by the trial investigators). Barratt 1997 reports skewed summary data (see Table 16) indicating no statistically significant difference between conditions for POMS anger‐hostility subscale scores at endpoint (six weeks) for both impulsive and non‐impulsive aggression subgroups (ANOVA, Geissner‐Greenhouse adjusted; completer analysis by the trial investigators). Stanford 2001 reports skewed summary data (see Table 16) indicating a statistically significant difference between conditions for POMS anger‐hostility subscale scores at endpoint (six weeks) (P = 0.011, ANOVA; completer analysis conducted by the trial investigators), favouring phenytoin.
9. Comparison 3: phenytoin/diphenylhydantoin versus placebo: anger‐hostility (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Barratt 1991 300 mg/day |
POMS anger‐hostility subscale, change from baseline, at 4 wks | 13 |
No data | 13 |
No data | Scores not significantly reduced from baseline (ANOVA, Geissner‐Greenhouse adjusted; no further details given) | Favours neither condition Completer analysis (no data available) |
||
|
Barratt 1991 100 mg/day |
POMS anger‐hostility subscale, change from baseline, at 4 wks | 13 |
No data | 13 |
No data | Scores not significantly reduced from baseline (ANOVA, Geissner‐Greenhouse adjusted; no further details given) | Favours neither condition Completer analysis (no data available) |
||
|
Barratt 1997 300 mg/day |
POMS anger‐hostility subscale, impulsive subgroup, at 6 wks | 30 | 20.4 | No data | 30 | 22.3 | No data | Scores not significantly reduced from baseline (ANOVA, Geissner‐Greenhouse adjusted; no further details given) | Favours neither condition (impulsive aggression subgroup) |
|
Barratt 1997 300 mg/day |
POMS anger‐hostility subscale, non‐impulsive subgroup, at 6 wks | 30 | 11.2 | No data | 30 | 12.5 | No data | Scores not significantly reduced from baseline (ANOVA, Geissner‐Greenhouse adjusted; no further details given) | Favours neither condition (non‐impulsive aggression subgroup) |
|
Stanford 2001 300 mg/day |
POMS anger‐hostility subscale, at 6 wks | 23 | 8.44 | 1.32 | 23 | 13.39 | 2.21 |
F2,42 = 5.78 (repeated measures ANOVA; P = 0.011; significant main effect for condition) Scores significantly reduced from baseline for phenytoin (F1,21 = 22.80; P < 0.001) but not for placebo condition |
Favours phenytoin |
POMS = Profile of Mood States; wks = weeks
3.6 Quality of life
No study set out to examine this as an outcome.
3.7 Satisfaction with care
No study set out to examine this as an outcome.
3.8 Non‐compliance
Only Stanford 2005 provides data on non‐compliance; these indicate no statistically significant difference between phenytoin and placebo conditions for the outcome of leaving the study early (OR 0.76; 95% CI 0.10 to 5.96, P = 0.80, Analysis 3.1). Neither Gottschalk 1973 nor Conners 1971 report any participant leaving the study early, although Gottschalk 1973 does note significant missing data with 21 participants failing to provide at least one of the six speech required samples. Rosenblatt 1976 reports five of 13 participants leaving the study early, but without indication on distribution of attrition between conditions.
3.1. Analysis.

Comparison 3 Phenytoin/diphenylhydantoin versus placebo, Outcome 1 Non‐compliance: leaving the study early, any reason.
3.9 Adverse events
Only Barratt 1997 provides data on adverse events; these indicate no statistically significant difference between phenytoin and placebo conditions for the presence of nausea (OR 1.00; 95% CI 0.06 to 16.76, P = 1.00, Analysis 3.2). Barratt 1997 also reports no significant side effects detectable via blood cell counts or liver enzyme tests. In terms of change in weight, none of the four studies provides data.
3.2. Analysis.
Comparison 3 Phenytoin/diphenylhydantoin versus placebo, Outcome 2 Adverse events, nausea.
4. Comparison 4: levetiracetam versus placebo
One study was included in this comparison: Mattes 2008 (outpatients with intermittent explosive disorder; dose mean 1738 mg/day; n = 40).
4.1 Aggression (observer‐reported)
No study set out to examine this as an outcome.
4.2 Aggression (self‐reported)
Mattes 2008, using the OAS‐M with slight revisions, reports skewed summary data (see Table 17) indicating no statistically significant difference between levetiracetam and placebo conditions at endpoint (10 weeks) for change in global overt aggression scores (P = 0.47), for change in total aggression (P = 0.51), for change in subjective irritability (P = 0.92), for change in verbal aggression (P = 0.94), for change in aggression against objects (P = 0.35) and for change in assault against others (P = 0.30). All tests were ANCOVAs (intention‐to‐treat analyses conducted by the trial investigators). Mattes 2008 also reports summary data (see Table 17) for change in relative rating of aggression (derived from the Rating Scale for Aggressive Behavior in the Elderly) indicating no statistically significant difference between conditions at endpoint (10 weeks) (ANCOVA; P = 0.70; intention‐to‐treat analysis conducted by the trial investigators).
10. Comparison 4: levetiracetam versus placebo: aggression, self‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in Global Overt Aggression score, at 10 wks | 19 | ‐0.84 | 0.94 | 20 | ‐0.93 | 0.98 | t = 0.74 (ANCOVA; P = 0.47) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in total aggression, at 10 wks | 19 | ‐4.68 | 5.54 | 20 | ‐4.48 | 4.70 | t = 0.67 (ANCOVA; P = 0.51) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in subjective irritability, at 10 wks | 19 | ‐0.66 | 0.80 | 20 | ‐0.68 | 0.95 | t = 0.11 (ANCOVA; P = 0.92) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
Mean change in relative rating of aggression, at 10 wks | 11 | ‐7.63 | 11.75 | 14 | ‐7.14 | 7.75 | t = ‐0.39 (ANCOVA; P = 0.70) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
Patient‐rated global improvement, at 10 wks | 19 | 1.74 | 1.45 | 20 | 1.60 | 1.39 | t = 0.39 (ANCOVA; P = 0.71) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in verbal aggression, at 10 wks | 19 | ‐3.00 | 3.30 | 20 | ‐2.75 | 2.69 | t = ‐0.08 (ANCOVA; P = 0.94) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in aggression against objects, at 10 wks | 19 | ‐1.42 | 2.22 | 20 | ‐1.28 | 1.23 | t = 0.95 (ANCOVA; P = 0.35) (see note 1) |
Favours neither condition |
|
Mattes 2008 mean 1738 mg/day |
OAS‐M‐revised, change in assault against others, at 10 wks | 19 | ‐0.13 | 0.64 | 20 | ‐0.43 | 1.05 | t = 1.04 (ANCOVA; P = 0.30) (see note 1) |
Favours neither condition |
1. Covariance analysis comparing levetiracetam versus placebo on change scores (final ‐ initial), covarying out the relationship between initial scores and change scores.
OAS = Overt Aggression Scale; wks = weeks
4.3 Impulsivity
No study set out to examine this as an outcome.
4.4 Hostility
Mattes 2008 reports skewed summary data (see Table 18) for change in the hostility subscale of the BPRS at endpoint (10 weeks) indicating no statistically significant difference between levetiracetam and placebo conditions (P = 0.89, ANCOVA, intention‐to‐treat analysis conducted by the trial investigators).
11. Comparison 4: levetiracetam versus placebo: hostility (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Mattes 2008 mean 1738 mg/day |
Change in BPRS hostility subscale, at 10 wks | 19 | ‐1.00 | 0.94 | 20 | ‐0.85 | 0.95 | t = ‐0.14 (ANCOVA; P = 0.89) (see note 1) |
Favours neither condition |
1. Covariance analysis comparing levetiracetam versus placebo on change scores (final ‐ initial), covarying out the relationship between initial scores and change scores; negative change = good.
BPRS = British Psychiatric Rating scale; wks = weeks
4.5 Anger
No study set out to examine this as an outcome.
4.5a Anger‐hostility
No study set out to examine this as an outcome.
4.6 Quality of life
No study set out to examine this as an outcome.
4.7 Satisfaction with care
No study set out to examine this as an outcome.
4.8 Non‐compliance
Mattes 2008 provides data on non‐compliance indicating no statistically significant difference between levetiracetam and placebo conditions for the outcome of leaving the study early (OR 6.33; 95% CI 0.67 to 60.16, P = 0.11, Analysis 4.1).
4.1. Analysis.
Comparison 4 Levetiracetam versus placebo, Outcome 1 Non‐compliance: leaving the study early (before 4 weeks), any reason.
4.9 Adverse events
Mattes 2008 reports the most frequently occurring adverse events as sedation, dizziness and headache. Data reported by Mattes 2008 indicate no statistically significant difference between conditions for presence of sedation (OR 1.52; 95% CI 0.43 to 5.43, P = 0.52, Analysis 4.2), dizziness (OR 4.75; 95% CI 0.48 to 46.91, P = 0.18, Analysis 4.3) or headache (OR 3.00; 95% CI 0.51 to 17.74, P = 0.23, Analysis 4.4). In terms of change in weight, Mattes 2008 offers no data on numbers of participants experiencing weight gain or weight loss, but provides summary data on weight loss indicating no statistically significant difference between conditions (mean weight loss: 0.22 lb levetiracetam group, 0.35 lb placebo group; t = 0.05, not significant).
4.2. Analysis.
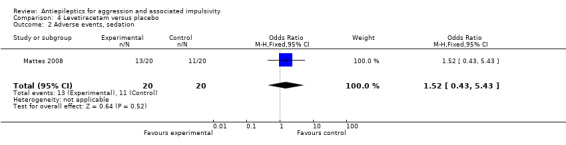
Comparison 4 Levetiracetam versus placebo, Outcome 2 Adverse events, sedation.
4.3. Analysis.

Comparison 4 Levetiracetam versus placebo, Outcome 3 Adverse events, dizziness.
4.4. Analysis.

Comparison 4 Levetiracetam versus placebo, Outcome 4 Adverse events, headache.
4.10 Other outcomes
Mattes 2008 additionally provide data on patient‐rated global improvement (see Table 17). Analysis provided by the trial investigators indicates no statistically significant difference between conditions on this outcome at 10 weeks (ANCOVA t = 0.11, P = 0.92). We report this as a post‐hoc analysis since patient‐rated global improvement is not a prospectively stated outcome for this review.
5. Comparison 5: oxcarbazepine versus placebo
One study was included in this comparison: Mattes 2005 (outpatients with intermittent explosive disorder; dose mean 1500 mg/day; n = 48).
5.1 Aggression (observer‐reported)
No study set out to examine this as an outcome.
5.2 Aggression (self‐reported)
Mattes 2005 reports data indicating a statistically significant difference between conditions for number of responders (identified as > 49% reduction in Global Overt Aggression score of the OAS‐M revised) at endpoint (10 weeks) (OR 4.88; 95% CI 1.36 to 17.47, P = 0.02, Analysis 5.1), favouring oxcarbazepine. Mattes 2005, using the OAS‐M with slight revisions, reports skewed summary data (see Table 19) indicating a statistically significant difference between conditions at endpoint (10 weeks) for change in global overt aggression scores (P = 0.035, t‐test), change in total aggression (P = 0.011, ANCOVA), change in subjective irritability (P = 0.049, t‐test), change in verbal aggression (P = 0.005, ANCOVA), and for change in aggression against objects (P = 0.028, ANCOVA), all favouring oxcarbazepine. All tests were by intention‐to‐treat analysis, and conducted by the trial investigators. Mattes 2005 also reports skewed summary data (see Table 19) indicating no statistically significant difference between conditions at endpoint (10 weeks) for change in assault against others (P = 0.251, t‐test, intention‐to‐treat analysis conducted by the trial investigators).
5.1. Analysis.

Comparison 5 Oxcarbazepine versus placebo, Outcome 1 Aggression (self‐reported): OAS‐M‐revised, number of responders as > 49% reduction in Global Overt Aggression score, at 10 wks.
12. Comparison 5: oxcarbazepine versus placebo: aggression, self‐reported (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in Global Overt Aggression score, at 10 wks | 21 | ‐0.98 | 0.83 | 24 | ‐0.44 | 0.83 | t = 2.18 (t‐test; P = 0.035) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
Patient‐rated global improvement, at 10 wks | 21 | 2.24 | 1.18 | 23 | 1.22 | 1.35 | t = 2.68 (t‐test; P = 0.010) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in total aggression, at 10 wks (see note 1) |
21 | ‐5.93 | 4.81 | 24 | ‐4.08 | 7.65 | t = 0.98 (t‐test; P = 0.33, but P = 0.011 with covariance analysis; see note 2) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in subjective irritability, at 10 wks | 21 | ‐0.86 | 0.73 | 24 | ‐0.44 | 0.65 | t = 2.03 (t‐test; P = 0.049) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in verbal aggression, at 10 wks (see note 1) |
21 | ‐3.79 | 3.00 | 24 | ‐2.42 | 4.91 | t = 1.14 (t‐test; P = 0.26, but P = 0.005 with covariance analysis; see note 2) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in aggression against objects, at 10 wks (see note 1) |
21 | ‐1.81 | 2.02 | 24 | ‐1.08 | 2.03 | t = 1.20 (t‐test; P = 0.24, but P = 0.028 with covariance analysis; see note 2) | Favours oxcarbazepine |
|
Mattes 2005 mean 1500 mg/day |
OAS‐M‐revised, change in assault against others, at 10 wks (see note 1) |
21 | ‐0.12 | 0.22 | 24 | ‐0.29 | 1.00 | t = 1.16 (t‐test; P = 0.251) | Favours neither condition |
1. Calculated multiplying severity x frequency; in calculating total aggression, verbal aggression and aggression against objects weighted equally, but assault given extra weighting based on the severity of the assault.
2. ANCOVA, covarying out the relationship between initial scores and change.
OAS = Overt Aggression Scale; wks = weeks
5.3 Impulsivity
No study set out to examine this as an outcome.
5.4 Hostility
Mattes 2005 reports skewed summary data (see Table 20) for change in the hostility subscale of the BPRS at endpoint (10 weeks) indicating a statistically significant difference between conditions (P = 0.018, intention‐to‐treat analysis conducted by the trial investigators), favouring oxcarbazepine.
13. Comparison 5: oxcarbazepine versus placebo: hostility (skewed data).
| Study | Outcome | n(Exp) | Mean(Exp) | SD(Exp) | n(Cntrl) | Mean(Cntrl) | SD(Cntrl) | Statistic | Notes |
|
Mattes 2005 mean 1500 mg/day |
Change in BPRS hostility subscale, at 10 wks | 21 | ‐1.29 | 0.90 | 23 | ‐0.57 | 1.04 | t = 2.46 (t‐test; P = 0.018) | Favours oxcarbazepine |
BPRS = British Psychiatric Rating scale
5.5 Anger
No study set out to examine this as an outcome.
5.5a Anger‐hostility
No study set out to examine this as an outcome.
5.6 Quality of life
No study set out to examine this as an outcome.
5.7 Satisfaction with care
No study set out to examine this as an outcome.
5.8 Non‐compliance
Mattes 2005 provides data on non‐compliance indicating no statistically significant difference between oxcarbazepine and placebo conditions for the outcome of leaving the study early for any reason (OR 0.51; 95% CI 0.16 to 1.61, P = 0.25, Analysis 5.2) or for leaving the study early due to adverse events (Peto OR 2.33; 95% CI 0.51 to 10.69, P = 0.28, Analysis 5.3). A statistically significant difference between conditions was indicated for the outcome of leaving the study early due to ineffectiveness (OR 0.24; 95% CI 0.06 to 0.90, P = 0.03, Analysis 5.4), with attrition due to ineffectiveness significantly more likely in the placebo group.
5.2. Analysis.
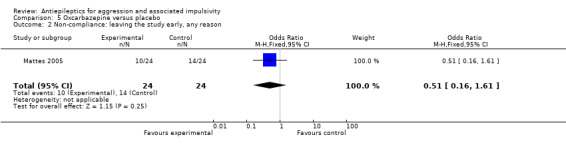
Comparison 5 Oxcarbazepine versus placebo, Outcome 2 Non‐compliance: leaving the study early, any reason.
5.3. Analysis.

Comparison 5 Oxcarbazepine versus placebo, Outcome 3 Non‐compliance: leaving the study early, due to adverse events.
5.4. Analysis.
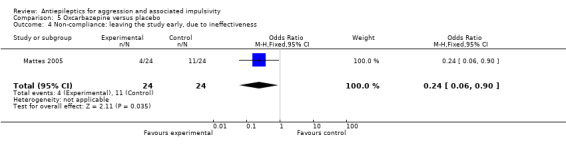
Comparison 5 Oxcarbazepine versus placebo, Outcome 4 Non‐compliance: leaving the study early, due to ineffectiveness.
5.9 Adverse events
Mattes 2005 reports no data on adverse events (other than noting they were generally minor and that hyponatraemia did not occur), or on change in weight.
5.10 Other outcomes
Mattes 2005 additionally provides data on patient‐rated global improvement (see Table 19). Analysis provided by the trial investigators indicates a statistically significant difference between conditions on this outcome at 10 weeks (ANCOVA t = 2.68, P = 0.01), favouring oxcarbazepine. We report this as a post‐hoc analysis since patient‐rated global improvement is not a prospectively stated outcome for this review.
Discussion
Summary of main results
Effects of antiepileptic drugs on primary outcome (aggression)
With regards the primary outcome (aggression), much of the quantitative data available from the studies included in this review met our criteria for skewed data as described in the section on Measures of treatment effect. This arises in part because most studies used measurements of aggression (such as the Overt Aggression Scale (OAS)) which are based on the number of observed aggressive acts over a given period. The minimum value of such a measurement is zero (i.e. no aggressive incidents) but there is no maximum value since the number of aggressive acts that an individual can perpetuate in a given time interval has no obvious limit. Consequently, in the absence of raw data from the trial investigators, we have had no alternative but to present much of the primary outcome data in separate tables. A further consequence is that we have often had to report statistics on primary outcomes as calculated by the trial investigators rather than performing our own analysis. This absence of effect size data prevented any synthesis of primary outcome data via meta‐analysis. The summaries that follow below are therefore essentially descriptive.
The focus of this review is relatively broad since it seeks evidence on effectiveness of any antiepileptic drug in the treatment of aggression. Considerable heterogeneity of the 14 included studies might therefore be anticipated. We found considerable difference between the studies in terms of participants, size of sample, intervention drug and choice of outcome measures. There were also differences between dosage regimes, although all doses were within the recommended range for prophylactic treatment of epilepsy according to the British National Formulary.
We found four antiepileptics (valproate/divalproex, carbamazepine, oxcarbazepine and phenytoin) which were effective, compared to placebo, in reducing aggression in at least one study, although it is important to note also that for three of these (valproate, carbamazepine and phenytoin) at least one other study showed no statistically significant difference between treatment and control conditions.
Valproate/divalproex was superior to placebo at a dose of 750 mg/day on OAS aggression scores in outpatient men (Stanford 2005, using sodium valproate) (no data on adverse effects), but was not significantly different from placebo on OAS total scores for children and adolescents with pervasive developmental disorder (PDD) (Hellings 2005, using sodium valproate). Hellings 2005 found increased appetite significantly more likely to occur in the valproate than the placebo group. This medication was also superior to placebo at up to 30 mg/kg/day on OAS‐M scores in impulsively aggressive adults with cluster B personality disorders in a relatively large trial (n = 246) in which treatment continued for 10 weeks (Hollander 2003, using divalproex). However, Hollander 2003 also reports that weight gain, nausea, and any side effect were significantly more common in the divalproex than the placebo condition. Divalproex was also superior to placebo at a dose of 750 to 1500 mg/day on modified‐MOAS scores in youths with conduct disorder (Donovan 2000, using divalproex) although this study provided no data on adverse effects. Meta‐analysis of data from Hellings 2005 and Hollander 2003 indicated that participants receiving valproate/divalproex were significantly more likely to report an adverse effect (any) and to report weight gain compared to those receiving placebo.
Carbamazepine was superior to placebo at a mean dose of 820 mg/day in reducing acts of self‐directed aggression in a small study of women with borderline personality disorder (Gardner 1986) (no data on adverse effects), but was not significantly different from placebo on OAS scores for children with conduct disorder (Cueva 1996) nor on CPRS aggression scores in outpatient men (Stanford 2005).
Oxcarbazepine was superior to placebo at a mean dose of 1500 mg/day over 10 weeks on revised OAS‐M scores for verbal aggression and aggression against objects in adult outpatients (Mattes 2005), although the proportion of participants lost to follow up was statistically significant (52.5%). This study provides no data on adverse events other than noting they were generally minor and that hyponatraemia did not occur.
Phenytoin, at a dose of 300 mg/day, was superior to placebo on the frequency of aggressive acts in a small study of male prisoners (Barratt 1991) (no data on adverse effects), and in male prisoners with impulsive (but not premeditated) aggression (Barratt 1997) with no significant difference reported between conditions for the presence of nausea. Phenytoin was also found superior to placebo on OAS scores in two studies of outpatient men (Stanford 2001) and of outpatient men with personality disorder (Stanford 2005) (neither study providing data on adverse effects). Phenytoin was, however, not superior to placebo on the frequency of 'behavioural incidents' in boys (Conners 1971) during a brief study lasting only two weeks.
The protocol required that we report these comparisons classified into three specific follow up periods: (1) within the first month, (2) between one and six months, and (3) between six and 12 months (see section on Measures of treatment effect). Because we were unable to perform any meta‐analysis of data on the primary outcome measure (aggression), we simply provide the following descriptive summary. In the 16 comparisons of an antiepileptic drug against placebo, only two reported endpoint results within the first month (Barratt 1991 after four weeks of treatment with phenytoin; Conners 1971 after two weeks of treatment with phenytoin), and none reported endpoint results at a time period greater than six months. Fourteen of the 16 comparisons thus fell within the one‐to‐six‐month period.
We identified three studies in which the beneficial effect of an antiepileptic drug appeared confined to a specific type of aggression:
In the small study by Gardner 1986, carbamazepine was found to reduce acts of aggression towards others, objects and self significantly compared to placebo, but not towards others and objects alone. Similar effects have been observed elsewhere. For example, in a non‐randomised pre‐post study by Lawson 2008 for divalproex in prisoners, the authors report: “surprisingly, reduction of behaviors directed against self reached significance, but disruptive behaviors directed towards others did not”. This finding may be a reflection of the aetiological heterogeneity of aggression. Although there may be contextual factors that are common to self‐directed aggression and aggression directed at others, it is likely that there will be distinct processes that influence how aggressive urges are manifest in behaviour.
In the study by Barratt 1997, where phenytoin was found to significantly reduce acts of impulsive aggression, but not premeditated aggression, compared to placebo. This is in line with evidence from the wider literature on aggression which suggests that different forms of aggression to others are underpinned by different mechanisms. The differences between impulsive or reactive aggression and premeditated or instrumental aggression have been well documented (e.g. Blair 2001). We suggest therefore that studies evaluating the effectiveness of interventions for aggression should use outcome measures that enable distinctions to be made according to the evidence‐based typologies of aggression.
In the study by Mattes 2005, where oxcarbazepine had a statistically significant effect on verbal aggression and aggression against objects but not on aggression against others. This finding would suggest that, as well as accounting for different types of aggression, outcome measures should be employed that allow differential effects by severity to be tested.
We identified one study in which the beneficial effect of an antiepileptic drug appeared confined to participants with a particular diagnosis.
In the study by Hollander 2003, divalproex was found to significantly reduce aggression in participants with cluster B personality disorder, but not with post traumatic stress disorder or intermittent explosive disorder. One possibility is that cluster B personality disorders are more likely to be associated with both low‐anxiety premeditated aggression (e.g. DeBrito 2009, p.143) and high‐anxiety impulsive/reactive aggression, whereas post‐traumatic stress disorder (PTSD) and intermittent explosive disorder (IED) are more likely to be associated with only high‐anxiety reactive impulsive aggression. This may arise from hypervigilance in the case of PTSD, and from sudden emotional outbursts in the case of IED.
Treatment compliance and attrition
There were eight comparisons in which compliance was reported, in each case as the proportion of participants leaving the study early: valproate/divalproex versus placebo (Donovan 2000; Hellings 2005; Hollander 2003; Stanford 2005); carbamazepine versus placebo (Stanford 2005); oxcarbazepine versus placebo (Mattes 2005); phenytoin (Stanford 2005). In none of these did the difference between intervention and control reach statistical significance. However, in their study of oxcarbazepine versus placebo, Mattes 2005 found attrition due to ineffectiveness to be significantly more likely in the placebo group.
The protocol required that we consider separately any studies where the rate of attrition exceeded 50%. We identified one study (Mattes 2005) where the proportion of participants lost to follow up compared to the number randomised was 52.5%, but here the results are already presented separately since this was the only study on oxcarbazepine. Statistics on rates of attrition in the 14 studies are given in the section on Incomplete outcome data.
Adverse events
The information on adverse events from the studies included in this review is relatively sparse with only five of the fourteen trials providing data. This is of concern because absence of such information does not necessarily mean that the treatment is safe, nor that the potential gains from the medication necessarily balance the risk of an adverse event occurring.
In this review we have included data on the three most common adverse effects reported for each trial, and have additionally included data on weight gain/loss where this has been presented by the trial investigators. Data on the occurrence of leucopenia has also been included where available on account of the potential seriousness of this adverse event. Statistically significant differences between conditions emerged in two studies with side effects more commonly noted for the intervention group in each case: increased appetite was more common in the valproate condition (Hellings 2005), and weight gain, nausea and any side effect were more common in the divalproex condition (Hollander 2003). Meta‐analysis of data from Hellings 2005 and Hollander 2003 indicated that participants receiving valproate/divalproex were significantly more likely to report an adverse effect (any) and to report weight gain compared to those receiving placebo.
A detailed side effect profile is, however, available for each of the drugs investigated here since each is (or has been) licensed for use in epilepsy.
Other secondary outcomes
No studies reported on quality of life or satisfaction with care. Two studies reported on impulsivity (Conners 1971; Rosenblatt 1976) and two reported on anger (Conners 1971; Gardner 1986) although none detected any statistically significant difference between treatment conditions. Mattes 2005 reported on British Psychiatric Rating scale (BPRS) hostility, with results favouring oxcarbazepine over placebo. Stanford 2001 reported on Profile of Moods Scale (POMS) anger‐hostility, with results favouring phenytoin over placebo.
Overall completeness and applicability of evidence
Whilst the evidence obtained from the identified studies is relevant to the review question, it is incomplete. In terms of interventions, the included studies addressed only five types of antiepileptic drug, whereas 13 are currently listed in the British National Formulary; these include a significant number of so‐called 'second‐generation' antiepileptic drugs that are purported to have better tolerability and a lower potential for interactions with other drugs. However, most of the studies identified in this review are of first‐generation antiepileptics (i.e. sodium valproate/divalproex, carbamazepine, phenytoin), and so the second‐generation antiepileptics are under‐represented.
In terms of participants, the differences between the studies in terms of sex and age were considerable (for example, six studies were male‐only, one was female‐only, and four involved children or youths). In terms of outcomes, there was inconsistency in the way primary and secondary outcomes were measured and reported, and the different types of aggression were poorly differentiated. We conclude that the studies identified are insufficient to address all the objectives of the review. This limits the review's external validity.
Quality of the evidence
We identified 14 studies that met the criteria for inclusion in this review, involving a total of 672 participants. We judged the overall quality of the evidence from these trials to be relatively poor for the following reasons.
The mean number of participants per study was only 54.4 (median 34.0) which is small compared to most drug trials.
The studies were carried out in a number of very different settings, including prisons, hospitals and in the community, and there was considerable heterogeneity of participants in terms of age and clinical diagnosis.
None of the studies followed up participants beyond the end of the intervention period.
There was considerable variation in how the studies were reported, and the information provided by the trial investigators in their published reports was often insufficient to allow a valid judgement to be made on the quality of their investigations. Attempts to contact the investigators for additional information that would allow decisions to be made on the risk of bias were not always successful.
Rates of attrition were significant in some studies, and ranged from zero to 52.5%.
A significant proportion of the primary (aggression) outcome data from these studies appeared not to be normally distributed, which prevented use of meta‐analysis to pool results and provide a quantitative summary.
The authors consider that the body of evidence summarised in this review is insufficient to allow any conclusion to be drawn about the use of antiepileptic medication in the treatment of aggression and associated impulsivity.
Potential biases in the review process
None known.
Agreements and disagreements with other studies or reviews
We were unable to identify any recent systematic review covering precisely the area defined in our protocol. However, two systematic reviews focusing more broadly on interventions for aggression have been published within the last three years, and we compare our findings with these.
Goedhard 2006 reviewed 35 studies of various classes of medication (including antiepileptics) in the treatment of outward‐directed aggressive behaviour within general adult psychiatry. As with the current review, only randomised controlled trials were included and applications in acute or emergency situations were excluded. They found six studies where an antiepileptic drug was the experimental condition, with the antiepileptic medication superior to placebo in three studies, each involving outpatients with cluster B personality disorders (divalproex in two studies; topiramate in one study). In comparison, our current review identified a greater number of relevant studies. This arises partly because additional relevant studies have been published in the last three years and partly because we searched more electronic databases with a more expansive search strategy. We identified the same six studies as Goedhard 2006, but excluded five of these because the two reviews differed in their inclusion and exclusion criteria. We required that study participants had displayed recurrent aggression, whereas Goedhard 2006 included studies of “psychiatric patients in whom aggression might be an ongoing problem”. Additionally, we included only placebo‐controlled studies whereas Goedhard 2006 did not have this restriction. Goedhard 2006 concluded that there was “weak evidence of efficacy in the management of aggression in Cluster B personality disordered outpatients” with “no serious adverse effects observed or reported,” but no strong evidence of efficacy overall. Our findings are similar in that we also found no strong evidence of efficacy, but we have been able to report on eight comparisons involving five different drugs where medication was superior to placebo.
Connor 2006 conducted a systematic review of both drug treatments and psychosocial interventions for juvenile aggression using an age limit of 18 years. They found four randomised studies where an antiepileptic drug was the experimental condition, with divalproex superior to placebo in two of these. We identified the same four studies as Connor 2006, but excluded one that was not placebo‐controlled. Connor 2006 concluded that divalproex sodium (and lithium) had been found effective in reducing aggression in children and adolescents with conduct disorder, but that it was predominately reactive, impulsive affective aggression that responded to medication. Our conclusions, based on a single study, are similar for individuals in this age group, and we have identified additional evidence to suggest that it is impulsive rather than premeditated aggression that appears more responsive to antiepileptic medication. Connor 2006 also concluded, as do we, that most studies do not distinguish the different forms of aggression and that this is an important area for future research.
Authors' conclusions
Implications for practice.
There is insufficient evidence upon which to base recommendations for practice. Current use of antiepileptic drugs to treat aggression in clinical practice remains a matter for the prescribing physician who will wish to weigh the limited evidence of effectiveness against any risk of possible harm; it should ideally be based on consultation with the multi‐disciplinary team involved in individual care.
Implications for research.
Further well‐designed, placebo‐controlled randomised trials are needed. Studies with positive findings reported here require replicating to confirm apparent efficacy, and further work is needed to explain the inconsistent findings reported for valproate/divalproex, carbamazepine and phenytoin (where at least one other study showed no statistically significant difference between treatment and control conditions). The design of such trials should take into account that aggression is a phenomenon for which there are likely to be many causal mechanisms and manifestations (McGuire 2008). We suggest attempts be made in future research to delineate different forms of aggression as well as measuring its severity and recording the diagnostic profile of the participant. Examples of different forms of aggression would include: directed at self versus others, reactive versus instrumental, relationship domain within which aggression displayed, presence versus absence of intoxication, and sexual versus non‐sexual.
Notes
We note that terms such as 'episodic dyscontrol syndrome' and 'intermittent aggression' are now being used more frequently in the literature. Selection of electronic search terms in any update of this review may wish to take this into account.
Acknowledgements
We gratefully acknowledge: David Fearnley's contribution in preparing an earlier version of the protocol; Toby Lasserson (Cochrane Airways Group, UK) for translation of a study in German; Celia Almeida (University of the West of England, UK) for translation of a study in Spanish; Jane Dennis (Cochrane DPLPG, UK) for translation of a study in Swedish; Jo Abbott (Cochrane DPLPG) for running the electronic searches, and Cathy Bennett for advice and helpful comments on an early draft of this review.
Appendices
Appendix 1. MEDLINE search strategy
Searched via OVID
(exp Homicide/ OR homicid$.tw. OR murder$.tw. OR manslaughter$.tw. OR infanticid$.tw. OR parricid$.tw. OR Torture/ OR tortur$.tw. OR (intent$ adj3 (kill$ or harm$)).tw. OR (bodily adj3 (harm or assault$)).tw. OR assail$.tw. OR attacker$.tw. OR child abuse/ or spouse abuse/ OR (physical adj3 abus$).tw. OR (spous$ adj3 abus$).tw. OR (partner adj3 abus$).tw. OR (child$ adj3 neglect$).tw. OR (child$ adj3 abus$).tw. OR (elder$ adj3 abus$).tw. OR Rape/ OR (rape$ or rapist$).tw. OR (sexual$ adj3 abus$).tw. OR bugger$.tw. OR Sex Offenses/ OR sodom$.tw. OR molest$.tw. OR (pedophil$ or paedophil$).tw. OR indecen$.tw. OR (masturbat$ adj3 public).tw. OR exhibitionis$.tw. OR lewd$.tw. OR sadis$.tw. OR sadomasochis$.tw. OR abduct$.tw. OR kidnap$.tw. OR aggress$.tw. OR violen$.tw. OR exp Anger/ OR malic$.tw. OR hostil$.tw. OR ((dangerous$ or disrupt$) adj3 (behav$ or histor$ or conduct$)).tw. OR ((destruct$ not self‐destruct$) adj3 (behav$ or histor$ or conduct$)).tw. OR cruel$.tw. OR delinquen$.tw. OR threaten$.tw. OR disorderly.tw. OR affray$.tw. OR (breach$ adj3 peace).tw. OR felon$.tw. OR unlawful$.tw. OR penal$.tw. OR penol$.tw. OR Crime/ OR crim$.tw. OR offen$.tw. OR Prisons/ OR prison$.tw. OR inmate$.tw. OR correctional$.tw. OR firearm$.tw. OR weapon$.tw. OR Firearms/ OR (gun or guns).tw. OR (agitat$ not (cancer or carcinoma)).tw. OR Impulsive Behavior/ OR impulsiv$.tw.)
AND
(Anticonvulsants/ OR (epilep$ adj3 drug$).tw. OR anti‐epileptic$.tw. OR antiepileptic$.tw. OR anti‐convulsant.tw. OR anticonvulsant$.tw. OR Carbamazepine/ OR carbamazepine.tw. OR Valproic Acid/ OR (sodium adj valproate).tw. OR gabapentin.tw. OR lamotrigine.tw. OR topiramate.tw. OR ethosuxamide.tw. OR Primidone/ OR primidone.tw. OR Vigabatrin/ OR vigabatrin.tw. OR Phenobarbital/ OR (phenobarbital or phenobarbitone).tw. OR Piracetam/ OR piracetam.tw. OR Acetazolamide/ OR acetazolamide.tw. OR levetiracetam.tw. OR pregabalin.tw. OR oxcarbazepine.tw. OR tiagabine.tw. OR zonisamide.tw. OR clobazam.tw. OR Clonazepam/ OR clonazepam.tw.)
MEDLINE was searched in combination with the Cochrane Collaboration’s search strategy for identifying reports of controlled trials as detailed in Section 6.4.11 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008).
Appendix 2. CENTRAL search strategy
Searched via the Cochrane Library
#1 MeSH descriptor Homicide explode all trees #2 homicid* #3 murder* #4 manslaughter* #5 infanticid* #6 parricid* #7 MeSH descriptor Torture explode all trees #8 tortur* #9 (intent* near/3 kill*) #10 (intent* near/3 harm*) #11 (bodily near/3 harm) #12 (bodily near/3 assault*) #13 (assail*) #14 (attacker*) #15 MeSH descriptor Child Abuse explode all trees #16 MeSH descriptor Spouse Abuse explode all trees #17 (physical near/3 abus*) #18 (spous* near/3 abus*) #19 (partner* near/3 abus*) #20 (child* near/3 neglect*) or (child* near/3 abus*) or (elder* near/3 abus*) #21 MeSH descriptor Rape explode all trees #22 (rape or rapist) #23 (sexual* near/3 abus*) or (bugger*) or (sodom*) or (molest*) or (pedophil* or paedophil*) #24 MeSH descriptor Sex Offenses explode all trees #25 (indecen*) or (masturbat* near/3 public) or (exhibitionis*) or (lewd*) or (sadis*) #26 (sadomasochis*) or (abduct*) or (kidnap*) or (aggress*) or (violen*) #27 MeSH descriptor Anger explode all trees #28 (malic*) or (hostil*) or (dangerous* near/3 behav*) or (dangerous* near/3 histor*) or (dangerous* near/3 conduct*) #29 (disrupt* near/3 behav*) or (disrupt* near/3 histor*) or (disrupt* near/3 conduct*) or (cruel*) or (delinquen*) #30 (destruct* near/3 behav*) #31 (destruct* near/3 histor*) or (destruct* near/3 conduct*) #32 (threaten*) or (disorderly) or (affray*) or (breach* near/3 peace) or (felon*) #33 (unlawful*) or (penal*) or (penol*) or (crim*) or (offen*) #34 MeSH descriptor Crime explode all trees #35 MeSH descriptor Prisons explode all trees #36 (prison*) or (inmate*) or (correctional*) or (firearm*) or (weapon*) #37 MeSH descriptor Firearms explode all trees #38 (gun or guns) #39 (agitat* not cancer) or (agitat* not carcinoma) #40 MeSH descriptor Impulsive Behavior explode all trees #41 (impulsiv*) #42 (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41) #43 MeSH descriptor Anticonvulsants explode all trees #44 (epilep* near/3 drug*) or (anti epileptic*) or (antiepileptic*) or (anti convulsant*) or (anticonvulsant*) #45 MeSH descriptor Carbamazepine explode all trees #46 (Carbamazepine) #47 MeSH descriptor Valproic Acid explode all trees #48 (sodium valporate) #49 (gabapentin) or (lamotrigine) or (topiramate) or (ethosuxamide) #50 MeSH descriptor Primidone explode all trees #51 (primidone) #52 MeSH descriptor Vigabatrin explode all trees #53 (vigabatrin) or (phenobarbital or phenobarbitone) or (piracetam) or acetazolamide or (levetiracetam) #54 MeSH descriptor Piracetam explode all trees #55 MeSH descriptor Acetazolamide explode all trees #56 (pregabalin) or (oxcarbazepine) or (tiagabine) or (zonisamide) or (clobazam) #57 MeSH descriptor Clonazepam explode all trees #58 (Clonazepam) #59 (#43 OR #44 OR #45 OR #46 OR #47 OR #48 OR #49 OR #50 OR #51 OR #52 OR #53 OR #54 OR #55 OR #56 OR #57 OR #58) #60 (#42 AND #59)
Appendix 3. EMBASE search strategy
Searched via OVID
1 exp Homicide/ 2 homicid$.tw. 3 murder$.tw. 4 manslaughter$.tw. 5 infanticid$.tw. 6 parricid$.tw. 7 Torture/ 8 tortur$.tw. 9 (intent$ adj3 (kill$ or harm$)).tw. 10 (bodily adj3 (harm or assault$)).tw. 11 assail$.tw. 12 attacker$.tw. 13 child abuse/ or spouse abuse/ 14 (physical adj3 abus$).tw. 15 (spous$ adj3 abus$).tw. 16 (partner adj3 abus$).tw. 17 (child$ adj3 neglect$).tw. 18 (child$ adj3 abus$).tw. 19 (elder$ adj3 abus$).tw. 20 Rape/ 21 (rape$ or rapist$).tw. 22 (sexual$ adj3 abus$).tw. 23 bugger$.tw. 24 Sexual Crime/ 25 sodom$.tw. 26 molest$.tw. 27 (pedophil$ or paedophil$).tw. 28 indecen$.tw. 29 (masturbat$ adj3 public).tw. 30 exhibitionis$.tw. 31 lewd$.tw. 32 sadis$.tw. 33 sadomasochis$.tw. 34 abduct$.tw. 35 kidnap$.tw. 36 aggress$.tw. 37 violen$.tw. 38 exp Anger/ 39 malic$.tw. 40 hostil$.tw. 41 ((dangerous$ or disrupt$) adj3 (behav$ or histor$ or conduct$)).tw. 42 ((destruct$ not self‐destruct$) adj3 (behav$ or histor$ or conduct$)).tw. 43 cruel$.tw. 44 delinquen$.tw. 45 threaten$.tw. 46 disorderly.tw. 47 affray$.tw. 48 (breach$ adj3 peace).tw. 49 felon$.tw. 50 unlawful$.tw. 51 penal$.tw. 52 penol$.tw. 53 Crime/ 54 crim$.tw. 55 offen$.tw. 56 Prison/ 57 prison$.tw. 58 inmate$.tw. 59 correctional$.tw. 60 firearm$.tw. 61 weapon$.tw. 62 Firearm/ 63 (gun or guns).tw. 64 (agitat$ not (cancer or carcinoma)).tw. 65 or/1‐64 66 Anticonvulsants/ 67 (epilep$ adj3 drug$).tw. 68 anti‐epileptic$.tw. 69 antiepileptic$.tw. 70 anti‐convulsant.tw. 71 anticonvulsant$.tw. 72 Carbamazepine/ 73 carbamazepine.tw. 74 Valproic Acid/ 75 (sodium adj valporate).tw. 76 gabapentin.tw. 77 lamotrigine.tw. 78 topiramate.tw. 79 ethosuxamide.tw. 80 Primidone/ 81 primidone.tw. 82 Vigabatrin/ 83 vigabatrin.tw. 84 Phenobarbital/ 85 (phenobarbital or phenobarbitone).tw. 86 Piracetam/ 87 piracetam.tw. 88 Acetazolamide/ 89 acetazolamide.tw. 90 levetiracetam.tw. 91 pregabalin.tw. 92 oxcarbazepine.tw. 93 tiagabine.tw. 94 zonisamide.tw. 95 clobazam.tw. 96 Clonazepam/ 97 clonazepam.tw. 98 or/66‐97 99 Impulsive Behavior/ 100 impulsiv$.tw. 101 65 or 99 or 100 102 divalproex.tw. 103 98 or 102 104 101 and 103 105 clin$.tw. 106 trial$.tw. 107 (clin$ adj3 trial$).tw. 108 singl$.tw. 109 doubl$.tw. 110 trebl$.tw. 111 tripl$.tw. 112 blind$.tw. 113 mask$.tw. 114 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$)).tw. 115 randomi$.tw. 116 random$.tw. 117 allocat$.tw. 118 assign$.tw. 119 (random$ adj3 (allocat$ or assign$)).tw. 120 crossover.tw. 121 120 or 119 or 115 or 114 or 107 122 exp Randomized Controlled Trial/ 123 exp Double Blind Procedure/ 124 exp Crossover Procedure/ 125 exp Single Blind Procedure/ 126 exp RANDOMIZATION/ 127 122 or 123 or 124 or 125 or 126 or 121 128 104 and 127
Appendix 4. CINAHL search strategy
Searched via EBSCO
S106 S73 and S105 S105 S74 or S75 or S76 or S77 or S78 or S79 or S80 or S81 or S82 or S83 or S84 or S85 or S86 or S87 or S88 or S89 or S90 or S91 or S92 or S93 or S94 or S95 or S96 or S97 or S98 or S99 or S100 or S101 or S102 or S103 or S104 S104 divalproex S103 clonazepam S102 (MH "Clonazepam") S101 clobazam S100 zonisamide S99 tiagabine S98 oxcarbazepine S97 pregabalin S96 levetiracetam S95 acetazolamide S94 (MH "Acetazolamide") S93 piracetam S92 phenobarbital or phenobarbitone S91 (MH "Phenobarbital") S90 vigabatrin S89 primidone S88 (MH "Primidone") S87 ethosuxamide S86 topiramate S85 lamotrigine S84 gabapentin S83 (sodium valporate) S82 (MH "Valproic Acid") Database ‐ CINAHL Display S81 carbamazepine S80 (MH "Carbamazepine") S79 anticonvulsant S78 anti‐convulsant S77 antiepileptic* S76 anti‐epileptic* S75 epilep* N3 drug* S74 (MH "Anticonvulsants") S73 S43 or S72 S72 S70 or S71 S71 S41 or S42 S70 S67 or S68 or S69 S69 S44 or S45 or S46 or S47 or S48 or S49 or S50 or S51 or S52 or S53 or S54 or S55 or S56 or S57 or S58 or S59 or S60 or S61 or S62 or S63 or S64 or S65 or S66 S68 S21 or S22 or S23 or S24 or S25 or S26 or S27 or S28 or S29 or S30 or S31 or S32 or S33 or S34 or S35 or S36 or S37 or S38 or S39 or S40 S67 S1 or S2 or S3 or S4 or S5 or S6 or S7 or S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 or S16 or S17 or S18 or S19 or S20 S66 impulsiv* S65 ( agitat* not cancer ) or ( agitat* not carcinoma ) S64 gun or guns S63 (MH "Firearms") S62 weapon* S61 firearm* S60 correctional* S59 inmate* S58 prison* S57 (MH "Correctional Facilities") S56 offen* S55 crim* S54 (MH "Crime") S53 penol* S52 penal* S51 unlawful* S50 felon* S49 breach* N3 peace S48 affray* S47 disorderly S46 threaten* S45 delinquen* S44 cruel* S43 destruct* N3 behav* or destruct* N3 histor* or destruct* N3 conduct* S42 disrupt* N3 behav* or disrupt* N3 histor* or disrupt* N3 conduct* S41 dangerous* N3 behav* or dangerous* N3 histor* or dangerous* N3 conduct* S40 hostil* S39 malic* S38 (MH "Anger") S37 violen* S36 aggress* S35 kidnap* S34 abduct* S33 sadomasochis* S32 sadis* S31 lewd* S30 exhibitionis* S29 masturbat* N3 public S28 indecen* S27 pedophil* or paedophil* S26 molest* S25 sodom* S24 bugger* S23 sexual* N3 abus* S22 rape* or rapist* S21 (MH "Rape") S20 elder* N3 abus* S19 child* N3 abus* S18 child* N3 neglect* S17 partner N3 abus* S16 spous* N3 abus* S15 physical N3 abus* S14 (MH "Intimate Partner Violence") S13 (MH "Child Abuse") S12 attacker* S11 assail* S10 bodily N3 harm* or bodily N3 assault* I S9 intent* N3 kill* or intent* N3 harm* S8 tortur* S7 (MH "Torture") S6 parricid* S5 infanticid* S4 manslaughter* S3 murder* S2 homicid* S1 (MH "Homicide+")
Appendix 5. PsycINFO search strategy
Searched via OVID
1 exp Homicide/ 2 homicid$.tw. 3 murder$.tw. 4 manslaughter$.tw. 5 infanticid$.tw. 6 parricid$.tw. 7 Torture/ 8 tortur$.tw. 9 (intent$ adj3 (kill$ or harm$)).tw. 10 (bodily adj3 (harm or assault$)).tw. 11 assail$.tw. 12 attacker$.tw. 13 child abuse/ or spouse abuse/ 14 (physical adj3 abus$).tw. 15 (spous$ adj3 abus$).tw. 16 (partner adj3 abus$).tw. 17 (child$ adj3 neglect$).tw. 18 (child$ adj3 abus$).tw. 19 (elder$ adj3 abus$).tw. 20 Rape/ 21 (rape$ or rapist$).tw. 22 (sexual$ adj3 abus$).tw. 23 bugger$.tw. 24 Sex Offenses/ 25 sodom$.tw. 26 molest$.tw. 27 (pedophil$ or paedophil$).tw. 28 indecen$.tw. 29 (masturbat$ adj3 public).tw. 30 exhibitionis$.tw. 31 lewd$.tw. 32 sadis$.tw. 33 sadomasochis$.tw. 34 abduct$.tw. 35 kidnap$.tw. 36 aggress$.tw. 37 violen$.tw. 38 exp Anger/ 39 malic$.tw. 40 hostil$.tw. 41 ((dangerous$ or disrupt$) adj3 (behav$ or histor$ or conduct$)).tw. 42 ((destruct$ not self‐destruct$) adj3 (behav$ or histor$ or conduct$)).tw. 43 cruel$.tw. 44 delinquen$.tw. 45 threaten$.tw. 46 disorderly.tw. 47 affray$.tw. 48 (breach$ adj3 peace).tw. 49 felon$.tw. 50 unlawful$.tw. 51 penal$.tw. 52 penol$.tw. 53 Crime/ 54 crim$.tw. 55 offen$.tw. 56 Prisons/ 57 prison$.tw. 58 inmate$.tw. 59 correctional$.tw. 60 firearm$.tw. 61 weapon$.tw. 62 Firearms/ 63 (gun or guns).tw. 64 (agitat$ not (cancer or carcinoma)).tw. 65 or/1‐64 66 Anticonvulsive Drugs/ 67 (epilep$ adj3 drug$).tw. 68 anti‐epileptic$.tw. 69 antiepileptic$.tw. 70 anti‐convulsant.tw. 71 anticonvulsant$.tw. 72 Carbamazepine/ 73 carbamazepine.tw. 74 Valproic Acid/ 75 (sodium adj valporate).tw. 76 gabapentin.tw. 77 lamotrigine.tw. 78 topiramate.tw. 79 ethosuxamide.tw. 80 Primidone/
81 primidone.tw. 82 vigabatrin.tw. 83 Phenobarbital/ 84 (phenobarbital or phenobarbitone).tw. 85 Piracetam/ 86 piracetam.tw. 87 Acetazolamide/ 88 acetazolamide.tw. 89 levetiracetam.tw. 90 pregabalin.tw. 91 oxcarbazepine.tw. 92 tiagabine.tw. 93 zonisamide.tw. 94 clobazam.tw. 95 Clonazepam/ 96 clonazepam.tw. 97 Impulsiveness/ 98 impulsiv$.tw. 99 65 or 97 or 98 100 divalproex.tw. 101 (or/66‐96) or 100 102 99 and 101 103 Treatment Effectiveness Evaluation/ 104 exp Treatment Outcomes/ 105 Psychotherapeutic Outcomes/ 106 PLACEBO/ 107 exp Followup Studies/ 108 placebo$.tw. 109 random$.tw. 110 comparative stud$.tw. 111 randomi#ed controlled trial$.tw. 112 (clinical adj3 trial$).tw. 113 (research adj3 design).tw. 114 (evaluat$ adj3 stud$).tw. 115 (prospectiv$ adj3 stud$).tw. 116 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$)).tw. 117 control$.tw. 118 117 or 109 or 107 or 115 or 114 or 110 or 103 or 108 or 104 or 116 or 112 or 106 or 105 or 113 or 111 119 118 and 102
Appendix 6. Cochrane Schizophrenia Group's register of trials on aggression
The terms used to search the Cochrane Schizophrenia Group's register of trials on aggression are as follows:
anticonvuls* OR antiepileptic* OR epilep* OR carbamazepine OR valproic OR valporate OR gabapentin OR lamotrigine OR topiramate OR ethosuxamide OR primidone OR vigabatrin OR phenobarbital OR phenobarbitone OR piracetam OR acetazolamide OR levetiracetam OR pregabalin OR oxcarbazepine OR tiagabine OR zonisamide OR clobazam OR clonazepam
Appendix 7. National Research Register
#1. HOMICIDE single term (MeSH) #2. homicid* #3. murder* #4. manslaughter* #5. infanticid* #6. parricid* #7. TORTURE single term (MeSH) #8. tortur* #9. (intent* near kill*) #10. (intent* near harm) #11. (bodily near harm) #12. (bodily near assault*) #13. assail* #14. attacker* #15. CHILD ABUSE explode all trees (MeSH) #16. SPOUSE ABUSE single term (MeSH) #17. (physical* near abuse*) #18. (spous* near abus*) #19. (partner* near abus*) #20. (child* near neglect*) #21. (child* near abus*) #22. (elder* near abus*) #23. RAPE single term (MeSH) #24. (rape* or rapist*) #25. (sexual* next near3 next abus*) #26. bugger* #27. SEX OFFENSES explode all trees (MeSH) #28. sodom* #29. molest* #30. (pedophil* or paedophil*) #31. indecen* #32. (masturbat* near public) #33. exhibitionis* #34. lewd* #35. sadis* #36. sadomasochis* #37. abduct* #38. kidnap* #39. aggress* #40. violen* #41. ANGER explode all trees (MeSH) #42. malic* #43. hostil* #44. (dangerous* or disrupt*) #45. destruct* #46. cruel* #47. delinquen* #48. threaten* #49. disorderly #50. affray* #51. (breach* near peace) #52. felon* #53. unlawful* #54. penal* #55. penol* #56. CRIME single term (MeSH) #57. crim* #58. offen* #59. PRISONS single term (MeSH) #60. prison* #61. inmate* #62. correctional* #63. firearm* #64. weapon* #65. FIREARMS single term (MeSH) #66. (gun or guns) #67. agitat* #68. (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54 or #55 or #56 or #57 or #58 or #59 or #60 or #61 or #62 or #63 or #64 or #65 or #66 or #67) #69. ANTICONVULSANTS single term (MeSH) #70. (epilep* near drug*) #71. anti‐epileptic* #72. antiepileptic* #73. anti‐convulsant* #74. anticonvulsant* #75. CARBAMAZEPINE single term (MeSH) #76. carbamazepine #77. VALPROIC ACID single term (MeSH) #78. (sodium next valporate) #79. gabapentin #80. lamotrigine #81. topiramate #82. ethosuxamide #83. PRIMIDONE single term (MeSH) #84. primidone #85. VIGABATRIN single term (MeSH) #86. vigabatrin #87. (phenobarbital or phenobarbitone) #88. PHENOBARBITAL single term (MeSH) #89. PIRACETAM single term (MeSH) #90. piracetam #91. acetazolamide #92. ACETAZOLAMIDE single term (MeSH) #93. levetiracetam #94. pregabalin #95. oxcarbazepine #96. tiagabine #97. zonisamide #98. clobazam #99. CLONAZEPAM single term (MeSH) #100. clonazepam #101. (#69 or #70 or #71 or #71 or #72 or #73 or #74 or #75 or #76 or #77 or #78 or #79 or #80 or #81 or #82 or #83 or #84 or #85 or #86 or #87 or #88 or #89 or #90 or #91 or #92 or #93 or #94 or #95 or #96 or #97 or #98 or #99 or #100) #102. (#68 and #101)
Data and analyses
Comparison 1. Valproate/divalproex versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall clinical response: numbers classed as 'responders', at endpoint (8 weeks) | 1 | 30 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.48, 10.60] |
| 2 Adverse events, any | 2 | 276 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.07 [1.42, 6.65] |
| 3 Adverse events, rash | 1 | 30 | Odds Ratio (M‐H, Fixed, 95% CI) | 7.80 [0.80, 75.64] |
| 4 Adverse events, headache | 2 | 276 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.44, 1.32] |
| 5 Adverse events, weight gain | 2 | 276 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.42 [1.10, 5.31] |
| 6 Adverse events, increased appetite | 1 | 30 | Odds Ratio (M‐H, Fixed, 95% CI) | 7.71 [1.28, 46.36] |
| 7 Non‐compliance: leaving the study early, any reason | 4 | 316 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.77, 1.96] |
| 8 Aggression: number improved as > 69% reduction on MOAS + SCL‐90 'irritability', at 6 wks | 1 | 15 | Odds Ratio (M‐H, Fixed, 95% CI) | 18.0 [1.27, 255.74] |
| 9 Adverse events, nausea | 1 | 246 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.81 [1.44, 5.47] |
| 10 Adverse events, somnolence | 1 | 246 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.94, 2.87] |
| 11 Non‐compliance: leaving the study early, any reason; Cluster B PD subgroup | 1 | 96 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.48, 2.41] |
Comparison 2. Carbamazepine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse events, any | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 10.0 [0.94, 105.92] |
| 2 Adverse events, rash/dermatitis | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.57 [0.84, 87.83] |
| 3 Adverse events, headache | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 19.93 [0.97, 408.44] |
| 4 Adverse events, dizziness | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 26.54 [1.30, 543.78] |
| 5 Adverse events, stomach ache | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.13, 5.09] |
| 6 Adverse events, weight loss | 1 | 21 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.05, 3.12] |
| 7 Adverse events, weight gain | 1 | 21 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.32, 19.53] |
| 8 Non‐compliance: leaving the study early, any reason | 1 | 20 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.10, 5.96] |
| 9 Aggression (self‐reported): number with any aggression to others/objects, over last 3 wks of intervention | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.01, 1.29] |
| 10 Aggression (self‐reported): number with any aggression to others/objects/self, over last 3 wks of intervention | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.06 [0.01, 0.63] |
| 11 Adverse events, leucopenia | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.57 [0.84, 87.83] |
| 12 Anger: more than one angry outburst, over 6 weeks | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.01, 1.29] |
| 13 Anger: any angry outburst, over 6 weeks | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.02, 1.92] |
Comparison 3. Phenytoin/diphenylhydantoin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐compliance: leaving the study early, any reason | 1 | 20 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.10, 5.96] |
| 2 Adverse events, nausea | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.06, 16.76] |
Comparison 4. Levetiracetam versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐compliance: leaving the study early (before 4 weeks), any reason | 1 | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.33 [0.67, 60.16] |
| 2 Adverse events, sedation | 1 | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.52 [0.43, 5.43] |
| 3 Adverse events, dizziness | 1 | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.75 [0.48, 46.91] |
| 4 Adverse events, headache | 1 | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.51, 17.74] |
Comparison 5. Oxcarbazepine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Aggression (self‐reported): OAS‐M‐revised, number of responders as > 49% reduction in Global Overt Aggression score, at 10 wks | 1 | 45 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.88 [1.36, 17.47] |
| 2 Non‐compliance: leaving the study early, any reason | 1 | 48 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.16, 1.61] |
| 3 Non‐compliance: leaving the study early, due to adverse events | 1 | 48 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.51, 10.69] |
| 4 Non‐compliance: leaving the study early, due to ineffectiveness | 1 | 48 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.06, 0.90] |
Comparison 6. Any epileptic drug versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐compliance: leaving the study early, any reason | 6 | 444 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.77, 1.70] |
| 2 Adverse events, any | 3 | 300 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.48 [1.68, 7.21] |
6.1. Analysis.
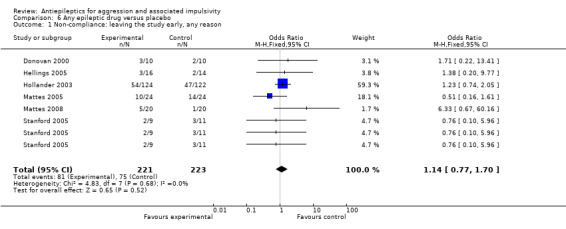
Comparison 6 Any epileptic drug versus placebo, Outcome 1 Non‐compliance: leaving the study early, any reason.
6.2. Analysis.
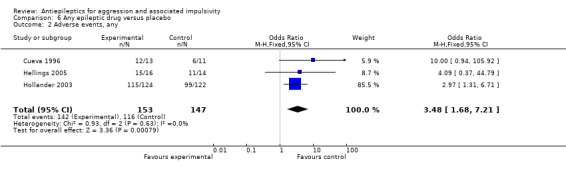
Comparison 6 Any epileptic drug versus placebo, Outcome 2 Adverse events, any.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Barratt 1991.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: male inmates in maximum security prison with impulsive aggressive behaviour Sex: male only Age: adults; age not reported Unit of allocation: individual participant Number randomised: 19 Number completing: 13 Setting: maximum security prison; USA (Texas) Inclusion criteria: violation of a prison rule involving an impulsive aggressive act (see note 1) at least one month preceding entry into the study and at least 3 times in the 3 months preceding entry Exclusion criteria: history or evidence of epilepsy of other central nervous system disorder; major psychiatric disorder under DSM‐III (Axis I) assessed using the Diagnostic Interview Schedule; IQ below 80; taking other medication Ethnicity: not reported Baseline characteristics: no further details reported |
|
| Interventions | Three conditions: phenytoin 100 mg/day / phenytoin 300 mg/day / placebo (see note 2)
Duration of intervention: 4 weeks Duration of trial: 9 weeks (cross‐over trial; 2 phases, 1‐week washout period between phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: not reported |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): frequency of aggressive acts (using same criteria of aggression as in study’s inclusion criteria) Secondary outcomes Hostility: Profile of Mood States anger‐hostility subscale scores |
|
| Notes | 1. Impulsive aggression defined as "spontaneous 'hair‐trigger' acts that are not premeditated and/or the result of gross psychopathology or other medical disorders" 2. In this cross‐over trial, participants were randomly allocated to 1 of 6 sequences of the 3 experimental conditions 3. Trial investigators noted that blood serum levels of phenytoin were relatively low at the 300mg/day dose and considered this “most likely related to poor medication compliance, but there may also be individual differences in metabolism” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that participants “were randomly assigned to one of six counterbalanced sequences of the three conditions” (p.388, col. 2) suggesting that the order of treatments was randomised in this cross‐over trial. No further details reported. Clarification about method of sequence generation has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Allocation concealment? | Unclear risk | No details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of participants | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of personnel | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of outcome assessors | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Investigators report that 19 participants volunteered and 13 completed the study, but no further details given. Unclear whether investigators carried out intention‐to‐treat analysis, but probably not. Insufficient reporting to permit judgement of ‘Yes’ or ‘No’. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. In this review, data from 13 participants completing the study were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes. The Profile of Mood States (POMS) outcome measure has three additional subscales (vigour, fatigue‐inertia, confusion‐bewilderment). Scores on these subscales are not reported. However, they were not mentioned prospectively and they are unlikely to be directly related to impulsive aggression. |
| Free of other bias? | Low risk | The study appeared to be free of other sources of bias. There was a 1‐week placebo washout period between phases in this cross‐over trial. Investigators report no detectable blood levels of phenytoin under the placebo conditions, suggesting that the trial was not biased from carry‐over effects. |
Barratt 1997.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: male prisoners with recurrent aggressive behaviour Sex: male only Age: adults; age not reported Unit of allocation: individual participant Number randomised: 150 Number completing: 126; results reported for 60 (30 with primarily impulsive aggression and 30 with primarily premeditated aggression; remaining 66 had committed mixed types of aggression and were not included) (see note 2) Setting: prisons (number not reported); USA (Texas) Inclusion criteria: at least 3 documented aggressive acts (see note 1) committed while in prison in the 3 months preceding entry to the study Exclusion criteria: IQ less than 80; presence of DSM‐III‐R Axis I disorder as measured with PDI‐R; taking medication; presence of neurological or other serious medical disorder; presence of 'medical aggression' Ethnicity: not reported Baseline characteristics: aggressive behaviours preceded incarceration (98%); DSM‐III‐R antisocial PD (100%); lifetime, but not current, drug abuse problem (55%) |
|
| Interventions | Two conditions: phenytoin / placebo
Duration of intervention: 6 weeks Duration of trial: 13 weeks (cross‐over trial; 2 phases, 1‐week washout period between phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: not reported |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): intensity of aggressive acts (Overt Aggression Scale); frequency of aggressive acts Secondary outcomes Hostility: Profile of Mood States anger‐hostility subscale scores |
|
| Notes | 1. Aggressive acts as defined in the handbook prepared by the Texas Department of Criminal Justice given to all prisoners 2. Aggressive acts classified as impulsive or non‐impulsive based on brief semi‐structured interview and written prison reports. An impulsive aggressive act defined as: "a 'hair‐trigger' non premeditated response to a stimulus that results in an immediate aggressive act or an agitated state that culminates in an aggressive act”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that participants “were randomly assigned to an initial drug/placebo condition” (p.3) suggesting that the order of treatments was randomised in this cross‐over trial. No further details reported. Clarification about method of sequence generation has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Allocation concealment? | Unclear risk | No details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of participants | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of personnel | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of outcome assessors | Unclear risk | Investigators describe the study as “double‐blind”. No further details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Insufficient reporting of attrition to permit judgement of ‘Yes’ or ‘No’. Appears that 24 did not complete study. A subgroup of 66 participants with 'mixed' type of aggression was excluded by investigators. Clarification has been requested from the trial investigators but no further information was available at the time this review was prepared. In this review, data from the subgroup of 60 completers were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes. |
| Free of other bias? | Unclear risk | There was a 1‐week placebo washout period between phases in this cross‐over trial. Trial investigators report no significant cross‐over effects for the aggression measures for the combined groups suggesting the study was not biased by carry‐over effects. However, of 150 randomised, results reported for only 60 of 126 completers (30 of which committed primarily impulsive and 30 of which committed primarily premeditated aggression; the remaining 66 had committed mixed types of aggression and were not included). Thus there is the possibility of bias through excluding the ‘mixed aggression’ group, although it is unclear what effect this would have on the results. |
Conners 1971.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: boys at a residential facility for delinquent offenders Sex: male only Age: mean 12.0 years; range 9 to 14 years Unit of allocation: individual participant Number randomised: 43 (30 to antiepileptic or placebo arms) Number completing: no attrition reported Setting: inpatient (residential); USA (Maryland) Inclusion criteria: not formally reported; participants described as “young juvenile offenders . . . . selected from 52 cases nominated by the training school staff as being the most aggressive or disturbed in their cottages” Exclusion criteria: any current medication; previous psychotropic medication; abnormal white blood cell count Ethnicity: not reported Baseline characteristics: not reported |
|
| Interventions | Three conditions: diphenylhydantoin sodium / methylphenidate / placebo (note that methylphenidate is not an antiepileptic drug)
Duration of intervention: 2 weeks Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: no information reported |
|
| Outcomes |
Primary outcomes None Secondary outcomes Impulsivity: Porteus Maze scores (see note 1) Anger: Rosenweig Picture Frustration Test categories (negative responses indicated ‘anger, aggressiveness or vengeful feeling’) Other outcomes Mean symptom scores (care staff/teachers ratings) Behavioural incidents (care staff ratings) |
|
| Notes | 1. Trial investigators report that the Porteus Maze test is “presumed to measure impulsiveness and ability to plan and organize response” (page 157), but later raise concerns about the degree to which scores on this test are a good measure of impulsivity. The Porteus Maze test was administered on last day of treatment one hour after medication given. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that “boys within each cottage were randomly assigned to three treatment groups“ (p.156, col 2). No further details given. Due to age of study, unable to contact trial investigators for more information. |
| Allocation concealment? | Unclear risk | No details reported. Due to age of study, unable to contact trial investigators for more information. |
| Blinding? of participants | Low risk | Investigators report that medication “was supplied to cottage parents in identical matched capsules, with the staff and investigators blind to the assignment condition” (p.156, col. 2). Review authors judge that blinding of participants was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators report that medication “was supplied to cottage parents in identical matched capsules, with the staff and investigators blind to the assignment condition” (p.156, col. 2). Review authors judge that blinding of personnel was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators report that Rosenweig Pictures were “scored blindly . . . by two independent raters” (p.157, col. 1). Review authors judge that blinding of outcome assessors was adequate and that it was unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Low risk | No attrition or exclusions were reported; no missing outcome data. In this review, data from 30 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Low risk | The study appeared to be free of other sources of bias |
Cueva 1996.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: children with DSM‐III‐R conduct disorder (solitary aggressive type) Sex: mixed Age: mean 8.97 years; range 5.33 to 11.7 years Unit of allocation: individual participant Number randomised: 24 Number completing: 22 (20 boys; 2 girls) Setting: inpatient; USA (New York) Inclusion criteria: aged 5 to 12 years; currently inpatient; normal intellectual functioning; diagnosis of DSM‐III‐R conduct disorder (solitary aggressive type); long history of severe explosive affect and aggressiveness with failure to respond to outpatient therapies including pharmacotherapy leading to hospitalisation; minimum of 3 aggressive episodes (physical aggression against others and/or objects) per week during the placebo baseline period as measured by the Overt Aggression Scale (OAS) Exclusion criteria: autistic disorder; schizophrenic disorder; major depression; mental retardation; cardiac or renal disease; seizure disorder; hypo‐/hyperthyroidism; history of other endocrine abnormality; urgency of discharge from hospital; psychoactive medication within 4 weeks prior to study; concurrent medication; failure to exhibit aggressive behaviour during 2‐week placebo baseline period Ethnicity: of 22 completers, 10 Hispanic, 9 black, 2 white, 1 Asian Baseline characteristics: IQ mean 85.3; range 59 to 111 |
|
| Interventions | Two conditions: carbamazepine / placebo
Duration of intervention: 6 weeks Duration of trial: 9 weeks (treatment preceded by 2‐week washout period and followed by 1‐week post‐treatment placebo period) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: dosage individually regulated for each child. Starting dose 200 mg carbamazepine, increased stepwise on individual basis to max of 1000 mg/day and/or until therapeutic or untoward effects observed. Each child then maintained on his/her optimal dose for four weeks. Carbamazepine tablets supplemented by matching placebo tablets to reach total number of 6. |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): scores on the Overt Aggression Scale (OAS); Children’s Psychiatric Rating Scale (CPRS) aggression subscale scores Secondary outcomes Hostility: Children’s Psychiatric Rating Scale (CPRS) hostility subscale scores Adverse events: Checklist of untoward effects Other outcomes Clinical Global Impressions (CGI); Global Clinical Judgements (consensus) Scale; Cognitive Battery Ratings; laboratory studies (ECG, bloods, urinalysis, carbamazepine levels) |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Sequence generation achieved using a random numbers table |
| Allocation concealment? | Low risk | Allocation concealed through use of identical drug containers, each sequentially assigned a letter of the alphabet |
| Blinding? of participants | Low risk | Blinding of participants achieved through use of identical drug containers for carbamazepine and placebo tablets Placebo and carbamazepine tablets were identical in appearance (p.483, col 1). Review authors judge that blinding of participants was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Blinding of personnel achieved through use of identical drug containers for carbamazepine and placebo tablets. Placebo and carbamazepine tablets were identical in appearance. Investigators report that medication was administered by a research nurse "on a blind basis at fixed times" (p.483, col 1). Review authors judge that blinding of personnel was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators report that all research staff "were blind to the treatment condition" (p.482, col 2). Review authors judge that blinding of outcome assessors was adequate and that it was unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Trial investigators report "three dropouts in terminal days of the study" (p.487, col 1) (1 due to accidental disclosure of their carbamazepine serum level, 1 due to pyrexia and decrease of white blood cells, 1 for reasons not given). No details given about how this missing data was distributed between experimental groups. Investigators provide an analysis of completers for CPRS outcome data, but provide insufficient details on numbers in each group. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. An intention‐to‐treat analysis is provided for OAS outcome data. In this review, data from 24 participants were included in the analysis of OAS data, and from 22 in the analysis of CPRS data. |
| Free of selective reporting? | Unclear risk | Investigators report that Cognitive Battery Ratings were made, but that "the results of these tests will be reported elsewhere" (p.487, col 2). No further information given. It might reasonably be assumed that these constituted one of the secondary outcomes, although this is not formally stated. |
| Free of other bias? | Low risk | The study appeared to be free of other sources of bias |
Donovan 2000.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: youths with conduct disorder or oppositional defiant disorder Sex: mixed (16 boys; 4 girls) Age: mean 13.8 (SD 2.4) years; range 10 to 18 years Unit of allocation: individual participant Number randomised: 20 Number completing: 17 (phase one); 15 (phases one and two) Setting: outpatient; USA (New York) Inclusion criteria: DSM‐IV disruptive behaviour disorder (oppositional defiant disorder or conduct disorder) (SCID; Diagnostic Inventory Scale for Children) plus:
Exclusion criteria: significant medical problems; IQ < 70; major depression; PTSD; head trauma; history of bipolar I or II disorder Ethnicity: 12 Hispanic, 5 African American, 3 Caucasian Baseline characteristics: truant or in special education classes classed as ‘emotionally disturbed’ (n = 18). DSM‐IV diagnoses: ADHD (n = 4), marijuana abuse (n = 6), disruptive behaviour disorder (n = 20). |
|
| Interventions | Two conditions: divalproex / placebo
Duration of intervention: 6 weeks in phase 1 Duration of trial: 12 weeks (cross‐over trial; 2 phases, no washout period between phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: divalproex increased over 2 weeks to 10 mg/lb/day. If blood level < 90 μg/ml at end week 2, additional 250 mg/day added. Placebo dose increases matched on an equal number of participants in the divalproex group. Final dose range 750 to 1500 mg/day. |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): Modified Overt Aggression Scale (MOAS) in combination with SCL90 anger‐hostility scale items, and dichotomised to ‘response’ (improved) and ‘no response’ (not improved) Secondary outcomes Irritability: scores on 6 items from the anger‐hostility subscale of SCL‐90, dichotomised to ‘response’ (improved) and ‘no response’ (not improved) Non‐compliance: proportion of participants discontinuing treatment |
|
| Notes | 1. Marijuana and prescribed stimulants were the only other psychotropic medications used by the participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Investigators report a “double‐blind cross‐over design, with patients randomly assigned to phase 1" (p.819, col 1). Information obtained from trial investigators indicated that sequence generation was achieved by use of a computer generated random numbers table. |
| Allocation concealment? | Low risk | Information obtained from trial investigators suggests central allocation such that allocation status was adequately concealed |
| Blinding? of participants | Low risk | A “matching placebo” was used (p.820, col 2). Appropriate care appears to have been taken to ensure blinding of participants. Unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators report “to preserve the blind, dose increases were allowed on an equal number of patients in the placebo phase” (p.819, col 2). Information obtained from trial investigators suggests appropriate care was taken to ensure blinding of personnel. Unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators report “independent evaluators blind to group assignment assessed response at the end of each phase” (p.818, col 2). Appropriate care appears to have been taken to ensure blinding of outcome assessors. Unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Low risk | For results from both phases of this cross‐over trial, at completion of intervention (6 weeks): 2/10 missing from intervention (divalproex) group (1 jailed for parole violation, 1 did not commence phase 2 for reasons not given); 3/10 missing from control (placebo) group (2 citing lack of efficacy, 1 did not commence phase 2 for reasons not given). Missing outcome data balance in numbers across intervention groups. Investigators report an analysis of data for the completers. In this review, data from 15 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Unclear risk | The study appeared to be free of other sources of bias However, the trial investigators do not report a placebo washout period between phases in this cross‐over trial which may have resulted in carry‐over effects. |
Gardner 1986.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: women with borderline personality disorder and extensive history of behavioural dyscontrol Sex: female only Age: mean 31.7 years; range 23 to 42 years Unit of allocation: individual participant Number randomised: 16 Number completing: 11 Setting: outpatient; USA (Maryland) Inclusion criteria: meeting DSM‐III criteria for BPD (average of 6.7 criteria met); meeting DIB criteria for BPD (average scale score 8.9); extensive history of behavioural dyscontrol (see note 1) Exclusion criteria: DSM‐III diagnosis of schizophrenia, major depressive disorder, or alcoholism or substance abuse disorder Ethnicity: not reported Baseline characteristics: recurrent rage episodes (88%), multiple overdoses (69%), multiple episodes of self‐burning (19%), multiple wrist, arm or abdomen cuts (56%), episodes of violence (44%), previous suicide attempt with intention of dying (56%), history of episodes of major depression (50%) |
|
| Interventions | Two conditions: carbamazepine / placebo (see note 2)
Duration of intervention: 6 weeks Duration of trial: 14 weeks (cross‐over trial; 2 phases for carbamazepine/placebo comparison, 1‐week dose tapering, then 1‐week washout period between phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: initial 2‐week dose adjustment period, followed by 4 weeks of steady dose administration |
|
| Outcomes |
Primary outcomes Aggression (self‐reported): individual data, plus ratings of behaviour dyscontrol incidents (see note 4) Secondary outcomes Anger: numbers with angry outbursts Non‐compliance: Attrition data? Other outcomes Ratings of mood (no results presented) |
|
| Notes | 1. Although an extensive history of behavioural dyscontrol was an inclusion criterion, it is unclear whether every subject had demonstrated episodes of either rage or violence. Nonetheless, those that may not will have experienced episodes of aggression to the self (self‐harm). 2. Participants randomly assigned to carbamazepine/placebo as part of a cross‐over study involving 4 active medications plus placebo 3. Data from the 11 paired comparisons are considered in this review 4. Here, ‘behaviour dyscontrol incidents’ includes aggressive acts and angry outbursts, but also suicide attempts and gestures and incidents of self‐harm |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that “the trials were randomized to avoid any sequence effects” (p.520 col. 1). No further details given. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Allocation concealment? | Unclear risk | No details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of participants | Low risk | Investigators describe the study as “a double‐blind cross‐over trial” (Abstract) and report that “the patients and physicians, who were blind to the medication, met weekly . . .” (p.520 col. 1). Review authors judge that blinding of participants was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators describe the study as “a double‐blind cross‐over trial” (Abstract) and report that “the patients and physicians, who were blind to the medication, met weekly . . .” (p.520 col. 1). Review authors judge that blinding of personnel was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators describe the study as “a double‐blind cross‐over trial” (Abstract) and report that “the patients and physicians, who were blind to the medication, met weekly to complete ratings of mood and behavior” (p.520 col. 1). Review authors judge that blinding of outcome assessors was adequate and that it was unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | In this cross‐over trial, 5/16 missing at completion of intervention (6 weeks). Two participants excluded by investigators (1 due to a seizure during rapid alprazolam withdrawal, 1 due to requiring adjunctive thioridazine). Three participants dropped out after completing a carbamazepine phase but not a placebo phase (reasons not given). Insufficient information to decide if reasons for missing outcome data likely to be related to true outcome. Trial investigators report a completer analysis on the 11 that had completed both phases. In this review, data from 11 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Reported as part of the complete study involving cross‐over trials with placebo and 4 active medications (only one of which was an antiepileptic). This study was limited to: “the preliminary findings regarding behavioral dyscontrol during the carbamazepine and placebo trials” (p.520 col. 1). Thus focus was on behaviour and not mood states, and so absence of mood results (mood ratings) was not relevant. Thus although study protocol is not available, it seems clear that the published report includes all expected outcomes, including those that were pre‐specified. |
| Free of other bias? | Unclear risk | An extensive history of behavioural dyscontrol (which the investigators take to include self‐harm) was an inclusion criterion for this study. The study is included in this review since the participants were not recruited solely on the basis of having self‐harmed. However, it is possible that 2 of the participants had not demonstrated episodes of either rage or violence and so may not have been aggressive other than to themselves. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. The study appeared to be free of other sources of bias, and the trial investigators report a 1‐week placebo washout period between phases in this cross‐over trial which will have reduced the possibility of carryover effects. |
Gottschalk 1973.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: male prisoners at an institution for dangerous and emotionally unstable recidivists Sex: male only Age: mean 25.36 (SD 6.15) years Unit of allocation: individual participant Number randomised: 42 Number completing: 42 Setting: inpatient (forensic); USA (Maryland) Inclusion criteria: violation of the institution’s discipline rules over last 6 months Exclusion criteria: none reported; however, the importance of abstaining from all illegally obtained drugs or alcohol was stressed at the screening interview Ethnicity: not reported Baseline characteristics: mean 8.25 (SD 1.90) years education |
|
| Interventions | Two conditions: diphenylhydantoin / placebo
Duration of intervention: 6 months Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: no details reported |
|
| Outcomes |
Primary outcomes None Secondary outcomes Hostility: 3 of 4 subscales on speech content analysis (Hostility‐out; Hostility‐in; Ambivalent hostility: see note 2) Other outcomes Anxiety scale content analysis of 5‐minute speech samples |
|
| Notes | 1. Placebo contained very low dose of the experimental drug (reported as ˜24 mg/day). Trialists reported that “a placebo with a few milligrams of DPH was given rather than a simple placebo to avoid informing subjects that a placebo was going to be used and then having to conceal from them whether they received it or not”. 2. Samples obtained at approx 1‐month intervals; first sample taken on day prior to commencing medication; up to 6 further speech samples taken post‐drug; transcripts of tape‐recorded samples scored independently by technicians using Gottschalk‐Gleser content analysis method |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that “subjects were randomly assigned” (p.505). No further details given. Due to age of study, unable to contact investigators for more information. |
| Allocation concealment? | Unclear risk | No details reported. Due to age of study, unable to contact investigators for more information. |
| Blinding? of participants | Low risk | Investigators report that “medication was administered in a syrup, uniform in taste and appearance for both preparations and packed in individually coded bottles at a hospital pharmacy” (p.505). Review authors judge that blinding of participants was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators describe study as a “double‐blind design” and state that “medication was administered in a syrup, uniform in taste and appearance for both preparations and packed in individually coded bottles at a hospital pharmacy” (page 505). Review authors judge that blinding of personnel was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators report that transcripts of recorded speech samples “were scored independently” by technicians “who were uninformed about the details and design of the study” (page 505). Review authors judge that blinding of outcome assessors was adequate and that it was unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | No attrition reported. Investigators report that over the 6 sampling periods at least 21 participants (of 42 randomised) missed at least 1 speech sample raising possibility of significant missing data, but no further details given; however, trial investigators' provide a fitted slopes analysis summarising results over the whole study period. In this review, data from 42 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Unclear risk | Participants had all violated the disciplinary code of 'an institution dedicated to the diagnosis and treatment of dangerous and emotionally unstable recidivists'. However, no further details are given leading to uncertainty about the extent to which all participants met the review's criteria for having exhibited recurrent aggression. The investigators acknowledge and defend questions about the choice and relevance of the outcome measure, content analysis of speech. The study appeared to be free of other sources of bias. |
Hellings 2005.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: children and adolescents with pervasive developmental disorder and significant aggressive behaviour Sex: mixed; 12 boys, 6 girls (valproate group); 14 boys, 4 girls (placebo group) Age: mean 10.3 (SD 3.7) years (valproate group); mean 12.1 (SD 4.8) years (placebo group) Unit of allocation: individual participant Number randomised: 30 (20 boys; 10 girls) Number completing: 25 Setting: outpatient; USA (Kansas) Inclusion criteria: aged 6 to 20 years; significant aggression to self, others, or property at least 3 times a week; presence of a pervasive developmental disorder (PDD) Exclusion criteria: Tourette’s Disorder (DSM‐IV); previous adequate trial of valproate within past year for any indication or for clinical seizures; history of degenerative neurological changes or metabolic disorders; history of thrombocytopenia, hepatitis, pancreatitis, pregnancy or polycystic ovarian syndrome; concomitant psychotropic or antiseizure medication (see note 1) Ethnicity: 27 Caucasian, 2 African‐American, 1 Hispanic Baseline characteristics: autistic disorder (n = 27); pervasive developmental disorder NOS (n = 1); Asperger’s disorder (n = 2); mean IQ = 54 (range 20 to 137); average or above‐average IQ (n = 2); borderline intellectual functioning (n = 2); mental retardation (n = 26) |
|
| Interventions | Two conditions: valproate / placebo
Duration of intervention: 8 weeks Duration of trial: 11 to 13 weeks (treatment preceded by washout period of 2 weeks (for tricyclics) or 4 weeks (for all other psychotropic medication) plus 1‐week placebo run‐in prior to randomisation) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: valproate gradually introduced from day 1 adding 250 mg every third day, replacing the equivalent amount of placebo liquid, to achieve a dosage of 20 mg/kg/day. Adequate blood levels within the therapeutic range were achieved and maintained from at least week 4 to week 8. |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): Overt Aggression Scale(OAS) completed by parents and teachers for each aggressive outburst. Aggression recurring after 30 minutes of non‐aggressive behaviour was documented as a separate episode. Secondary outcomes Non‐compliance: proportion of participants discontinuing treatment Adverse events: rated via checklist derived from the Physicians’ Desk Reference Other outcomes Clinical Global Impressions‐Improvement subscale (CGI‐I); Aberrant Behavior Checklist‐Community scale (ABC‐C) ‐ irritability subscale |
|
| Notes | 1. Stimulant medications stopped the day before placebo run‐in commenced | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report "two parallel groups of subjects, randomized to VPA or PBO by the study pharmacist" (p.685, col 1). No further details given. Insufficient information to permit judgement on adequacy of sequence generation. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Allocation concealment? | Unclear risk | No details reported. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of participants | Low risk | Investigators report that participants and parents were blinded regarding allocation status and that “mock dosage adjustment was made for the placebo group” (p.685, col 2). Appropriate care appears to have been taken to ensure blinding of participants. Unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators report that “investigators, parents and teachers were blinded regarding medication or PBO status” (p.685, col 2). Appropriate care appears to have been taken to ensure blinding of personnel. Unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Investigators report that “investigators, parents and teachers were blinded regarding medication or PBO status” (p.685, col 2). Appropriate care appears to have been taken to ensure blinding of outcome assessors. Unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Low risk | At completion of intervention (8 weeks): 3/16 missing from intervention (valproate) group; 2/14 missing from control (placebo) group. Investigators do not provide a breakdown by intervention group of reasons for non‐completion. However, review authors judge that missing outcome data was balanced in numbers across intervention groups and reasons for missing outcome data were unlikely to be related to true outcome. Trial investigators provided an intention‐to‐treat analysis. In this review, data from 30 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Low risk | The study appeared to be free of other sources of bias. It would however have been helpful if the investigators had clarified whether the use of diphenhydramine as a ‘rescue medication’ was a possible confounder. |
Hollander 2003.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: adults with impulsive aggression subgrouped by diagnosis (cluster B personality disorder; intermittent explosive disorder; post‐traumatic stress disorder) (see note 1) Sex: mixed (169 men; 64 women) (see note 2) Age: mean 40.3 years; range 19 to 67 years Unit of allocation: individual participant Number randomised: 246 (124 divalproex; 122 placebo) (see note 2) Number randomised by diagnostic group: cluster B PD 96 (47 divalproex; 49 placebo); IED 116 (59 divalproex; 57 placebo); PTSD 34 (18 divalproex; 16 placebo) Number analysed (overall): 233 (116 divalproex; 117 placebo) (see note 2) Number analysed by diagnostic group: cluster B PD 91 (43 divalproex; 48 placebo); IED 109 (55 divalproex; 54 placebo); PTSD 33 (18 divalproex; 15 placebo) Number completing: 145 Setting: outpatient (19 sites); USA Inclusion criteria: aged 18 to 65 years; diagnosis of cluster B personality disorder (see note 3) or intermittent explosive disorder (IED), or PTSD (see note 4); average of 2 episodes of physical or verbal aggressive outbursts per week for at least a month prior to screening, causing marked distress or impairment in occupational or interpersonal function where the aggressive behavior was judged to be neither premeditated nor committed to achieve a tangible objective; minimum score of 15 on OAS at first screening visit and at either the second screening visit or at randomisation; if receiving psychotherapy, have a stable psychotherapy schedule for at least 3 months prior to screening and maintained throughout the study Exclusion criteria: lifetime bipolar I disorder; bipolar II disorder with hypomania in the last year or a baseline Mania Syndrome Scale Score >= 12; major depressive disorder > 15 on HAM‐D; history of schizophrenia or other psychotic disorder; symptoms of dementia; serious homicidal or suicidal ideation; impulsive aggression resulting from previous head trauma or other medical condition; pregnant or lactating females; clinically abnormal laboratory data; unstable medical condition; any underlying condition that would confound the interpretation of study results; concurrent use of psychotropic medication, with exception of SSRIs, tricyclic antidepressants and stimulants if taken at a stable dose for at least 2 months prior to screening and continued at same dose throughout the study; participants specifically prohibited from use of benzodiazepines, mood stabilisers, anticonvulsants, MAOIs and antipsychotic agents (see note 5) Ethnicity: 195 Caucasian, 26 black, 12 other (see note 2) Baseline characteristics: at least one psychiatric hospitalisation (n = 36); history of alcohol abuse/dependence (n = 75); history of drug abuse/dependence (n = 38); history of incarceration (n = 52); median OAS‐M aggression score (43.7 divalproex group; 33.7 placebo group) |
|
| Interventions | Within the 3 diagnostic groups (cluster B PD; IED; PTSD), random allocation to 2 conditions: divalproex sodium / placebo (see note 6)
Duration of intervention: 12 weeks Duration of trial: 15 weeks (treatment preceded by screening period not exceeding 14 days and followed by 1‐week tapering period) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: initiated at 500 mg/day, and increased by 250 mg every 3 to 7 days during first 3 weeks of treatment, based on individual clinical response and tolerance. Maximum dose 30 mg/kg/day. |
|
| Outcomes |
Primary outcomes Aggression (self‐reported): Overt Aggression Scale‐Modified (OAS‐M) Secondary outcomes Non‐compliance: proportion of participants discontinuing treatment Adverse events: via self‐report and clinical monitoring Other outcomes Clinical Global Impression (CGI) |
|
| Notes | 1. Clinician at the investigative site specified which diagnosis most closely associated with the symptoms of impulsive aggression 2. Although 246 participants were randomised and received at least one dose of the study drug, figures reported are for 233 participants included in the intention‐to‐treat analyses of efficacy after excluding 13 because they did not have a post‐baseline OAS‐M Aggression score 3 Cluster B personality disorder comprised AsPD, BPD, HisPD, NarPD, or cluster B PD NOS (cluster B PD NOS required presence of at least 5 features from more than one specific cluster B disorder in the absence of full criteria for any one cluster B PD) 4. PTSD as confirmed by the SCID‐IV conducted during screening and modified for this study 5. Zolpidem tartrate (up to 10 mg/day up to 4 days/week) allowed for control of insomnia but not within 8 hours prior to efficacy ratings 6. Participants were randomly allocated to divalproex sodium/placebo within the 3 diagnostic groups (cluster B PD; IED; PTSD) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report “patients were randomised in equal numbers, within each of the three diagnostic groups, to receive either divalproex sodium delayed‐release tablets . . . or matching placebo” (col 1, page 1188). No further details given. Insufficient information to permit judgement on adequacy of sequence generation. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Allocation concealment? | Unclear risk | Insufficient information to permit judgement on adequacy of allocation concealment. Clarification has been requested from the trial investigators, but no further information was available at the time this review was prepared. |
| Blinding? of participants | Low risk | Investigators describe study throughout as “double‐blind” and that participants received a “matching placebo”. Review authors judge that blinding of participants was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Investigators report “An unblinded person from the central laboratory reported serum valproate levels . . . to the investigators, so that the dose of the study drug could be adjusted appropriately. In order to preserve the study blind, sham valproate levels were reported for selected placebo patients” (p.1188, col 1). Review authors judge that blinding of personnel was adequate and that it was unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Unclear risk | Insufficient information to permit judgement on adequacy of blinding of outcome assessors |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Investigators excluded 13 from analysis (no OAS‐M score post baseline). For all participants at completion of intervention (12 weeks): 54/124 missing from intervention (valproate) group (reasons not given); 47/122 missing from control (placebo) group reasons not given). Unclear whether reason for missing outcome data is likely to be related to true outcome. Trial investigators report an intention‐to‐treat analysis, although precise nature of this is unclear. For subgroup of participants with cluster B PD at completion of intervention (12 weeks): 22/47 missing from intervention (valproate) group (reasons not given); 22/49 missing from control (placebo) group reasons not given). Unclear whether reason for missing outcome data is likely to be related to true outcome. Trial investigators report an intention‐to‐treat analysis, although precise nature of this is unclear. In this review, data from 233 participants were included in the analysis of the whole sample, data from 91 participants were included in the analysis of the cluster B PD subgroup, and data from 33 participants were included in the analysis of the PTSD subgroup. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Low risk | The investigators note that the mean final valproate serum level was 64.2 μg/ml, which is well below possible therapeutic range (80 to 120 μg/ml) based on previous studies. They also draw attention to the heterogeneity of aggressive behaviours across whole sample (and recommend that future studies discriminate between aggression subtypes). The study appeared to be free of other sources of bias. |
Mattes 2005.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: adults meeting Coccaro‐revised criteria for intermittent explosive disorder (see note 1) Sex: mixed (39 men; 9 women) Age: mean 41.7 (SD 8.8) years Unit of allocation: individual participant Number randomised: 48 Number completing: 24 completed 10 weeks; 45 completed 4 weeks Setting: outpatient; USA Inclusion criteria: aged 18 to 65 years; in generally good health; practicing effective contraception (women of childbearing potential); meeting Coccaro‐revised criteria for intermittent explosive disorder:
Exclusion criteria: schizophrenia; bipolar disorder; epilepsy; dementia; mental retardation; substance abuse in last 6 months; need for treatment with antipsychotics, anticonvulsants or mood stabilisers, or any recent change (within 3 months) in psychotropic medication; significant risk of severely injuring others or self; any current psychiatric or neurological conditions which required specific treatment Ethnicity: not reported Baseline characteristics: mean 13.5 years of education; married (n = 36); physically abused as a child (n = 14); history of ‘road rage’ (n = 38); arrested previously (n = 13); previously in jail because of aggressiveness (n = 7); restraining orders instituted against them (n = 6); prior psychiatric treatment (n = 34); prior psychiatric hospitalisation (n = 6); attempted suicide (n = 4), experience of military combat (n = 4); history of perinatal trauma (n = 4); taking other psychotropic medication (n = 3, all on SSRIs) Age aggressiveness began: in childhood (n = 26), at puberty (n = 9), in adulthood (n = 13). In family history (first‐degree relatives), impulsive aggression (n = 32), depression (n = 15), alcoholism (n = 10). Four diagnoses occurred frequently enough (at least 10 participants) to warrant analysis: ADHD (n = 15), prior alcohol or drug abuse (n = 15), prior major depression (n = 13), and intermittent explosive disorder by DSM‐IV criteria (n = 10). No participants had antisocial or borderline PD, nor a neurological condition that seemed related to aggressiveness. |
|
| Interventions | Two conditions: oxcarbazepine / placebo
Duration of intervention: 10 weeks Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: initial dose oxcarbazepine 150 mg (evening) increased by 150 to 300 mg/day (given in 2 divided doses) after 2 to 4 days on each dose, to at least 1200 mg/day (if tolerated), with a maximum of 2400 mg/day by day 25 if needed. Dose could be administered more slowly, and more could be given at bedtime, if adverse events (assessed by psychiatrist). |
|
| Outcomes |
Primary outcomes Aggression (self‐reported): OAS‐M (revised; see note 2) Secondary outcomes Hostility: BPRS hostility rating Non‐compliance: proportion of participants discontinuing treatment Other outcomes Patient‐rated global improvement; BPRS total score |
|
| Notes | 1. All participants met the Coccaro‐revised criteria for intermittent explosive, although only 10 of the 48 randomised met DSM‐IV criteria for intermittent explosive disorder 2. OAS‐M was “slightly revised” by the trial investigators, to “improve face validity and limit variability and skewness” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Information obtained from lead author indicated that sequence generation was achieved by the throwing of a dice |
| Allocation concealment? | Low risk | Further details provided by lead author indicated that concealment achieved by use of, effectively, central allocation by a person not otherwise involved in the study, so that participants and any investigator enrolling participants could not foresee assignment |
| Blinding? of participants | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of participants. |
| Blinding? of personnel | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of personnel. |
| Blinding? of outcome assessors | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of outcome assessors. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | At completion of intervention (10 weeks): 10/24 missing from intervention (oxcarbazepine) group (6 due to adverse events, 4 due to lack of effectiveness); 14/24 missing from control (placebo) group (3 due to adverse events, 11 due to lack of effectiveness). Unclear whether reason for missing outcome data likely to be related to true outcome. However, trial investigators provide an intention‐to‐treat analysis. In this review, data from 48 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Unclear risk | Several small revisions to the OAS‐M instrument are described and justified, but it is unclear whether the revised scale has been formally tested for validity and reliability. In addition, the investigator states that funding was “provided by Novartis Pharmaceuticals Group” (p.579, col 1) who are the manufacturers of Trileptal, the proprietary name for oxcarbazepine. “Novartis also provided oxcarbazepine tablets and matching placebo” (p.579, col 2). However, the trial is described as “an investigator‐initiated study” and the report clearly states that “this manuscript was written without assistance from Novartis” (p.579, col 1). In addition, the authors describe small revisions to the OAS‐M. The study appeared to be free of other sources of bias. |
Mattes 2008.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: adults meeting Coccaro‐revised criteria for intermittent explosive disorder (see note 1) Sex: mixed (35 men; 5 women) Age: mean 45.38 (SD 11.2) years; range 21 to 64 years Unit of allocation: individual participant Number randomised: 40 Number completing: 19 completed full 10 weeks; 34 completed first 4 weeks Setting: outpatient; USA Inclusion criteria: aged 18 to 65 years; in general good health; women of childbearing potential had to be practicing effective contraception; meeting Coccaro‐revised criteria for intermittent explosive disorder (see note 1):
Exclusion criteria: schizophrenia; bipolar disorder; epilepsy; dementia; mental retardation; substance abuse in last 6 months; need for treatment with antipsychotics, anticonvulsants or mood stabilisers, or any recent change (within 3 months); significant risk of severely injuring others or self; any current psychiatric or neurological conditions which required specific treatment Ethnicity: not reported Baseline characteristics: currently married (n = 23); never been married (n = 8); mean 12.9 (SD 2.2) years of education; history of perinatal trauma (n = 9); physically abused as a child (n = 13); history of ‘road rage’ (n = 34); arrested previously (n = 10); previously in jail because of aggressiveness (n = 9); restraining orders instituted against them (n = 10); prior psychiatric treatment (n = 30); attempted suicide (n = 2); in family history (first‐degree relatives), ‘a bad temper’ (n = 30), depression (n = 10), alcoholism (n = 9) Four diagnoses occurred frequently enough (at least 8 participants) to warrant analysis: ADHD (residual or in remission) (n = 13), prior alcohol or drug abuse/dependence (n = 12), prior major depression (n = 11), and intermittent explosive disorder by DSM‐IV criteria (n = 8) |
|
| Interventions | Two conditions: levetiracetam / placebo
Duration of intervention: 10 weeks Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: initial dose levetiracetam 250 mg twice daily, increased by 250 mg twice daily after 1 week of treatment with each dose to at least 1000 mg/day if tolerated, with a maximum of 3000 mg/day by week 6 if needed. Due to tolerability, the dose could be escalated more slowly, and more could be given at bedtime. |
|
| Outcomes |
Primary outcomes Aggression (self‐reported): OAS‐M scores (see note 2); Relative Rating of Aggressive Behavior (see note 3) Secondary outcomes Hostility: BPRS hostility scale Non‐compliance: proportion of participants discontinuing treatment Adverse events: incidence of overall adverse events Other outcomes Patient‐rated global improvement |
|
| Notes | 1. All participants met the Coccaro‐revised criteria for intermittent explosive, although only eight of the 49 randomised met DSM‐IV criteria for intermittent explosive disorder 2. The OAS‐M was “slightly revised”, in part to “improve face validity and limit variability and skewness” as described in Mattes (2005) 3. Derived from the rating Scale for Aggressive Behaviour in the Elderly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Investigators report “at baseline, patients were randomly assigned to either levetiracetam or placebo in a 1:1 ratio” (p.312, col 1). Further details obtained from lead author indicated that sequence generation was achieved by the throwing of a dice. |
| Allocation concealment? | Low risk | Further details provided by lead author indicated that concealment achieved by use of, effectively, central allocation by a person not otherwise involved in the study, so that participants and any investigator enrolling participants could not foresee assignment |
| Blinding? of participants | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of participants. Unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of personnel. Unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | Information provided by lead author indicated that this was a double‐blind study. Appropriate care appears to have been taken to ensure blinding of outcome assessors. Unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | After 4 weeks of treatment (by which time trial investigators considered participants had received an 'adequate trial'): 5/20 missing from intervention (levetiracetam) group (suicidal ideation (n = 2), impaired co‐ordination (n = 1), increased irritability (n = 1), sedation and dizziness (n = 1); 1/20 missing from control (placebo) group (depressed mood). At completion of intervention (10 weeks), 21/40 missing (breakdown by treatment group not available). Unclear whether reasons for missing outcome data were likely to be related to true outcome. Trial investigators carried out intention‐to‐treat analysis. In this review, data from 40 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Unclear risk | Several small revisions to the OAS‐M instrument are described and justified, but it is unclear whether the revised scale has been formally tested for validity and reliability. The study is described as having funding support from UCB Pharma (the manufacturers of Keppra, the proprietary name for levetiracetam) but that “Dr Mattes reports no additional financial or other relationship relevant to the subject of this article” (p.310, col 1). The study appeared to be free of other sources of bias. |
Rosenblatt 1976.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: adults who had complained of difficulty in controlling physically punitive impulses towards (or were suspected of physical assault on) their own children Sex: mixed (11 women; 2 men) Age: adults, age not reported Unit of allocation: individual participant Number randomised: 13 (11 women; 2 men) (see note 1) Number completing: 8 completed phase 1 (6 women; 2 men) (see note 1) Setting: outpatient; USA (New York) Inclusion criteria: suspected of physical assault on (or complaining of difficulty in controlling physically punitive impulses towards) own children Exclusion criteria: not reported Ethnicity: not reported Baseline characteristics: not reported |
|
| Interventions | Two conditions: diphenylhydantoin / placebo
Duration of intervention: 8 weeks Duration of trial: 16 weeks (cross‐over trial; 2 phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: not reported |
|
| Outcomes |
Primary outcomes None Secondary outcomes Aggressive impulsiveness: subscale of Q‐Sort A, a self‐rating scale (developed by the authors) Hostility: subscale of Q‐Sort A, a self‐rating scale (developed by the authors); subscale of Q‐Sort B, a self‐rating scale (developed by the authors) Non‐compliance: proportion of participants discontinuing treatment |
|
| Notes | 1. n = 13 randomised, but information on number allocated to each intervention at start of trial was not provided. Results for phase 1 completers only (5 in diphenylhydantoin group; 3 in placebo group). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Investigators report that participants were allocated “on a random, double‐blind basis . . “ (p.333). No further details given. Due to age of study, unable to contact trial investigators for more information. |
| Allocation concealment? | Unclear risk | No details reported. Due to age of study, unable to contact trial investigators for more information. |
| Blinding? of participants | Unclear risk | Investigators report that participants were allocated “on a random, double‐blind basis . . “ (p.333). No further details given. Due to age of study, unable to contact trial investigators for more information. |
| Blinding? of personnel | Unclear risk | Investigators report that participants were allocated “on a random, double‐blind basis . . “ (p.333). No further details given. Due to age of study, unable to contact trial investigators for more information. |
| Blinding? of outcome assessors | Unclear risk | Investigators report that participants were allocated “on a random, double‐blind basis . . “ (p.333). No further details given. Due to age of study, unable to contact trial investigators for more information. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Investigators report attrition overall (5/13 in phase 1) but not by treatment condition. Insufficient reporting of attrition to permit judgement of ‘Yes’ or ‘No’. In this review, data from 8 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes. |
| Free of other bias? | Unclear risk | Although this was a 16‐week cross‐over trial, results reported for first week only. Investigators report that after the first week of the trial, “confounding factors such as the interviews, changes in life situation etc., strongly influence the dependent variable”. Insufficient information to assess whether an important risk of bias exists. |
Stanford 2001.
| Methods | Design: placebo‐controlled cross‐over trial | |
| Participants |
Participants: men with DSM‐IV personality disorder and impulsive aggressive behaviour Sex: male only Age: mean 45.1 (SD 6.8) years Unit of allocation: individual participant Number randomised: 46 Number completing: 29 (phases 1 and 2); 23 (phase 1 only) Setting: outpatient; USA Inclusion criteria: over past 6 months, several discrete participant‐identified episodes of failure to resist aggressive impulses resulting in serious assaultative acts or destruction of property; degree of aggressiveness expressed during the episodes was grossly out of proportion to any precipitating psychosocial stressor; at least 2 such episodes during the month prior to entering the study; score of 8 or higher on the Irritability sub scale of the Buss‐Durkee Hostility Inventory; must have identified an individual willing to document any impulsive‐aggressive outbursts that occurred during the study Exclusion criteria: female (due to potential teratogenic effects of phenytoin); verbal IQ < 80; diagnosis of a DSM‐IV‐TR Axis I psychiatric disorder; present use of medication; medical/neurological problems (including seizures); liver enzymes not within normal limits Ethnicity: not reported Baseline characteristics: mean verbal IQ 105.8 (SD 10.7); mean 14.3 (SD 2.4) years education; DSM‐IV personality disorder diagnoses for phase one completers: obsessive‐compulsive PD (n = 12), antisocial PD (n = 10), narcissistic PD (n = 1) |
|
| Interventions | Two conditions: phenytoin / placebo
Duration of intervention: 6 weeks Duration of trial: 16 weeks (cross‐over trial; 2 phases, 2‐week placebo baseline period, and 2‐week placebo washout period between phases) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: no details reported |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): Overt Aggression Scale (OAS) Secondary outcomes Hostility: Profile of Mood States anger‐hostility subscale scores Other outcomes Psychophysiological recordings (including evoked potentials) |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Investigators report that “subjects were randomly assigned” (p.195, col. 2) suggesting that the order of treatments was randomised in this cross‐over trial. Further details obtained from trial investigators (Stanford 2009) indicated that sequence generation was achieved by use of computer generated random numbers. |
| Allocation concealment? | Low risk | Response from trial investigators suggests that appropriate care was taken to ensure blinding of participants |
| Blinding? of participants | Low risk | Investigators describe the study as “double‐blind”. Response from trial investigators suggests that appropriate care was taken to ensure blinding of participants |
| Blinding? of personnel | Low risk | Investigators describe the study as “double‐blind”. Response from trial investigators suggests that appropriate care was taken to ensure blinding of personnel. |
| Blinding? of outcome assessors | Low risk | Investigators describe the study as “double‐blind”. Response from trial investigators suggests that appropriate care was taken to ensure blinding of outcome assessors. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | In this cross‐over trial, 23/46 missing by end of phase 2. Breakdown by intervention group not reported and unclear whether reason for missing outcome data likely to be related to true outcome. Insufficient information to permit judgement of ‘Yes’ or ‘No’. 17/46 discontinued and 6/46 excluded by investigators (2 because serum phenytoin levels were zero and 4 because they reported no outbursts throughout the trial). Investigators report completer analysis for those 23 participants (11 phenytoin; 12 placebo) completing both phases. In this review, data from 23 participants were included in the analysis. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes |
| Free of other bias? | Unclear risk | The investigators declare their research sponsored by the Dreyfus Health Foundation, which is focused on phenytoin and was established “to study, collect, and disseminate information and sponsor collaborative, clinical, and basic health research on its benefits”. The authors have insufficient information to assess whether this constitutes a risk of bias. The trial investigators report a 2‐week placebo washout period between phases in this cross‐over trial which will have reduced the possibility of carryover effects and the study appeared to be free of other sources of bias. |
Stanford 2005.
| Methods | Design: placebo‐controlled parallel trial | |
| Participants |
Participants: men with recurrent impulsive aggressive behaviour Sex: male only Age: mean 28.7 (SD 6.7) years (phenytoin group); mean 34.9 (SD 12.8) years (carbamazepine group); mean 33.6 (SD 4.6) years (valproate group); mean 34.8 (SD 9.7) years (placebo group) Unit of allocation: individual participant Number randomised: 38 Number completing: 29 Setting: outpatient; USA (vicinity of New Orleans) Inclusion criteria: over past 6 months, several discrete episodes of failure to resist aggressive impulses resulting in serious assaultative acts or destruction of property; degree of aggressiveness expressed during the episodes was grossly out of proportion to any precipitating psychosocial stressor; at least 2 such episodes during the month prior to entering the study; score of 8 or higher on the Irritability subscale of the Buss‐Durkee Hostility Inventory; must have identified an individual willing to document any impulsive‐aggressive outbursts that occurred during the study Exclusion criteria: female (due to potential teratogenic effects of phenytoin); verbal IQ < 80; current bipolar disorder; current thought disorder; present use of psychoactive medication; history of medical/neurological problems (including seizures); non‐native English speaker; liver enzymes not within normal limits Ethnicity: not reported Baseline characteristics: for 29 completers overall: at least one Axis I diagnosis (n = 12); major depression (n = 5); alcohol abuse (n = 7); substance abuse (n = 4); at least one Axis II diagnosis (n = 24); antisocial personality disorder (n = 17); borderline personality disorder (n = 3) |
|
| Interventions | Four conditions: phenytoin / carbamazepine / valproate / placebo
Duration of intervention: 6 weeks Duration of trial: 8 weeks (treatment preceded by 2‐week placebo‐baseline period) Length of follow up: participants were not followed up beyond the end of the intervention period Dose adjustment: Not reported. Serum blood levels measured after sixth week of administration. |
|
| Outcomes |
Primary outcomes Aggression (observer‐reported): OAS scores, averaged over four 2‐week periods (placebo‐baseline, 0‐2 weeks, 2‐4 weeks, 4‐6 weeks) Secondary outcomes Non‐compliance: proportion of participants discontinuing treatment |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Sequence generation achieved using a random numbers table |
| Allocation concealment? | Low risk | Investigators state “anticonvulsants and placebo were administered in identical, unmarked capsules obtained from a local pharmacy” (p.74, col 1). The lead author [MS] “was responsible for the random assignment and the maintenance/administrations of all study medication. He was not involved in participant assessment subsequent to the placebo‐baseline” (p.73, col 2). |
| Blinding? of participants | Low risk | Investigators state “anticonvulsants and placebo were administered in identical, unmarked capsules obtained from a local pharmacy” (p.74, col 1). Appropriate care appears to have been taken to ensure blinding of participants. Unlikely that this blinding could have been broken. |
| Blinding? of personnel | Low risk | Appropriate care appears to have been taken to ensure blinding of personnel. Unlikely that this blinding could have been broken. |
| Blinding? of outcome assessors | Low risk | The lead author [MS] “was responsible for the random assignment and the maintenance/administrations of all study medication. He was not involved in participant assessment subsequent to the placebo‐baseline” (p.73, col 2). Appropriate care appears to have been taken to ensure blinding of outcome assessors. Unlikely that this blinding could have been broken. |
| Incomplete outcome data addressed? All outcomes | Low risk | At completion of intervention (6 weeks): 2/9 missing from phenytoin group (reasons not given); 2/9 missing from valproate group (reasons not given); 2/9 missing from carbamazepine group (reasons not given); and 3/11 missing from control (placebo) group (reasons not given). Missing outcome data balance in numbers across intervention groups. Trial investigators provide a completer analysis. In this review, data from 15 participants were included in the analysis of the phenytoin intervention, data from 15 participants were included in the analysis of the valproate intervention, and data from 15 participants were included in the analysis of the carbamazepine intervention. |
| Free of selective reporting? | Low risk | Study protocol is not available but it seems clear that the published report includes all expected outcomes, including those that were pre‐specified |
| Free of other bias? | Unclear risk | Investigators report that this study was sponsored by the Dreyfus Health Foundation which is focused on phenytoin and, according to its website, was established “to study, collect, and disseminate information and sponsor collaboration, clinical, and basic health research into its [phenytoin’s] benefits”. This raises the potential for bias in a study such as this which compares phenytoin with other anticonvulsants as well as against placebo. In addition, the authors note the relatively low serum blood levels of carbamazepine and the possibility that these may arise from poor compliance with taking this medication as well as from individual differences in metabolism of the drug. |
ADHD = attention‐deficit hyperactivity disorder
AsPD = antisocial personality disorder
BPD = borderline personality disorder
BPRS = British Psychiatric Rating scale
CGI = Clinical Global Impressions
CPRS = Children's Psychiatric Rating Scale
DPH = diphenylhydantoin
DSM = Diagnostic and Statistical Manual of Mental Disorders
ECG = electrocardiogram
HisPD = histrionic personality disorder
IED = intermittent explosive disorder
MOAS = Modified Overt Aggression Scale
NarPD = narcissistic personality disorder
OAS = Overt Aggression Scale
PBO = placebo
PD = personality disorder
PD NOS = personality disorder not otherwise specified
PDD = pervasive developmental disorder
POMS = Profile of Mood States
PTSD = post‐traumatic stress disorder
SCID = Structured Clinical Interview for DSM
SD = standard deviation
VPA = valproate
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Akhondzadeh 2008 | Contact with trial investigators confirmed that participants had not been selected on basis of having displayed recurrent aggression |
| Al‐Kaisi 1974 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Barzman 2006 | Comparative study of quetiapine versus divalproex in adolescents with co‐occurring bipolar disorder and disruptive behaviour disorder; no control group |
| Bowden 1994 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| De la Fuente 1994 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| DeVogelaer 1981 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Donovan 2003 | Pre‐treatment analysis of data from participants in the trial reported in Donovan 2000; no additional data |
| Dose 1998 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Esser 1984 | Study was reported in German. Following discussion with the translator, we found reasonable grounds for concluding that participants were not selected on basis of having displayed recurrent aggression, and that results of any aggressive subgroup are not reported separately. |
| Frankenburg 2002 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Hollander 2001 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Hollander 2005 | Further analysis of data from a subset of participants in the trial reported in Hollander 2003; no additional data |
| Killaspy 2006 | This is a brief summary of Loew 2006; participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Kivalo 1968 | Study was reported in Swedish. Following discussion with the translator, we found reasonable grounds for concluding that the participants were not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup were not reported separately. |
| Klein 1984 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Labiner 2009 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately. Study compared adjunctive levetiracetam + existing antiepileptic drug therapy with adjunctive lamotrigine + existing antiepileptic drug therapy, and so lacked a control arm. |
| Lefkowitz 1969 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Loew 2006 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Lorr 1961 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| McElroy 1991 | Participants not selected on basis of having displayed recurrent aggression; study compared ‘responders’ and ‘non‐responders’ from a previous trial |
| Monroe 1975 | Overview summarising findings from several studies completed without random allocation |
| Neppe 1982 | Participants not selected on basis of having displayed recurrent aggression (no quantitative data provided for a subgroup which had displayed recurrent aggression); one participant suffered ‘post traumatic dementia’; unclear if allocation was randomised |
| Nickel 2004 | Contact made with trial investigators who confirmed that participants were not selected on basis of having displayed recurrent aggression. This study recruited participants with borderline personality disorder, although recurrent aggression is not a necessary criterion for this diagnosis. Aggression was not measured as an outcome even though the phrase "treatment of aggression" is included in the title of the paper. |
| Nickel 2005a | Contact made with trial investigators who confirmed that participants were not selected on basis of having displayed recurrent aggression. This study recruited participants with borderline personality disorder, although recurrent aggression is not a necessary criterion for this diagnosis. Aggression was not measured as an outcome even though the investigators conclude "topiramate was more effective than placebo in treating the aggressive component of borderline psychopathology" (p.1517, col 1). |
| Nickel 2005b | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Nickel 2008 | Participants not selected on basis of having displayed recurrent aggression. This is an 18‐month follow‐up to the study reported by Nickel 2005a |
| Okuma 1988 | Japanese paper describing study reported in greater detail in Okuma 1989. Quasi‐randomised (alternate allocation) |
| Okuma 1989 | Quasi‐randomised (alternate allocation) |
| Rojo Sierra 1972 | Study was reported in Spanish. Following discussion with the translator, we found reasonable grounds for concluding that the study had no placebo arm |
| Simopoulos 1971 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately (study described in greater detail in Simopoulos 1974) |
| Simopoulos 1974 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
| Steiner 2003 | Comparative study of high dose versus low dose divalproex sodium for youths with conduct disorder; no control group |
| Tritt 2005 | Contact made with trial investigators who confirmed that participants were not selected on basis of having displayed recurrent aggression. This study recruited participants with borderline personality disorder, although recurrent aggression is not a necessary criterion for this diagnosis. Aggression was not measured as an outcome even though the investigators conclude ‘lamotrigine being specifically more effective than placebo in treating aggression in borderline psychopathology’ (p.290, col 1) |
| Wasserman 2006 | Participants not selected on basis of having displayed recurrent aggression, and results of any aggressive subgroup are not reported separately |
Differences between protocol and review
The review includes two additional outcomes not specified in the original protocol. These were:
number of overall clinical responders (reported by Hellings 2005 in a trial of valproate for outpatient children and adolescents with pervasive developmental disorder);
patient‐rated global improvement (reported by Mattes 2008 in a trial of levetiracetam and by Mattes 2005 in a trial of oxcarbazepine, both for outpatients with intermittent explosive disorder).
We chose to include them because none of the prespecified outcomes provided information on global change.
The review omits two analyses specified in the original protocol because of insufficient data. These were:
subgroup analysis of effect on primary outcome of participants' age, diagnosis, setting and class of drug.
sensitivity analysis to investigate the robustness of findings concerning concealment of allocation, blinding of outcome assessors, and extent of dropouts.
Contributions of authors
RN, NH and MF helped prepare the protocol; HJ, MF and NH examined results of searches and independently assessed studies; NH & MF extracted data and assisted in writing the report; HJ assisted in editing the report; RN provided adjudication and assisted in selecting studies and in writing the report.
Sources of support
Internal sources
Nottinghamshire Healthcare NHS Trust, UK.
External sources
NHS Cochrane Collaboration Programme Grant Scheme, UK.
Declarations of interest
None known.
New
References
References to studies included in this review
Barratt 1991 {published data only}
- Barratt ES, Kent TA, Bryant SG, Felthous AR. A controlled trial of phenytoin in impulsive aggression. Journal of Clinical Psychopharmacology 1991;11(6):388‐9. [DOI] [PubMed] [Google Scholar]
Barratt 1997 {published data only}
Conners 1971 {published data only}
- Conners CK, Kramer R, Rothschild GH, Schwartz L, Stone A. Treatment of young delinquent boys with diphenylhydantoin sodium and methylphenidate: a controlled comparison. Archives of General Psychiatry 1971;24(2):156‐60. [DOI] [PubMed] [Google Scholar]
Cueva 1996 {published data only}
- Cueva JE, Overall JE, Small AM, Armenteros JL, Perry R, Campbell M. Carbamazepine in aggressive children with conduct disorder: a double‐blind and placebo‐controlled study. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(4):480‐90. [DOI] [PubMed] [Google Scholar]
Donovan 2000 {published data only}
- Donovan SJ, Stewart JW, Nunes EV, Quitkin FM, Parides M, Daniel W, et al. Divalproex treatment for youth with explosive temper and mood lability: a double‐blind, placebo‐controlled crossover design [erratum appears in 2000 Jul;157(7):1192]. American Journal of Psychiatry 2000;157(5):818‐20. [DOI] [PubMed] [Google Scholar]
Gardner 1986 {published data only}
- Gardner DL, Cowdry RW. Positive effects of carbamazepine on behavioral dyscontrol in borderline personality disorder. American Journal of Psychiatry 1986;143(4):519‐22. [DOI] [PubMed] [Google Scholar]
Gottschalk 1973 {published data only}
- Gottschalk LA, Covi L, Uliana R, Bates DE. Effects of diphenylhydantoin on anxiety and hostility in institutionalized prisoners. Comprehensive Psychiatry 1973;14(6):503‐11. [DOI] [PubMed] [Google Scholar]
Hellings 2005 {published data only}
- Hellings JA, Weckbaugh M, Nickel EJ, Cain SE, Zarcone JR, Reese RM, et al. A double‐blind, placebo‐controlled study of valproate for aggression in youth with pervasive developmental disorders. Journal of Child & Adolescent Psychopharmacology 2005;15(4):682‐92. [DOI] [PubMed] [Google Scholar]
Hollander 2003 {published data only}
- Hollander E, Tracy KA, Swann AC, Coccaro EF, McElroy SL, Wozniak P, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology 2003;28(6):1186‐97. [DOI] [PubMed] [Google Scholar]
Mattes 2005 {published data only}
- Mattes JA. Oxcarbazepine in patients with impulsive aggression: a double‐blind, placebo‐controlled trial. Journal of Clinical Psychopharmacology 2005;25(6):575‐9. [DOI] [PubMed] [Google Scholar]
Mattes 2008 {published data only}
- Mattes JA. Levetiracetam in patients with impulsive aggression: a double‐blind placebo‐controlled trial. Journal of Clinical Psychiatry 2008;69(2):310‐5. [DOI] [PubMed] [Google Scholar]
Rosenblatt 1976 {published data only}
- Rosenblatt S, Schaeffer D, Rosenthal JS. Effects of diphenylhydantoin on child‐abusing parents: a preliminary report. Current Therapeutic Research, Clinical and Experimental 1976;19(3):332‐6. [PubMed] [Google Scholar]
Stanford 2001 {published and unpublished data}
- Stanford 2009. Email from M Stanford to J Dennis clarifying trial methods and providing SPSS file with raw POMS scores for each arm of the crossover 2009.
- Stanford MS, Houston RJ, Mathias CW, Greve KW, Villemarette‐Pittman NR, Adams D. A double‐blind placebo‐controlled crossover study of phenytoin in individuals with impulsive aggression. Psychiatry Research 2001;103(2‐3):193‐203. [DOI] [PubMed] [Google Scholar]
Stanford 2005 {published data only}
- Stanford MS, Helfritz LE, Conklin SM, Villemarette‐Pittman NR, Greve KW, Adams D, et al. A comparison of anticonvulsants in the treatment of impulsive aggression. Experimental and Clinical Psychopharmacology 2005;13(1):72‐7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Akhondzadeh 2008 {published data only}
- Akhondzadeh S, Tajdar H, Mohammadi MR, Mohammadi M, Nouroozinejad GH, Shabstari OL, et al. A double‐blind placebo controlled trial of piracetam added to risperidone in patients with autistic disorder. Child Psychiatry and Human Development 2008;39:237‐45. [DOI] [PubMed] [Google Scholar]
Al‐Kaisi 1974 {published data only}
- Al Kaisi A, McGuire R. The effect of sulthiame on disturbed behaviour in mentally subnormal patients. British Journal of Psychiatry 1974;124(578):45‐9. [DOI] [PubMed] [Google Scholar]
Barzman 2006 {published data only}
- Barzman DH, DelBello MP, Adler CM, Stanford KE, Strakowski SM. The efficacy and tolerability of quetiapine versus divalproex for the treatment of impulsivity and reactive aggression in adolescents with co‐occurring bipolar disorder and disruptive behavior disorder(s). Journal of Child and Adolescent Psychopharmacology 2006;16(6):665‐70. [DOI] [PubMed] [Google Scholar]
Bowden 1994 {published data only}
- Bowden C, Brugger A, Swann A, Calabrese J, Janicak P. Efficacy of divalproex, lithium and placebo in treatment of mania. JAMA. 93/94‐2309, 1994; Vol. 271, issue 12:918‐24. [PubMed]
De la Fuente 1994 {published data only}
DeVogelaer 1981 {published data only}
- DeVogelaer J. Carbamazepine in the treatment of psychotic and behavioral disorders: a pilot study. Acta Psychiatrica Belgica 1981;81(6):532‐41. [PubMed] [Google Scholar]
Donovan 2003 {published data only}
- Donovan SJ, Nunes EV, Stewart JW, Ross D, Quitkin FM, Jensen PS, et al. "Outer‐directed irritability": a distinct mood syndrome in explosive youth with a disruptive behavior disorder. Journal of Clinical Psychiatry 2003;64(6):698‐701. [DOI] [PubMed] [Google Scholar]
Dose 1998 {published data only}
- Dose M, Hellweg R, Yassouridis A, Theison M, Emrich HM. Combined treatment of schizophrenic psychoses with haloperidol and valproate. Pharmacopsychiatry 1998;31(4):122‐5. [DOI] [PubMed] [Google Scholar]
Esser 1984 {published data only}
- Esser G, Schmidt MH, Witkop HJ. Effectiveness of carbamazepine in hyperkinetic children. Zeitschrift fuer Kinder und Jugendpsychiatrie 1984;12:275‐83. [Google Scholar]
Frankenburg 2002 {published data only}
- Frankenburg F, Zanarini MC. Divalproex sodium treatment of women with borderline personality disorder and bipolar II disorder: a double‐blind placebo‐controlled pilot study. Journal of Clinical Psychiatry 2002;63(5):442‐6. [DOI] [PubMed] [Google Scholar]
Hollander 2001 {published data only}
- Hollander E, Allen A, Lopez RP, Bienstock CA, Grossman R, Siever LJ, et al. A preliminary double‐blind, placebo‐controlled trial of divalproex sodium in borderline personality disorder. Journal of Clinical Psychiatry 2001;62(3):199‐203. [DOI] [PubMed] [Google Scholar]
Hollander 2005 {published data only}
- Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. American Journal of Psychiatry 2005;162(3):621‐4. [DOI] [PubMed] [Google Scholar]
Killaspy 2006 {published data only}
- Killaspy H. Topiramate improves psychopathological symptoms and quality of life in women with borderline personality disorder. Evidence‐Based Mental Health 2006;9(3):74. [DOI] [PubMed] [Google Scholar]
Kivalo 1968 {published data only}
- Kivalo E, Seppalainen AM, Lydecken K. Treatment of the agitated and most severe cases of cerebral injury and cerebral palsy with carbamazepine (Tegretol). [Swedish]. Nordisk Psykiatrisk Tidsskrift 1968;22(1):44‐52. [DOI] [PubMed] [Google Scholar]
Klein 1984 {published data only}
Labiner 2009 {published data only}
- Labiner DM, Ettinger AB, Fakhoury TA, Chung SS, Shneker B, Tatum WO, et al. Effects of lamotrigine compared with levetiracetam on anger, hostility, and total mood in patients with partial epilepsy. Epilepsia 2009;50(3):434‐42. [DOI] [PubMed] [Google Scholar]
Lefkowitz 1969 {published data only}
- Lefkowitz M. Effects of diphenylhydantoin on disruptive behavior: study of male delinquents. Archives of General Psychiatry 1969;20(6):643‐51. [DOI] [PubMed] [Google Scholar]
Loew 2006 {published data only}
- Loew TH, Nickel MK, Muehlbacher M, Kaplan P, Nickel C, Kettler C, et al. Topiramate treatment for women with borderline personality disorder: a double‐blind, placebo‐controlled study. Journal of Clinical Psychopharmacology 2006;26(1):61‐6. [DOI] [PubMed] [Google Scholar]
Lorr 1961 {published data only}
- Lorr M, McNair DM, Weinstein GJ, Michaux WW, Raskin A. Meprobamate and chlorpromazine in psychotherapy: some effects on anxiety and hostility of outpatients. Archives of General Psychiatry 1961;4:381‐9. [DOI] [PubMed] [Google Scholar]
McElroy 1991 {published data only}
- McElroy SL, Keck PEJ, Pope HGJ, Hudson JI, Morris D. Correlates of antimanic response to valproate. Psychopharmacology Bulletin 1991;27(2):127‐33. [PubMed] [Google Scholar]
Monroe 1975 {published data only}
- Monroe RR. Anticonvulsants in the treatment of aggression. Journal of Nervous and Mental Disease 1975;160(2):119‐26. [DOI] [PubMed] [Google Scholar]
Neppe 1982 {published data only}
Nickel 2004 {published data only}
- Nickel M, Nickel C, Mitterlehner FO, Tritt K, Lahmann C, Leiberich PK, et al. Topiramate treatment of aggression in female borderline personality disorder patients: a double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2004;65(11):1515‐9. [DOI] [PubMed] [Google Scholar]
Nickel 2005a {published data only}
- Nickel M, Nickel C, Kaplan P, Lahmann C, Mühlbacher M, Tritt K, et al. Treatment of aggression with topiramate in male borderline patients: a double‐blind, placebo‐controlled study. Biological Psychiatry 2005;57(5):495‐9. [DOI] [PubMed] [Google Scholar]
Nickel 2005b {published data only}
- Nickel C, Lahmann C, Tritt K, Muehlbacher M, Kaplan P, Kettler C, et al. Topiramate in treatment of depressive and anger symptoms in female depressive patients: a randomized, double‐blind, placebo‐controlled study. Journal of Affective Disorders 2005;87(2‐3):243‐52. [DOI] [PubMed] [Google Scholar]
Nickel 2008 {published data only}
- Nickel MK, Loew TH. Treatment of aggression with topiramate in male borderline patients, part II: 18‐month follow‐up. European Psychiatry 2008;23:115‐7. [DOI] [PubMed] [Google Scholar]
Okuma 1988 {published data only}
- Okuma T, Yamashita I, Takahashi R, Itoh H, Otsuki S, Watanabe S, et al. Comparison of the therapeutic effect of carbamazepine and placebo in schizophrenia and atypical psychosis by double‐blind controlled study. Rinsho Hyoka (Clinical Evaluation). 0, 1988; Vol. 16, issue 2:327‐76.
Okuma 1989 {published data only}
- Okuma T, Yamashita I, Takahashi R, Itoh H, Otsuki S, Watanabe S, et al. A double‐blind study of adjunctive carbamazepine versus placebo on excited states of schizophrenic and schizoaffective disorders. Acta Psychiatrica Scandinavica 1989;80(3):250‐9. [DOI] [PubMed] [Google Scholar]
Rojo Sierra 1972 {published data only}
- Rojo Sierra M, Giner J, Diez M, Lloret E. Aggressiveness in juvenile character disorders and therapeutic psychopharmacologic results. [Spanish]. Archivos de Neurobiologia 1972;35(4):293‐322. [PubMed] [Google Scholar]
Simopoulos 1971 {published data only}
- Simopoulos A, Pinto A, Uhlenhuth E, Mcgee J. Old wine in new bottles: decreased hostility in chronic psychotic inpatients treated with diphenylhydantoin (Dilantin). World Congress of Psychiatry (5). Ciudadde, Mexico, 9999; Vol. 5:8.
Simopoulos 1974 {published data only}
- Simopoulos A, Pinto A, Uhlenhuth EH, McGee JJ, DeRosa ER. Diphenylhydantoin effectiveness in the treatment of chronic schizophrenics: a double‐blind controlled study with a placebo. Archives of General Psychiatry 1974;30(1):106‐11. [DOI] [PubMed] [Google Scholar]
Steiner 2003 {published data only}
- Steiner H, Petersen ML, Saxena K, Ford S, Matthews Z. Divalproex sodium for the treatment of conduct disorder: a randomized controlled clinical trial. Journal of Clinical Psychiatry 2003;64(10):1183‐91. [DOI] [PubMed] [Google Scholar]
Tritt 2005 {published data only}
- Tritt K, Nickel C, Lahmann C, Leiberich PK, Rother WK, Loew TH, et al. Lamotrigine treatment of aggression in female borderline‐patients: a randomized, double‐blind, placebo‐controlled study. Journal of Psychopharmacology 2005;19(3):287‐91. [DOI] [PubMed] [Google Scholar]
Wasserman 2006 {published data only}
- Wasserman S, Lyengar R, Chaplin WF, Watner D, Waldoks SE, Anagnostou E, et al. Levetiracetam versus placebo in childhood and adolescent autism: a double‐blind placebo‐controlled study. International Clinical Psychopharmacology 2006;21(6):363‐7. [DOI] [PubMed] [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewedness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
American Psychiatric Association 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). Washington DC: American Psychiatric Association, 2000. [Google Scholar]
Anderson 1999
- Anderson SW, Bechara A, Damasio H, Tranel D, Damasio AR. Impairment of social and moral behavior related to early damage in the human prefrontal cortex. Nature Neuroscience 1999;2:1032‐7. [DOI] [PubMed] [Google Scholar]
Arseneault 2000
- Arseneault L, Moffitt TE, Caspi A, Taylor PJ, Silva PA. Mental disorders and violence in a total birth cohort. Results from the Dunedin Study. Archives of General Psychiatry 2000;57:979‐86. [DOI] [PubMed] [Google Scholar]
Attkisson 1982
- Attkisson CC, Zwick R. The client satisfaction questionnaire. Psychometric properties and correlations with service utilization and psychotherapy outcome. Evaluation and Program Planning 1982;5:233‐7. [DOI] [PubMed] [Google Scholar]
Barratt 1997
- Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. Journal of Clinical Psychopharmacology 1997;17(5):341‐9. [DOI] [PubMed] [Google Scholar]
Barratt 1999
- Barratt ES, Stanford MS, Dowdy L, Liebman MJ, Kent TA. Impulsive and premeditated aggression: a factor analysis of self‐reported acts. Psychiatry Research 1999;86(2):163‐73. [DOI] [PubMed] [Google Scholar]
Berkowitz 1993
- Berkowitz L. Aggression: its causes, consequences and control. Philadelphia PA: Temple University Press, 1993. [Google Scholar]
Blair 2001
- Blair RJR. Neurocognitive models of aggression, the antisocial personality disorders, and psychopathy. Journal of Neurology, Neurosurgery and Psychiatry 2001;71:727‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brennan 2000
- Brennan PA, Mednick SA, Hodgins S. Major mental disorders and criminal violence in a Danish birth cohort. Archives of General Psychiatry 2000;57(5):494‐500. [DOI] [PubMed] [Google Scholar]
Buss 1992
- Buss AH, Perry M. The Aggression Questionnaire. Journal of Personality and Social Psychology 1992;63:452‐9. [DOI] [PubMed] [Google Scholar]
Coccaro 1991
- Coccaro EF, Harvey PH, Kupshaw‐Lawrence E, Herbert JL, Bernstein DP. Development of neuropharmacologically based behavioral assessments of impulsive aggressive behavior. Journal of Neuropsychiatry and Clinical Neurosciences 1991;3 (Suppl):44‐51. [PubMed] [Google Scholar]
Connor 1996
- Connor DF, Steingard RJ. A clinical approach to the pharmacotherapy of aggression in children and adolescents. Annals of the New York Academy of Sciences 1996;794:290‐307. [DOI] [PubMed] [Google Scholar]
Connor 2006
- Connor DF, Carlson GA, Chong KD, Daniolos PT, Ferziger R, Findling RL, et al. Juvenile maladaptive aggression: a review of prevention, treatment, and service configuration and a proposed research agenda. Journal of Clinical Psychiatry 2006;67(5):808‐20. [PubMed] [Google Scholar]
DeBrito 2009
- Brito SA, Hodgins S. Antisocial personality disorder. In: McMurran M, Howard RC editor(s). Personality, personality disorder and violence. Chichester, UK: Wiley‐Blackwell, 2009:133‐53. [Google Scholar]
Derogartis 1973
- Derogartis LR, Lipman RS, Covi L. SCL‐90: an outpatient psychiatric rating scale; preliminary report. Psychopharmacology Bulletin 1973;9:13‐28. [PubMed] [Google Scholar]
Donner 2001
- Donner A, Piaggio G, Villar J. Statistical methods for the meta‐analysis of cluster randomized trials. Statistical Methods in Medical Research 2001;10:325‐38. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
EuroQoL group 1990
- EuroQoL Group. EuroQoL: a new facility for measurement of health‐related quality of life. Health Policy 1990;16:199‐208. [DOI] [PubMed] [Google Scholar]
Fazel 2006
- Fazel S, Grann M. The population impact of severe mental illness on violent crime. American Journal of Psychiatry 2006;163(8):1397‐403. [DOI] [PubMed] [Google Scholar]
Follmann 1992
- Follmann D, Elliot P, Suh I, Cutler J. Variance imputation for overviews of clinical trials with continuous response. Journal of Clinical Epidemiology 1992;45(7):769‐73. [DOI] [PubMed] [Google Scholar]
Gardner 1986
- Gardner DL, Cowdry RW. Positive effects of carbamazepine on behavioral dyscontrol in borderline personality disorder. American Journal of Psychiatry 1986;143(4):519‐22. [DOI] [PubMed] [Google Scholar]
Gobbi 2006
- Gobbi, G, Gaudreau P, Leblanc N. Efficacy of topiramate, valproate and their combination on aggression/agitation behaviour in patients with psychosis. Journal of Clinical Psychopharmacology 2006;26(5):467‐73. [DOI] [PubMed] [Google Scholar]
Goedhard 2006
- Goedhard LE, Stolker JJ, Heerdink ER, Nijman HL, Olivier B, Egberts TC. Pharmacotherapy for the treatment of aggressive behavior in general adult psychiatry: a systematic review. Journal of Clinical Psychiatry 2006;67(7):1013‐24. [DOI] [PubMed] [Google Scholar]
Haller 2006
- Haller J, Kruk MR. Normal and abnormal aggression: human disorders and novel laboratory models. Neuroscience and Biobehavioral Reviews 2006;30(3):292‐303. [DOI] [PubMed] [Google Scholar]
Hawkins 2000
- Hawkins KA, Tobst KK. Frontal lobe dysfunction and aggression: conceptual issues and research findings. Aggressive and Violent Behavior 2000;5(2):147‐57. [Google Scholar]
Higgins 2008
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Hollander 2003
- Hollander E, Tracy KA, Swann AC, Coccaro EF, McElroy SL, Wozniak P, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology 2003;28(6):1186‐97. [DOI] [PubMed] [Google Scholar]
Hollander 2005
- Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. American Journal of Psychiatry 2005;162(3):621‐4. [DOI] [PubMed] [Google Scholar]
Kanemoto 1999
- Kanemoto K, Kawasaki J, Mori E. Violence and epilepsy: a close relation between violence and postictal psychosis. Epilepsia 1999;40(1):107‐9. [DOI] [PubMed] [Google Scholar]
Kavoussi 1989
- Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. Journal of Clinical Psychiatry 1989;59(12):676‐80. [DOI] [PubMed] [Google Scholar]
Kay 1989
- Kay SR, Opler LA, Lindenmayer JP. The Positive and Negative Syndrome Scale (PANSS): rationale and standardisation. British Journal of Psychiatry 1989;155 (Suppl):59‐65. [PubMed] [Google Scholar]
Keele 2005
- Keele NB. The role of serotonin in impulsive and aggressive behaviors associated with epilepsy‐like neuronal hyperexcitability in the amygdala. Epilepsy and Behavior 2005;73(3):325‐35. [DOI] [PubMed] [Google Scholar]
Kessler 2006
- Kessler R, Coccaro E, Fava M, Jaeger S, Jin R, Walters E. The prevalence and correlates of DSM‐IV intermittent explosive disorder in the National Comorbidity Survey replication. Archives of General Psychiatry 2006;63(6):669‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lawson 2008
- Lawson WB, Nanos J. Effects of divalproex on disruptive behavior of jail inmates (letter). Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2008;32:909‐10. [DOI] [PubMed] [Google Scholar]
Lindenmayer 2000
- Lindenmayer JP, Kotsaftis AT. Use of sodium valproate in violent and aggressive behaviors: a critical review. Journal of Clinical Psychiatry 2000;61(2):123‐8. [DOI] [PubMed] [Google Scholar]
Malone 1994
- Malone RP, Luebbert RP, Pena‐Ariet M, Biesecker K, Delaney MA. The Overt Aggression Scale in the study of lithium in aggressive conduct disorder. Psychopharmacology Bulletin 1994;30:215‐8. [PubMed] [Google Scholar]
Marson 2009
- Marson T. Personal correspondence (email from Prof Marson to Dr C Bennet on 5.04.09). Personal correspondence via email 2009.
McGuire 2008
- McGuire J. A review of effective interventions for reducing aggression and violence. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 2008;363:2577‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miczek 2002
- Miczek KA, Fish EW, Bold JF, Almeida RM. Social and neural determinants of aggressive behavior: pharmacotherapeutic targets at serotonin, dopamine and gamma‐aminobutyric acid systems. Psychopharmacology (Berl). 2002;163(3‐4):434‐58. [DOI] [PubMed] [Google Scholar]
Nelson 2007
- Nelson RJ, Trainor BC. Neural mechanisms of aggression. Nature Reviews Neuroscience 2007;8(7):536‐46. [DOI] [PubMed] [Google Scholar]
Nestor 2002
- Nestor PG. Mental disorder and violence: personality dimensions and clinical features. American Journal of Psychiatry 2002;159(12):1973‐8. [DOI] [PubMed] [Google Scholar]
Patton 1995
- Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. Journal of Clinical Psychology 1995;51(6):768‐74. [DOI] [PubMed] [Google Scholar]
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre. The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre. The Cochrane Collaboration, 2008.
Siever 2008
- Siever LJ. Neurobiology of aggression and violence. American Journal of Psychiatry 2008;165(4):429‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spielberger 1999
- Spielberger CD. STAXI‐2: State‐Trait Anger Expression Inventory‐2. Odessa, FL: Psychological Assessment Resources, Inc., 1999. [Google Scholar]
Staller 2007
- Staller JA. Psychopharmacologic treatment of aggressive preschoolers: a chart review. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2007;31(1):131‐5. [DOI] [PubMed] [Google Scholar]
Stanford 2001
- Stanford MS, Houston RJ, Mathias CW, Greve KW, Villemarette‐Pittman NR, Adams D. A double‐blind placebo‐controlled crossover study of phenytoin in individuals with impulsive aggression. Psychiatry Research 2001;103(2‐3):193‐203. [DOI] [PubMed] [Google Scholar]
Stanford 2009
- Stanford M. Email from M Stanford to J Dennis clarifying trial methods and providing SPSS file with raw POMS scores for each arm of the cross‐over 2009.
Stein 1992
- Stein G. Drug treatment of the personality disorders. British Journal of Psychiatry 1992;161:167‐82. [DOI] [PubMed] [Google Scholar]
Tyrer 1988
- Tyrer P, Seivewright N. Pharmacological treatment of personality disorders. Clinical Neuropharmacology 1988;11(6):493‐9. [DOI] [PubMed] [Google Scholar]
van Elst 2000
- Elst LT, Woermann FG, Lemieux L, Thompson PJ, Trimble MR. Affective aggression in patients with temporal lobe epilepsy: a quantitative MRI study of the amygdala. Brain 2000;123(2):234‐43. [DOI] [PubMed] [Google Scholar]
Vitiello 1997
- Vitiello B, Stoff DM. Subtypes of aggression and their relevance to child psychiatry. American Academy of Child and Adolescent Psychiatry 1997;36(3):307‐15. [DOI] [PubMed] [Google Scholar]
World Health Organization 1993
- World Health Organization. The ICD‐10 Classification of Mental and Behavioral Disease. Geneva: World Health Organization, 1993. [Google Scholar]
World Health Organization 2002
- World Health Organization. World Report on Violence and Health. Geneva: World Health Organization, 2002. [Google Scholar]