

Abstract
The epidermal growth factor receptor (EGFR) is a kind of receptor tyrosine kinase (RTK) that plays a critical role in the initiation and development of malignant tumors via modulating downstream signaling pathways. In non-small cell lung cancer (NSCLC), the activating mutations located in the tyrosine kinase domains of EGFR have been demonstrated in multiple researches as the “Achilles’ heel” of this deadly disease since they could be well-targeted by epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs). However, it’s still too early to celebrate since the first-generation EGFR-TKIs such as gefitinib and erlotinib have only achieved limited clinical benefits and acquired resistance to this kind of drugs occurred inevitably in almost all the NSCLC patients. In order to make the most of EGFR-TKIs and develop more effective regimens for the NSCLC patients, researchers majoring in different aspects start a battle against EGFR-TKI resistance. Challenging as it is, we still progress stably and step firmly toward the final victory. This review will summarize the major mechanisms of acquired resistance to EGFR-TKIs, and then discuss the development of rationally designed molecular target drugs in accordance with each mechanism, in the hope of shedding light on the great achievements we have obtained and tough obstacles we have to overcome in the battle against this deadly disease.
Keywords: Non-small cell lung cancer, epithelial growth factor receptor, tyrosine kinase inhibitors, drug resistance, molecular targeted therapy
Introduction
Cancers of the lung have long been the leading cause of cancer-related death all over the world. According to the statistic announced by the American Cancer Society, in the year 2014, the Unit States is projected to witness 224,210 new cases of lung cancer, ranking the second frequent cancer in both male and female. 159,260 cases are estimated to finally die of lung cancer, accounting for 27.2% of all the cancer-related death [1]. Non-small cell lung cancer (NSCLC), the major object of this review, is the largest subgroup of lung cancer, occurring at the frequency of about 80% [2]. Generally speaking, the therapeutic effect of NSCLC is far from satisfactory. Although great progress has been made in the chemotherapy, almost no more than 10 months median overall survival (OS) can be achieved even by the most effective platinum-based chemotherapeutic regimens [3].
EGFR and EGFR-TKIs in lung cancer
With deeper digging into the molecular events underlying the oncogenesis and progression of NSCLC, epidermal growth factor receptor (EGFR), a kind of tyrosine kinase receptor, became one of the landmark targets of NSCLC therapy. It is a member of the HER family, which also includes HER2 (ErbB2), HER3 (ErbB3), HER4 (ErbB4) [4]. When EGFR’s extracellular domain binds to its ligand, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), it forms dimers with other EGFR or other HER family members to get itself auto-phosphorylated at the key tyrosine residues. Subsequently, the phosphorylated EGFR further activates several downstream signaling pathways such as PI3K/AKT/mTOR, RAS/RAF/MAPK, JAK/STAT, which play the critical roles in regulating multiple cellular processes, including proliferation, survival and apoptosis [4,5]. The constitutive activation of EGFR signaling pathway, caused by gene mutations or by gene amplification or both, has been demonstrated to have close connection with the initiation, progression and poor prognosis of NSCLC [6]. EGFR activating mutations, majorly located in the tyrosine kinase domains and in the form of a base-pair deletion at exon 19 (delE746_A750, account for about 54%) or a point mutation at exon 21 (L858R, account for about 43%), occur at about 20% of NSCLC patients, with significantly increased proportions in the subgroup of adenocarcinoma histology (40%), female sex (42%), Asian ethnicity (30%), and never-smoker status (51%) [7]. They enable the EGFR to activate, without the ligand binding, the downstream molecules [8]. As such, the tumor cells are consequently addicted to the EGFR signal pathway. The first-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs), gefitinib and erlotinib, designed to reversibly compete for the ATP binding sites and thus block EGFR-induced downstream signaling activation, have and are being extensively investigated in NSCLC treatment. The landmark Iressa Pan-Asia Study (IPASS) showed gefitinib could significantly prolong median progression free survival (PFS) compared with carboplatin/paclitaxel in the subgroup of patients with EGFR mutation-positive tumors (median PFS 9.5 months versus 6.3 months; hazard ratio [HR] 0.48, 95% CI 0.36 to 0.64, p<0.001), together with much improved qualification of life (QoL) and delayed deterioration of symptoms [9,10]. Similar benefits of EGFR-TKIs in EGFR mutation-positive NSCLC patients were shown in several other large-scale studies, such as WJTOG3405 comparing gefitinib with cisplatin/docetaxel as first-line treatment in Asian patients (median PFS 9.2 months versus 6.3 months; HR 0.489, 95% CI 0.336 to 0.710, p<0.0001), OPTIMAL comparing erlotinib with chemotherapy as first-line treatment in Asian patients (median PFS 13.1 months versus 4.6 months; HR 0.16, 95% CI 0.10 to 0.26, p<0.0001), EURTAC comparing erlotinib with standard chemotherapy as first-line treatment in European patients (median PFS 9.7 months versus 5.2 months; HR 0.37, 95% CI 0.25 to 0.54, p<0.0001) [11-13]. However, the superiority of gefitinib in patients with high EGFR-gene-copy number over docetaxel was not proven in the INTEREST study [14]. In addition, the carboplatin/paclitaxel combination demonstrated superior efficiency than gefitinib in the EGFR-gene-amplified but EGFR mutation-negative subgroup (median PFS: 5.5 months versus 1.5 months; HR 2.85, 95% CI 2.05 to 3.98, p<0.001) [9]. Therefore, it is plausible that it’s the EGFR mutation status that determines the tumor responses to EGFR-TKIs. This conclusion is widely recognized and dramatically promotes the clinical practices of detecting EGFR mutation status and applying the EGFR-TKIs to the subset of EGFR mutant patients. However, problems arose that almost all the patients with initial dramatic responses to gefitinib or erlotinib ultimately underwent tumor progression and inevitably became resistant to them mostly within 6-12 months, which has been defined as “acquired resistance” [15,16]. Several mechanisms underlying have been discovered, but more arduous efforts should be made since 30% of the required resistant cases remain unexplainable. In addition, a better understanding of the mechanisms is only the first step and coping rationally with them is the critical next step to win the battle against EGFR-TKI resistance.
Mechanisms of EGFR-TKI resistance in NSCLC
In a long period of time, we had no idea about the mechanisms leading to the EGFR-TKI resistance until 2005, when Susumu Kobayashi and his colleagues firstly discovered the T790M mutation after sequencing the EGFR gene of a patient with acquired resistance to gefitinib [17]. After that, a series of tremendous successes have been achieved in this field (Figure 1).
Figure 1.
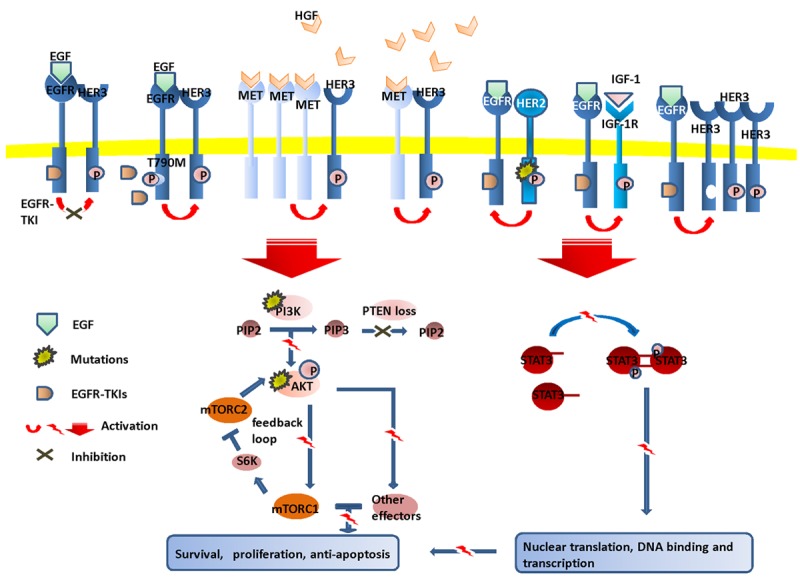
Various mechanisms of EGFR-TKI resistance. Despite of constitutive existence of first-generation of EGFR-TKIs, the tumor cells manage to survive and proliferate via EGFR-T790M mutation, upregulation of MET/HGF, HER2 mutations, overexpression of HER3, persistent activation of IGF-1R, mutations of PIK3CA/AKT, loss or downregulation of PTEN and abnormal dimerization of STAT3.
Gatekeeper mutation in EGFR: T790M mutation
T790, located in the ATP binding pocket, is named “the gatekeeper residue” as it determines the affinity of ATP-competitive EGFR-TK inhibitors to EGFR-TK. Substitution of Threonine 790 with Methionine (T790M) increases the ATP’s affinity to EGFR and attenuates the binding efficacy of gefitinib and erlotinib consequently [18]. Approximately 50% of the acquired resistance developed to erlotinib or gefitinib is linked to T790M mutation and the proportion could be underestimated as more accurate prevalence of 68% was achieved using LNA-PCR/sequencing assay [19]. While the reason remains enigmatic, the good news is that the patients with T790M at the time of TKI failure tend to have longer post-progression survival (PPS) than those without such a mutation [20].
Compensatory contribution of other RTKs
c-MET
MET receptor, a trans-membrane tyrosine kinase encoded by proto-oncogene MET, has been highlighted as an important cause for acquired resistance of NSCLC to gefitinib or erlotinib. As the ligand for MET receptor, hepatocyte growth factor (HGF, also known as scatter factor), once binding to MET receptor, will promote the phosphorylation of MET tyrosine kinase and subsequently trigger the activation of downstream PI3K/AKT/mTOR pathway, which is the key signaling pathway for cell proliferation, survival and anti-apoptosis [21,22]. A lot of preclinical trials have shown that uncontrolled activation of MET was oncogenic and facilitated the cells to become malignant, invasive, metastatic and EGFR-TKI resistant. Mechanisms underlying are multiple, such as MET and HGF overexpression, MET gene amplification or mutation [23,24]. The MET gene amplification emerges as one of the most relevant mechanisms, and is correlated with poor clinical outcomes [25]. About 22% of the EGFR-TKI acquired resistant specimens have been demonstrated to possess MET gene amplification [26,27]. Of note, the MET over-activation in most circumstances occurs via increased transcription and expression of MET protein instead of MET amplification [28]. In addition, over 20 mutations have been identified in MET and the majority of them were found to be germline mutations [29]. They are oncogenic in a large variety of human cancers, including NSCLC. The most frequent mutations locate in the semaphorin domain (affecting HGF binding), the juxtamembrane domain (affecting the actin cytoskeleton, cell motility and migration) and the TK domain (activating MET even in the absence of HGF) [30]. The occurrence rates of MET gene mutations vary along ethnic and racial lines, with the highest frequency occurring in East Asian [29]. Generally speaking, the MET gene mutation in NSCLC patients was less frequently reported (8~13%) and their correspondence with acquired EGFR-TKI resistance needs to be interpreted with cautions [31]. Moreover, HGF overexpression is a much more frequent event in acquired resistant NSCLC tumors (61% in a Japanese research) than in the sensitive ones (10%), supporting the role for HGF overexpression in promoting drug resistance [32]. The high-level of HGF can be secreted by lung cancer cells themselves and stromal fibroblasts in the tumor microenvironment as well [33]. How HGF contributes to the resistance formation can be elucidated by two known mechanisms. Firstly, HGF can stimulate the activation of PI3K signal pathway by means of binding and activating MET receptor without the involvement of EGFR, which attenuates the addiction of tumor cells at EGFR signal pathway and thus eliminates the anti-tumor potency of EGFR-TKIs [23]. Secondly, the presence of HGF can induce a tyrosine kinase-independent function of EGFR, which is interacting with other tumor relevant proteins such as CDCP1, AXL, EohE2 et al. [34]. Taken together, the aberrant activation of HGF-MET pathway is one of the main obstacles to fight against EGFR-TKI resistance in NSCLC and greater efforts are warrant to make steps further in this aspect.
IGF-1 Receptor
Growing evidences have emerged for the involvement of the IGF-1 Receptor (IGF-1R) pathway in the acquisition of resistance to EGFR-TKIs. Constitutive activation of IGF-1R pathway has been detected in multiple gefitinib or erlotinib resistant lung cancer lines. This connection was further proven by testing primary NSCLC samples using immunohistochemistry (IHC) that higher IGF-1R expression level was detected in acquired gefitinib resistant patients than those who were sensitive [35]. The way IGF-1R interfering the anti-tumor activity of EGFR-TKIs seems to be complicated. Amandine Hurbin et al. suggested IGF-1R managed it by inhibiting apoptosis via amphiregulin [36]. Besides, Floriana Morgillo and his colleagues also found that IGF-1R could be activated via forming a heterodimer with EGFR after erlotinib treatment. The activated IGF-1R keeps transmitting extracellular survival signals to downstream mediators such as PI3K/AKT and MAPK to stimulate mammalian target of rapamycin (mTOR), which mediates the synthesis of EGFR and anti-apoptotic survivin proteins [37]. How IGF-1R signaling is activated in TKI resistant cells remains largely unknown. Evidences arose that it was at least partially induced by the down-regulation of major IGF carrier protein IGFBP3 [38]. Co-treatment of IGF-1R inhibitors such as α-IR3, AG1024, R1507 with EGF-TKIs enhanced TKI-induced growth inhibition and apoptosis, offering a potential approach to overcome the primary resistance of EGFR-TKIs in NSCLC [39,40].
HER2
HER2 mutation occurred at about 2% of patients with NSCLC, significantly more frequent in never smokers, adenocarcinoma histology, oriental ethnicity and female gender [41]. Almost all HER2 mutations locate in exon 20, encoding the kinase domain of HER2 protein [42]. Distinct from the other family members, HER2 has strong kinase activity but has no identified ligand binding domain. Thus, it has to form heterodimers with the other family members including activated EGFR to get trans-phosphorylated [43]. The dependence of HER2 activation on the trans-phosphorylation of EGFR determines the strong inhibition of EGFR-TKIs on the wide type HER2. However, when HER2 mutates in the kinase domain, it becomes EGFR-independent and in turn trans-phosphorylates EGFR even in the presence of EGFR-TKIs [44]. Consequently, NSCLC cells holding the mutant HER2 are more potent in activating downstream signal transducers and exert resistance to EGFR-TKIs and knockdown of the mutant HER2 succeed in restoring sensitivity to EGFR-TKIs. Moreover, EGFR-TKI resistance can also be induced by HER2 gene amplification or protein overexpression [45]. Therefore, it provides a rationale to detect the HER2 status and add HER2-targeted agents such as lapatinib, trastuzumab and dacomitinib to EGFR-TKIs in the treatment of certain NSCLC patients [46-48].
HER3
While EGFR and HER2 have been ranked as two of the most heated targets in NSCLC targeting treatment, it was not rare that even effective blockage of EGFR and HER2 in EGFR-driven or HER2-driven xenografts could not durably suppress AKT signaling. Growing evidences have shown the involvement of HER3, another HER family member, in this phenomenon [49]. The persistent activation of HER3 was detected in human cancer cells and essential for the binding activated EGFR, HER2, MET to PI3K/AKT signaling. HER3 lies upstream the PI3K signaling pathway and functions as an accessory partner of EGFR and HER2 [50]. When HER3 forms a dimer with EGFR or HER2, it gets phosphorylated by intrinsic tyrosine kinase activity of EGFR or HER2 and then couples them to the PI3K signaling pathway since EGFR and HER2 lack the tyrosine-phosphorylated Tyr-X-X-Met motif necessary for docking PI3K while HER3 contains 7 copies [49,51]. HER3 is dependent on EGFR and HER2 to get tyrosine-phosphorylated, and meanwhile, EGFR and HER2 have to rely on HER3 to recruit and trans-phosphorylate the PI3K molecular. Ideally, EGFR-TKIs can disassociate the EGFR-HER3 dimers and consequently block the downstream PI3K/AKT pathway. However, continuous EGFR-TKI exposure often triggers the overexpression of HER3 as a result of the loss of AKT-mediated negative feedback signaling. The overexpressed HER3 promotes the forward shift in the equilibrium of the HER3 phosphorylation-dephosphorylation reactions and results in superphosphorylated state of HER3 and AKT, which requires much higher concentration of RTK-TKIs to fully disassociate the heterodimers or much more potent HER3-targeted drugs to completely dephosphorylate HER3 [52].
Activation of compensatory signaling pathways
PI3K/AKT/mTOR signaling pathway
Just as mentioned before, the PI3K/AKT/mTOR signaling pathway downstream the RTKs (EGFR, MET, HER2, HER3) plays a key role in promoting the proliferation, survival, drug resistance of cancer cells [53]. When an extracellular ligand binding to the receptor (such as EGF binding to EGFR) leads to phosphorylation and activation of the RTK, PI3K is triggered to catalyze the phosphorylation of phosphatidylinositol bisphosphate (PIP2) into phosphatidylinositol triphosphate (PIP3). Accumulation of PIP3 localizes AKT to the plasma membrane, where AKT gets phosphorylated by 3-phosphoinositide-dependent kinase 1 (PDK-1) indirectly or by PIP3 directly. The activated AKT regulates the phosphorylation of downstream effectors and thus leads to changes in gene expression and cell behavior. The major downstream effector is the mammalian target of rapamycin complex 1 (mTORC1), which regulates its downstream molecules such as the eIF4E-binding proteins (4E-BPs) and S6 kinases (S6K1 and S6K2) [54]. PI3K pathway regulation appears to be quite complicated. It’s negatively regulated by an important tumor suppressor, phosphatase and tensin homolog (PTEN), which converts active PIP3 to inactive PIP2, thus inactivating PI3K/AKT signaling. In addition, S6K can attenuate PI3K signaling via the S6K-IRS1 feedback loop and AKT phosphorylation via inhibiting mammalian target of rapamycin complex 2 (mTORC2) which is necessary for full AKT activation together with PIP3 [55]. Therefore, it’s rational to raise the hypothesis that there is a negative connection between the activation of this pathway and the susceptibility to EGFR-TKIs. Indeed, AKT activation and mTOR phosphorylation were frequently present in NSCLC patients (43-90% and 60-90%, respectively) [56]. They can be induced by multiple mechanisms, including AKT gene mutation, mutations and amplifications of PIK3CA (the gene encoding the main catalytic subunit of PI3K) as well as loss or reduced expression of PTEN [57-59]. Although the clinical data about the prevalence of PI3K/AKT/mTOR pathway induced EGFR-TKIs resistance is rare, the involvement of PI3K/AKT/mTOR pathway induced EGFR-TKIs resistance was repeatedly confirmed by preclinical researches. For instance, Jeffery A. Engelman et al. transfected a NSCLC cell line with P110α E545K, a PIK3CA oncogenic mutation, resulting in dramatically suppressed sensitivity to gefitinib compared with the control ones [60]. Horimasa Takeda et al. reported a 5-fold increased IC50 value of gefitinb in NSCLC cells when PTEN was knocked-down using a vector containing short heparin RNA against PTEN [61]. Reversely, reconstitution of PTEN can restore the tumor cell killing potency of TKIs [59].
JAK2/STAT3 pathway
The STAT (Signal Transducer and Activator of Transcription) protein, especially the STAT3, was reported to be another critical downstream signal transducer of activated EGFR besides the RAS/RAF/MAPK and PI3K/ARK [62]. Inappropriate activation of STAT3 was observed at high frequency (50%) in lung cancer patients [63]. Activated STAT3 acts as a transcriptional factor transferred into the nucleus and regulating the transcription of target-genes which mediate survival (survivin, bcl-xl, mcl-1, cellular FLICE-like inhibitory protein), proliferation (c-fos, c-myc, cyclin D1), invasion (matrix metalloproteinase-2), and angiogenesis (vascular endothelial growth factor) [64]. The activation of STAT3 is a complex event. It begins when the extracellular proteins such as cytokines (IL-6, interferon), growth factors (EGF, PDGF) bind to the corresponding receptors. The ligand bound receptor recruits and phosphorylates the tyrosine kinases JAK2 (JAK family: JAK1, JAK2, JAK3 and Tyk2. JAK2 is most frequently involved in oncogenesis), which eventually leads to STAT3 protein phosphorylation, dimerization and activation. In addition, Several other kinases can activate STAT3 directly without the mediation of JAK, such as the Src family (Lck, Src), Abl family (BCR-Abl), EGFR, IGF-1R, protein kinase C (PKC) and so on [65]. Apart from the remarkable role of JAK2/STAT3 in oncogenesis, clues have emerged recently that aberrant JAK2/STAT3 activation was partially involved in attenuated sensitivity of NSCLC to the first generation EGFR-TKIs. In an erlotinib resistant NSCLC cell line, the phosphorylation of STAT3 was remarkably increased although EGFR and MAPK were markedly suppressed by the existing erlotinib [66]. When adding the JAK inhibitor JSI-124 together with erlotinib, the sensitivity to erlotinib was restored both in vitro and in vivo [67]. However, abundance of relevant questions remain unrecognized and warrant for further investigation, such as the mechanisms underlying the aberrant JAK2/STAT3 activation, the safety and anti-tumor potency of JAK or STATs inhibitors, how to make the best of EGFR-TKIs via drugs-combination and so on.
SCLC phenotypic transforming
Although little is known about the phenotypic transforming of NSCLC histology to small cell lung cancer (SCLC), increasing attention has been drawn to this phenomenon. Lecia V. Sequist and her colleagues recently reported 5 in 37 (14%) NSCLC patients were diagnosed of SCLC after developing resistance to EGFR-TKIs. This SCLC transforming was confirmed with positive immunohistochemical staining in the post-resistant biopsy specimen for synaptophysin which specially exists in the SCLC [68]. Besides, genetic analysis showed the newly-emerged SCLCs harbored the same EGFR mutations with the original NSCLC ones, and this was surprising since it was an exceedingly rare event for SCLC to harbor EGFR mutations (about 4%) [68,69]. Actually, the SCLC transforming from NSCLC has been previously reported as a series of individual cases [70-72]. Those cases shared the same characteristics of never-smoking, female, transforming from adenocarcinoma to SCLC after developing TKI resistance. No exact mechanism underlying this phenomenon has been launched. Probably, SCLC cells originate from the minor pre-existent cells under the selection pressure of EGFR-TKIs, or trans-differentiate from the adenocarcinoma cells, or arise from the multi-potent stem cells [72]. Analyzing the genetic and phenotypic biopsy specimens all along the course of NSCLC diagnosis and treatment can help us for the better understanding this drug resistant mechanism, and more importantly, help us to make wiser clinical decisions throughout the course of the disease [68].
EMT phenotypic transforming
Epithelial to mesenchymal transition (EMT) refers to a complex program by which close-connected and polar-ranged epithelial cells turn into spindle-shape mesenchymal cells with significantly increased motility, invasiveness, and resistance to apoptosis [73]. Loss of epithelial cell junction proteins such as E-cadherin and the gain of mesenchymal markers such as vimentin or fibronectin are the distinctive molecular evens that are frequently accompanied with EMT [74]. Increasing evidences have emerged to correlate EMT with increased resistance of NSCLC cells to EGFR-TKIs, and oppositely, the NSCLC cells with lower degree of EMT shown sensitivity to EGFR-TKIs even without EGFR activating mutations both in vitro and in xenografts [75,76]. Clinically, EMT has been proven to contribute about 5% to EGFR-TKI resistance via biopsy paired specimens achieved pre- and post-resistance [68]. However, how EMT promotes TKI resistance remains unknown. Nevertheless, some common mechanisms such as EGFR T790m mutation or MET amplification are unlikely the culprit.
Strategies to reverse TKI resistance
Knowledge of mechanisms underlying the drug resistance keeps updating and this consequently stimulates the development of diverse new drugs and combinational regimens to overcome EGFR-TKIs resistance. For the next part, we will focus on those newly emerged drugs and regimens in terms of their action modes as well as their anti-tumor potency both in vivo and in vitro (Figure 2 and Table 1).
Figure 2.
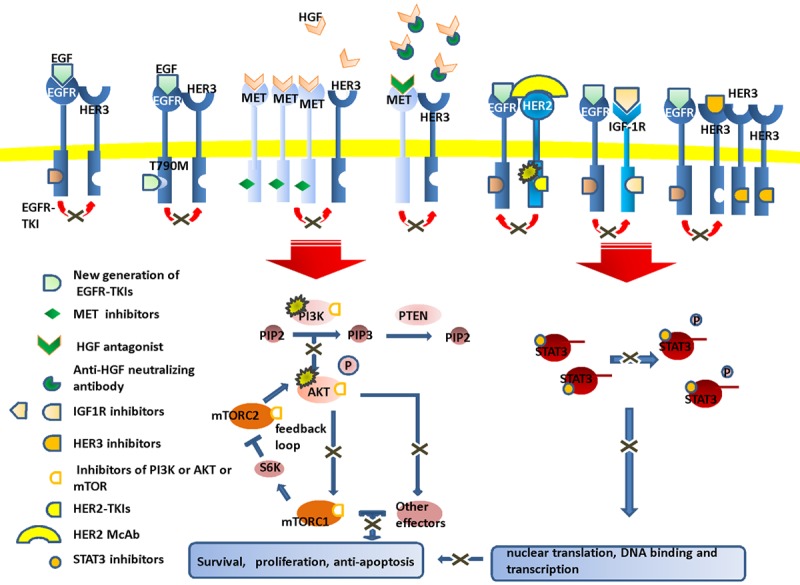
New agents to overcome EGFR-TKI resistance. To cope with the molecular events responsible for the resistance, multiple targeted agents are developed including the new-generation EGFR-TKIs, MET/HGF inhibitors, HER2 inhibitors, HER3 inhibitors, IGF-1R inhibitors, PIK3CA/AKT inhibitors and STAT3 inhibitors.
Table 1.
Summary of the targeted agents and clinical trials to overcome resistance to EGFR-TKI
| Target | classification | Agents | Clinical trials finished | Clinical trials ongoing |
|---|---|---|---|---|
| T790M | Covalent EGFR-TKI inhibitor | Afatinib | LUX-lung 1 [78], LUX-lung 2 [163], LUX-lung 3 [80], LUX-lung 4 [79], LUX-lung 6 [81] | NCT01085136, NCT01466660, NCT01523587 |
| T790M-selective inhibitor | CO-1686, PKC412, AZD9291 | Phase I studies of PKC412 [89,164] | NCT01526928, NCT02147990 (TIGER-2), NCT01802632 (AURA), NCT02094261 (AURA2) | |
| HGF | HGF antagonist | NK4 | ||
| Anti-HGF antibody | TAK-701, ficlatuzumab | Phase I study of TAK 701 [94] | NCT01039948 | |
| Phase II trial comparing gefitinib with and without ficlatuzumab [165] | ||||
| MET | MET tyrosine kinase inhibitors | Tivantinib, cabozantinib, INC280 | Phase II study comparing erlotinib with or without tivantinib [97] | NCT01580735, NCT02049060, NCT01377376, NCT00596648, NCT01866410, NCT01639508, NCT01610336 |
| Phase III study comparing erlotinib with and without tivantinib [29] | ||||
| MET mono-antibody | Onartuzumab | phase II trial comparing erlotinib with and without onartuzumab [166] | NCT01456325 (MetLung) NCT01887886, NCT02031744 | |
| HER3 | Anti-HER3 agents | Pertuzumab | Phase II study exploring pertuzumab plus erlotinib [107] | |
| HER2 | Anti-HER2 agents | Lapatinib, trastuzumab, neratinib | Phase II trial evaluating lapatinib as monotherapy [167] | NCT01827267, NCT01184482, NCT01306045, NCT00004883, NCT00758134, NCT01827267, NCT00266877 |
| Phase II trial evaluating neratinib [168] | ||||
| IGF-1R | IGF-1R mono-antibody | R1507 | Phase II study exploring erlotinib plus placebo or R1507 [169] | NCT00773383 |
| PI3K | PI3K inhibitors | BAY 80-6946, LY294002 | NCT01460537, NCT01411410, NCT01404390, NCT00962611 | |
| mTOR | mTORC1 inhibitors | Everolimus, temsirolimus, sirolimus | Phase II exploring everolimus plus gefitinib [111] | NCT00079235, NCT01827267, NCT01737502, NCT01050985, NCT01482156 |
| Phase II study of temsirolimus monotherapy [170] | ||||
| Dual PI3K/mTOR inhibitors | BEZ235, PF-04691502, PKI-402 | Phase I study of PF-04691502 monotherapy [122] | NCT01482156, NCT00620594, NCT01343498, NCT01508104 | |
| AKT | AKT inhibitors | MK-2206, Enzastaurin, | Phase I trial exploring MK-2206 plus carboplatin/paclitaxel, docetaxel or erlotinib [124] | NCT01147211, NCT01294306, NCT00452413 |
| Phase I trials exploring enzastaurin plus gemcitabine/cisplatinin [126] | ||||
| Phase II study, exploring enzastaurin plus cisplatin/pemetrexed [127] | ||||
| HSP90 | Hsp90 inhibitors | Ganetespib, AUY-922, DS-2248, retaspimycin, 17-DMAG | Phase II study evaluating IPI-504 monotherapy [140] | NCT01348126, NCT01031225, NCT01259089, NCT01784640, NCT01288430, NCT01362400, NCT01427946 |
| Phase II study evaluating ganetespib monotherapy [141] |
Targeting EGFR T790M
Secondary generation EGFR-TKIs
In view of the fact that resistant tumor cells are still addicted to the EGFR signaling pathway, new drugs which can irreversibly block EGFR-TK via the formation of covalent bonds in the pocket of the catalytic site should be able to increase the potency of EGFR-TK inhibition. One such inhibitor, the second generation EGFR-TKI afatinib (BIBW2992), designed to bind covalently with Cys-797 at the gatekeeper pocket, can potently and selectively block both wild-type and mutant forms of ErbB family receptors (EGFR, HER2, ErbB3 and ErbB4) [18]. It has been confirmed of a sustained and potential antineoplastic activity in a battery of cell lines and xenograft models. Compared with erlotinib, gefitinib or lapatinib, it showed 100-fold more sufficient ability to inhibit the enzymatic activity of the L858R-T790M EGFR and comparable potency against HER2 [77]. Therefore, a series of clinical trials have been conducted to evaluate clinical benefits of afatinib. LUX-lung1 explored afatinib versus placebo in NSCLC patients who failed in prior treatment with at least 1 line of platinum-based chemotherapy and disease progressed after at least 12 weeks of treatment of erlotinib or gefitinib. Among them, 83% were EGFR-mutant positive, including those progressed after a short complete response (CR)/partial response (PR) to prior EGFR-TKIs and/or were treated for ≥48 weeks with prior EGFR-TKIs. Although the primary endpoint overall survival (OS) showed no benefit from afatinib (afatinib versus placebo: 10.8 months versus 12.0 months; HR 1.08, 95% CI 0.86-1.35, p=0.74), the afatinib group experienced a prolonged progress-free survival (PFS) (3.3 months versus 1.1 months; HR 0.38, 95% CI 0.31-0.48; p<0.0001), better confirmed objective response rate (ORR) (7% versus <1%), better disease control rate for ≥8 weeks (58% versus 18%) and improved life quality in the overall population than that of placebo. Especially in the subgroup of patients meeting the Jackman criteria of acquired resistance to EGFR-TKI, afatinib showed a pronounced PFS benefit over placebo (4.5 months versus 1.0 months) [78]. This encouraging result was further confirmed by LUX-lung 4, which was conducted in patients failed prior erlotinib and/or gefitinib [79]. Furthermore, first-line afatinib has been compared with standard combination chemotherapeutic regimens in LUX-lung 3 (afatinib versus combination chemotherapy of cisplatin and pemetrexed) and LUX-lung 6 (afatinib versus combination chemotherapy of cisplatin and gemcitabine) [80,81]. The pooled analysis of those two large open-label phase III studies announced in the 2014 ASCO Annual Meeting proved the favorable anti-tumor activity of afatinib. Median OS was prolonged from 24.3 months in chemotherapy group to 27.3 months in the afatinib group (HR=0.81; CI 0.66 to 0.99; p=0.037). Especially in the Del19 subgroup, the HR was 0.59 (CI 0.45 to 0.77; p<0.001), preferable for the afatinib group [82]. Besides, LUX-lung 7 and LUX-lung 8 directly comparing afatinib with gefitinib or erlotinib are still recruiting patients.
Despite of the benefits on the T790M-positive NSCLC patients achieved from the utility of second generation EGFR-TKIs, the improvement seems to be rather limited. It is largely caused by the insufficient drug concentrations as the toxicity of this drug limits the blood concentrations under the level required to overcome the EGFR T790M mutation [83]. In the clinical trial of LUX-lung 1, adverse events such as skin rash, acne and diarrhea were observed with at least 10% higher incidence than the placebo group, including 150 in 390 (38%) patients needed a dose reduction and 70 (18%) patients discontinued afatinib because of those adverse events [78]. The mechanism underlying goes to the potent blockage of afatinib against both the wild-type EGFR and the mutant ones [77].
Third generation of EGFR-TKIs
In view of this, the third generation EGFR-TKIs that selectively target the mutant EGFR, in particular the T790M mutation, but exhibiting minimal potency toward the wild-type receptor emerged in quick succession. CO-1686 is one of them and exhibits potent inhibition of EGFR T790M but circumvents wild-type EGFR [84]. The phase I/II clinical trial evaluating the safety, pharmacokinetic and preliminary efficacy in patients with previously treated EGFR mutant NSCLC of CO-1686 has been half done (NCT01526928). Some promising discoveries have been released in the 4th European lung cancer conference (ELCC) in 2014. It reported overall response rate (ORR) of 80%, PFS of over 6 months in T790M-positive NSCLC patients who administered CO-1686 following the development of resistance to erlotinib. Another phase II study exploring CO-1686 as a second-line therapy in the T790M mutant patients who failed the previous EGFR-TKI treatment, TIGER-2, just started recruiting patients this year (NCT02147990). WZ4002 is another mutation-selective EGFR inhibitor that displays high degree of selectivity against EGFR T790M even at low concentrations, and no significant inhibition of wild-type EGFR was accompanied in preclinical studies [85]. Several studies are further evaluating its anti-EGFR T790M activity and one of them showed pronounced anti-tumor capacity and tolerable toxicity in NSCLC bearing mice when combined with MET inhibitor crizotinib [86]. However, no study assessing WZ4002 in human beings is available yet. Midostaurin (PKC412), the multi-target tyrosine kinase inhibitor (targeting PKC, FLT3, AKT, c-kit, PDGFR et al.), is deeply studied and widely utilized to treat patients with AML or MDS [87]. It has been proven in T790M mutation positive NSCLC cell lines and tumor models of a novel function of potent and selective inhibition of EGFR T790M other than FLT3. Meanwhile, it showed no significant blockage of wide-type EGFR activity which means much less toxicity and better tolerance of PKC412 versus first- and second-generation TKIs [83]. Clinical evaluation of PKC412 as a PKC, FLT3 inhibitor against AML and MDS is under investigation. No obviously toxicity against normal cells was observed despite of its multi-targeting capacity [88]. A phase I trial conducted in 23 patients with advanced NSCLC showed good effectiveness of PKC412 at the dosage of 50 mg/day combined with gemcitabine and cisplatin and no significant side reactions including myelosuppression were observed [89]. However, further studies are warranted to assess its efficacy in overcoming T790m induced TKI resistance.
Targeting HGF-MET pathway
HGF antagonist
NK4, composed of the N-terminal hairpin and amino-terminal four kringle domains of hepatocyte growth factor (HGF), acts as the competitive antagonist of HGF as well as an angiogenesis inhibitor against VGFR and FGF. The two functions of NK4 are mutually independent but interactional with each other, making it potential in suppressing malignant tumors in both tumor growth and spreading [90,91]. NK4 over-expressed in the established lung cancer xenograft models dramatically suppressed the tumor growth, angiogenesis, and metastases without obvious side effects [92]. However, the relevance of NK4 to overcome TKI resistance is not yet investigated.
Anti-HGF neutralizing antibody
TAK-701 is a potent humanized monoclonal antibody to HGF. It works by suppressing the HGF binding to MET receptor and thus restrains the proliferation effects of MET pathway. Wataru Okamoto et al. treated the HGF overexpression induced TKI- resistant NSCLC cells with TAK-701 and gefitinib and observed significant suppression on the activation of MET, ERK, AKT and cell growth [93]. This promising outcome indicated that the addition of TAK-701 to gefitinib was a feasible strategy to abrogate EGFR-TKI resistance induced by HGF. NCT00831896 is the first clinical trial to determine the safety, tolerability, and pharmacokinetics profile of TAK-701 in adult patients with advanced non-hematologic malignancies. It showed good tolerance at the dosage of up to 20 mg/kg every other week and preferable negative conversion ratio of free HGF (71.4%) [94].
MET tyrosine kinase inhibitors
Tivantinib is a non-ATP-competitive small molecule MET inhibitor. It works by stabilizing the inactive conformation of MET, and thus hinders the activation of downstream signaling pathway. Given the well-tolerance and potential ability of tivantinib both as single-agent therapy and in combination with erlotinib announced in several preclinical and phase I clinical trials, a series of work have and are being carried out to evaluate its antitumor efficacy [95,96]. An international randomized phase II study conducted on 167 NSCLC patients suggested benefits from the combination of tivantinib and erlotinib, compared with erlotinib alone (PFS 3.8 months versus 2.3 months; adjusted HR 0.68, 95% CI 0.47-0.98, p=0.04). Especially in the patients of non-squamous histology, a subgroup with higher possibility to be MET-positive (75% in non-squamous histology versus 12% in squamous histology), a significant prolongation has been demonstrated in median PFS (4.4 months versus 2.3 months, adjusted HR 0.61, 95% CI 0.47-0.98, p<0.05) and median OS (9.9 months versus 6.8 months; adjusted HR 0.58, 95% CI 0.34-0.99, p<0.05) [97,98]. Based on this encouraging result, a phase III randomized study of erlotinib plus tivantinib in pretreated but TKI-naive advanced non-squamous NSCLC patients was carried out [98]. Unfortunately, this trial was recommended to cease since the interim analysis shown the achieved PFS benefit could not carry over into OS prolongation [29]. Anyway, a variety of clinical trials alike are underway, including a phase II single-arm study (NCT01580735) investigating tivantinib plus erlotinib in EGFR-TKI resistant locally advanced or metastatic NSCLC subjects and a phase III randomized placebo-controlled study (NCT01377376) exploring tivantinib plus erlotinib versus erlotinib monotheray in advanced and EGFR mutation negative non-squamous NSCLC. Cabozantinib (XL-184) is a novel tyrosine kinase inhibitor that is under evaluation in preclinical and clinical studies. The characteristics of inhibiting MET and VEGFR-2 make it the powerful drug in inhibiting tumor growth and survival, as well as blood vessel formation, invasiveness and metastasis [99]. It also displays inhibitory activity against several other RTKs including RET, KIT, FLT3 and TEK, which are all critical molecules in oncogenesis [100]. Several studies preliminarily explored the potential of cabozantinib combining EGFR-TKI in gefitinib- or erlotinib-resistant NSCLC cell lines in vitro and xenograft tumors in vivo. The outcomes were quite encouraging since extensive tumor shrinkage and decreased tumor invasiveness and metastasis were observed by this combinational regimen [99,100]. The results of a phase I/II randomize clinical trial (NCT00596648) assessing cabozantinib, either alone or in combination with erlotinib in acquired resistant NSCLC patients are expected in the near future. Recently, a novel MET inhibitor, INC280 caught our attention. In the 2014 ASCO Annual Meeting, Yi-Long Wu et al. disclosed the latest phase Ib results of the ongoing phase Ib/II study exploring INC280 plus gefitinib in NSCLC patients who were EGFR mutated, MET-amplified and failed in the prior EGFR inhibitor treatment. 6 in 41 (15%) evaluable patients obtained partial responses and all those responders had high MET status [101]. The phase II trial to further verify the efficiency of this combination regimen is currently ongoing (NCT01610336).
MET monoclonal antibody
Onartuzumab (MetMAb) is a newly developed humanized monoclonal antibody targeting MET. It blocks the HGF binding to MET, and thus attenuates the activation of its downstream transducers and effectors [102]. It was evaluated in a randomized phase II trial comparing erlotinib with and without onartuzumab in advanced NSCLC patients. Despite of no statistically significant differences between the two arms in the overall population, the combinational arm showed 47% reduction in the risk of disease progression and significant prolongation in median PFS (2.9 months versus 1.5 months; HR 0.53, 95% CI 0.283-0.99, p=0.04) and median OS (12.6 months versus 3.8 months; HR 0.37, 95% CI 0.19-0.72, p=0.002) in the subset of MET-positive, which was confirmed to be associated with bad prognosis. In contrast, MET-negative patients experienced earlier progression in the combinational arm (median PFS loss of 1.3 months; HR 1.82, 95% CI 0.99-3.32, p=0.05) and shorter survival (median OS loss of 7.2 months; HR 1.78, 95% CI 0.79 to 3.99, p=0.16), indicating its potential to reverse or prevent MET induced TKI resistance [103]. Unfortunately, the inspiring outcome could not carry over to the phase III study (MetLung, NCT01456325), which enrolled 499 MET-positive advanced NSCLC patients. It was recommended by an independent data review committee to cease since no improved OS (6.8 months versus 9.1 months; HR 1.27, p=0.068), PFS (2.7 months versus 2.6 months; HR 0.99, p=0.92), and overall response rate (8.4% versus 9.6%; p=0.63) was observed in the onartuzumab plus erlotinib arm [104].
Targeting HER3 pathway
Anti-HER3 agents
Several measures can overcome acquired resistance induced by HER3 activation. One strategy is to increase the dosage of EGFR-TKIs to fully inactivating concentration or develop more potent anti -HER drugs since studies have convicted TKI-refractory HER3 phosphorylation relays on unsuppressed HER2 [52]. Another strategy is to develop anti-HER3 agents blocking HER3 activation, such as antibodies against HER3 [105]. Pertuzumab is a novel HER2/HER3 dimerization inhibitor [106]. The outcome of a phase II study exploring the combination of pertuzumab and erlotinib in 41 relapsed NSCLC patients was published recently. In contrast to the original intention of greater activity as a result of a broader HER family blockage, this combination regimen shown only modest anti-tumor potency but generally poor tolerance [107]. Thus, until more powerful anti-HER drugs are developed, multi-targeting treatment by combining EGFR-TKI with inhibitors on other targets such as mTOR or PI3K is still the first choice [49].
Targeting PI3K/AKT/mTOR pathway
PI3K inhibitors
BAY 80-6946, developed as an intravenous drug, can inhibit pan-class I PI3K with high potency and selectivity. Sustained response was observed in animal models bearing patient-derived NSCLC xenografts when co-treated with BAY 80-6946 and paclitaxel [108]. Till now, at least 4 phase I studies are further evaluating the clinical value of BAY 80-6946 in multiple advanced tumors including NSCLC (NCT01460537, NCT01411410, NCT01404390, NCT00962611).
mTORC1 inhibitors
Rapamycin and its analogues are the most developed inhibitors targeting PI3K/AKT/mTOR pathway. Everolimus (RAD001), an orally bioavailable rapamycin derivative, is a potent inhibitor of mTORC1. It showed promising efficacy in restoring sensitivity of gefitinib resistant NSCLC cell lines which harbor PIK3CA mutation or lose PTEN [109,110]. Clinical studies have been carried out to assess the efficacy of combining everolimus with EGFR-TKIs or chemotherapies. In contrast to the promising outcomes of preclinical trials, the efficacy of those combination regimens in NSCLC patients seem rather modest. A phase II combination study conducted by Katharine A. Price et al in EGFR-TKI naive patients with advanced NSCLC only witnessed response rate of 13% (8 of 62 patients), which did not meet the prespecified response threshold to pursue further study of combining everolimus and gefitinib [111]. Another phase I study carried out by Jean-Charles Soria et al showed better disease-control rate (DCR), CR or PR rate, median duration of stable disease in the group of advanced NSCLC patients received with the combination of everolimus and erlotinib (50%, 12%, 9.3 months) as second- or third-line therapy than in patients with erlotinib alone (45%, 8.9%, 7.9 months) [112]. This result suggests that the combination treatment with erlotinib and everolimus may be superior to treatment with erlotinib alone in the molecularly unselected population. Many trials on various combinations are ongoing, such as everolimus plus cetuximab, everolimus plus capecitabine and everolimus plus cisplatin, in the hope of finding more effective treatment combinations [113-115].
Of note, the success achieved by rapamycin and its analogues, such as everolimus appears to be limited, and many preclinical trials have noticed the abnormal AKT phosphorylation even in the existence of those agents [116]. This phenomenon can be explained by their nature of merely inhibiting mTORC1, which leaves AKT constitutive activated via the abrogation of S6K-IRS1-PI3K feedback loop or via mTORC2 [55,116]. Thus, it’s rational to develop new agents that target both mTORC1 and mTORC2, or novel regimens of combining different kinds of PI3K/AKT/mTOR inhibitors to maximize pathway inactivation and overcome TKI resistance.
Dual PI3K /mTORC1 /mTORC2 inhibitors
The inhibitors co-targeting PI3K /mTORC1 /mTORC2 were designed on the basis of high-level structural homology of the catalytic site of PI3K and mTOR. Their nature of dual inhibition makes them superior than mere mTORC1 inhibitors by attenuating PI3K/mTORC2 induced AKT activation. They strongly reduced the proliferation rate as well as inducing a dramatic apoptotic response [117]. Although the dual PI3K/mTORC1/mTORC2 targeting compounds are still in early development, pronounced anti-tumor capacity have been observed in many preclinical researches. BEZ235, a novel ATP-competitive PI3K/mTOR dual inhibitor, has displayed striking anti-proliferative effects in HGF induced EGFR resistant lung cancer cell lines and a xenograft model even as monotherapy [118]. Furthermore, when BEZ235 was combined with everolimus, marked synergy was achieved in the inhibition of NSCLC cell growth both in vitro and in vivo [119]. BEZ235 is under investigation in phase I/II trials either by itself alone or in combination with other agents in patients with various types of cancer including NSCLC (NCT01482156, NCT00620594, NCT01343498, NCT01508104 et al). Since inhibition of mTOR signaling can induce autophagy to enable cell survival under unfavorable conditions, combining autophagy-blocking drugs may enhance the anti-tumor capacity of BEZ235 [120]. PF-04691502 is another dual PI3K and mTOR inhibitor. It has entered phase I clinical trials as it pronouncedly reduced AKT phosphorylation in PTEN null or PIK3CA mutated cells, and induced tumor shrinking in NSCLC xenografts resistant to first-generation EGFR-TKI [121]. According to the phase I clinical study in patients with advanced solid tumors, oral administration of PF-04691502 reduced phosphorylated AKT and STAT3, although no objective responses were observed [122].
AKT inhibitors
AKT is the critical downstream effector of PI3K. Aberrant phosphorylation of AKT signaling results in reduced sensitivity of NSCLC to receptor TKIs. MK-2206, an oral AKT inhibitor, is undergoing varies of in vitro and in vivo studies. The co-treatment with MK-2206 and erlotinib has shown a synergistic effect in erlotinib-insensitive NSCLC cell lines and tumors [123]. The combination effects could be explained by the blockage of both AKT and ERK pathways. On this background, a phase I trial was carried out to evaluate the effect of MK-2206 plus carboplatin and paclitaxel, docetaxel or erlotinib on patients with advanced solid tumors including 13 NSCLC patients. Well-tolerance was observed as well as early evidence of antitumor activity. One NSCLC patient treated with MK-2206 and docetaxel after 2 prior lines of platinum-based chemotherapy and erlotinib treatment obtained additional partial response (PR) [124]. Based on those promising outcomes, following trials are underway to further investigate the combination of MK-2206 with other standard cytotoxic or targeted treatments (NCT01147211, NCT01294306). Enzastaurin is initially developed as an anti-tumor agent via anti-angiogenesis activity. But later it was proven to directly suppress tumor cell proliferation and induce tumor cell death owing to the newly discovered potential of blocking AKT pathway [125]. Enzastaurin has been investigated in a phase I clinical trials in combination with gemcitabine and cisplatinin in patients with advanced solid tumors. This regimen was well-tolerated and resulted in clinical benefits in 16 out of 33 patients [126]. In a phase II randomized study, the combining regimen of cisplatin/pemetrexed plus enzastaurin was evaluated in patients with advanced NSCLC. Despite of the pleasant outcome of 7 PR and 2 SD in 13 patients in the lead-in phase, this clinical trial was suspended due to the negative result of the other two peer phase II studies [127].
Inhibiting epithelial-mesenchymal transition
Histone deacetylase (HDAC) inhibitors
Since the loss of E-cadherin is the specific event of EMT, and has been associated with poor clinical outcome as well as bad response to EGFR-TKIs in NSCLC, attempts have been made to restore the E-cadherin expression using histone deacetylase (HDAC) inhibitors. Entinostat (MS-275) is one of such drugs. It succeeded in restoring E-cadherin expression, growth-inhibitory and apoptosis-promoting effects of erlotinib and gefitinib preclinically [128]. Furthermore, a randomized phase II trial evaluating the combination of erlotinib with entinostat, a kind of oral HDAC inhibitor, in advanced NSCLC patients who failed in prior chemotherapy have completed with acceptable adverse events and prolonged OS (9.4 months versus 5.4 months; HR 0.3, 95% CI 0.13 to 0.92, p=0.03) as well as PFS (3.68 months versus 1.88 months; HR 0.55, 95% CI 0.22 to 1.37, p=0.19) in the subset of patients with high E-cadherin levels [74]. Certainly, HDAC inhibitors can activate the expression of many silenced tumor suppressors, leading to tumor inhibition independent of EMT reversion [129]. In addition, the molecular events underlying EMT are so complicated that more breakthroughs are needed to develop EMT-targeting drugs overcoming TKI-resistance.
Newly emerged multi-targeted agents
HSP90 inhibitors
The heat shock protein 90 (Hsp90) is a kind of molecular chaperones responsible for the conformational maturation and stabilization of its substrate proteins [130]. Elevated levels of Hsp90 have been demonstrated in NSCLCs and they protected the tumor cells against unfavorable conditions via stabilizing proteins necessary for tumor survival [131]. Multiple well-known oncogenetic drivers and drug-resistance causing proteins of NSCLC including EGFR, HER2, MET and AKT are the substrates of Hsp90 [132,133]. When one of them is blocked, the tumor cells are smart enough to signal through the alternative kinases via oncogenic switching frequently induced by Hsp90. Furthermore, the mutated or overexpressed forms of those oncogenetic proteins preferentially are more dependent on Hsp90 to remain stable than their wild-type counterparts [133]. All mentioned above suggest the increasing virtue of Hsp90 inhibitors as multi-targeted agents in the treatment of oncogene addicted NSCLC [134]. Multiple preclinical NSCLC models containing one or more mutant oncogenes have been established in vitro and in vivo. Following the exposure to HSP 90 inhibitors, the expression of these oncogenetic proteins were compromised and cell growth was abrogated [135-137]. Several newly-developed Hsp90 inhibitors such as STA-9090 (Ganetespib), AUY-922, DS-2248, IPI-504 (retaspimycin) and 17-DMAG are in active clinical evaluation either as monotherapy or in combination with other agents (NCT01348126, NCT01031225, NCT01259089, NCT01784640, NCT01288430, NCT01362400, NCT01427946 et al)[138-141].
Targeting miRNAs
miRNAs are a class of 18-24 nt small noncoding RNAs that negatively regulate the target genes expression either by inhibiting mRNA translation or by promoting mRNA degradation [142]. Emerging evidences have suggested their master regulatory roles in oncogenesis either as oncogenes or as tumor suppressor genes [143]. In NSCLC, upregulated miRNA30b, miRNA30c, miRNA221, miRNA222 are associated with resistance to gefitinib treatment through the regulation of PTEN and APAF-1 expression, while miRNA103 and miRNA203 induce apoptosis in gefitinib resistant cells and promote mesenchymal to epithelial transformation via the down-regulation of PKC-ε, SRC and Dicer [144]. Since miRNA expression in gefitinib and erlotinib resistant NSCLC cell lines were profiled, various relevance of miRNAs to TKI resistance have been gradually clarified (Table 2). Particularly, miRNAs typically target a cluster of genes rather than one specific gene, modulating miRNAs by introduction of suppressive miRNAs or inhibition of oncogenetic miRNAs might bring a powerful therapeutic strategy to overcome EGFR-TKIs resistance [143]. This idea is not inconceivable since studies in vivo have succeeded in delivering anti-sense oligonucleotides against targeted miRNAs into laboratory animals with up-regulated levels of corresponding miRNAs [145,146]. In addition, new technologies able to raise the suppressive miRNA levels in tumor cells via introducing synthetic miRNA mimics keep arising one after another [147]. Besides, oncogenetic miRNAs and suppressor miRNAs can be either down-regulated or up-regulated by a much safer approach using several “natural agents” such as isoflavone, 3,3’-diinodolylmethane (DIM) [148]. Although several key issues of this approach remains unsolved such as the delivery methods, stability and safety in humans, miRNA-based treatment will finally take their place in the battle against EGFR-TKIs resistance.
Table 2.
Summary of miRNAs involved in EGFR-TKI resistance in NSCLC
| miRNA name | Expression in resistant NSCLC | Targets | References |
|---|---|---|---|
| miR-21 | Up | PDCD4, MDR1, PTEN and bcl-2 | [171-173] |
| miR-103 and -203 | Down | PKC-ε, SRC, Dicer. | [174,175] |
| miR-126 | Down | PIK3R2, CRK, VEGF | [176-178] |
| miR-128b | Down | EGFR | [179] |
| miR-214 | Up | PTEN, p38, MAPK | [180,181] |
| miR-221, miR-222 | Up | PTEN, APAF-1, BIM, TIMP3 | [174,182] |
| MiR-145 | Down | c-MYC, AKT, ERK, OCT4 | [183-185] |
Conclusions and perspectives
EGFR activating mutations have been overoptimistically recognized as the Achilles’ heel of NSCLC after some clinical successes have been achieved. Unfortunately, almost all patients initially responding to gefitnib or erlotinib would inevitably progress to develop acquired resistance. To our relief, the constantly updating knowledge of the mechanisms underlying has already been translated to the development of novel targeted agents, rational combination regimens and improved survival benefits to NSCLC patients.
In order to make use of these molecular tools to full extend, many hurdles lying ahead must be overcome. Firstly, there remains the requirement of exploring unknown mechanisms of resistance since nearly 30% of the acquired resistance cannot be explained by the mechanisms recognized. New potential mechanisms are keeping proposed such as CRKL gene amplification, AXL kinase over-activation, acquisition of stem cell-like properties, loss of an EGFR-amplified chromosome 7 [149-154]. Certainly, they need to be confirmed in clinical researches with large sample sizes before the development of novel drugs targeting these synthetically lethal vulnerabilities. Secondly, too many failures in the battle against NSCLC have told us that the tumors are much smarter than we thought. They outsmart single-target drugs to escape from the fate of death by various mechanisms. And it is not rare to see several different resistant mechanisms existing in one patient synchronously or continuously. In regard of that, the exploration of potential and well-tolerated combinational regimens and novel “one target multiple” drugs should be the hotspots of future drug development. Meanwhile, the profiles of different EGFR-TKI resistant subgroups classified by different mechanisms should be described to provide individualized anti-tumor regimens such as combination of various drugs targeting different targets. Thirdly, biopsy specimen for the genetic profiles of cancer patients should be performed all along the course of the disease since resistant clones can emerge up to 10 months before radiological changes [155]. An early discovery of new genetic aberrations in NSCLC patients such as T790M mutation, PI3KCA mutation or MET amplification makes it possible for patients to cease expensive and toxic EGFR-TKIs treatment regimens and start potentially successful changes in accordance to real-time genetic status as soon as possible. The traditional tissue biopsy is not always accessible, and only one third patients enrolled in the IPASS trial were available for EGFR-mutation status examination [10]. In addition, single biopsy is sometimes impossible to represent heterogeneous landscape of the tumor [156]. “Liquid biopsy” screening for circulating tumor DNA (ctDNA) within a blood sample drawn from NSCLC patients has emerged and is under active evolution to reflect tumor genetics and tumor dynamics [156-158]. T790M mutation, KRAS or BRAF mutations, PIK3CA mutation, HER2 gene amplification have already been successfully detected in NSCLC, colorectal cancer or breast cancer patients by this technology [159-162]. The information of all tumor sites provided by the “Liquid biopsy” will surely better monitor the EGFR-targeted therapies although many critical obstacles are lying ahead. Nevertheless, the battle against acquired resistance to EGFR-TKI in EGFR-mutant NSCLC has come a long way, although there is still a long way ahead.
Acknowledgements
This work was supported by Ministry of Education (20110101110137), National Natural Science Foundation of China (81372178), Natural Science Foundation of Zhejiang Province (LR12H16001), and 973 project (No. 2012CB526600).
Disclosure of conflict of interest
We have no conflict of interest to claim.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Chunhacha P, Chanvorachote P. Review Article Roles of caveolin-1 on anoikis resistance in non small cell lung cancer. Int J Physiol Pathophysiol Pharmacol. 2012;4:149–155. [PMC free article] [PubMed] [Google Scholar]
- 3.Klastersky J, Awada A. Milestones in the use of chemotherapy for the management of non-small cell lung cancer (NSCLC) Crit Rev Oncol Hematol. 2012;81:49–57. doi: 10.1016/j.critrevonc.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 5.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okabe T, Okamoto I, Tamura K, Terashima M, Yoshida T, Satoh T, Takada M, Fukuoka M, Nakagawa K. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 2007;67:2046–2053. doi: 10.1158/0008-5472.CAN-06-3339. [DOI] [PubMed] [Google Scholar]
- 7.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 8.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 9.Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, Chao TY, Nakagawa K, Chu DT, Saijo N, Duffield EL, Rukazenkov Y, Speake G, Jiang H, Armour AA, To KF, Yang JC, Mok TS. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J. Clin. Oncol. 2011;29:2866–2874. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 10.Wu YL, Fukuoka M, Mok TS, Saijo N, Thongprasert S, Yang JC, Chu DT, Yang JJ, Rukazenkov Y. Tumor response and health-related quality of life in clinically selected patients from Asia with advanced non-small-cell lung cancer treated with first-line gefitinib: post hoc analyses from the IPASS study. Lung Cancer. 2013;81:280–287. doi: 10.1016/j.lungcan.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Muñoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica. Erlotinib versus standard chemotherapy as fi rst-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 12.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 13.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, Shimizu E, Saito H, Toyooka S, Nakagawa K, Fukuoka M West Japan Oncology Group. Gefiinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–28. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 14.Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, Li LY, Watkins CL, Sellers MV, Lowe ES, Sun Y, Liao ML, Østerlind K, Reck M, Armour AA, Shepherd FA, Lippman SM, Douillard JY. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372:1809–1818. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- 15.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 16.Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, Lynch T, Johnson BE, Miller VA. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.KobayashI S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 18.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, Kris MG, Pao W, Miller VA, Ladanyi M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, Fujita S, Kaji R, Imai Y, Monden K, Matsumoto T, Nagata K, Otsuka K, Tachikawa R, Tomii K, Kunimasa K, Iwasaku M, Nishiyama A, Ishida T, Nishimura Y. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119:4325–4332. doi: 10.1002/cncr.28364. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Sakai K, Nakamura T, Matsumoto K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol. 2011;26(Suppl 1):188–202. doi: 10.1111/j.1440-1746.2010.06549.x. [DOI] [PubMed] [Google Scholar]
- 22.Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J. Clin. Oncol. 2011;29:4837–4838. doi: 10.1200/JCO.2011.37.7929. [DOI] [PubMed] [Google Scholar]
- 23.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T, Yatabe Y, Nakamura T, Sone S. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 24.Onitsuka T, Uramoto H, Nose N, Takenoyama M, Hanagiri T, Sugio K, Yasumoto K. Acquired resistance to gefitinib: the contribution of mechanisms other than the T790M, MET, and HGF status. Lung Cancer. 2010;68:198–203. doi: 10.1016/j.lungcan.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 25.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M, Toschi L, Finocchiaro G, Destro A, Terracciano L, Roncalli M, Alloisio M, Santoro A, Varella-Garcia M. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 28.Krishnaswamy S, Kanteti R, Duke-Cohan JS, Loganathan S, Liu W, Ma PC, Sattler M, Singleton PA, Ramnath N, Innocenti F, Nicolae DL, Ouyang Z, Liang J, Minna J, Kozloff MF, Ferguson MK, Natarajan V, Wang YC, Garcia JG, Vokes EE, Salgia R. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res. 2009;15:5714–5723. doi: 10.1158/1078-0432.CCR-09-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39:793–801. doi: 10.1016/j.ctrv.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Cipriani NA, Abidoye O, Vokes E, Salgia R. MET as a target for treatment of chest tumors. Lung Cancer. 2009;63:169–179. doi: 10.1016/j.lungcan.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin Sattler RH, Mamatha M. Reddy, Tara Gangadhar and Ravi Salgia. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol. 2011;3:171–184. doi: 10.1177/1758834011408636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, Nokihara H, Ohe Y, Yokota J, Uramoto H, Yasumoto K, Kiura K, Higashiyama M, Oda M, Saito H, Yoshida J, Kondoh K, Noguchi M. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–6638. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 34.Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32:3846–3856. doi: 10.1038/onc.2012.396. [DOI] [PubMed] [Google Scholar]
- 35.Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C, Hirsch FR. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol (Dordr) 2013;36:277–288. doi: 10.1007/s13402-013-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J Biol Chem. 2002;277:49127–49133. doi: 10.1074/jbc.M207584200. [DOI] [PubMed] [Google Scholar]
- 37.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–10111. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 38.Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, Christensen JG, Wong KK, Gray NS, Janne PA. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73:834–843. doi: 10.1158/0008-5472.CAN-12-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, Zakowski MF, Frankel S, Peifer M, Thomas RK, Ladanyi M, Pao W. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507) PLoS One. 2009;4:e7273. doi: 10.1371/journal.pone.0007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi YJ, Rho JK, Jeon BS, Choi SJ, Park SC, Lee SS, Kim HR, Kim CH, Lee JC. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: a lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother Pharmacol. 2010;66:381–388. doi: 10.1007/s00280-009-1174-7. [DOI] [PubMed] [Google Scholar]
- 41.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, Fujisawa T, Minna JD, Gazdar AF. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 42.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, Moro-Sibilot D, Dansin E, Chouaid C, Wislez M, Diebold J, Felip E, Rouquette I, Milia JD, Gautschi O. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 43.Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–3503. doi: 10.1182/blood-2006-04-015487. [DOI] [PubMed] [Google Scholar]
- 44.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 45.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, Ladanyi M, Politi K, Pao W. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to Trastuzumab therapy in non-small cell lung cancer. N Engl J Med. 2006;354:2619–2621. doi: 10.1056/NEJMc060020. [DOI] [PubMed] [Google Scholar]
- 47.Kelly RJ, Carter C, Giaccone G. Personalizing therapy in an epidermal growth factor receptor-tyrosine kinase inhibitor resistant non-small cell lung cancer using P-00299804 and Trastuzumab. J. Clin. Oncol. 2010;28:e507–e510. doi: 10.1200/JCO.2010.29.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, Althaus IW, Gandhi L, Shapiro GI, Nelson JM, Heymach JV, Meyerson M, Wong KK, Janne PA. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 49.Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. 2007;97:453–457. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stephen SP, Soltoff KL 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013;14:322–332. doi: 10.1016/j.cllc.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 55.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR Pathway -beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heavey S, O’Byrne KJ, Gately K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat Rev. 2014;40:445–456. doi: 10.1016/j.ctrv.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Do H, Salemi R, Murone C, Mitchell PL, Dobrovic A. Rarity of AKT1 and AKT3 E17K mutations in squamous cell carcinoma of lung. Cell Cycle. 2010;9:4411–4412. doi: 10.4161/cc.9.21.13654. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Date H, Lam WL, Minna JD, Gazdar AF. PIK3CA Mutations and Copy Number Gains in Human Lung Cancers. Cancer Res. 2008;68:6913–6921. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M, Thomas RK. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, Tracy S, Zhao X, Heymach JV, Johnson BE, Cantley LC, Janne PA. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeda H, Takigawa N, Ohashi K, Minami D, Kataoka I, Ichihara E, Ochi N, Tanimoto M, Kiura K. Vandetanib is effective in EGFR-mutant lung cancer cells with PTEN deficiency. Exp Cell Res. 2013;319:417–423. doi: 10.1016/j.yexcr.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 62.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 63.Li YC, Zhang DR, Pan JH, Li CJ. Signal transducers and activators of transcription 3 function in lung cancer. J Cancer Res Ther. 2013;9:S67–73. doi: 10.4103/0973-1482.119100. [DOI] [PubMed] [Google Scholar]
- 64.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bowman T, Garcia R, Turkson J. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 66.Harada D, Takigawa N, Ochi N, Ninomiya T, Yasugi M, Kubo T, Takeda H, Ichihara E, Ohashi K, Takata S, Tanimoto M, Kiura K. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012;103:1795–1802. doi: 10.1111/j.1349-7006.2012.02363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Seibti SM. Discovery of JSI-124 (Cucurbitacin I), a selective Janus Kinase/Signal Transducer and Activator of Transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- 68.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tatematsu A, Shimizu J, Murakami Y, Horio Y, Nakamura S, Hida T, Mitsudomi T, Yatabe Y. Epidermal growth factor receptor mutations in small cell lung cancer. Clin Cancer Res. 2008;14:6092–6096. doi: 10.1158/1078-0432.CCR-08-0332. [DOI] [PubMed] [Google Scholar]
- 70.Zakowski MF, Ladanyi M, Kris MG Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355:213–215. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 71.Fukui T, Tsuta K, Furuta K, Watanabe S, Asamura H, Ohe Y, Maeshima AM, Shibata T, Masuda N, Matsuno Y. Epidermal growth factor receptor mutation status and clinicopathological features of combined small cell carcinoma with adenocarcinoma of the lung. Cancer Sci. 2007;98:1714–1719. doi: 10.1111/j.1349-7006.2007.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morinaga R, Okamoto I, Furuta K, Kawano Y, Sekijima M, Dote K, Satou T, Nishio K, Fukuoka M, Nakagawa K. Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer. 2007;58:411–413. doi: 10.1016/j.lungcan.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 73.Nurwidya F, Takahashi F, Murakami A, Takahashi K. Epithelial mesenchymal transition in drug resistance and metastasis of lung cancer. Cancer Res Treat. 2012;44:151–156. doi: 10.4143/crt.2012.44.3.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, Varella-Garcia M, Bunn PA Jr, Hirsch FR. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012;30:2248–2255. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin CY, O’Neill V, Amler LC. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 76.Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 77.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, Heo DS, Crino L, Tan EH, Chao TY, Shahidi M, Cong XJ, Lorence RM, Yang JC. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–38. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 79.Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, Nishio K, Seki Y, Ebisawa R, Shahidi M, Yamamoto N. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J. Clin. Oncol. 2013;31:3335–3341. doi: 10.1200/JCO.2012.45.0981. [DOI] [PubMed] [Google Scholar]
- 80.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, Schuler M. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 81.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y, Geater SL. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 82.Yang JCH, Sequist LV, Schuler MH, Mok T, Yamamoto N, O’Byrne KJ, Hirsh V, Geater SL, Zhou C, Massey D, Zazulina V, Wu YL. Overall survival (OS) in patients (pts) with advanced non-small cell lung cancer (NSCLC) harboring common (Del19/L858R) epidermal growth factor receptor mutations (EGFR mut): Pooled analysis of two large open-label phase III studies (LUX-Lung 3 [LL3] and LUX-Lung 6 [LL6] ) comparing afatinib with chemotherapy (CT) ASCO Meeting Abstracts. 2014;32:8004. [Google Scholar]
- 83.Lee HJ, Schaefer G, Heffron TP, Shao L, Ye X, Sideris S, Malek S, Chan E, Merchant M, La H, Ubhayakar S, Yauch RL, Pirazzoli V, Politi K, Settleman J. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 2013;3:168–181. doi: 10.1158/2159-8290.CD-12-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakagawa T, Takeuchi S, Yamada T, Nanjo S, Ishikawa D, Sano T, Kita K, Nakamura T, Matsumoto K, Suda K, Mitsudomi T, Sekido Y, Uenaka T, Yano S. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther. 2012;11:2149–2157. doi: 10.1158/1535-7163.MCT-12-0195. [DOI] [PubMed] [Google Scholar]
- 87.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, Klimek VM, Nimer SD, Gilliland DG, Dutreix C, Huntsman-Labed A, Virkus J, Giles FJ. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010;28:4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanz M, Burnett A, Lo-Coco F, Lowenberg B. FLT3 inhibition as a targeted therapy for acute myeloid leukemia. Curr Opin Oncol. 2009;21:594–600. doi: 10.1097/CCO.0b013e32833118fd. [DOI] [PubMed] [Google Scholar]
- 89.Monnerat C, Henriksson R, Le Chevalier T, Novello S, Berthaud P, Faivre S, Raymond E. Phase I study of PKC412 (N-benzoyl-staurosporine), a novel oral protein kinase C inhibitor, combined with gemcitabine and cisplatin in patients with non-small-cell lung cancer. Ann Oncol. 2004;15:316–323. doi: 10.1093/annonc/mdh052. [DOI] [PubMed] [Google Scholar]
- 90.Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamur T. HGF/NK4, a four-kringle antagonist of Hepatocyte Growth Factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mic. Cancer Res. 2000;60:6737–6743. [PubMed] [Google Scholar]
- 91.Matsumoto K, Nakamura T, Sakai K, Nakamura T. Hepatocyte growth factor and Met in tumor biology and therapeutic approach with NK4. Proteomics. 2008;8:3360–3370. doi: 10.1002/pmic.200800156. [DOI] [PubMed] [Google Scholar]
- 92.Maemondo M, Narumi K, Saijo Y, Usui K, Tahara M, Tazawa R, Hagiwara K, Matsumoto K, Nakamura T, Nukiwa T. Targeting angiogenesis and HGF function using an adenoviral vector expressing the HGF antagonist NK4 for cancer therapy. Mol Ther. 2002;5:177–185. doi: 10.1006/mthe.2002.0533. [DOI] [PubMed] [Google Scholar]
- 93.Okamoto W, Okamoto I, Tanaka K, Hatashita E, Yamada Y, Kuwata K, Yamaguchi H, Arao T, Nishio K, Fukuoka M, Janne PA, Nakagawa K. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther. 2010;9:2785–2792. doi: 10.1158/1535-7163.MCT-10-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:553–572. doi: 10.1517/14728222.2012.680957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goldman JW, Laux I, Chai F, Savage RE, Ferrari D, Garmey EG, Just RG, Rosen LS. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118:5903–5911. doi: 10.1002/cncr.27575. [DOI] [PubMed] [Google Scholar]
- 96.Rosen LS, Senzer N, Mekhail T, Ganapathi R, Chai F, Savage RE, Waghorne C, Abbadessa G, Schwartz B, Dreicer R. A phase I dose-escalation study of Tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin Cancer Res. 2011;17:7754–7764. doi: 10.1158/1078-0432.CCR-11-1002. [DOI] [PubMed] [Google Scholar]
- 97.Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, Ramlau R, Arthur S, Gorbachevsky I, Schwartz B, Schiller JH. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2011;29:3307–3315. doi: 10.1200/JCO.2010.34.0570. [DOI] [PubMed] [Google Scholar]
- 98.Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, von Pawel J, Schwartz B, Von Roemeling R, Sandler AB. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13:391–395. doi: 10.1016/j.cllc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 99.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou YC, Joly AH. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Y, Guessous F, Kofman A, Schiff D, Abounader R. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs. 2010;13:112–121. [PMC free article] [PubMed] [Google Scholar]
- 101.Wu YL, Yang JCH, Kim DW, Su WC, Ahn MJ, Lee DH, Vansteenkiste JF, Zhang L, Felip E, Peng B, Gong Y, Zhao S, Amagasaki T, Akimov M, Tan DSW. Safety and efficacy of INC280 in combination with gefitinib (gef) in patients with EGFR-mutated (mut), MET-positive NSCLC: A single-arm phase lb/ll study. ASCO Meeting Abstracts. 2014;32:8017. [Google Scholar]
- 102.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RA, Kasman I, Mai E, Young J, Zha J, Zhang Z, Ross S, Schwall R, Colbern G, Merchant M. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–4368. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 103.Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, Govindan R, Patel T, Orlov SV, Wertheim MS, Yu W, Zha J, Yauch RL, Patel PH, Phan SC, Peterson AC. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013;31:4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Spigel DR, Edelman MJ, O’Byrne K, Paz-Ares L, Shames DS, Yu W, Paton VE, Mok T. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. ASCO Meeting Abstracts. 2014;32:8000. [Google Scholar]
- 105.Noto A, De Vitis C, Roscilli G, Fattore L, Malpicci D, Marra E, Luberto L, D’Andrilli A, Coluccia P, Giovagnoli MR, Normanno N, Ruco L, Aurisicchio L, Mancini R, Ciliberto G. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits Non-small Cell Lung Cancer. Oncotarget. 2013;4:1253–1265. doi: 10.18632/oncotarget.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sakai K, Yokote H, Murakami-Murofushi K, Tamura T, Saijo N, Nishio K. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 2007;98:1498–1503. doi: 10.1111/j.1349-7006.2007.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hughes B, Mileshkin L, Townley P, Gitlitz B, Eaton K, Mitchell P, Hicks R, Wood K, Amler L, Fine BM, Loecke D, Pirzkall A. Pertuzumab and Erlotinib in Patients With Relapsed Non-Small Cell Lung Cancer: A Phase II Study Using 18F-Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography Imaging. Oncologist. 2014;19:175–176. doi: 10.1634/theoncologist.2013-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, Fracasso PR, Wilkie DP, Hentemann M, Wilhelm SM, Scott WJ, Mumberg D, Ziegelbauer K. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 109.Dong S, Zhang XC, Cheng H, Zhu JQ, Chen ZH, Zhang YF, Xie Z, Wu YL. Everolimus synergizes with gefitinib in non-small-cell lung cancer cell lines resistant to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Chemother Pharmacol. 2012;70:707–716. doi: 10.1007/s00280-012-1946-3. [DOI] [PubMed] [Google Scholar]
- 110.La Monica S, Galetti M, Alfieri RR, Cavazzoni A, Ardizzoni A, Tiseo M, Capelletti M, Goldoni M, Tagliaferri S, Mutti A, Fumarola C, Bonelli M, Generali D, Petronini PG. Everolimus restores gefitinib sensitivity in resistant non-small cell lung cancer cell lines. Biochem Pharmacol. 2009;78:460–468. doi: 10.1016/j.bcp.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 111.Price KA, Azzoli CG, Krug LM, Pietanza MC, Rizvi NA, Pao W, Kris MG, Riely GJ, Heelan RT, Arcila ME, Miller VA. Phase II trial of Gefitinib and Everolimus in advanced Non-small cell lung cancer. J Thorac Oncol. 2010;5:1623–1629. doi: 10.1097/JTO.0b013e3181ec1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Papadimitrakopoulou VA, Sori JC, Jappe A, Jehl V, Klimovsky J, Johnson EB. Everolimus and Erlotinib as second- or third-line therapy in patients with advanced Non-small cell lung cancer. J Thorac Oncol. 2012;7:1594–1601. doi: 10.1097/JTO.0b013e3182614835. [DOI] [PubMed] [Google Scholar]
- 113.Ciunci CA, Perini RF, Avadhani AN, Kang HC, Sun W, Redlinger M, Harlacker K, Flaherty KT, Giantonio BJ, Rosen MA, Divgi CR, Song HK, Englander S, Troxel A, Schnall M, O’Dwyer PJ. Phase 1 and pharmacodynamic trial of everolimus in combination with cetuximab in patients with advanced cancer. Cancer. 2014;120:77–85. doi: 10.1002/cncr.28294. [DOI] [PubMed] [Google Scholar]
- 114.Deenen MJ, Klumpen HJ, Richel DJ, Sparidans RW, Weterman MJ, Beijnen JH, Schellens JH, Wilmink JW. Phase I and pharmacokinetic study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced solid malignancies. Invest New Drugs. 2012;30:1557–1565. doi: 10.1007/s10637-011-9723-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fury MG, Sherman E, Haque S, Korte S, Lisa D, Shen R, Wu N, Pfister D. A phase I study of daily everolimus plus low-dose weekly cisplatin for patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;69:591–598. doi: 10.1007/s00280-011-1734-5. [DOI] [PubMed] [Google Scholar]
- 116.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martelli AM, Cappellini , Chiarini F, Buontempo F, Bressani D, Evangelisti C, Fini M, McCubrey A. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget. 2012;3:371–94. doi: 10.18632/oncotarget.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Herrera VA, Zeindl-Eberhart E, Andreas Jung, Huber RM, Bergner A. The dual PI3K/mTOR inhibitor BEZ235 is effective in lung cancer cell lines. Anticancer Res. 2011;31:849–854. [PubMed] [Google Scholar]
- 119.Xu CX, Li Y, Yue P, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PLoS One. 2011;6:e20899. doi: 10.1371/journal.pone.0020899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. Augmentation of NVP-BEZ235’s anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol Ther. 2011;12:549–555. doi: 10.4161/cbt.12.6.16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, Hallin J, Blasina A, Zhang E, Nguyen L, Sun M, Vogt PK, McHarg A, Cheng H, Christensen JG, Kan JL, Bagrodia S. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10:2189–2199. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]
- 122.Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, Pierce K, Wainberg ZA, Lorusso PM. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs. 2014;32:510–7. doi: 10.1007/s10637-013-0062-5. [DOI] [PubMed] [Google Scholar]
- 123.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 124.Molife LR, Yan L, Vitfell-Rasmussen J, Zernhelt AM, Sullivan DM, Chen PA, Biondo A, Tetteh E, Siu LL, Patnaik A, Papadopoulos KP, de Bono JS, Tolcher AW, Minton S. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7:1–12. doi: 10.1186/1756-8722-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, Banks C, Capen A, Goode R, Lewis JE, Sams L, Huss KL, Campbell RM, Iversen PW, Neubauer BL, Brown TJ, Musib L, Geeganage S, Thornton D. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615. HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462–7469. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- 126.Rademaker-Lakhai JM, Beerepoot LV, Mehra N, Radema SA, van Maanen R, Vermaat JS, Witteveen EO, Visseren-Grul CM, Musib L, Enas N, van Hal G, Beijnen JH, Schellens JH, Voest EE. Phase I pharmacokinetic and pharmacodynamic study of the oral protein kinase C beta-inhibitor enzastaurin in combination with gemcitabine and cisplatin in patients with advanced cancer. Clin Cancer Res. 2007;13:4474–4481. doi: 10.1158/1078-0432.CCR-06-2912. [DOI] [PubMed] [Google Scholar]
- 127.Vansteenkiste J, Ramlau R, von Pawel J, San Antonio B, Eschbach C, Szczesna A, Kennedy L, Visseren-Grul C, Chouaki N, Reck M. A phase II randomized study of cisplatin-pemetrexed plus either enzastaurin or placebo in chemonaive patients with advanced non-small cell lung cancer. Oncology. 2012;82:25–29. doi: 10.1159/000335268. [DOI] [PubMed] [Google Scholar]
- 128.Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, Chan Z, Baron A, Franklin W, Drabkin HA, Girard L, Gazdar AF, Minna JD, Bunn PA Jr. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–950. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 129.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 131.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pillai RN, Ramalingam SS. Hsp90 Inhibitors. J Thorac Oncol. 2012;7:S407–S408. doi: 10.1097/JTO.0b013e31826df2bb. [DOI] [PubMed] [Google Scholar]
- 133.Shimamura T, Shapiro GI. Heat Shock Protein 90 inhibition in Lung cancer. J Thorac Oncol. 2008;(Suppl 2):S152–S159. doi: 10.1097/JTO.0b013e318174ea3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 135.Koizumi H, Yamada T, Takeuchi S, Nakagawa T, Kita K, Nakamura T, Matsumoto K, Suda K, Mitsudomi T, Yano S. Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis. J Thorac Oncol. 2012;7:1078–1085. doi: 10.1097/JTO.0b013e3182519a2c. [DOI] [PubMed] [Google Scholar]
- 136.Xu L, Kikuchi E, Xu C, Ebi H, Ercan D, Cheng KA, Padera R, Engelman JA, Janne PA, Shapiro GI, Shimamura T, Wong KK. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72:3302–3311. doi: 10.1158/0008-5472.CAN-11-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jeong CH, Park HB, Jang WJ, Jung SH, Seo YH. Discovery of hybrid Hsp90 inhibitors and their anti-neoplastic effects against gefitinib-resistant non-small cell lung cancer (NSCLC) Bioorg Med Chem Lett. 2014;24:224–227. doi: 10.1016/j.bmcl.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 138.Ueno T, Tsukuda K, Toyooka S, Ando M, Takaoka M, Soh J, Asano H, Maki Y, Muraoka T, Tanaka N, Shien K, Furukawa M, Yamatsuji T, Kiura K, Naomoto Y, Miyoshi S. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer. 2012;76:26–31. doi: 10.1016/j.lungcan.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 139.Kobayashi N, Toyooka S, Soh J, Yamamoto H, Dote H, Kawasaki K, Otani H, Kubo T, Jida M, Ueno T, Ando M, Ogino A, Kiura K, Miyoshi S. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor. Lung Cancer. 2012;75:161–166. doi: 10.1016/j.lungcan.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 140.Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, Sweeney J, Walker JR, Fritz C, Ross RW, Grayzel D, Engelman JA, Borger DR, Paez G, Natale R. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J. Clin. Oncol. 2010;28:4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, Harper H, Huberman M, Ramalingam S, Wong KK, Teofilovici F, Guo W, Shapiro GI. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pacurari M, Addison JB, Bondalapati N, Wan YW, Luo D, Qian Y, Castranova V, Ivanov AV, Guo NL. The microRNA-200 family targets multiple non-small cell lung cancer prognostic markers in H1299 cells and BEAS-2B cells. Int J Oncol. 2013;43:548–560. doi: 10.3892/ijo.2013.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 146.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 147.Garofalo M, Croce CM. microRNAs: Master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol. 2011;51:25–43. doi: 10.1146/annurev-pharmtox-010510-100517. [DOI] [PubMed] [Google Scholar]
- 148.Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Furugaki K, Iwai T, Moriya Y, Harada N, Fujimoto-Ouchi K. Loss of an EGFR-amplified chromosome 7 as a novel mechanism of acquired resistance to EGFR-TKIs in EGFR-mutated NSCLC cells. Lung Cancer. 2014;83:44–50. doi: 10.1016/j.lungcan.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 150.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cheung HW, Du J, Boehm JS, He F, Weir BA, Wang X, Butaney M, Sequist LV, Luo B, Engelman JA, Root DE, Meyerson M, Golub TR, Janne PA, Hahn WC. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1:608–625. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, Vinarsky V, Cho JL, Breton S, Sahay A, Medoff BD, Rajagopal J. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–223. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Serizawa M, Takahashi T, Yamamoto N, Koh Y. Genomic aberrations associated with erlotinib resistance in non-small cell lung cancer cells. Anticancer Res. 2013;33:5223–523. [PubMed] [Google Scholar]
- 154.Corominas-Faja B, Oliveras-Ferraros C, Cuyas E, Segura-Carretero A, Joven J, Martin-Castillo B, Barrajon-Catalan E, Micol V, Bosch-Barrera J, Menendez JA. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: a novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle. 2013;12:3390–3404. doi: 10.4161/cc.26417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 157.Breitbach S, Tug S, Helmig S, Zahn D, Kubiak T, Michal M, Gori T, Ehlert T, Beiter T, Simon P. Direct quantification of cell-free, circulating DNA from unpurified plasma. PLoS One. 2014;9:e87838. doi: 10.1371/journal.pone.0087838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT, Lai PB, Chiu RW, Lo YM. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013;59:211–224. doi: 10.1373/clinchem.2012.196014. [DOI] [PubMed] [Google Scholar]
- 159.Mouliere F, El Messaoudi S, Gongora C, Guedj AS, Robert B, Del Rio M, Molina F, Lamy PJ, Lopez-Crapez E, Mathonnet M, Ychou M, Pezet D, Thierry AR. Circulating Cell-Free DNA from Colorectal Cancer Patients May Reveal High KRAS or BRAF Mutation Load. Transl Oncol. 2013;6:319–28. doi: 10.1593/tlo.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, Diehl F, Angenendt P, Huang P, Cope L, Argani P, Murphy KM, Bachman KE, Greshock J, Wolff AC, Park BH. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18:3462–3469. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kuang Y, Rogers A, Yeap BY, Wang L, Makrigiorgos M, Vetrand K, Thiede S, Distel RJ, Janne PA. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15:2630–2636. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bechmann T, Andersen RF, Pallisgaard N, Madsen JS, Maae E, Jakobsen EH, Bak Jylling AM, Steffensen KD, Jakobsen A. Plasma HER2 amplification in cell-free DNA during neoadjuvant chemotherapy in breast cancer. J Cancer Res Clin Oncol. 2013;139:995–1003. doi: 10.1007/s00432-013-1413-5. [DOI] [PubMed] [Google Scholar]
- 163.Yang JC, Shih JY, Su WC, Hsia TC, Tsai CM, Ou SH, Yu CJ, Chang GC, Ho CL, Sequist LV, Dudek AZ, Shahidi M, Cong XJ, Lorence RM, Yang PC, Miller VA. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13:539–548. doi: 10.1016/S1470-2045(12)70086-4. [DOI] [PubMed] [Google Scholar]
- 164.Propper DJ, McDonald AC, Man A, Thavasu P, Balkwill F, Braybrooke JP, Caponigro F, Graf P, Dutreix C, Blackie R, Kaye SB, Ganesan TS, Talbot DC, Harris AL, Twelves C. Phase I and Pharmacokinetic Study of PKC412, an Inhibitor of Protein Kinase C. J. Clin. Oncol. 2001;19:1485–1492. doi: 10.1200/JCO.2001.19.5.1485. [DOI] [PubMed] [Google Scholar]
- 165.D’Arcangelo M, Cappuzzo F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics. 2013;7:61–68. doi: 10.2147/BTT.S28908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, Govindan R, Patel T, Orlov SV, Wertheim MS, Yu W, Zha J, Yauch RL, Patel PH, Phan SC, Peterson AC. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013;31:4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ross HJ, Blumenschein GR Jr, Aisner J, Damjanov N, Dowlati A, Garst J, Rigas JR, Smylie M, Hassani H, Allen KE, Leopold L, Zaks TZ, Shepherd FA. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. 2010;16:1938–1949. doi: 10.1158/1078-0432.CCR-08-3328. [DOI] [PubMed] [Google Scholar]
- 168.Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, Eaton K, Zacharchuk C, Freyman A, Powell C, Ananthakrishnan R, Quinn S, Soria JC. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010;28:3076–3083. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 169.Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, Schneider CP, Novello S, Eberhardt WE, Crino L, Habben K, Liu L, Janne PA, Brownstein CM, Reck M. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2011;29:4574–4580. doi: 10.1200/JCO.2011.36.6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reungwetwattana T, Molina JR, Mandrekar SJ, Allen-Ziegler K, Rowland KM, Reuter NF, Luyun RF, Dy GK, Marks RS, Schild SE, Jett JR, Adjei AA. Brief report: A phase II “Window-of-Opportunity” frontline study of the mTOR Inhibitor, temsirolimus given as a single agent in patients with advanced NSCLC, an NCCTG study. J Thorac Oncol. 2012;7:919–22. doi: 10.1097/JTO.0b013e31824de0d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC) Clin Chim Acta. 2010;411:846–852. doi: 10.1016/j.cca.2010.02.074. [DOI] [PubMed] [Google Scholar]
- 172.Sarkar FH, Li Y, Wang Z, Kong D, Ali S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010;13:57–66. doi: 10.1016/j.drup.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 174.Garofalo M, Romano G, Di Leva G, Nuovo G, Jeon YJ, Ngankeu A, Sun J, Lovat F, Alder H, Condorelli G, Engelman JA, Ono M, Rho JK, Cascione L, Volinia S, Nephew KP, Croce CM. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18:74–82. doi: 10.1038/nm.2577. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 175.Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16:498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 176.Yang J, Lan H, Huang X, Liu B, Tong Y. MicroRNA-126 inhibits tumor cell growth and its expression level correlates with poor survival in non-small cell cung cancer patients. PLoS One. 2012;7:e42978. doi: 10.1371/journal.pone.0042978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Crawford M, Brawner E, Batte K, Yu L, Hunter MG, Otterson GA, Nuovo G, Marsh CB, Nana-Sinkam SP. MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem Biophys Res Commun. 2008;373:607–612. doi: 10.1016/j.bbrc.2008.06.090. [DOI] [PubMed] [Google Scholar]
- 178.Liu B, Peng XC, Zheng XL, Wang J, Qin YW. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer. 2009;66:169–175. doi: 10.1016/j.lungcan.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 179.Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, Varella-Garcia M, Bunn PA Jr, Haney J, Helfrich BA, Kato H, Hirsch FR, Franklin WA. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. 2008;19:1053–1059. doi: 10.1093/annonc/mdn006. [DOI] [PubMed] [Google Scholar]
- 180.Wang YS, Wang YH, Xia HP, Zhou SW, Schmid-Bindert G, Zhou CC. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pac J Cancer Prev. 2012;13:255–260. doi: 10.7314/apjcp.2012.13.1.255. [DOI] [PubMed] [Google Scholar]
- 181.Salim H, Akbar NS, Zong D, Vaculova AH, Lewensohn R, Moshfegh A, Viktorsson K, Zhivotovsky B. miRNA-214 modulates radiotherapy response of non-small cell lung cancer cells through regulation of p38MAPK, apoptosis and senescence. Br J Cancer. 2012;107:1361–1373. doi: 10.1038/bjc.2012.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zucali PA, Ruiz MG, Giovannetti E, Destro A, Varella-Garcia M, Floor K, Ceresoli GL, Rodriguez JA, Garassino I, Comoglio P, Roncalli M, Santoro A, Giaccone G. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19:1605–1612. doi: 10.1093/annonc/mdn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yin R, Zhang S, Wu Y, Fan X, Jiang F, Zhang Z, Feng D, Guo X, Xu L. microRNA-145 suppresses lung adenocarcinoma-initiating cell proliferation by targeting OCT4. Oncol Rep. 2011;25:1747–1754. doi: 10.3892/or.2011.1252. [DOI] [PubMed] [Google Scholar]
- 184.Zhong M, Ma X, Sun C, Chen L. MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem Biol Interact. 2010;184:431–438. doi: 10.1016/j.cbi.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 185.Chen Z, Zeng H, Guo Y, Liu P, Pan H, Deng A, Hu J. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res. 2010;29:151. doi: 10.1186/1756-9966-29-151. [DOI] [PMC free article] [PubMed] [Google Scholar]