

Abstract
Members of the Peroxiredoxin (Prx) family are major cellular antioxidants that scavenge hydrogen peroxide and play essential roles in oxidative stress and cell signaling. 2-Cys Prxs, including Prx1, 2, 3 and 4, have been indicated in multiple oncogenic signaling pathways and thus may contribute to various processes of cancer development. The significance of 2-Cys Prxs in lung cancer development and their biological function in signal transduction have not been fully investigated. In this study we analyzed the expression of 2-Cys Prxs in lung cancer, and examined their levels of expression in a variety of cell lines established from human lung normal or cancer tissues. We found that 2-Cys Prxs, in particular, Prx1 and Prx4, were preferentially expressed in cell lines derived from human lung cancer. Through isoform specific knockdown of individual Prx, we demonstrated that Prx1 and Prx4 (but not Prx3) were required for human lung cancer A549 cells to form soft agar colony and to invade through matrigel in culture. Knockdown of Prx1 or Prx4 significantly reduced the activation of c-Jun and repressed the AP-1 mediated promoter activity. In mouse xenograft models, knockdown of Prx4 in A549 cells reduced subcutaneous tumor growth and blocked metastasis formation initiated through tail vein injection. Moreover, overexpression of Prx1 or Prx4 further enhanced the malignancy of A549 cells both in culture and in mouse xenografts in vivo. These data provide an in-depth understanding of the contribution of Prx1 and Prx4 to lung cancer development and are of importance for future development of therapeutic methods that targeting 2-Cys Prxs.
Keywords: Peroxiredoxin, lung cancer, tumor invasion and metastasis, cell signaling
Introduction
Peroxiredoxins (Prxs), or thioredoxin-dependent peroxidases, are originally discovered as cellular antioxidants with peroxidase activity. They scavenge H2O2 to reduce oxidative stress and to protect the inactivation of multiple cellular enzymes including proteins such as glutamine synthetase [1]. There are six isoforms of Prxs in mammals. Based on the number and position of Cysteine residues involved in the peroxidase reaction, they are classified into three subgroups, i.e., 2-Cys, atypical 2-Cys and 1-Cys Prxs. The 2-Cys Prxs, including Prx1, 2, 3 and 4, are the predominant Prxs in mammalian cells [2]. Most Prxs are considered as ubiquitously expressed in various tissues and organs, and they varies in their subcellular locations, such as cytoplasm (Prx1, 2 and 6), nucleus (Prx1), mitochondria (Prx3, 5 and 6), peroxisomes (Prx5), endoplasmic reticulum (Prx4), or even secreted into extracellular matrix in certain type of cells (Prx4) [3,4]. Due to these variations and their structure characteristics, members of the Prx family may have non-redundant, distinct intracellular functions. For example, expression and activation of Prx1 are required for the growth and proliferation of human mammary epithelial cells [5]. Prx1 also interacts with oncogenic protein c-Abl to inhibit its tyrosine kinase activity [6]. Prx 2 is a negative regulator of platelet-derived growth factor (PDGF) receptor signaling, and depletion of Prx2 results in increased cell proliferation and migration in response to PDGF in mouse fibroblasts and human muscle cells [7]. Prx2 can also function as a negative regulator of NF-κB signaling in mouse fibroblasts [8]. Prx3 is a target of c-Myc activation in rat fibroblasts and is required for Myc-mediated cell proliferation and transformation [9]. Prx4 also acts as a negative regulator of NF-κB activation in HeLa cells [10]. These studies reveal that, in addition to be scavengers and mediators of hydrogen peroxide signaling, members of the Prx family involve not only cellular response to oxidative stress but also various physiological or pathological processes. Therefore, understanding the contribution and molecular basis of Prxs in cell signal transduction may be of important value for the development of novel strategies to prevent or treat human disease.
Previous studies report that certain Prxs are aberrantly activated and expressed in a variety of human cancer, including breast cancer (Prx 1, 2 and 3) [11], lung cancer (Prx1, 3 and 4) [12,13], bladder cancer (Prx1 and 6) [14], thyroid cancer (Prx1) [15] and oral squamous cell carcinoma (Prx1) [16]. However, the role of Prxs in tumorigenesis and cancer progression has not been fully investigated and understood. For example, the role of Prx1 in human cancer is still controversial in that it can either function as a tumor suppressor or a pro-oncogenic factor, depending on the cellular context. There are several lines of evidence support that Prx1 functions as a tumor suppressor. Firstly, elevated expression of Prx1 in Myc-transformed fibroblasts significantly reduces anchorage-independent colony formation and tumorigenesis in a mouse xenograft model [17]. Secondly, Prx1 knockout cells are much more susceptible to Ras transformation [18]. Thirdly, genetic loss of Prx1 in mice leads to spontaneous tumor formation in multiple organs [19]. On the other hand, the pro-oncogenic role of Prx1 in tumorigenesis and cancer progression is also well documented in literature. For example, the levels of Prx1 in specimens of bladder cancer have been found to be significantly higher than normal adjacent tissue, and the increased expression of Prx1 is associated with worse clinical staging, higher rate of recurrence and poor prognosis [14]. Expression of Prx1 in breast cancer facilitates cancer cell survival from oxidative stress induced cell death and promotes cancer cell malignancy [20,21]. The pro-survival effect of Prxs may be attributed to its function as a molecular chaperone to enhance resistance to stress as demonstrated in yeast and various mammalian cells [22,23]. The exact role of other 2-Cys Prxs in human cancer development is also not conclusive. Therefore, a systematic evaluation of Prxs may be critical for the understanding of their biological significance. In particular, the study of 2-Cys Prxs should be accomplished under the specific context of certain types of human cancer.
Lung cancer is the leading cause of cancer-related mortality in USA and worldwide. Although significant progress has been made over the past decade in the early detection and combined treatment of lung cancer, the five-year survival rate of patients with advanced lung cancer is less than 20% (cancer statistics, WHO). With more than fifty histological variants, lung cancer is extremely heterogeneous and adenocarcinoma accounts for more than 40% of its overall incidence. In this study, we examined the expression of 2-Cys Prxs in human lung cancer and explored the functional significance of each isoform of 2-Cys Prxs under the context of lung adenocarcinoma. Our data shed light on the differential function of individual member of the Prx family in lung cancer development and we identified the unique contribution of Prx1 and Prx4 in lung cancer development and intracellular signal transduction. Our findings may provide novel insights for the understanding of human lung cancer pathogenesis.
Materials and methods
Cell lines
HEK293T cells and all other cell lines were commercially obtained from NCI repository or ATCC. Three immortalized cell lines established from lung normal epithelium were used, including BEAS-2B (immortalized by SV40 T antigen), NL20 (immortalized by SV40 T antigen) and Nuli-1 (immortalized by HPV-E6/7 and hTERT). Two cell lines established from lung small cell carcinoma were used, including NCI-H69 and NCI-H82 cells. Three cell lines established from lung squamous cell carcinoma were used, including NCI-H520 (from primary tumor), NCI-H226 and SK-MES-1(from pleural effusion). Three cell lines established from lung adenocarcinoma were used, including A549 (from primary tumor), NCI-H2030 (from lymph node metastasis) and NCI-H2122 (from pleural effusion). Cells were cultured in provider’s suggested culture medium in an atmosphere of 5% CO2 at 37°C with 80~85% relative humidity.
Cell lysis and western blotting
Cultured cells were lysed in RIPA lysis buffer in the presence of proteinase inhibitors. Proteins were separated on a 4-12% gradient gel and transferred to PVDF membrane. Western blot was performed using standard protocol. Primary antibodies used include, rabbit anti-Prx1, 2 and 4 (Abcam), mouse anti-β-actin (Sigma-Aldrich), mouse anti-Prx3, c-Jun, and c-Myc (Santa Cruz), phosphor-c-Jun (p-63 and p-73) (Cell Signaling). Western blotting was performed following standard procedure.
Lentiviral ShRNA knockdown of Prx and establishment of stable knockdown cell lines
Strictly controlled ShRNA-based knockdown experiments were designed and performed according to previous published suggestions [24]. All ShRNA constructs including MISSION® pLKO.1-puro control vector (vector control), MISSION® Non-Target shRNA (ShNT) and ShRNAs specifically targeting either Prx1 (ShPrx1), 3 (ShPrx3), or 4 (ShPrx4) were commercially obtained (Sigma-Aldrich) and all sequences were confirmed by commercial sequencing. Lentiviral particles expressing ShRNAs were produced in HEK293T cells using the provider’s plasmid packaging system and Fugene 6 transfection reagent following suggested transfection and virus production procedures. The titer of virus-containing medium was determined by measuring the level of p24 using the ELISA and Lenti-X GoStix kits (Clontech). To establish stable knockdown, A549 cells were infected with lentiviral particles at a multiplicity of infection (MOI) around 10 in all experiments. Cells were maintained in puromycin containing medium to establish stable cells.
Cloning of human 2-Cys Prxs into lentiviral expression vector and establishment of overexpressing stable cells
To clone human Prx1, 2, 3 and 4, the total mRNAs of cultured HEK293T cells were extracted and purified following commercial kit protocol (Qiagen). Reverse transcriptase PCR (RT-PCR) was performed using gene specific primer and SuperScript™ reverse transcriptase kit (Invitrogen). The coding region of Prx gene was first cloned into the BamH I/Xho I sites of the pCDNA3.1-Myc vector to generate expression plasmid that encodes c-Myc tagged fusion protein. The coding sequences were confirmed by DNA sequencing and fusion protein expression was verified by western blot. The validated coding sequences of the fusion protein were then transferred into pLVX-IRES-Puro vector for expression in lentiviral vectors using restriction enzyme digestion and T4 DNA ligase. For lentivirus production, Fugene 6 and Clontech’s lentiviral packaging system were used to produce infectious particles that expressing pLVX-IRES-Puro empty vector (vector control) or c-Myc tagged Prx. A549 cells were then infected and maintained in puromycin containing medium as described above for stable cell selection.
Colony formation in soft agar and transwell matrigel cell invasion assay
For colony formation experiment, cells were suspended in 0.3% agar and 15,000 cells/well were seeded into 6-well plate pre-coated with 1.0 ml of 0.6% agar. Medium was changed every 5 days for four weeks. The number and size of colonies were examined and data were obtained by analyzing with Image J software. Transwell matrigel invasion assays using BD invasion chamber were performed following the manufacturer’s suggested protocol with 10% serum containing medium as chemo-attractants. Invaded cells were stained by Diff-QuikTM staining and images were taken under microscope (X4). Numbers of invaded cells were counted using the Image J software.
Proteome profiler human phospho-kinase array
The antibody based array kit, which is capable of simultaneously measuring the levels of 46 phosphorylated proteins (all are kinase substrates) in duplicates on the same membrane, was commercially obtained (R&D Systems). Cells were cultured in 100 mm2 dishes and starved for 24 hours, followed by stimulation with or without 10% serum-containing medium for 15 minutes. Cells were then harvested in RIPA lysis buffer and 700 μg of cell lysates were used for kinase array following the manufacturer’s suggested protocol. All array membranes were processed at the same time under the same conditions, and results were obtained by exposing membranes to a single X-ray film with exactly the same duration of exposure time. The intensity of each spot representing individual phosphorylated protein was determined using Image J software. The relative spot intensity was obtained by normalizing with the intensity of the internal positive control on each membrane.
The AP-1 luciferase reporter activity assay
A549 cells cultured in 96-well plate were transiently transfected with an AP-1 firefly luciferase reporter construct and a control renila luciferase construct using Lipofectamine 2000. Luciferase activities were measured at 48 hours after transfection using a dual luciferase assay kit (Promega) and a luminometer. Relative luciferase unit (RLU) was determined as the ratio of firefly luciferase to renila luciferase value.
Subcutaneous or tail-vein injection of A549 cells into SCID mice
The protocol for mouse xenograft experiments was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). All animal procedures were conducted following the Policy on Humane Care and Use of Laboratory Animals, and Guidelines of the Animal Care and Laboratory Animal Welfare (NIH). A double-blind experimental design was applied to eliminate potential subjective bias on protocol execution and data collection. Briefly, severe combined immunodeficiency (SCID) female mice at 5-week old were commercially obtained (NCI). Mice were exposed to food and water ad libitum and hosted in a 12/12 (hr/hr) light-dark cycle. At the age of 6-week, mice were randomly separated into six groups to receive either A549 ShNT, ShPrx4 or MycPrx4 cells. A total of 5 × 105 cells/mouse (in 100 μl of PBS) were injected either subcutaneously or through tail-vein. The growth of tumor in subcutaneous injection was measured with a calibre every other day, until the diameter at one dimension was equal or larger than 1.0 cm. Groups of mice injected with tumor cells through tail-vein were all euthanized at 8 weeks after injection due to the deterioration of health in the group receiving MycPrx4 cells. Primary tumors (subcutaneous injection) and mouse lung (tail-vein injection) were fixed in 4% paraformaldehyde and stored in 70% ethanol, and proceeded with standard paraffin-embedding, sectioning, H&E staining and histopathological examination.
Statistical analysis
Quantitative data were presented as means ± standard deviation (x̅ ± sd). Data were analyzed with indicated statistical methods using GraphPad Prism (Version 5.04). For calculation of the p value, parameters of two-tailed, 95% confidence interval were used for all analysis. A p value of less than 0.05 is considered statistically significant.
Results
Differential expression of Prxs in human lung cancer and cell lines
To date there are no reports in the literature that simultaneously evaluate the transcript and protein levels of 2-Cys Prxs in human lung cancer. Therefore we first examined the published repository of gene expression profiling for Prxs using Oncomine database. By focusing on the human lung cancer microarray data, we identified and summarized the expression profiling of Prx1, 2, 3 and 4 from at least two independent reports based on the array of patient specimens. Analysis of the first study [25] revealed that Prx1, 2 and 4 were highly expressed in tumors of lung adenocarcinoma and squamous cell carcinoma (SCC) when compared with those of normal lung epithelium or small/large cell carcinoma (Figure 1A); whereas increased expression of Prx3 was mainly found in tumor samples of lung adenocarcinoma. Analysis of the second study [26] revealed a very similar finding of increased Prxs in human lung adenocarcinoma and SCC, but with variations in normal lung epithelium (Figure 1B).
Figure 1.
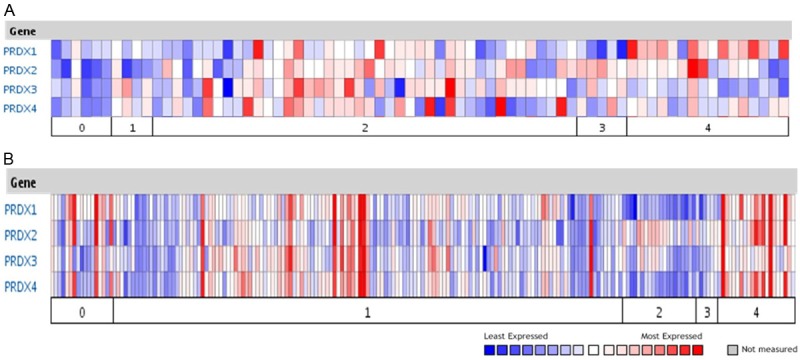
mRNA expression of 2-Cys Prxs in human lung normal and cancer tissues from published microarrays. Data were summarized using the Oncomine database (www.oncomine.com). A: Microarray results from Garber M.E., et al, PNAS USA 98:13784~9. Category and number of samples included: 0, normal lung (n = 6); 1, large cell lung carcinoma (n = 4); 2, lung adenocarcinoma (n = 42); 3, small cell lung carcinoma (n = 5); 4, squamous cell lung carcinoma (n = 16). B: Microarray results from Bhattacharjee A., et al, PNAS USA 98:13790~5. Category and number of samples included: 0, normal lung (n = 17); 1, lung adenocarcinoma (n = 139); 2, lung carcinoid tumor (n = 20); 3, small cell lung carcinoma (n = 6); 4, squamous cell lung carcinoma (n = 21).
To facilitate the study of Prxs in human lung cancer, we asked whether Prxs are differentially expressed in established cell lines of human lung normal epithelium or various tumor types. Western blot was used to measure the endogenous protein levels of Prxs in a total of eleven cell lines (Figure 2A). We found that Prx1 was expressed in all cell lines, and the levels of Prx1 were relatively low in normal cell lines (BEAS2B, NL-20 and Nuli-1), medium in cell lines of small cell carcinoma (H69 and H82) and relatively high in cell lines of lung SCC (H520, H226 and SKMES) and adenocarcinoma (A549, H2030 and H2122). Moreover, Prx2 was relatively low in normal cell lines and much higher in cell lines of small cell carcinoma and SCC. However, in some lung cancer cell lines including SKMES, A549 and H2030 cells, the levels of Prx2 was below the limit of detection or absent. On the other hand, Prx3 was found to be universally expressed in all cell lines tested at relatively high levels except lower expression was found in one of the normal cell lines (NL-20). Two of three normal cell lines (except Nuli-1) have relatively low expression of Prx4, and higher expression of Prx4 was found in cell lines of small cell carcinoma, SCC (except SKMES) and adenocarcinoma. Taken together, these data indicate that there’s an overall trend of higher expression of Prx1, 2 and 4 in cell lines of small cell carcinoma and SCC compared with those of normal cell lines, whereas in lung adenocarcinoma, the levels of Prx1 and 4 appear to be consistently higher than those of normal cells.
Figure 2.
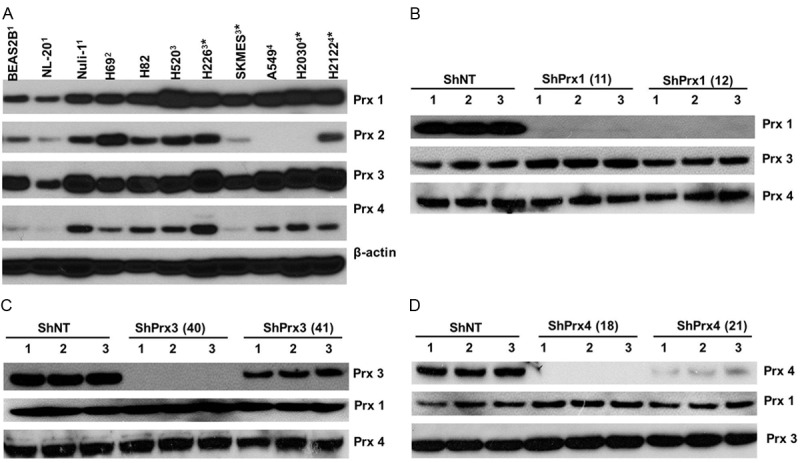
Differential expression of 2-Cys Prxs in various cell lines and knockdown of individual Prx in human lung adenocarcinoma A549 cells. (A) Expression of 2-Cys Prxs in cell lines established from human lung normal epithelium or different types of lung cancer. Cell lines were originally derived from: 1lung normal epithelium; 2small cell carcinoma; 3squamous cell carcinoma; 4adenocarcinoma; *cancer metastasis. For each cell line, cells were lysed in RIPA buffer at the concentration of 1× 107 cell/ml and equal volume of lysates were loaded to the gel for Western blot. (B-D) Knockdown of endogenous Prx1 (B), Prx3 (C) or Prx4 (D) in A549 cells using isoform specific ShRNAs targeting the correspondent protein coding regions. Two sets of unique ShRNA (indicated by the number in the parenthesis) were used for each specified Prx target. The endogenous Prx levels were measured by Western blot from triplicate dishes of stable cells established by lentivrial infection and antibiotic selection. Note that knockdown of targeted Prx does not interfere with the expression of other isoforms of 2-Cys Prxs.
Knockdown of Prx1 or Prx4 in A549 cells represses anchorage independent colony formation and matrigel invasion
A549 cells were chosen for the following experiments because there were no detectable, endogenous levels of Prx2 expression, which simplifies our efforts to characterize the contribution of individual Prx in these cells. A set of four different ShRNA constructs targeting the distinctive regions of the transcript of either Prx1, 2 or 3, were tested for their efficiency to knockdown the endogenous protein expression by lentiviral infection. After initial evaluation of knockdown efficiency in HEK293T cells, we selected two ShRNAs that target different coding regions of either Prx1, 3 or 4 with relatively higher efficiency to establish stable cells in A549. Our efforts of establishing stable knockdown cells were successful and we had stable cells completely depleted of Prx1 (Figure 2B), Prx3 (Figure 2C) or Prx4 (Figure 2D). Although 2-Cys Prxs share commonly conserved sequences and structural motifs, our ShRNA knockdown was very specific in that the ShRNA had knockdown effect only to the targeted Prx but had no off-targets effects to other Prx isoforms. The phenotypical features of these stable cells were then compared with control cells expressing a non-target ShRNA (ShNT cells). We found that knockdown of Prx1 or Prx4 led to the reduction of anchorage independent colony formation in soft agar in A549 cells (Figure 3A and 3B). Moreover, knockdown of Prx1 or Prx4 also significantly repressed serum-induced cell invasion in matrigel invasion assay (Figure 3C and 3D). However, knockdown of Prx3 in A549 cells did not produce any effects on either colony formation or cell invasion. Therefore, our data indicate that endogenously expressed Prx1 and Prx4 are required for the integrity of colony formation and invasion of A549 cells.
Figure 3.
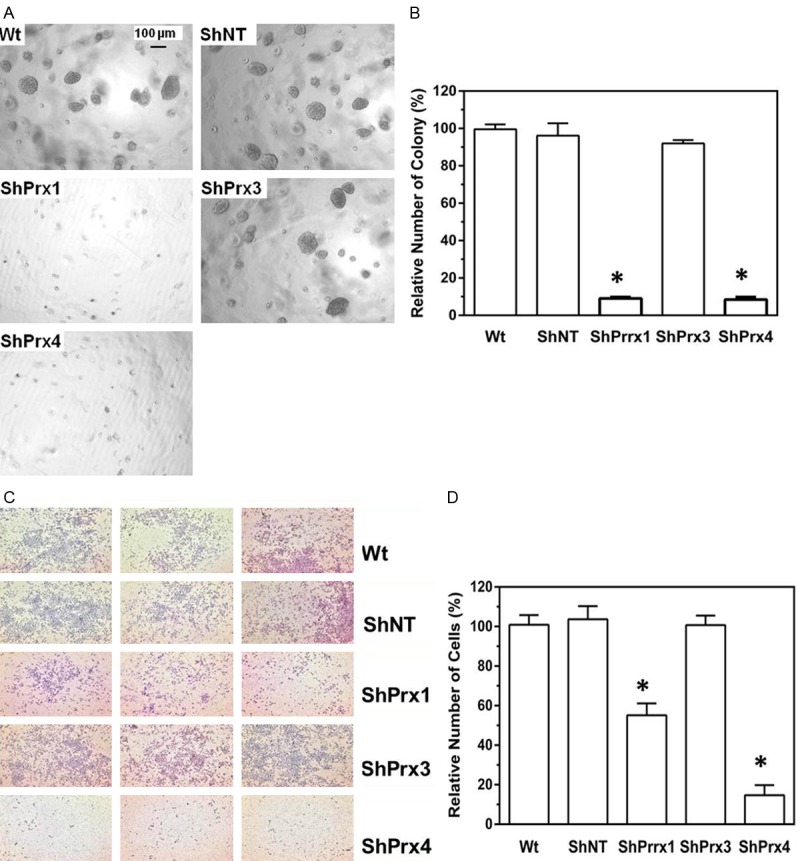
Knockdown of Prx1 or Prx4 in A549 cells abolishes their ability of to form anchorage independent colonies in soft agar and represses their capability of invading through matrigel. A, B: Anchorage independent colony formation in soft agar; C, D: Transwell cell invasion assay. Compared to wildtype parental A549 (Wt) or ShNT cells, *p<0.05 (n = 6, t test).
Overexpression of Prx1 or Prx4 in A549 cells further enhances anchorage independent colony formation and matrigel invasion
Next we asked whether ectopic expression of Prxs in A549 cells may have an opposite effect on colony formation and cell invasion. The coding regions of Human Prx1~4 were amplified from HEK293T cells and cloned into a mammalian expression vector for protein expression (Figure 4A). Stable cells overexpressing either Myc tagged Prx1, 2, 3 or 4 were established (Figure 4B). The phenotypical features of these stable cells were then compared with parental cells or control cells expressing an empty vector. We found that overexpression of MycPrx1 or MycPrx4 in A549 cells led to significant increases in anchorage independent colony formation in soft agar (Figure 4C and 4D) and cells invaded through matrigel (Figure 4E and 4F). However, overexpression of either MycPrx2 or MycPrx3 had no significant effects on colony formation in soft agar as well as cell invasion through matrigel. These data indicate that overexpression of Prx1 or Prx4, but not Prx2 or Prx3, is able to further promote the anchorage independent colony formation and matrigel invasion of human lung cancer A549 cells.
Figure 4.
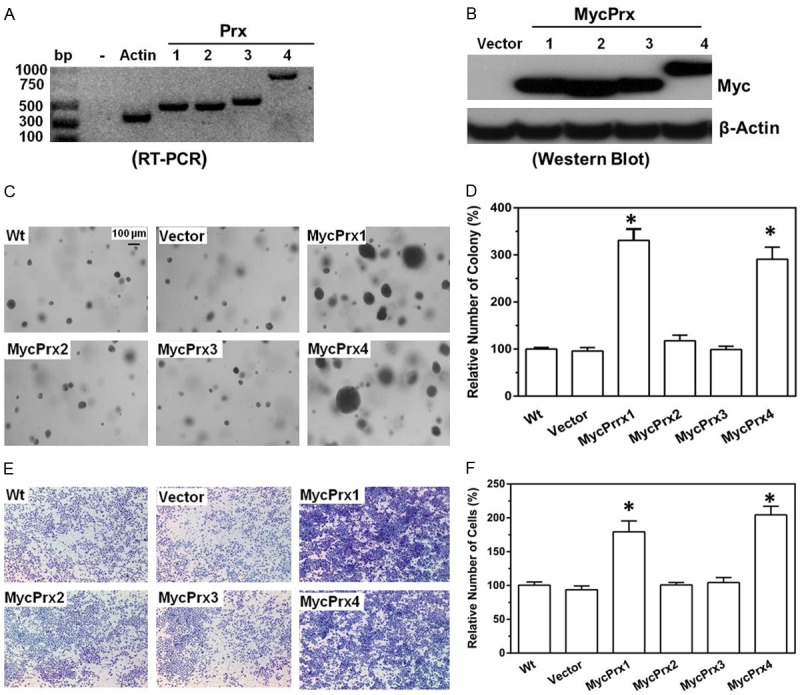
Overexpression of Prx1 or Prx4 in A549 cells enhances their ability to form anchorage independent colonies in soft agar and increase their capability of invading through matrigel. (A) The protein coding regions of human Prx1, Prx2, Prx3 or Prx4 gene in HEK293T cells were reverse transcribed and amplified by PCR, and then cloned into the pCDNA3.1-Myc vector for protein expression; (B) Myc tagged Prx1, 2, 3 or 4 expression in A549 stable cells; (C-F) Anchorage independent colony formation in soft agar (C, D) and transwell invasion assay (E, F) using cells with stably overexpression of individual Prx.
Prx1 and Prx4 are required for the sustained activation of AP-1 signaling
To understand the molecular basis of Prx1 and Prx4 mediated cancer cell phenotype changes, we examined the global phosphokinase signaling changes mediated by Prx1 or Prx4 in A549 cells. This phosphokinase assay simultaneously detected the levels of 46 phosphorylated proteins in duplicates, along with several pre-designed negative and positive controls. To identify proteins in which phosphorylation was affected by the manipulation of either Prx1 or Prx4 levels, multiple arrays were performed using stable cells expressing ShNT, ShPrx1 (ShPrx4) or MycPrx1 (MycPrx4). The following criteria were used to determine whether the phosphorylation of a particular protein was causally related with the levels of Prx1 or Prx4: (1) compared with no stimulation, the levels of phosphorylated protein in ShNT cells were induced in the presence of serum containing medium; (2) such induced activation was significantly repressed in ShPrx1 and ShPrx4 cells; and (3) the phosphorylation can be further enhanced in MycPrx1 and MycPrx4 cells. Following this criteria, we identified that the levels of phosphorylated c-Jun was positively correlated with the levels of Prx1 or Prx4 in A549 cells (Figure 5A and 5B show results from ShPrx4/MycPrx4 cells and similar results were obtained from ShPrx1/MycPrx1 cells).
Figure 5.
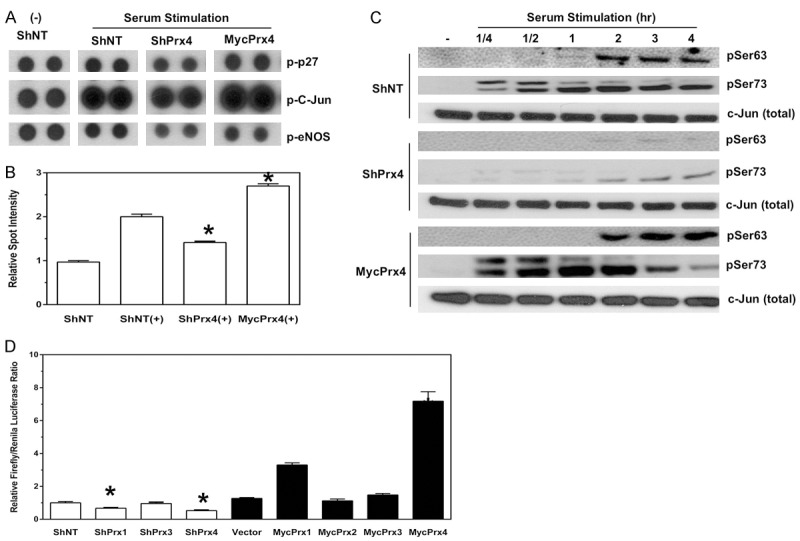
Knockdown of Prx4 represses, whereas overexpression of Prx4 activates c-Jun mediated AP-1 activation. (A, B) Proteome profiler human phosphokinase array (A) and quantification of phosphorylated c-Jun levels (B). -, no stimulation. +, serum stimulation. Compared to ShNT (+) cells, *p<0.05 (n = 4, t test). (C) Western blot of phosphorylated c-Jun after serum stimulation in A549 cells expressing ShNT, ShPrx4 or MycPrx4. (D) AP-1 luciferase reporter assay in A549 cells with or without expression of ShRNAs or MycPrxs. Compared to either ShNT or vector cells, *p<0.05 (n = 6, t test).
To confirm whether the activation/phosphorylation of c-Jun was indeed affected by depletion or overexpression of Prx1 or Prx4, Western blot was used to examine a serum-induced, time-dependent phosphorylation of c-Jun in these cells. Previous studies have shown that phosphorylation of Serine residues at 63 (Ser63) and 73 (Ser73) determines the activation of c-Jun, we thus examined the levels of phosphorylation at both sites with phospho-specific antibodies. As shown in Figure 5C, ShPrx4 cells showed a significant reduction in the phosphorylation levels of c-Jun at Ser63 and Ser73 residues at multiple time points after serum stimulation, and the levels of phosphorylation at both residues in MycPrx4 cells were significantly higher than those of ShNT cells (Figure 5C). C-Jun is one of the major components of the AP-1 transcription factor complex, whose activation contributes to multiple oncogenic processes including the stimulation of cell growth and proliferation, cell invasion and metastasis in various cancers. Therefore, we used an AP-1 luciferase reporter assay to test whether manipulation of Prx1 or Prx4 levels had any effect on the AP-1 mediated luciferase expression. As shown in Figure 5D, depletion of either Prx1 or Prx4 in A549 cells led to the reduction of the AP-1 luciferase activity, whereas depletion of Prx3 had no significant effect. Compared with vector control cells, overexpression of either MycPrx1 or MycPrx4 significantly stimulated the AP-1 luciferase reporter activity, but overexpression of MycPrx2 or MyxPrx3 did not affect the AP-1 luciferase activity. Therefore, through maintaining and promoting the oncogenic AP-1 activation, expression of Prx1 and Prx4 may contribute to the activation of AP-1 downstream signaling pathways that are critical for the malignancy of human lung cancer cells.
Prx4 expression is required for tumor xenograft growth and metastasis formation in vivo
From cell culture studies we found that both Prx1 and Prx4 were important for A549 cells to grow in soft agar and to invade through matrigel. Previous studies have shown that downregulation of Prx1 in A549 cells led to the reduced tumor xenograft growth and inhibition of metastasis in mouse xenograft experiments [27-29], which were consistent with our observation that knockdown of Prx1 led to reduced malignant phenotype in A549 cells. However, whether manipulating the levels of Prx4 in A549 cells may affect tumor growth and metastasis in vivo has not been reported. Therefore, we injected ShNT, ShSrx or MycPrx4 cells subcutaneously into groups of SCID mice to examine the ability of these cells to initiate/support tumor growth in vivo. Compared with mice injected with ShNT cells, subcutaneous tumor growth in mice injected with ShPrx4 cells was significantly reduced, whereas tumor growth in mice injected with MycPrx4 cells was significantly accelerated (Figure 6A). By the end of the sixth week after subcutaneous injection, all mice were euthanized and tumors were extracted from all mice, except one mouse in ShPrx4 group was free of tumor mass (Figure 6B). Compared with tumors from mice receiving ShNT cells, tumors from mice receiving MycPrx4 cells were much larger in size and heavier in weight, while tumors from mice receiving ShPrx4 cells were smaller in size and lighter in weight (Figure 6B and 6C). These data suggest that Prx4 positively contributes to tumor xenograft growth in vivo, which is consistent with the observation that knockdown (or overexpression) of Prx4 leads to reduced (or enhanced) colony formation of A549 cells in soft agar.
Figure 6.
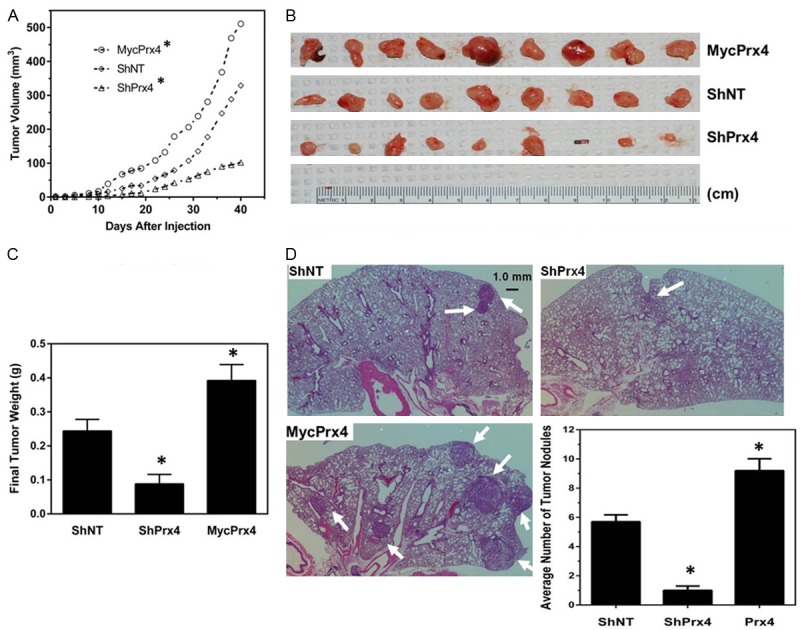
Knockdown of Prx4 represses, whereas overexpression of MycPrx4 enhances tumor xenograft growth and metastasis formation in mouse models in vivo. (A) Tumor growth curves of subcutaneously injected A549-ShNT, ShPrx4 or MycPrx4 cells into SCID mice; (B, C) Images (B) and average weight (C) of primary tumors extracted from injection sites 40 days post subcutaneous injection; (D) Lung tumor nodules found in SCID mice receiving tail vein injection of A549-ShNT, ShPrx4 or MycPrx4 cells. Arrow heads indicated tumor nodules. Bar graph shows the average number of tumor nodules found in each experimental group. Compared to mice receiving ShNT cells, *p<0.05 (n = 9, t test).
The malignancy of cancer cells can be evaluated by their invasiveness and their capability of initiating tumor metastasis in vivo. Based on the observation that Prx4 is required for A549 cells to invade through matrigel in culture, we hypothesized that Prx4 is also required for lung cancer metastasis. To test this hypothesis, A549 cells expressing ShNT, ShSrx or MycPrx4 were injected into the tail vein of SCID mice. At 8 weeks after injection, all mice were euthanized and mouse lung was extracted. Tumor nodules with diameter equal or larger than 1.0 mm were identified microscopically (as indicated by the arrow heads in Figure 6D) and data were analyzed. Compared with mice receiving ShNT cells, there’s a significant reduction in the number of tumor nodules in mice injected with ShPrx4 cells; whereas a robust increase in the number of tumor nodules was found in mice injected with MycPrx4 cells (Figure 6D). Therefore, knockdown of Prx4 in A549 cells represses, whereas overexpression of MycPrx4 enhances, their ability to form lung tumor metastasis in SCID mice.
Discussion
The Prx family of peroxidase provides critical defense against oxidative stress through scavenging H2O2 and thus protects cells from oxidative damages. Therefore, the abundance of Prxs is normally associated with attenuation of oxidative stress and increased rate of cell survival under various stress conditions. Due to an essential secondary messenger function of H2O2, Prxs are also considered as receptors for cellular H2O2 and thus play multiple roles in many physiological as well as pathological processes (for review, refer to [30]). In human lung cancer, Prx1 is frequently identified as one of the major cellular antioxidants that are preferentially expressed in cancer tissues but not in normal lung epithelium or nonmalignant tumors [13]. As a validated biomarker of lung adenocarcinoma [12,31,32], it mediates the pro-oxidants induced lung cancer cell growth and invasion [33] and is required for human lung cancer cells to grow as tumor xenograft and to establish cancer metastasis in mice [27,29]. Expression of Prx1 also confers human lung cancer cells resistance to ionizing radiation [27] and chemotherapeutic drugs [34]. Similar to Prx1, Prx2 is also identified as aberrantly increased in human lung cancer and its levels are positively correlated with high-grade lung carcinomas [32,35], but the molecular basis of Prx2 contribution to lung cancer development has not been investigated. In breast cancer, however, silencing of Prx2 leads to inhibition of cancer cell growth and reduced formation of lung metastasis in mice, which may be attributed to a novel function of Prx2 in regulating cellular metabolism [36]. Unlike Prx1 or Prx2, which are mainly localized in the cytosol, Prx3 is a mitochondria protein that is also overexpressed in human lung cancer [37]. Disruption of Prx3 in mitochondria may function as a novel mechanism of cellular response to cancer chemotherapeutics [38]. Prx4 is mainly localized in the endoplasmic reticulum and is involved in cellular inflammatory response [39]. However, the role of Prx4 in human cancer is much less studied compared with other 2-Cys Prxs. By loss- and gain-of-function experiments, our study reveals a critical role of Prx1 and Prx4 in human lung cancer pathogenesis.
To date there are no genetic mutations identified in the Prx family of proteins that may associate with human diseases including cancer. Therefore, the contribution of Prxs to human cancer development is mainly resulted from their aberrantly activated expression rather than any genetic gain or loss of functional mutations. Expression of Prxs can be regulated at multiple levels, in which the activation of gene transcription plays a major role. Carcinogens and tumor promoters, such as cigarette smoke, asbestos, 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), phorbol acetate and arsenate, have been shown to stimulate the expression of 2-Cys Prxs through activation of protein kinase C, mitogen activated protein kinase (MAPK) and P38MAPK pathways [32,40,41]. Hypoxia may also have some effect on the expression of 2-Cys Prx expression, such as activation of Prx 1 [42,43]. Among all the transcription factors, nuclear related factor 2 (Nrf2) plays a critical role in the activation of 2-Cys Prx expression [30]. In our study we identified that the levels of Prx1 and Prx4 were much higher in human lung cancer cells. However, we did not study the mechanisms of their up-regulation, in the future it may be of interesting to understand why Prx1 and Prx4 were aberrantly activated in human lung cancer cells.
Due to the complicated role of H2O2 in mediating cell signaling, it is not surprising that the function of Prxs is beyond the simple model of acting as a pro-oncogenic factor. In fact, members of the Prx family may also have tumor suppressor activities. In particular, genomic loss of Prx1 in mice leads to spontaneous tumor formation in multiple organs, which suggest that Prx1 may function as a tumor suppressor [19]. Interestingly, the effect of Prx1 depletion in mice may be strain dependent, since Prx1 null mice established from another group are completely normal and free of tumors. Other 2-Cys Prx null mice, including genomic knockout of Prx2, 3 or 4 [44-47], are also phenotypically normal and free of developmental defects. However, one of the common features of Prx knockout mice is that they are more sensitive to oxidative stress induced cell death in general. In our study we found that overexpression of Prx1 or Prx4 was able to further promote the malignancy of human lung cancer cells. Systematic overexpression of Prx4 in mice, however, is not able to drive spontaneous tumorigenesis under laboratory conditions [48]. It is not clear whether overexpression of both Prx1 and Prx4 in mice is sufficient to drive de novo tumorigenesis in vivo. Transgenic mice that overexpress Prx3 also develop normally, and cells from these mice have an increased resistance to stress-induced cell death [49]. In the future, it will be of interest to study whether overexpression of a single or any combination of 2-Cys Prx in mice may affect the process of tumorigenesis or malignant progression of lung tumors induced by carcinogens, tumor promoters or pre-existed, oncogenic mutations.
2-Cys Prxs are also involved in the activation of various signaling pathways [30]. In mechanistic study, our data demonstrated a critical role of Prx1 and Prx4 in mediating the sustained activation of c-Jun, a major component of the AP-1 transcription factor complex. The molecular mechanisms of why Prx1 and Prx4 are required for c-Jun phosphorylation and the AP-1 mediated promoter activity still remain elusive. The AP-1 activity is stimulated by numerous factors including growth factors, chemokines and environmental stress. As a heterodimer, the AP-1 complex contains components of c-Fos, FosB, FosL1 (Fra-1), FosL2 (Fra-2), c-Jun, JunB, JunD, etc. Among them, c-Jun is activated by the phosphorylation from upstream activated kinases including MAPK, RSK and JNK kinase systems [50]. Additionally, the transcriptional activity of AP-1 complex is also regulated by s-glutathionylation [51,52], s-nitrosylation [53] and oxidation [54]. Ref-1 is a redox sensitive protein that activates the transcriptional activity of AP-1 either through a direct reduction of the oxidized cysteine residue of c-Jun [55,56], or facilitating the nuclear translocation of thioredoxin [57-59]. In yeast, activation of Tpx1 (yeast homologue of Prx I) facilitates the reduction of oxidized cysteine in PAP1 (yeast homologue of c-Jun) and activates PAP1-dependent gene transcription [60,61]. Other possible mechanisms have also been reported in the literature. For example, Prx1 may affect JNK activities either through regulating the levels of intracellular hydrogen peroxide [62], or directly interacting with GSTpi-JNK complex to cause the dissociation of JNK and subsequent activation [28]. It will be of interest to investigate whether these potential mechanisms are also applied in mammalian cells for Prx1 or Prx4 to activate the AP-1 activity.
Activation of the AP-1 signaling is well documented in promoting the growth, proliferation, invasion and metastasis of human lung cancer cells [63,64]. On one hand, activation of the AP-1 complex can induce the expression of downstream targeted genes including matrix metalloproteinase MMP1, MMP2, MMP3 and MMP9, osteonectin, autotaxin, etc [65,66]. Expression of these genes promotes the remodeling of extracellular matrix, epithelial-mesenchymal transition and cell invasion. On the other hand, the AP-1 complex can also function as transcriptional repressor to repress genes that function as invasion suppressors such as TSC-36, fibronectin, Krp1 and other proteins [67,68]. In principle, the expression of these genes may be affected since we observed a reduced activation of c-Jun phosphorylation and an impaired AP-1 promoter activity in Prx1/Prx4 knockdown cells. As a result, these gene expression changes may explain the reduced cell invasion in culture and metastasis formation in mice. Future understanding of how these gene expression patterns are affected in human lung cancer cells in response to different levels of 2-Cys Prxs may be informative. Nevertheless, the contribution of 2-Cys Prxs to the AP-1 signaling pathway is unambiguously associated with cell invasion and metastasis of human lung cancer, targeting 2-Cys Prxs to develop novel therapeutic strategies in the future may provide novel thoughts for cancer prevention or drug discovery.
In summary, in this study we analyzed the expression of 2-Cys Prxs in lung cancer, and examined their levels of expression in a variety of cell lines including human lung normal and cancer cell lines. We found that Prx1 and Prx4 were preferentially expressed in cell lines established from human lung cancer including SCC and adenocarcinoma. We demonstrated that Prx1 and Prx4 (but not Prx3) were required for human lung cancer A549 cells to form soft agar colony and to invade through matrigel in culture. Knockdown of Prx1 or Prx4 significantly reduced the activation of c-Jun and thus repressed AP-1 mediated promoter activity, which may contribute to the changes of cancer cell phenotype. In mouse xenograft models in vivo, we found that knockdown of Prx4 reduced subcutaneous tumor growth and blocked metastasis formation. Furthermore, overexpression of Prx1 or Prx4 further enhanced the malignancy of A549 cells in culture and in mouse xenografts in vivo. Our data provide an in-depth understanding of the contribution of Prx1 and Prx4 to lung cancer development and provide valuable information for future development of therapeutic methods that targeting 2-Cys Prxs.
Acknowledgements
This work was partially supported by the National Institutes of Health, with funding from the National Cancer Institute [grant number R00CA149144 to Q. Wei] and a pilot project [to Q. Wei] of the National Institute of General Medical Sciences COBRE grant [number 5P20GM103486-10 to L. Hersh at the University of Kentucky].
Disclosure of conflict of interest
None.
References
- 1.Kim IH, Kim K, Rhee SG. Induction of an antioxidant protein of Saccharomyces cerevisiae by O2, Fe3+, or 2-mercaptoethanol. Proc Natl Acad Sci U S A. 1989;86:6018–6022. doi: 10.1073/pnas.86.16.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 3.Tavender TJ, Sheppard AM, Bulleid NJ. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem J. 2008;411:191–199. doi: 10.1042/BJ20071428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okado-Matsumoto A, Matsumoto A, Fujii J, Taniguchi N. Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J Biochem. 2000;127:493–501. doi: 10.1093/oxfordjournals.jbchem.a022632. [DOI] [PubMed] [Google Scholar]
- 5.Prosperi MT, Ferbus D, Karczinski I, Goubin G. A human cDNA corresponding to a gene overexpressed during cell proliferation encodes a product sharing homology with amoebic and bacterial proteins. J Biol Chem. 1993;268:11050–11056. [PubMed] [Google Scholar]
- 6.Prosperi MT, Ferbus D, Rouillard D, Goubin G. The pag gene product, a physiological inhibitor of c-abl tyrosine kinase, is overexpressed in cells entering S phase and by contact with agents inducing oxidative stress. FEBS Lett. 1998;423:39–44. doi: 10.1016/s0014-5793(98)00057-x. [DOI] [PubMed] [Google Scholar]
- 7.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 8.Han YH, Kwon JH, Yu DY, Moon EY. Inhibitory effect of peroxiredoxin II (Prx II) on Ras-ERK-NFkappaB pathway in mouse embryonic fibroblast (MEF) senescence. Free Radic Res. 2006;40:1182–1189. doi: 10.1080/10715760600868552. [DOI] [PubMed] [Google Scholar]
- 9.Wonsey DR, Zeller KI, Dang CV. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci U S A. 2002;99:6649–6654. doi: 10.1073/pnas.102523299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin DY, Chae HZ, Rhee SG, Jeang KT. Regulatory role for a novel human thioredoxin peroxidase in NF-kappaB activation. J Biol Chem. 1997;272:30952–30961. doi: 10.1074/jbc.272.49.30952. [DOI] [PubMed] [Google Scholar]
- 11.Noh DY, Ahn SJ, Lee RA, Kim SW, Park IA, Chae HZ. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001;21:2085–2090. [PubMed] [Google Scholar]
- 12.Chang JW, Jeon HB, Lee JH, Yoo JS, Chun JS, Kim JH, Yoo YJ. Augmented expression of peroxiredoxin I in lung cancer. Biochem Biophys Res Commun. 2001;289:507–512. doi: 10.1006/bbrc.2001.5989. [DOI] [PubMed] [Google Scholar]
- 13.Park JH, Kim YS, Lee HL, Shim JY, Lee KS, Oh YJ, Shin SS, Choi YH, Park KJ, Park RW, Hwang SC. Expression of peroxiredoxin and thioredoxin in human lung cancer and paired normal lung. Respirology. 2006;11:269–275. doi: 10.1111/j.1440-1843.2006.00849.x. [DOI] [PubMed] [Google Scholar]
- 14.Quan C, Cha EJ, Lee HL, Han KH, Lee KM, Kim WJ. Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J Urol. 2006;175:1512–1516. doi: 10.1016/S0022-5347(05)00659-2. [DOI] [PubMed] [Google Scholar]
- 15.Yanagawa T, Ishikawa T, Ishii T, Tabuchi K, Iwasa S, Bannai S, Omura K, Suzuki H, Yoshida H. Peroxiredoxin I expression in human thyroid tumors. Cancer Lett. 1999;145:127–132. doi: 10.1016/s0304-3835(99)00243-8. [DOI] [PubMed] [Google Scholar]
- 16.Yanagawa T, Iwasa S, Ishii T, Tabuchi K, Yusa H, Onizawa K, Omura K, Harada H, Suzuki H, Yoshida H. Peroxiredoxin I expression in oral cancer: a potential new tumor marker. Cancer Lett. 2000;156:27–35. doi: 10.1016/s0304-3835(00)00434-1. [DOI] [PubMed] [Google Scholar]
- 17.Mu ZM, Yin XY, Prochownik EV. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem. 2002;277:43175–43184. doi: 10.1074/jbc.M206066200. [DOI] [PubMed] [Google Scholar]
- 18.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 19.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 20.Chang R, Wang E. Mouse translation elongation factor eEF1A-2 interacts with Prdx-I to protect cells against apoptotic death induced by oxidative stress. J Cell Biochem. 2007;100:267–278. doi: 10.1002/jcb.20969. [DOI] [PubMed] [Google Scholar]
- 21.Bae JY, Ahn SJ, Han W, Noh DY. Peroxiredoxin I and II inhibit H(2)O(2)-induced cell death in MCF-7 cell lines. J Cell Biochem. 2007;101:1038–45. doi: 10.1002/jcb.21155. [DOI] [PubMed] [Google Scholar]
- 22.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Moon JC, Hah YS, Kim WY, Jung BG, Jang HH, Lee JR, Kim SY, Lee YM, Jeon MG, Kim CW, Cho MJ, Lee SY. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 24.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 25.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MF, Keng PC, Shau H, Wu CT, Hu YC, Liao SK, Chen WC. Inhibition of lung tumor growth and augmentation of radiosensitivity by decreasing peroxiredoxin I expression. Int J Radiat Oncol Biol Phys. 2006;64:581–591. doi: 10.1016/j.ijrobp.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66:7136–7142. doi: 10.1158/0008-5472.CAN-05-4446. [DOI] [PubMed] [Google Scholar]
- 29.Chen MF, Chen WC, Wu CT, Lin PY, Shau H, Liao SK, Yang CT, Lee KD. p53 status is a major determinant of effects of decreasing peroxiredoxin I expression on tumor growth and response of lung cancer cells to treatment. Int J Radiat Oncol Biol Phys. 2006;66:1461–1472. doi: 10.1016/j.ijrobp.2006.07.1372. [DOI] [PubMed] [Google Scholar]
- 30.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 31.Deng B, Ye N, Luo G, Chen X, Wang Y. Proteomics analysis of stage-specific proteins expressed in human squamous cell lung carcinoma tissues. Cancer Biomark. 2005;1:279–286. doi: 10.3233/cbm-2005-1603. [DOI] [PubMed] [Google Scholar]
- 32.Rostila A, Puustinen A, Toljamo T, Vuopala K, Lindstrom I, Nyman TA, Oksa P, Vehmas T, Anttila SL. Peroxiredoxins and tropomyosins as plasma biomarkers for lung cancer and asbestos exposure. Lung Cancer. 2012;77:450–459. doi: 10.1016/j.lungcan.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 33.Kinnula VL, Paakko P, Soini Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung. FEBS Lett. 2004;569:1–6. doi: 10.1016/j.febslet.2004.05.045. [DOI] [PubMed] [Google Scholar]
- 34.Hwang KE, Park C, Seol CH, Hwang YR, Hwang JS, Jung JW, Choi KH, Jeong ET, Kim HR. Elevated prx1 provides resistance to docetaxel, but is not associated with predictive significance in lung cancer. Tuberc Respir Dis (Seoul) 2013;75:59–66. doi: 10.4046/trd.2013.75.2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehtonen ST, Svensk AM, Soini Y, Paakko P, Hirvikoski P, Kang SW, Saily M, Kinnula VL. Peroxiredoxins, a novel protein family in lung cancer. Int J Cancer. 2004;111:514–521. doi: 10.1002/ijc.20294. [DOI] [PubMed] [Google Scholar]
- 36.Stresing V, Baltziskueta E, Rubio N, Blanco J, Arriba MC, Valls J, Janier M, Clezardin P, Sanz-Pamplona R, Nieva C, Marro M, Petrov D, Sierra A. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene. 2013;32:724–735. doi: 10.1038/onc.2012.93. [DOI] [PubMed] [Google Scholar]
- 37.Kim YS, Lee HL, Lee KB, Park JH, Chung WY, Lee KS, Sheen SS, Park KJ, Hwang SC. Nuclear factor E2-related factor 2 dependent overexpression of sulfiredoxin and peroxiredoxin III in human lung cancer. Korean J Intern Med. 2011;26:304–313. doi: 10.3904/kjim.2011.26.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newick K, Cunniff B, Preston K, Held P, Arbiser J, Pass H, Mossman B, Shukla A, Heintz N. Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS One. 2012;7:e39404. doi: 10.1371/journal.pone.0039404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada S, Ding Y, Sasaguri Y. Peroxiredoxin 4: critical roles in inflammatory diseases. J UOEH. 2012;34:27–39. doi: 10.7888/juoeh.34.27. [DOI] [PubMed] [Google Scholar]
- 40.Chaudhary N, Bhatnagar S, Malik S, Katare DP, Jain SK. Proteomic analysis of differentially expressed proteins in lung cancer in Wistar rats using NNK as an inducer. Chem Biol Interact. 2013;204:125–134. doi: 10.1016/j.cbi.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Li B, Ishii T, Tan CP, Soh JW, Goff SP. Pathways of induction of peroxiredoxin I expression in osteoblasts: roles of p38 mitogen-activated protein kinase and protein kinase C. J Biol Chem. 2002;277:12418–12422. doi: 10.1074/jbc.M111443200. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Chae HZ, Kim YJ, Kim YH, Hwangs TS, Park EM, Park YM. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biol Toxicol. 2003;19:285–298. doi: 10.1023/b:cbto.0000004952.07979.3d. [DOI] [PubMed] [Google Scholar]
- 43.Fajardo I, Svensson L, Bucht A, Pejler G. Increased levels of hypoxia-sensitive proteins in allergic airway inflammation. Am J Respir Crit Care Med. 2004;170:477–484. doi: 10.1164/rccm.200402-178OC. [DOI] [PubMed] [Google Scholar]
- 44.Iuchi Y, Okada F, Tsunoda S, Kibe N, Shirasawa N, Ikawa M, Okabe M, Ikeda Y, Fujii J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem J. 2009;419:149–158. doi: 10.1042/BJ20081526. [DOI] [PubMed] [Google Scholar]
- 45.Li L, Shoji W, Takano H, Nishimura N, Aoki Y, Takahashi R, Goto S, Kaifu T, Takai T, Obinata M. Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS-induced oxidative stress. Biochem Biophys Res Commun. 2007;355:715–721. doi: 10.1016/j.bbrc.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 46.Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, Dho SH, Kwon KS, Kwon HJ, Han YH, Jeong S, Kang SW, Shin HS, Lee KK, Rhee SG, Yu DY. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101:5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem. 2003;278:25179–25190. doi: 10.1074/jbc.M302706200. [DOI] [PubMed] [Google Scholar]
- 48.Ding Y, Yamada S, Wang KY, Shimajiri S, Guo X, Tanimoto A, Murata Y, Kitajima S, Watanabe T, Izumi H, Kohno K, Sasaguri Y. Overexpression of peroxiredoxin 4 protects against high-dose streptozotocin-induced diabetes by suppressing oxidative stress and cytokines in transgenic mice. Antioxid Redox Signal. 2010;13:1477–1490. doi: 10.1089/ars.2010.3137. [DOI] [PubMed] [Google Scholar]
- 49.Chen L, Na R, Gu M, Salmon AB, Liu Y, Liang H, Qi W, Van Remmen H, Richardson A, Ran Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell. 2008;7:866–878. doi: 10.1111/j.1474-9726.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 51.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. Faseb J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 52.Klatt P, Molina EP, Lamas S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274:15857–15864. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- 53.Tabuchi A, Sano K, Oh E, Tsuchiya T, Tsuda M. Modulation of AP-1 activity by nitric oxide (NO) in vitro: NO-mediated modulation of AP-1. FEBS Lett. 1994;351:123–127. doi: 10.1016/0014-5793(94)00839-6. [DOI] [PubMed] [Google Scholar]
- 54.Abate C, Patel L, Rauscher FJ 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 55.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688–6695. [PubMed] [Google Scholar]
- 58.Ordway JM, Eberhart D, Curran T. Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol Cell Biol. 2003;23:4257–4266. doi: 10.1128/MCB.23.12.4257-4266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bozonet SM, Findlay VJ, Day AM, Cameron J, Veal EA, Morgan BA. Oxidation of a eukaryotic 2-Cys peroxiredoxin is a molecular switch controlling the transcriptional response to increasing levels of hydrogen peroxide. J Biol Chem. 2005;280:23319–23327. doi: 10.1074/jbc.M502757200. [DOI] [PubMed] [Google Scholar]
- 62.Kang SW, Chang TS, Lee TH, Kim ES, Yu DY, Rhee SG. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J Biol Chem. 2004;279:2535–2543. doi: 10.1074/jbc.M307698200. [DOI] [PubMed] [Google Scholar]
- 63.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 64.Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. The activator protein-1 transcription factor in respiratory epithelium carcinogenesis. Mol Cancer Res. 2007;5:109–120. doi: 10.1158/1541-7786.MCR-06-0311. [DOI] [PubMed] [Google Scholar]
- 65.Black EJ, Clair T, Delrow J, Neiman P, Gillespie DA. Microarray analysis identifies Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity, as a specific target of cell transformation by v-Jun. Oncogene. 2004;23:2357–2366. doi: 10.1038/sj.onc.1207377. [DOI] [PubMed] [Google Scholar]
- 66.Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15:519–526. doi: 10.1016/s0945-053x(97)90026-3. [DOI] [PubMed] [Google Scholar]
- 67.Liu S, Wang L, Wang W, Lin J, Han J, Sun H, Guo H, Sun R, Wu Q. TSC-36/FRP inhibits vascular smooth muscle cell proliferation and migration. Exp Mol Pathol. 2006;80:132–140. doi: 10.1016/j.yexmp.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 68.Spence HJ, Johnston I, Ewart K, Buchanan SJ, Fitzgerald U, Ozanne BW. Krp1, a novel kelch related protein that is involved in pseudopod elongation in transformed cells. Oncogene. 2000;19:1266–1276. doi: 10.1038/sj.onc.1203433. [DOI] [PubMed] [Google Scholar]