

Abstract
Dual oxidase 1 (DUOX1), which is the main sources for reactive oxygen species (ROS) production in the airway, are frequently silenced in human lung cancer. In poorly differentiated follicular thyroid carcinoma, a high expression of DUOX1 was associated with a reduced risk of death. However, the role of DUOX1 in human hepatocellular carcinoma (HCC) is still not clear. Here, we investigated DUOX1 expression and its promoter methylation status in primary HCC. To date, We found that expression of DUOX1 was decreased significantly in 76.9% (60/78) human hepatocellular carcinoma and 66.7% (6/9) liver cancer cell lines, compared with the paired adjacent non-tumor tissues and immortalized normal cell line. Moreover, which was well correlated with its promoter methylation status. Methylation was further detected in primary HCC, but none or occasionally in paired adjacent non-tumor tissues. Detailed methylation analysis of 35 CpG sites at a 324-bp promoter region by bisulfi te genomic sequencing (BGS) confi rmed its methylation. DUOX1 silencing could be reversed by chemical demethylation treatment with 5-aza-2’-deoxycytidine (5-Aza-dC), indicating direct epigenetic silencing. Restoring DUOX1 expression in lowly expressed cancer cells signifi cantly inhibited cancer cells growth and colony formation ability through the induction of G2/M phase cell cycle arrest and an increase in ROS generation, while knockdown of DUOX1 could markedly promote cancer cells proliferation. In conclusion, we demonstrate that epigenetic silencing of DUOX1 via promoter hypermethylation is common in human liver cancer cells and primary HCC and DUOX1 appears to be a functional tumor suppressor involved in liver carcinogenesis.
Keywords: DUOX1, methylation, ROS, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) remains the fifth leading cause of cancer-related death worldwide and the second most common malignancy in China. Moreover, HCC is often diagnosed at a late stage and the prognosis is poor. Recent studies highlight new molecular mechanisms involved in HCC pathogenesis, including consequence of cumulative genetic and epigenetic events. CpG hypermethylation acts as an alternative mechanism to gene inactivation and it is now recognized as an important mechanism during tumor initiation and progression, including liver cancer [1,2]. It is important and intriguing to identify new genes silenced by promoter hypermethylation in liver cancer because aberrant tumor suppressor gene (TSG) hypermethylation is both a mechanism and a biomarker for tumorigenesis [3].
DUOX1, dual oxidases 1, is a key phenotype of NADPH-oxidases (NOXs) family. The main function of such genes is reactive oxygen species (ROS) production. Moreover, NOXs are major non-mitochondrial sources of ROS in human cells [4]. DUOX1 is predominantly found in thyroid, which is involved in the synthesis of thyroid hormones [5]. It is also highly expressed in normal epithelial cells in airway, pancreas, placenta, prostate, testis, and salivary gland [6-9]. Recent researches indicated that DUOX1 may function as a selective TSG during tumor initiation and progression. In lung cancer cells, DUOX1 is frequently silenced by its promoter hypermethylation [10]. In Poorly differentiated follicular thyroid carcinoma, a high expression of DUOX1 was associated with a reduced risk of death [11]. However, the functional role of DUOX1 in liver tumorigenesis remains unknown. The expression of DUOX1 and its regulatory mechanisms during the development of liver cancer needs to be determined.
Reactive oxygen species (ROS), chemically-reactive molecules containing oxygen, including oxygen ions and peroxides, are the key mediators of cellular oxidative stress and redox dysregulation involved in cancer initiation and progression. For a long time, ROS were considered oncogenic since it was implicated in cancer progression and metastasis. Organisms living in aerobic conditions are constantly subjected to ROS. Increased ROS levels contribute to genetic instability and cancer initiation and progression [12,13]. The high tumor cell resistance to cytolysis by hydrogen peroxide and other types of ROS can be explained by upregulation of elements of the antioxidant system. It is now widely accepted that constitutively elevated levels of cellular oxidative stress and dependence on mitogenic and anti-apoptotic ROS-signaling in cancer cells are involved in the carcinogenesis. Paradoxically, apart from being involved in proliferative, anti-apoptotic, metastatic, and angiogenic signaling, ROS may also exert cytotoxic and proapoptotic functions that would limit tumorigenicity and malignant progression [14,15].
Here, we demonstrated that DUOX1 underwent promoter CpG hypermethylation-associated silencing in human liver cancer. The analysis of liver cancer and cell lines showed that DUOX1 hypermethylation was a common event in the liver carcinogenesis. We also showed that DUOX1 functions as a TSG to suppress tumor cell growth through the induction of G2/M phase cell cycle arrest and an increase in ROS generation. Epigenetic changes in cellular redox homeostasis and ROS levels will affect viability through redox modulation of the mitochondrial permeability transition pore opening leading to cytochrome C release, apoptosome assembly and activation of executioner caspases, which is supportive for ROS-directed cancer chemotherapeutics.
Materials and methods
Cell lines and cell culture
HCC cell lines HepG2, Hep3B, Huh7, HCC-LM3, BEL-7402, SMMC-7721, Sk-Hep1, MHCC-97H and MHCC-97L cells were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), and 1% penicillin-streptomycin, maintained in a humidified atmosphere of 5% CO2 at 37°C. One immortalized normal human liver cell line L02 was used as “normal” controls for HCCs analysis.
Primary tumor tissues
Primary HCCs were obtained from liver cancer patients in Huashan Hospital at the time of surgery, matched non-tumor samples from liver cancer patients were also obtained at least 2 cm distant from the tumor. Non-tumor portions were trimmed off from the frozen tumor blocks and the selected tumor areas had more than 80% tumor cells as confirmed by histology. All HCC patients had not received chemotherapy, percutaneous ethanol injection, radio frequency ablation or any other anti-tumor treatment before the operation. All patients gave informed consent for obtaining the study specimens. The basic demographic and clinical characteristics of HCC patients are listed in Table 1. Experiments and procedures were in accordance with the Helsinki Declaration of 1975, and approved by the Human Ethics Committee of Shanghai Fudan University.
Table 1.
Correlation of DUOX1 mRNA expression with clinical characteristics in 78 patients with HCC
| Characteristics | No. of patients | Relative Duox1 mRNA levels(log10) (Mean ± SEM) | P value |
|---|---|---|---|
| Gender | 0.442 | ||
| Male | 59 | 0.3456 ± 0.0801 | |
| Female | 19 | 0.4362 ± 0.1128 | |
| Age (years) | 0.062 | ||
| ≤ 50 | 33 | 0.4706 ± 0.1173 | |
| > 50 | 45 | 0.2922 ± 0.0755 | |
| HBsAg | 0.123 | ||
| Positive | 66 | 0.3037 ± 0.7350 | |
| Negative | 12 | 0.7198 ± 0.1063 | |
| HBeAg | 0.585 | ||
| Positive | 25 | 0.2425 ± 0.1088 | |
| Negative | 53 | 0.4267 ± 0.0824 | |
| Tumor size (cm) | 0.323 | ||
| ≤ 5 cm | 18 | 0.4131 ± 0.1195 | |
| > 5 cm | 35 | 0.4869 ± 0.1063 | |
| Edmondson stage | 0.677 | ||
| I | 13 | 0.3987 ± 0.1145 | |
| II | 50 | 0.3960 ± 0.0852 | |
| III | 15 | 0.2465 ± 0.1737 | |
| TNM stage | 0.049 | ||
| I | 20 | 0.1644 ± 0.1149 | |
| II | 26 | 0.6145 ± 0.1212 | |
| III | 16 | 0.3574 ± 0.1438 | |
| IV | 15 | 0.2333 ± 0.1410 |
HCC hepatocellular carcinoma, HBsAg hepatitis B surface antigen, HBeAg hepatitis B e antigen, TNM tumor–node–metastasis.
5-aza-2’-deoxycytidine treatment and RNA/DNA extraction
For pharmacological demethylation, cells were treated with 5uM 5-aza-2’-deoxycytidine (5-AzadC) (Sigma, St. Louis, Mo. USA) for 3 consecutive days. Culture medium was changed every 24 hours. An equivalent concentration of vehicle (DMSO) was used as the control.
Total RNA and genomic DNA were extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Total RNA and DNA concentrations were quantified by NanoDrop 1000 (Nanodrop, Wilmington, Del., USA).
Reverse transcription and quantitative real-time PCR
A reverse transcription reaction was performed using 1 μg of total RNA with High Capacity cDNA Reverse Transcription kit (SYBR qPCR RT Mix, FSQ-101, TOYOBO). The mRNA expression levels of DUOX1 were determined by real-time PCR using THUNDERBIRD™ SYBR® qPCR Mix (TOYOBO) and ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, Calif, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. Real-time PCR was performed in triplicate. Primers used for DUOX1 were: DUOX1-F 5’-CCACCAGGAGTGGCATAAGT-3’ and DUOX1-R 5’-CAGCTGACGGATGACTTGAA-3’ (110 bp product).
Bisulfite treatment of DNA and methylation-specific PCR
Genomic DNA was bisulfite-treated with Zymo DNA Modification Kit (Zymo Research, Orange, Calif., USA) according to the protocol provided. Methylation-specific PCR (MSP) was carried out for 40 cycles with annealing temperature at 60°C. Methylation-specific primers were: DUOX1-MF 5’-ACGGAATATTTTTATTTGCGTTTC-3’ and DUOX1-MR 5’-GTCCGATACCTCTACAA CTCTACG-3’, and unmethylation-specific primers were: DUOX1-UF 5’-TGGAATATTTTTATT TGTGTTTTGG-3’ and DUOX1-UR 5’-ATCCAATACCTCTACAACTCTACACC-3’.
Bisulfite genome sequencing
Bisulfite-treated genomic DNA was amplified using bisulfite genome sequencing (BGS) primers, DUOX1-BF: 5’-TTTAGTTTTATGGGATTTGTGAAGG-3’ and DUOX1-BR: 5’-AAAAAACTA ACATTCCCCTTTCTTC-3’. PCR products were purified with Illustra GFXTMPCR and gel band purification kit (GE Healthcare Life Science, Uppsala, Sweden) and cloned into pCR4-TOPO vector for sequencing (Invitrogen). At least four colonies were randomly chosen for plasmid extraction and sequencing analysis using the ABI PRISM BigDye Terminator Cycle Sequencing Kit in the ABI 3100 sequencer (Applied Biosystems).
Colony formation assay
The pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector are a gift kindly provided by Dr. Xavier De Deken from the Institute de Recherche Interdisciplinaire en Biologie Humaine et Mole’culaire of Universite’ Libre de Bruxelles in Brussels. Human liver cancer cells transfected with pcDNA3 empty vector or co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector were used for the monolayer colony formation assay to evaluate cellular growth in vitro. Cells were cultured overnight in a 12-well plate (5 × 105 cells/well) and transfected with pcDNA3 empty vector or co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector using lipofectamine 2000 (Invitrogen). Forty-eight hours later, the transfectants were replated in triplicate and cultured for 10 ~ 15 days in complete DMEM medium containing G418 (400 ug/ml). Surviving colonies were stained with Gentian Violet after methanol fixation and visible colonies (≥50 cells) were counted. The experiments were repeated three times.
Small interfering RNA (siRNA) preparation and transfection of cells
Predesigned human DUOX1 siRNAs (No. 24969) were purchased from Ambion (Singapore). The 21-nt sequences of DUOX1 (siRNA no. 24969) were (sense) GGACUUAUCCUGGCUAGAGtt and (antisense) CUCUAGCCAGGAUAAGUCCtg. Silencer Negative Control no. 1 siRNA (Ambion) was used as a nonspecific siRNA. SiRNA transfection into Hep3B cells was carried out by using the DharmaFECT 4 Transfection Reagent (Dharmacon, T-2001-02) according to the manufacturer’s protocol. Specific silencing of DUOX1 was confirmed by using real-time quantitative PCR at 72 h after transfection.
Cell growth assay
Cells growth were determined by a non-ra-dioactive proliferation assay based on the ability of metabolically active cells to convert 3-(4,5-dimethylthiazol-2-yl)-5 (3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Promega, Madison, WI, USA) into formazan. Briefly, the transfected cells were transferred into a 96-well plate with different cell number and cultured in complete DMEM medium overnight. The quantity of formazan was measured at 490 nm absorbance after one hour incubation with CellTiter 96 AQueous One Solution Reagent following instructions provided.
Western blotting
The transfected cells were lysed in M2 lysis buffer (20 mM Tris at pH 7, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM glycerol phosphate, 1 mM sodium vanadate and proteinase inhibitor cocktail). Equal amount of protein was fractionated on SDS-PAGE and transferred onto PVDF membrane (Bio-Rad). After blocked with 5% nonfat milk, the membrane was probed with designated antibodies p21 (Santa Cruz), phosphor-cdc25c (Ser216), cyclin D1 (Cell Signaling Technology) and developed with enhanced chemiluminescence method (Pierce) and visualized by Kodak Image Station 440CF (Kodak). The band density was quantified using image processing program and normalized to that of the control group.
Cell cycle analysis and determination of ROS production
The transfected cells were harvested, washed in phosphate-buffered saline and fixed in ice-cold 70% ethanol-phosphate-buffered saline. After washing out ethanol, the fixed cells were treated with 0.01% RNase (10 mg/ml, Sigma, St. Louis, MO, USA) for 10 min at 37°C and then stained with 0.05% propidium iodide for 20 min at 4°C in dark. The cell cycle distribution was determined using a FACScan flow cytometry (Becton Dickinson, Mountain View, CA, USA) and analyzed with Modfit software (Phoenix, San Diego, CA, USA).
ROS levels were measured by using 2’-7’Dichlorodihydrofluorescein diacetate (DCFH-DA) (Invitrogen) in a flow cytometry assay as described [16].
Statistic analysis
The results were expressed as mean ± SEM. Student t test was used to compare the differences of DUOX1 expression on the effect of colony formation and cell proliferation. All statistical calculations were done using SPSS version 11.0 for windows (SPSS, Inc., Chicago, IL). Value of P < 0.05 was taken as statistical significance.
Results
Downregulation of DUOX1 in human liver cancer cells
The expression of DUOX1 was dramatically downregulated in 66.7% (6/9) human liver cancer cell lines including HepG2, Hep3B, HuH7, HCC-LM3, BEL-7402, SMMC-7721, SK-Hep1, MHCC-97H and MHCC-97L (Figure 1A) when compared to human normal liver cells L02. In order to investigate whether downregulation of DUOX1 should be attributed to DNA methylation, the expression of DUOX1 in human liver cancer cell lines before and after 5-Aza-dC treatment were determined by real-time PCR. The expression of DUOX1 was significantly upregulated in liver cancer cell lines HepG2, Huh7, BEL-7402, SMMC-7721, MHCC-97H and MHCC-97L (6/9, 66.7%) after 5-Aza-dC treatment (Figure 1B), indicating that DUOX1 is likely downregulated through promoter hypermethylation in human liver cancer.
Figure 1.
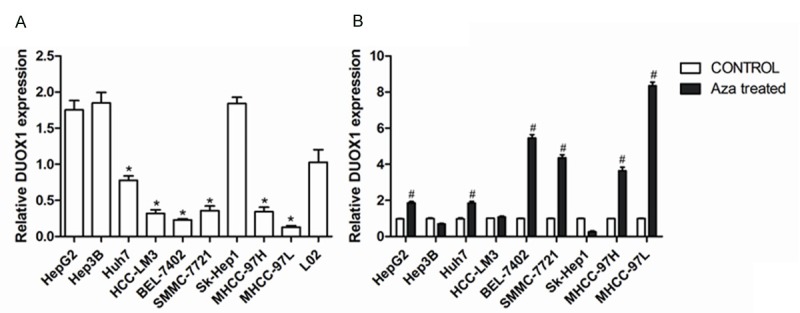
Pharmacological demethylation reversed DUOX1 downregulation in human liver cancers. A: DUOX1 expression in human liver cancer cell lines and normal control was determined by real-time PCR. B: Relative DUOX1 expression level in cancer cells before and after 5-Aza-dC treatment was determined by real-time PCR. Data are mean ± SEM from three independent experiments. *P < 0.05, versus L02; #P < 0.05, versus CONTROL.
Methylation of DUOX1 promoter in human liver cancer cells
In order to confirm the earlier findings, we directly measured the methylation status of DUOX1 promoter. Indeed one typical CpG Islands (CGI) were found around DUOX1 exon 1 using the following criteria: GC content > 55%, Obs CpG/Exp CpG > 0.65, and length > 500bp (Figure 2A). The methylation status of this CGI in human liver cancer cells was determined by methylation-specific PCR (MSP). As shown in Figure 2B, methylation of DUOX1 promoter was readily detected in liver cancer cell lines Huh7, BEL-7402, HCC-LM3, SMMC-7721, MHCC-97H and MHCC-97L which all showed down-regulation of DUOX1, but partly or not in HepG2, Hep3B and SK-Hep1. Besides, BGS also revealed that DUOX1 promoter was heavily methylated in SMMC-7721 and MHCC-97L cells (Figure 2C).
Figure 2.
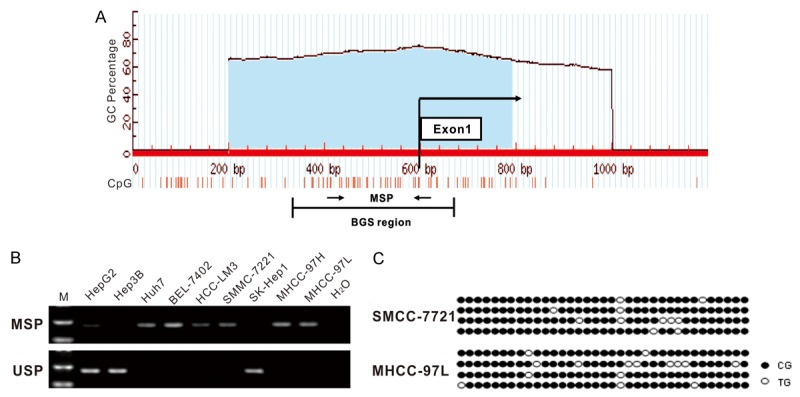
Methylation of DUOX1 promoter in human liver cancers. (A) Schematic structure of the DUOX1 CGI, with the exon 1 and MSP and BGS region indicated. Each short vertical line represents one CpG site. The position of MSP primers were marked as arrows. The methylation status of the DUOX1 CGI was analyzed by MSP (B) and BGS (C). MSP = Methylation-specific PCR; USP = Unmethylation-specific PCR, the gels have been run under the same experimental conditions. For BGS (C), each circle indicates one CpG site and circles filled in black represent methylated CpG sites. One row of circles represents a single colony.
Downregulation and promoter hypermethylation of DUOX1 in human primary hcc tissues
To further confirm the relevance of DUOX1 promoter CGI hypermethylation in mediating its silencing in human liver cancer, DUOX1 expression and promoter methylation in primary hepatocellular carcinoma and adjacent non-tumor tissues were analyzed by real-time PCR and MSP, respectively. DUOX1 expression was significantly downregulated in 76.9% (60/78) human primary HCC tissues (Figure 3A). When compared with adjacent non-tumor tissues. The expression of Duox1 mRNA was not related to the age of the patient at the time of surgery, to the gender, the serum HBsAg, HBeAg, nor to the size of tumor and histological grade (p >0.05; Table 1, Figure 3B). But it seems to have correlation with TNM stage of HCCs (p = 0.049; Table 1, Figure 3C). 20 patients, whose tumors showed significant down-regulation of DUOX1 were chosen for MSP and unmethylation-specific PCR (USP) analysis. The promoter hypermethylation of DUOX1 was detected in 18/20 tumors tissues (Figure 3D). Methylated alleles of DUOX1 were seen in 5/20 adjacent non-tumor tissues, indicating that DUOX1 is mainly downregulated by promoter hypermethylation in HCCs and it may involved in human liver carcinogenesis.
Figure 3.
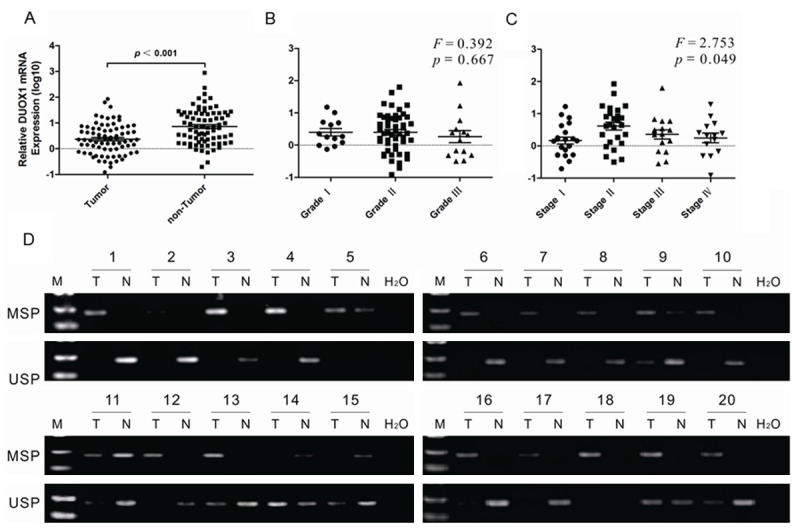
DUOX1 was downregulated and hypermethylated in primary HCC tissues. The expression of DUOX1 in HCC tissues and adjacent non-tumor tissues was determined by real-time PCR. A: 78 pairs of samples were from liver tissue, including tumor tissue and adjacent non-tumor tissue. B: The expression of DUOX1 mRNA in different Edmondson stage of primary HCC tissues. C: The expression of DUOX1 mRNA in different TNM stage of primary HCC tissues. D: The methylation status of DUOX1 promoter in primary HCC tissues and adjacent non-tumor tissues was detected by MSP. Representative results were shown. “T” indicates tumor tissues and “N” represents adjacent non-tumor tissues. The gels have been run under the same experimental conditions.
DUOX1 inhibited cancer cell growth
Whether DUOX1 was a tumor suppressor in cancer cells, cell growth was determined using various assays. The effect of exogenous DUOX1 expression on the growth of human cancer cells was investigated by a monolayer colony formation assay. To further investigate the potential role of DUOX1 in tumor suppression, SMMC-7721 cells were transfected with pcDNA3 empty vector or co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 (DUOXA1, an identified maturation factor for DUOX1, which is located in head-to-head arrangement with DUOX1 on chromosome 15q15) [17] expressing vector and the cell colony formation ability was examined under the selection of G418. Compared with control cells transfected with empty vector pcDNA3, cancer cells co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector showed decreased colony formation ability (Figure 4A). These data suggest that DUOX1 may play a role in tumor suppression. Besides, DUOX1 expression level in the transfected SMMC-7721 cells was also confirmed by western blot as shown in Figure 4B.
Figure 4.
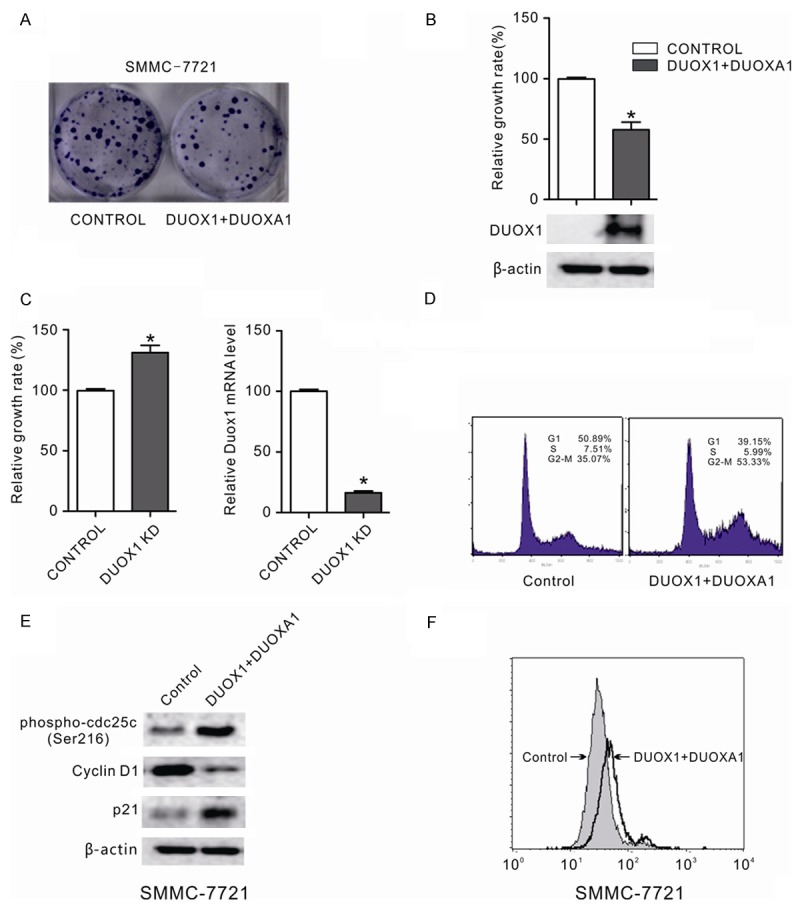
DUOX1 suppressed the growth of cancer cells by inducing G2/M phase cell cycle arrest and increasing ROS production. A: The effect of ectopic DUOX1 expression on cancer cell growth was investigated by the monolayer colony formation assay. The scanned colony formation in six-well plates was shown in the panel. B: The growth of cancer cells after DUOX1 overexpression was determined by MTS cell growth assay. Ectopic expression of DUOX1 suppressed the growth of cancer cells. DUOX1 expression level in transfected SMMC-7721 cells was confi rmed by western blot. C: Knockdown of DUOX1 promoted Hep3B cells proliferation. Hep3B cells were transfected with DUOX1 siRNA (100 nM) or NC siRNA (100 nM). After 48h, the cell growth was determined by MTS and knockdown efficiency was confirmed by real-time PCR. D: Effect of Duox1 on the cell cycle in liver cancer cell. The cell cycle distribution of liver cancer cell line SMMC-7721 with and without DUOX1 expression was evaluated by flow cytometry analysis. E: Effect of Duox1 on cell cycle regulators (phospho-cdc25 (Ser216), p21 and cyclin D1) were determined using western blotting. β-actin was used as the internal control. F: Exogenous DUOX1 increased ROS level in liver cancer cells. The effect of ectopic DUOX1 expression on oxidative stress was determined by flow cytometry assay as shown. Stable cells were stained with DCFH-DA and then the redox state of cells was measured by flow cytometry. Data are mean ± SEM from three independent experiments. The gels have been run under the same experimental conditions.
When pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector were co-transfected into SMMC-7721 cells, the growth inhibitory function of DUOX1 in these cells was analyzed by MTS cell growth assay. The growth rate of those cells dramatically slowed down upon DUOX1 overexpression (p < 0.05; Figure 4B), while knock down of DUOX1 markedly promoted Hep3B cells proliferation (p < 0.05; Figure 4C), showing a growth-suppressive effect of DUOX1 on cancer cells.
Ectopic expression of duox1 induced cell cycle arrest
To determine the molecular mechanism that DUOX1 suppressed colony formation and cell proliferation, we investigated the effect of DUOX1 on cell cycle. After propidium iodide staining, fluorescence-activated cell sorting analysis of SMMC-7721 cells co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector revealed an increase in the number of G2/M phase cells (Figure 4D). To prove these findings, major mediators in cell cycle process were further assessed. Our results showed that overexpression of DUOX1 and DUOXA1 in the SMMC-7721 cells induced phosphorylation of the protein phosphatase cdc25c (Ser216) which is the key regulator in the G2/M phase; the protein level of G0/G1 phase regulator p21 was increased while G1/S phase reulator cyclin D1 was reduced in the SMMC-7721 cells co-transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 (Figure 4E).
DUOX1 overexpression increased intracellular ROS generation
The NADPH oxidases including DUOX1 were linked to generation of reactive oxygen species, in particular superoxide anion-radical and hydrogen peroxide [6,7]. The ROS level was measured in cancer cells transfected with pcDNA3-DUOX1 and pcDNA3.1-DUOXA1 expressing vector. Exogenous DUOX1 clearly increased intracellular ROS level in SMMC-7721 cells (Figure 4F). Therefore, DUOX1 could increase intracellular ROS production which exerts cytotoxic and proapoptotic functions and limits tumorigenicity and malignant progression.
Discussion
More and more novel tumor suppressor genes have been found to be inactivated by promoter hypermethylation. Promoter methylation was thus proposed as an important marker for the identification of novel tumor suppressor genes. In the current study, we identified that DUOX1 was frequently down-regulated in 66.7% HCC cell lines and in 76.9% (60/78) primary HCCs (Figure 1A, 1B), Hypermethylation was further detected in 100% HCC cell lines (Figure 2B) and also in 90% primary HCC tissues with down-regulation of DUOX1 (Figure 3C). In contrast, DUOX1 hypermethylation was occasionally detected in paired adjacent non-tumor tissues, suggesting that DUOX1 may function as a novel tumor suppressor candidate with its downregulation through promoter hypermethylation in the carcinogenesis of hepatocellular carcinoma. We further demonstrated that treatment with the demethylation reagent 5-Aza-dC upregulated DUOX1 expression in lowly expressed liver cancer cells (Figure 1B) and the methylation status was verified by genome sequencing, indicating that DNA hypermethylation mediated DUOX1 inactivation. This is also the first report to show that DUOX1 was epigenetically silenced in human liver carcinogenesis.
Although promoter methylation frequently inactivated DUOX1 in human liver cells, we cannot exclude the involvement of other mechanisms responsible for the DUOX1 downregulation, such as defects in histone remodeling. For instance, in Hep3B, HCC-LM3 and Sk-Hep1 DUOX1 expression failed to be fully upregulated after 5-Aza-dC treatment. Nevertheless, we found that DUOX1 expression was upregulated after 5-Aza-dC in combination with and TSA treatment, one histone deacetylase inhibitor (data not shown).
In addition, to serve as a new marker to define novel tumor suppressor genes, promoter hypermethylation can also be used as a sensitive marker for cancer diagnosis and prognosis prediction [18,19]. As shown in Figure 3, We determined DUOX1 expression in 78 pairs of primary HCCs and methylation status in 20 pairs of primary HCCs, suggesting that DUOX1 is frequently downregulated by promoter hypermethylation in HCCs. Furthermore, the expression of DUOX1 seems to have correlation with TNM stage of HCCs as shown in Table 1 and Figure 3C. Which indicating down-regulation of DUOX1 might be a potential biomarker for HCC diagnosis and prognosis prediction.
As shown in Figure 4, overexpression of DUOX1 markedly reduced colony formation abilities and suppressed cancer cells growth. However, the underlying molecular mechanism responsible for DUOX1 as a tumor suppressor remains unknown. Cell cycle checkpoints are important control mechanism that ensures the proper execution of cell cycle events. The growth suppression induced by ectopic DUOX1 expression seems to be caused by increasing G2/M phase cell number. In addition to inducing cell cycle arrest, importantly in this study, the growth inhibitory effect of DUOX1 as a tumor suppressor may also be mediated through enhancing the production of intracellular ROS (Figure 4F). Generally, the increase in ROS production in epithelial cells is mainly attributed to mitochondrial superoxide production [20]. However, coordinated expression of DUOX1 oxidase and its maturation factor DUOXA1 in some epithelial cancer cells suggests that the intracellular level of ROS (superoxide and hydrogen peroxide) in epithelial cells might be partially controlled by the dual oxidases. Regardless of ROS’s role in cancer initiation and progression, a recent report linked intracellular ROS accumulation to the establishment of senescence, thereby connecting ROS to tumor suppression [21]. Moreover, numerous studies have demonstrated the causative involvement of ROS formation in the mediation of cancer cell apoptosis induced by various standard chemotherapeutic agents including paclitaxel, cisplatin, bortezomib and etoposide. Remarkably, it has been demonstrated that cisplatin apoptogenicity depends on formation of ROS and occurs independent of nuclear DNA damage, suggesting that apoptogenic oxidative stress is the crucial mechanism of cisplatin-induced cancer cell death [22].
In summary, we found that DUOX1 is frequently silenced by promoter hypermethylation in most liver cancer cells and primary HCC tissues. Our results suggested that epigenetic inactivation of DUOX1 was an important factor in the tumorigenesis of liver cancers. We also demonstrated that promoter hypermethylation-mediated silencing of DUOX1 could be reversed by pharmacologic demethylation and overexpression of DUOX1 suppressed tumor cell growth through inducing G2/M phase cell cycle arrest and increasing ROS production, providing direct evidence that DUOX1 functions as a tumor suppressor. Therefore, it will be valuable to explore the possible application of DUOX1 as a molecular marker for the detection and treatment of these malignancies.
Acknowledgements
This work was supported by grants from the National Science Foundation of Shanghai Municipal Health Bureau Project (Grant No. 20114270), by the Shanghai Science and Technology Research Program (Grant No. 12ZR1304300), by the National Natural Science Foundation of China (NSFC) Project (Grant No. 81101240 and 81371821).
Disclosure of conflict of interest
None.
References
- 1.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 3.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 4.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 5.De Deken X, Wang D, Many MC, Costaqliola S, Libert F, Vassart G, Dumont JE, Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233. doi: 10.1074/jbc.M000916200. [DOI] [PubMed] [Google Scholar]
- 6.Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, Edens HA, Tang X, Sullards C, Flaherty DB, Benian GM, Lambeth JD. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol. 2001;154:879–891. doi: 10.1083/jcb.200103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forteza R, Salathe M, Miot F, Forteza R, Conner GE. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;32:462–469. doi: 10.1165/rcmb.2004-0302OC. [DOI] [PubMed] [Google Scholar]
- 8.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 9.Schwarzer C, Machen TE, Illek B, Fischer H. NADPH oxidase-dependent acid production in airway epithelial cells. J Biol Chem. 2004;279:36454–36461. doi: 10.1074/jbc.M404983200. [DOI] [PubMed] [Google Scholar]
- 10.Luxen S, Belinsky SA, Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008;68:1037–1045. doi: 10.1158/0008-5472.CAN-07-5782. [DOI] [PubMed] [Google Scholar]
- 11.Pulcrano M, Boukheris H, Talbot M, Cailou B, Dupuy C, Virion A, De Vathaire F, Schlumberger M. Poorly differentiated follicular thyroid carcinoma: prognostic factors and relevance of histological classification. Thyroid. 2007;17:639–646. doi: 10.1089/thy.2007.0029. [DOI] [PubMed] [Google Scholar]
- 12.Sablina AA, Budanov AV, llyinskaya GV, Aqapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 14.Cabello CM, Bair WB 3rd, Wondrak GT. Experimental therapeutics: targeting the redox Achilles heel of cancer. Curr Opin Investig Drugs. 2007;8:1022–1037. [PubMed] [Google Scholar]
- 15.Fruehauf JP, Meyskens FL. Reactive oxygen species: A breath of life or death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 16.Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 17.Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272. doi: 10.1074/jbc.C600095200. [DOI] [PubMed] [Google Scholar]
- 18.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 19.Jones PA, Baylin SB. The epigenetics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73C:26–33. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Ramsey MR, Sharpless NE. ROS as a tumour suppressor? Nat Cell Biol. 2006;8:1213–1215. doi: 10.1038/ncb1106-1213. [DOI] [PubMed] [Google Scholar]
- 22.Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer. 2007;120:175–180. doi: 10.1002/ijc.22132. [DOI] [PubMed] [Google Scholar]