

Abstract
Monepantel (MPL) is a new anthelmintic agent approved for the treatment of nematode infections in farm animals. As a nematicide, it acts through a nematode-specific nicotinic receptor subtype which explains its exceptional safety in rodents and mammals. In the present study, we evaluated its potential as an anticancer agent. In vitro treatment of epithelial ovarian cancer cells with MPL resulted in reduced cell viability, inhibition of cell proliferation and suppression of colony formation. Proliferation of human ovarian surface epithelial cells and other non-malignant cells were however minimally affected. MPL-induced inhibition was found to be independent of the acetylcholine nicotinic receptor (nAChR) indicating that, its target in cancer cells is probably different from that in nematodes. Analysis of MPL treated cells by flow cytometry revealed G1 phase cell cycle arrest. Accordingly, MPL treated cells expressed reduced levels of cyclins D1 and A whereas cyclin E2 expression was enhanced. Consistent with a G1 phase arrest, cellular levels of cyclin dependent kinases (CDKs) 2 and 4 were lower, whereas expression of CDK inhibitor p27kip was increased. In cells expressing the wild-type p53, MPL treatment led to increased p53 expression. In line with these results, MPL suppressed cellular thymidine incorporation thus impairing DNA synthesis and inducing cleavage of poly (ADP-ribose) polymerase (PARP-1). Combined these pre-clinical findings reveal for the first time the anticancer potential of monepantel.
Keywords: Monepantel, ovarian cancer, cell cycle, cyclins, PARP-1
Introduction
Monepantel [N-[(1S)-1-Cyano-2-(5-cyano-2-trifluoromethyl-phenoxy)-1-methyl-ethyl]-4-trifluoromethylsulfanyl-benzamide; MPL] is a novel anthelmintic drug from a new class of synthetic chemicals known as Amino-Acetonitrile Derivatives (AADs). Due to its favourable pharmacological profile, MPL was selected from a pool of several hundred AADs as the best candidate for development as a nematicide [1]. It has been shown to be efficacious against various species of livestock-pathogenic nematodes [2] and has thus been approved for certain veterinary uses [1,3]. However, it has recently been shown to be ineffective in human soil-transmitted helminthiasis [4] hence hindering its development as a human anthelmintic. Interestingly, MPL has been shown to be very well tolerated in rodents and mammals, indicating that its molecular target is either absent or inaccessible in the host [5]. In susceptible nematodes, MPL affect ligand-gated ion channels leading to interference of signal transduction, possibly at their neuromuscular synapses [1,5]. The affected parasites then experience deregulation in muscle contraction, paralysis, necrosis and finally expulsion from the host. So far, three nicotinic acetylcholine receptors (nAChR) related genes have been identified as the primary targets of MPL [1,6]. Interestingly, all of the three genes encode for the proteins representing the DEG-3 subfamily of nAChR subunits that are only present in nematodes [3,7] which may well explain the exceptional safety of MPL in mammals [1]. The realization that MPL is a very well tolerated agent in mammals coupled with the fact that the other major antiparasitic treatments such as the macrocyclic lactones, imidazothiazoles and in particular the benzimidazole carbamates have all shown some degree of anticancer activity encouraged us to evaluate MPL in laboratory based anticancer tests. Our work on benzimidazole carbamates (BZD) has shown potent anticancer properties for this group of agents and in particular for albendazole in ovarian cancer [8,9]. Similarly, macrocyclic lactones and imidazothiazoles have also been shown to exert anticancer activity in pre-clinical models of cancer [10,11]. We therefore placed MPL in a step by step progressive series of tests starting from visualization of its microscopic effects on cells, to cellular and molecular. Results obtained conclusively demonstrate that MPL has anticancer properties. The effects are characterised by inhibition of cancer cell proliferation and colony formation, induction of cell cycle arrest, interference with the expression of cyclins and cyclin dependent kinases (CDKs), inhibition of DNA synthesis and cleavage of PARP-1. This is the first report to describe the anticancer properties of monepantel.
Materials and methods
Chemical compounds
Monepantel was kindly gifted by Novartis, Basel, Switzerland or purchased from a contract manufacturer. Unless otherwise stated, all other chemicals or reagents used in this study were purchased from Sigma-Aldrich (Sydney, Australia). The following primary antibodies were used throughout this study: mouse monoclonal antibodies specific for cyclin D1 (DCS6), cyclin A (BF683) and rabbit antibodies specific for cyclin E, p27Kip1 and PARP and secondary antibody, anti-rabbit IgG, HRP-linked (Cell signalling Technology, Sydney, Australia), mouse monoclonal anti-GAPDH (Sigma-Aldrich, Sydney, Australia) and rabbit polyclonal antibodies specific for CDK2 (H-298), CDK4 (C-22) and p53 (FL-393) and secondary antibody, anti-mouse IgG conjugated with horseradish peroxidise (Santa Cruz Biotechnology, Sydney, Australia).
Cell lines
The human ovarian cancer cell lines A2780 (p53 wild-type [12]), IGROV-1 (p53 wild-type [13]), OVCAR-3 (p53 mutant [12]), and SKOV-3 (p53 null [12]) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in RPMI 1640 medium with 2 mM l-glutamine, 2 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, 1 mM sodium pyruvate (Invitrogen, Sydney, Australia) supplemented with 10% heat inactivated fetal bovine serum (FBS) and penicillin-streptomycin (50 U/ml) at 37°C in a humidified atmosphere containing 5% CO2. Together with human ovarian surface epithelial (HOSEpiC) in OEpiCM (SienCell, CA, USA). The culture media used were all supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin mixture (Invitrogen, CA, USA), primary human umbilical vein endothelial cells, primary human umbilical vein endothelial cells (HUVEC), Chinese hamster ovarian (CHO) and human embryonic kidney (HEK) cells, used in this study were originally obtained from the American Type Culture Collection (ATCC) and maintained according to their instructions.
Morphology
Staining with Giemsa (Sigma-Aldrich) was used to look at morphology of OVCAR-3 and A2780 cell after treatment with desired amount of MPL (0, 5, 10 and 25 µM) for 24, 48 and 72 h. Briefly; cells (104 cells/well) seeded in 6-well plates and allowed to adhere overnight on cover sleeps followed by treatment with MPL for indicated time points. Cells were then fixed with methanol for 10 min. followed by staining with Giemsa for 15 min. and washed with tap water. The images were captured with Zeiss, AxioCam, AxioSkop microscope, West Germany with 100 × magnifications. Experiment repeated twice with the same result.
Cell viability and cell proliferation assays
The effect of MPL on cell viability of cancer cell lines and normal cells of different origin was measured using the Trypan blue assay. Cells were seeded in 6-well plates and exposed to increasing concentrations of MPL (0-25 μmol/L) for 72 h. MPL was initially dissolved in ethanol and diluted with cell culture media with the final ethanol concentration of 1% (v/v). At the end of treatment period, cells were washed with PBS, trypsinized and counted using trypan blue and a hemocytometer. Each concentration was tested in replicates of 8 and each experiment was repeated twice. Effect of MPL on in vitro cell proliferation was assessed using the sulforhodamine B (SRB) assay [14]. Briefly, cells seeded in 96-well plates (2000-3000 cells/ well) were treated with increasing concentrations of MPL (0-100 μmol/L) for 72 h. Cells were then fixed, washed and stained with 100 μL of 0.4% (w/v) SRB dissolved in 1% acetic acid. Unbound dye was removed by five washes with 1% acetic acid before air drying. Bound SRB was solubilized with 100 μL of 10 mM Tris base (pH 10.5) and the absorbance read at 570 nm. Each drug concentration was tested in replicates of 8 and each experiment was repeated twice. Pooled data from at least two experiments are presented as % control (mean ± SEM) where vehicle treated cells is taken to represent 100% proliferation.
Clonogenic assay
In order to determine the effect of MPL on cell integrity following exposure and then withdrawal of the drug from the media, colony formation assay was performed. Briefly, 5 × 106 cells were plated in 100 mm Petri dishes and allowed to attach overnight. Media were aspirated off and exponentially growing cells were incubated with various concentrations of MPL for 72 h. At this point, the medium was aspirated, the dishes were washed with PBS, and drug free medium was added to each plate. Plates were then incubated for two weeks under standard cell culture conditions in an incubator at 37°C. Following this, plates were gently washed with PBS and cells were fixed with 100% ethanol and stained with a 0.1% solution of filtered crystal violet (w/v). Colonies consisting of more than 50 cells were counted manually. Results are presented as mean ± SEM relative to vehicle treated controls.
Cell cycle analysis
The effect of MPL on the cell cycle was determined using flow cytometry analysis. Briefly, 0.7 × 106 cells seeded in 25 cm3 flasks and allowed to adhere overnight were treated with MPL for 48 h. Cells were collected with trypsinization and pooled with the cells floating in the medium. The cell suspensions were centrifuged, washed with PBS and fixed with methanol. Washed cells were resuspended in propodium iodide and ribonuclease-A (diluted in PBS) for 30 min at room temperature (22°C) and analysed by flow cytometry (Becton Dickinson FACS Calibur).
Western blot analysis
Treated cells were washed in ice-cold PBS and extracted for 30 min with a RIPA buffer containing 10% phosphatase inhibitor and protease inhibitor cocktail (Sigma, St. Louis, MO). Lysates were cleared by centrifugation at 13,000 × g for 30 min and protein concentrations were determined using Bio-red protein assay. Equivalent amounts of whole cell extracts were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore Corporation, MA, USA). The membranes were then probed with specific antibodies. Immune-complexes were detected using horseradish peroxidase conjugated with either anti-mouse or anti-rabbit followed by chemiluminescence detection (Perkin Elmer Cetus, Foster City, CA, USA). To demonstrate equal protein loading, blots were stripped and re-probed with a specific antibody recognizing GAPDH.
3H-thymidine incorporation assay
3H-thymidine assay was used to determine the effect of MPL on cellular thymidine incorporation (DNA synthesis) [15]. Briefly, cells (104) were seeded onto 24-well plates and treated with increasing concentrations of MPL (0-100 μmol/L) for 72 h. For the last 4 h of the incubation, 1 μCi of 3H-thymidine (60 Ci/mM; ICN Biochem) was added to each well. The amount of radioactivity incorporated into cells was determined using a β-scintillation counter. Scintillation fluid was added and counting was performed on a Beckman LS 6000 scintillation counter. Results (mean ± SEM) are expressed as counts per minute (CPM).
Statistical analysis
All data are reported as the mean ± SEM (standard error of the mean). In vitro quantitative variables were compared using the Student’s t-test. Significant statistical difference was defined at p values of < 0.05.
Results
MPL decreases cell viability and proliferation of human ovarian cancer cells
As this is the first report on the effect of MPL (or any other AAD) on the growth of cancer cell-lines, the chemical structure of MPL is depicted in Figure 1. On the same basis we initially examined the effect of MPL on the growth of a range of human cancer and non-cancerous (normal) cells before deciding to investigate in detail its effects on human ovarian cancer cell lines. The cell lines were chosen in a way to represent a wide variety of cancers. The ovarian cancer cell lines (OVCAR-3, A2780, SKOV-3 and IGROV-1) were selected on the basis of their different molecular characteristics and clinical behaviour. In these cells, addition of MPL to media bathing the cells led to concentration-dependent reduction of cell viability, while minimally affecting the normal cells and in particular the HOSE cells. As presented in Figures 2 and 3, MPL treatment profoundly impaired the survival capacity of cancer cells. Trypan blue exclusion assay was used to quantitatively determine the effect of MPL on cell viability. The above listed 4 ovarian cancer cell lines together with normal (non-malignant) human cells (HOSE, CHO, HEK and HUVEC) were all examined for their response to MPL (Figure 3). As demonstrated by the IC50 values, clear difference between the effects of MPL on cancer versus normal non-malignant cells was found. The treatment of cancer cells with MPL resulted in concentration-dependent reduction of cell viability, whereas the viability of normal cells was either hardly affected (HOSE) or far less (CHO) affected by MPL. Comparison of the IC50 values demonstrates that HOSE cells are almost 10 fold less sensitive to MPL than human epithelial ovarian cancer cells such as OVCAR-3 (74.8 ± 7.7 µM for HOSE compared to 7.2 ± 0.2 µM for OVCAR-3). Based on these results, we next examined the effect of MPL on cell proliferation. Treatment of cells with MPL suppressed proliferation of all 4 ovarian cancer cell-lines used in this study (Figure 4). The inhibitory effect of MPL on proliferation was concentration-dependent with IC50 values ranging from 4.4 ± 0.27 μM to 31.18 ± 0.76 μM. It is evident from these results that the ovarian cancer cell lines are quite sensitive to the antiproliferative effects of MPL whereas, normal cells and in particular the HOSE cells are minimally affected (p < IC50 value for MPL in HOSE compared to OVCAR-3 using student’s t-test). Whereas the IC50 value (10.5 ± 0.4 μM) for A2780 with wild-type p53 was quite similar to that for p53 mutant OVCAR-3. The highly chemoresistant p53 null SKOV-3 cells with an IC50 value of 31.2 ± 0.7 μM were found to be the least sensitive of the ovarian cancer cells to MPL which is some 5 to 7 fold higher than values for the other cells.
Figure 1.

Chemical structure of monepantel (MPL).
Figure 2.

MPL inhibits growth of epithelial ovarian cancer cells. Human ovarian cancer cell lines OVCAR-3 and A2780 were grown in 6 well tissue culture plates under standard cell culture conditions in the presence of MPL (0, 5, 10, 25 μmol/L) for 72 h. Cells were then stained with Giemsa, washed and photographed (40 × magnification; using Leica DM IRB light microscope). Experiment repeated twice with the same result.
Figure 3.

Effect of MPL on cell viability is cell specific. Effect of MPL on cell viability was determined using trypan blue dye exclusion assay. Treatment of ovarian cancer cells (top panel) with MPL (0, 5, 10, 25 μmol/L) for 72 h, reduced cell viability and induced cell-death in a concentration-dependent manner. Normal (non-malignant) cells exposed to the same concentrations of MPL over the same period of time were far less susceptible (bottom panel). Effect of MPL on the viability of human ovarian surface epithelial (HOSE) cells was minimal (p < 0.001 at all concentrations compared to OVCAR-3 cells). Each concentration was tested in replications of 8 and each experiment was repeated twice. Data represent mean ± SEM from two independent experiments combined. Population of live and dead cells at the end of treatment period are presented as percentage control (mean ± SEM).
Figure 4.

MPL suppresses proliferation of ovarian cancer cells. The impact of MPL (0, 5, 10, 25, 50 and 100 μmol/L) on cell proliferation was assessed using the SRB assay. Control (vehicle treated) cells were taken to present 100% proliferation and values for the MPL treated groups are expressed as percentage of control (mean ± SEM). Each concentration was tested in replications of 8 and each experiment was repeated twice. Each drug concentration was tested in quadruplicate and each experiment was repeated at least twice. For statistical comparisons, each drug treated group was compared with the control group using Student’s t-test. Then, to confirm concentration dependency of the drug effect, ANOVA was used to compare the between group values.
MPL inhibits colony formation
In order to assess the effect of MPL on the reproductive integrity of cell lines to establish colonies, we next investigated the clonogenic activity of MPL treated cells (OVCAR-3 and A2780). Following 72 h exposure to varying concentrations of MPL (0, 5, 10 and 25 μM), cells seeded in agar plates were then incubated with drug free media and left for 2 weeks in an incubator at 37°C. It was found that MPL exposure leads to profound suppression of colony formation by these cells. Higher concentrations of MPL (> 10 µM) led to almost complete loss (p < 0.001 versus control) of their clonogenic capacity (Figure 5).
Figure 5.
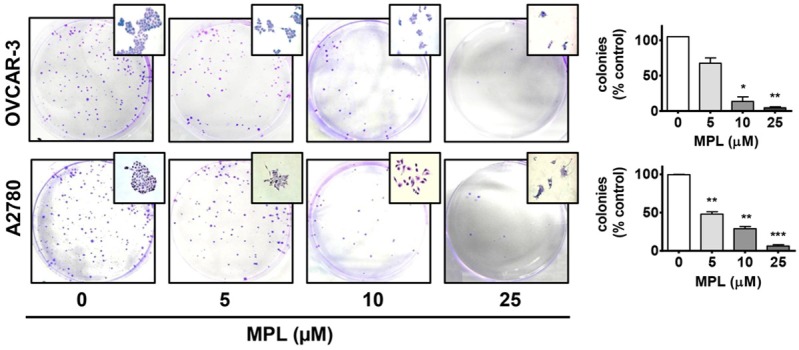
MPL effects on the colony formation activity of ovarian cancer cells. Following incubation of OVCAR-3 and A2780 cells with MPL (0, 5, 10, 25 μmol/L) for 72 h, cells were washed and then transferred to agar plates, cultured with RPMI growth medium (drug free) and incubated under standard conditions for 14 days. Cells were then fixed with 100% methanol and stained with 0.1% crystal violet. Colonies (Cluster of cells greater than 50) were counted manually. Number of colonies counted for different experimental groups (MPL treated) is expressed as percentage of the control.* = p < 0.05; ** = p < 0.01 and ***= p < 0.001 as compared to control (vehicle treated) group using Student t-test.
Antiproliferative activity of MPL is independent of acetylcholine signaling
The nematicidal effect of MPL has been linked to its interaction with the nicotinic subtype MPTL-1 nematode receptor. To determine if the MPL antiproliferative effects are also mediated via acetylcholine receptors, cells were pre-treated with a range of cholinergic/anticholinergic agents. On this basis, cells were initially treated with nicotine and the stable long acting synthetic nicotinic agonist carbachol. Compared to control vehicle treated cells, these nicotinic receptor agonists did not exert an effect of their own nor did they alter the extent of MPL antiproliferative activity (Figure 6). Activity at nematode nicotinic acetylcholine receptors (nAChRs) has been considered to account for a major part of the mechanism of action of several classical anthelmintic agents [16]. To extend these findings, in the next series of experiments, cells were pre-treated with cholinergic receptor antagonists ranging from the broad cholinergic antagonist atropine and then followed by the more nicotinic selective mecamylamine, tubocurarine and progressed to finally using the highly selective irreversible nAChR7α antagonist α-bungarotoxin [17]. The antagonist concentrations used were selected from literature where a positive receptor mediated effect had been reported. It is evident from our results that unlike the nematicidal effects, the antiproliferative activity of MPL in cancer cells is not mediated through the nicotinic signalling pathway. This notion is further supported by the finding that the nematode inactive R-enantiomer of MPL demonstrated equipotent antiproliferative activity in cancer cells (data not shown).
Figure 6.

MPL-antiproliferative effect is not mediated through acetylcholine nicotinic receptor. Pre-treatment (30 min) of OVCAR-3 cells with nicotinic agonist (nicotine, carbachol) or nicotinic antagonists (atropine, mecamylamine, tubocurarine or α-Bungarotoxin) at the indicated concentrations did not change the a ntiproliferative effects of MPL under the cell culture conditions. Proliferation was assessed using SRB assay. Each concentration was tested in replicates of 8 and each experiment was repeated twice. Data represent mean ± SEM from two independent experiments combined.
MPL induces cell cycle arrest through down regulating the expression of cyclins and cyclin-dependent kinases
To find out the mechanism/s through which MPL inhibits growth of cancer cells, using flow cytometry (FACS) we next examined the effects of MPL on cell cycle progression, using the same concentrations as above (0, 5, 10 and 25 µM). It was found that MPL interferes with the cell cycle progression resulting in higher number of cells in the G1 phase in time and concentration-dependent manner (Figure 7). Accumulation of cells in the G1 phase was accompanied by sharp decline of percentage of cells in the S phase. In OVCAR-3 cells the percentage of cells in the G1 phase significantly (p < 0.05) increased from 65.8 ± 6.5 (control) to 87.7 ± 2.2 (MPL 25 µM). Consistent with this, the percentage of cells in the S phase declined from 8.3 ± 1.9 in vehicle treated cells to 2.5 ± 0.5 in 25 µM MPL treated cells (p < 0.05). The effects of various MPL concentrations on the cell cycle and percentage of cells in each phase are also summarized in Figure 7. As the treatment of these cells with MPL induces G1 arrest, we next assessed the effect of MPL on cell cycle regulatory molecules notably cyclins D1, A and E2 together with their associated cyclin-dependent kinases CDK2 and CDK4 and the CDK inhibitors p27Kip1 and p53 (Figure 8A).
Figure 7.

MPL effects on cell cycle progression of human ovarian cancer cells. OVCAR-3 and A2780 cells treated with either MPL or the vehicle (control group) for 48 h were harvested, washed, digested and stained with propidium iodide and analysed by flow cytometry. Distribution of cells in the various phases of the cell cycle (G1, S and G2/M) are presented as percentage, with values (mean ± SEM) representing mean of two independent experiments.
Figure 8.

MPL modulates expression of the G1 cell cycle regulatory proteins. A. Western blot of lysates prepared from cells treated with MPL (0, 5, 10, 25 μmol/L for 48 h) were analysed for the expression of cyclin D1, CDK4, cyclin E2, cyclin A, CDK2, p27kip and p53 proteins. The house-keeping gene (GAPDH) was used to confirm similar protein loading and blot transfer. B. Thymidine incorporation in MPL (0, 5, 10, 25, 50 and 100 μmol/L) treated cells. The y axis presents the actual counts per minute (CPM). These values are mean ± SEM) of two independent experiments. C. Immunoblot analysis for the detection of PARP-1 and cleaved PARP-1 in cells treated for the indicated period of time (24-72 h) with MPL (0, 5, 10, 25 μmol/L).
MPL inhibits thymidine incorporation
Impairment of the cell cycle machinery leading to cell cycle arrest at G1 inevitably leads to reduced DNA synthesis which in turn attenuates cellular thymidine incorporation. Besides this, cyclins, their kinases together with their inducers and inhibitors are all known to regulate thymidine incorporation which is an essential process for the cell proliferation machinery to operate. To determine the effect of MPL on DNA synthesis in these cells, we measured thymidine incorporation. As depicted in Figure 8B, MPL treatment conferred profound reduction in 3H-thymidine incorporation as judged by reduced counts per minute (CPM) produced by the scintillation counter. Under the influence of 25 µM MPL, CPM was reduced by 75.6% in OVCAR-3 and 84.2% in A2780 cells (in both cases p < 0.001 when compared to vehicle treated cells). The results confirm the above findings on MPL-induced cell cycle arrest and inhibition of proliferation in ovarian cancer cell lines.
MPL induces PARP-1 cleavage
PARP help cells to maintain their viability and hence cleavage of PARP facilitates cellular disassembly and serves as marker of cell death [18]. Therefore to determine if the MPL-induced loss of viability, proliferation and clonogenic activity is accompanied by cleavage of PARP-1, using western blot analysis we examined lysates of MPL-treated cells for the expression of cleaved PARP-1 after 24, 48 and 72 h of drug treatment. Under cell culture conditions, MPL induced cleavage of PARP in both OVCAR-3 and A2780 cells. This observation is in line with the above presented data showing inhibition of cell proliferation and colony formation activity together with suppression of thymidine uptake by these cells. MPL-induced PARP-1 cleavage was more evident in A2780 than in OVCAR-3 cells. Whereas in the former, cleaved PARP-1 was clearly detectable at 24 h, in OVCAR-3 cells, cleaved PARP-1 was only detected in 25 µM MPL treated cells in 48 and 72 h samples (Figure 8C). PARP-1 participates in DNA repair, genomic integrity, and cell death [19].
Discussion
Findings from this study reveal for the first time the anticancer properties of monepantel. As an anthelmintic, MPL has been shown to be a safe agent with high selectivity towards nematodes where it is thought to act as an agonist on an acetylcholine nicotinic receptor related structure designated ACR-23/MPTL-1 [3,6]. Here we present the first report to show that the novel AAD compound, MPL significantly inhibits viability, proliferation and colony formation of ovarian cancer cell lines under in vitro cell culture conditions. To find out if the MPL effect is selective towards malignant cells, a number of normal cells including HOSE, CHO, HUVEC and HEK were also treated with MPL. Comparisons of the IC50 values indicate that, MPL is some × 10 more toxic to the ovarian cancer cells than it is to the normal human epithelial cells (HOSE). These results are consistent with literature showing the exceptional safety of MPL in animals [20]. Subsequent experiments revealed the robust G1 cell cycle arrest in cancer cells by MPL. Inhibition of cellular thymidine incorporation followed by the cleavage of PARP-1 confirmed that MPL inhibits growth and proliferation of cancer cells. At this stage, these observations can perhaps be best explained by the effect of MPL on the cell cycle where progression from the G1 phase is halted. Cancer is one of several diseases considered to be a cell cycle related phenomenon. In the normal cell, the transition from one phase to another occurs in an orderly fashion well regulated by various proteins. The cell cycle is tightly controlled at specific points by CDKs, which play a crucial role in cell cycle progression. Of the various CDKs identified so far, CDK2 and CDK4 seem essential for entry in G1 and G1-S transition [21]. For activation, the CDKs require different cyclins at different phases of the cycle. For the G1 transition and progression of the cell cycle, cyclins A, D and E are required [22]. Cyclins A and E bind to CDK2 while cyclin D1 binds to CDK4 and CDK6 [23]. As evidenced by increased percentage of cells in the G1 phase accompanied by the sharp decline of cells present in the S phase, it is quite obvious that in these epithelial ovarian cancer cells, MPL induces cell cycle arrest at the G1 phase. This is confirmed by depressed expression of essential cell cycle regulatory proteins cyclin D1 and cyclin A and their CDKs, CDK4 and CDK2 respectively. However the cyclin E2 results are not so obvious. Whereas in OVCAR-3 cells there was no detectable change in E2 expression with increasing MPL concentrations, in A2780 cells which normally express low levels of E2, a profound increase in cyclin E2 expression is seen at the highest MPL concentration used (25 µM). This basically reveals the very complex nature of the interaction between the cell cycle regulatory apparatus. Based on previous reports on the interaction of the cyclins and their kinases in ovarian cancer, it seems that while both D1 and E2 cyclins act as positive regulators, suppression of cyclin D1 expression drives the cell to produce more of the downstream cyclin E2 to compensate for the D1 loss. However, it has been shown that, suppression of cyclin D1 can still cause G1 cell cycle arrest. Bowe and colleagues have shown that in mice with growing mammary tumors, cyclin D1 deficiency is compensated by cyclin E2 expression [24]. More recently, Masamha and Benbrook have elegantly demonstrated that in ovarian cancer cells irrespective of p53 status, loss of D1 and increased cyclin E2 expression still cause cell cycle arrest at the G1 phase. Thus suggesting that in ovarian cancer cells, attenuation of cyclin D1 availability is sufficient to induce G1 cell cycle arrest [25]. Additionally, consistent with the inhibition of cell proliferation, cyclin A levels, which positively correlate with cell proliferation [26] were reduced in MPL treated cells. The cell cycle progression is negatively regulated by CDK inhibitors such as p27Kip1. Thus, increased cellular CDK inhibitor levels contribute towards G1 cell cycle arrest [27]. In MPL treated cells up-regulation of p27Kip1 was observed.
Blocking progression of cancer cells through both the G1 and the G2-M phases of the cell cycle represents a major area for oncology drug development. Drugs targeting the G2-M phase of the cell cycle have been in common practice as highly effective anticancer agents for decades. These drugs which also include the taxanes, the vinca alkaloids and the epothilones bind to and interfere with the microtubule dynamics [microtubule disrupting agents (MDAs)]. The MDAs arrest the cell cycle progression at the G2-M phase [28]. However, mutations eventually lead to development of resistance to these drugs thus rendering them in-effective. Novel drugs which target the G1 phase of the cycle may therefore end up being used concomitantly with the MDAs to defer development of resistance or alternatively be employed as a treatment option in MDA-resistant patients. Additionally, the frequent loss of G1 regulation in human cancers has turned several components of the G1 phase machinery to potential drug targets. Drugs inhibiting the cyclins and in particular those reducing cyclin D1 levels and activity or the CDKs are therefore highly sought. Cyclin D1 over-expression in many tumors is believed to shorten the G1 phase and to be closely linked to cell cycle progression where it binds and activates its catalytic partner CDK4 leading to out of control cell proliferation [29]. Cell cycle arrest accordingly causes a reduction in lower thymidine uptake and DNA synthesis. Depending on various other factors, events associated with cell cycle arrest can lead to cleavage of PARP. PARP expression is frequently up regulated in ovarian serous carcinomas and may serve as a marker of aggressive behaviour with prognostic value [30]. It is well established that, PARP help cells to maintain their viability and hence cleavage of PARP facilitates cellular disassembly and serves as marker of cells undergoing cell death as its cleavage prevents survival [24]. Our results show that MPL exerts time and concentration-dependent cleavage of PARP-1. Western blot analysis of MPL-treated cells suggests that the A2780 cells are probably more sensitive to the MPL-induced PARP cleavage than OVCAR-3 cells. This may be partly related to the wild-type p53 status of A2780 cells compared to mutated expression of p53 in OVCAR-3 cells. PARP-1 regulates the stability of the wild type p53 protein [31]. Furthermore, p53 can regulate both necrotic and apoptotic cell death. Therefore, mutations or deletions in this tumor-suppressor protein may be selected by cancer cells to provide not only their resistance to apoptosis but also to necrosis, and explain resistance to chemotherapy and radiation even when it kills via non-apoptotic mechanisms [32]. These results provide adequate explanation for the MPL-induced inhibition of cell proliferation and tumor growth. However, the mechanism through which MPL precisely affects the cell cycle regulatory machine remains to be elucidated through further studies. The anthelmintic activity of MPL has been related to its effects on the nematode nAChR α-subunit analogue that holds resemblance to the second transmembrane domain of nAChRα7 subunit and studies on C. elegans indicate that a choline-activated ion channel which is known to be partially permeable to Ca2+ might be involved in the MPL mediated nematicidal activity [33,34]. Here in our study, we found that neither acetylcholine nicotinic receptor antagonist (including the selective α7-nAChRs antagonist, α-bungarotoxin [35]), nor calcium channel blockers (data not shown) have the capacity to prevent the antiproliferative efficacy of MPL in epithelial ovarian cancer cells. These observations suggest that at least in these cells, MPL-induced cytotoxicity is not mediated via nAChRs and are rather the result of the drug’s interaction with the cell cycle regulatory proteins.
In summary, we present the first report on the anticancer capacity of an aminoacetonitrile derivative (monepantel) in pre-clinical models of ovarian cancer. We have shown that monepantel modulates cell cycle regulatory proteins D1, A and E2, reduces the expression of associated cyclin-dependent kinases CDK2 and CDK4 accompanied by up-regulation of p27Kip1 culminating in G1 cell cycle arrest and cleavage of PARP-1. Further studies aimed at determining the effect of monepantel on other types of cancers is currently being extensively pursued.
Acknowledgements
We thank Samina Badar for technical support and Novartis Animal Health Inc. (Basel, Switzerland), for providing us with monepantel.
Disclosure of conflict of interest
FB: No competing interest exist; DLM & MHP: Own stocks in PharmAust and were the inventors of the use of MPL in cancer; LR: is an employee of Novartis, who developed monepantel for intestinal parasites.
References
- 1.Kaminsky R, Ducray P, Jung M, Clover R, Rufener L, Bouvier J, Weber SS, Wenger A, Wieland-Berghausen S, Goebel T, Gauvry N, Pautrat F, Skripsky T, Froelich O, Komoin-Oka C, Westlund B, Sluder A, Maser P. A new class of anthelmintics effective against drug-resistant nematodes. Nature. 2008;452:176–180. doi: 10.1038/nature06722. [DOI] [PubMed] [Google Scholar]
- 2.Kaminsky R, Mosimann D, Sager H, Stein P, Hosking B. Determination of the effective dose rate for monepantel (AAD 1566) against adult gastro-intestinal nematodes in sheep. Int J Parasitol. 2009;39:443–446. doi: 10.1016/j.ijpara.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Rufener L, Keiser J, Kaminsky R, Maser P, Nilsson D. Phylogenomics of ligand-gated ion channels predicts monepantel effect. PLoS Pathog. 2010;6:e1001091. doi: 10.1371/journal.ppat.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tritten L, Silbereisen A, Keiser J. In vitro and in vivo efficacy of Monepantel (AAD 1566) against laboratory models of human intestinal nematode infections. PLoS Negl Trop Dis. 2011;5:e1457. doi: 10.1371/journal.pntd.0001457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rufener L, Baur R, Kaminsky R, Maser P, Sigel E. Monepantel allosterically activates DEG-3/DES-2 channels of the gastrointestinal nematode Haemonchus contortus. Mol Pharmacol. 2010;78:895–902. doi: 10.1124/mol.110.066498. [DOI] [PubMed] [Google Scholar]
- 6.Rufener L, Maser P, Roditi I, Kaminsky R. Haemonchus contortus acetylcholine receptors of the DEG-3 subfamily and their role in sensitivity to monepantel. PLoS Pathog. 2009;5:e1000380. doi: 10.1371/journal.ppat.1000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mongan NP, Jones AK, Smith GR, Sansom MS, Sattelle DB. Novel alpha7-like nicotinic acetylcholine receptor subunits in the nematode Caenorhabditis elegans. Protein Sci. 2002;11:1162–1171. doi: 10.1110/ps.3040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pourgholami MH, Yan Cai Z, Lu Y, Wang L, Morris DL. Albendazole: a potent inhibitor of vascular endothelial growth factor and malignant ascites formation in OVCAR-3 tumor-bearing nude mice. Clin Cancer Res. 2006;12:1928–1935. doi: 10.1158/1078-0432.CCR-05-1181. [DOI] [PubMed] [Google Scholar]
- 9.Pourgholami MH, Cai ZY, Badar S, Wangoo K, Poruchynsky MS, Morris DL. Potent inhibition of tumoral hypoxia-inducible factor 1alpha by albendazole. BMC Cancer. 2010;10:143. doi: 10.1186/1471-2407-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013–1021. [PubMed] [Google Scholar]
- 11.Naito S, Koike K, Ono M, Machida T, Tasaka S, Kiue A, Koga H, Kumazawa J. Development of novel reversal agents, imidazothiazole derivatives, targeting MDR1- and MRP-mediated multidrug resistance. Oncol Res. 1998;10:123–132. [PubMed] [Google Scholar]
- 12.Brüning A, Vogel M, Burger P, Rahmeh M, Gingelmaier A, Friese K, Lenhard M, Burges A. Nelfinavir induces TRAIL receptor upregulation in ovarian cancer cells. Biochem Biophys Res Commun. 2008;377:1309–1314. doi: 10.1016/j.bbrc.2008.10.167. [DOI] [PubMed] [Google Scholar]
- 13.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profi les. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 15.de Fries R, Mitsuhashi M. Quantification of mitogen induced human lymphocyte proliferation: comparison of alamarBlue assay to 3H-thymidine incorporation assay. J Clin Lab Anal. 1995;9:89–95. doi: 10.1002/jcla.1860090203. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz-Lancheros E, Viau C, Walter TN, Francis A, Geary TG. Activity of novel nicotinic anthelmintics in cut preparations of Caenorhabditis elegans. Int J Parasitol. 2011;41:455–461. doi: 10.1016/j.ijpara.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Rao TS, Correa LD, Lloyd GK. Effects of lobeline and dimethylphenylpiperazinium iodide (DMPP) on N-methyl-D-aspartate (NMDA)-evoked acetylcholine release in vitro: evidence for a lack of involvement of classical neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1997;36:39–50. doi: 10.1016/s0028-3908(96)00162-1. [DOI] [PubMed] [Google Scholar]
- 18.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 19.Pagano A, Metrailler-Ruchonnet I, Aurrand-Lions M, Lucattelli M, Donati Y, Argiroffo CB. Poly(ADP-ribose) polymerase-1 (PARP-1) controls lung cell proliferation and repair after hyperoxia-induced lung damage. Am J Physiol Lung Cell Mol Physiol. 2007;293:L619–629. doi: 10.1152/ajplung.00037.2007. [DOI] [PubMed] [Google Scholar]
- 20.Hosking BC, Griffiths TM, Woodgate RG, Besier RB, Le Feuvre AS, Nilon P, Trengove C, Vanhoff KJ, Kaye-Smith BG, Seewald W. Clinical field study to evaluate the efficacy and safety of the amino-acetonitrile derivative, monepantel, compared with registered anthelmintics against gastrointestinal nematodes of sheep in Australia. Aust Vet J. 2009;87:455–462. doi: 10.1111/j.1751-0813.2009.00511.x. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka S, Araki H. Regulation of the initiation step of DNA replication by cyclin-dependent kinases. Chromosoma. 2010;119:565–574. doi: 10.1007/s00412-010-0291-8. [DOI] [PubMed] [Google Scholar]
- 22.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 23.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowe DB, Kenney NJ, Adereth Y, Maroulakou IG. Suppression of Neu-induced mammary tumor growth in cyclin D1 deficient mice is compensated for by cyclin E. Oncogene. 2002;21:291–298. doi: 10.1038/sj.onc.1205025. [DOI] [PubMed] [Google Scholar]
- 25.Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009;69:6565–6572. doi: 10.1158/0008-5472.CAN-09-0913. [DOI] [PubMed] [Google Scholar]
- 26.Khan AA, Abel PD, Chaudhary KS, Gulzar Z, Stamp GW, Lalani EN. Inverse correlation between high level expression of cyclin E and proliferation index in transitional cell carcinoma of the bladder. Mol Pathol. 2003;56:353–361. doi: 10.1136/mp.56.6.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 28.Risinger AL, Giles FJ, Mooberry SL. Microtubule dynamics as a target in oncology. Cancer Treat Rev. 2009;35:255–261. doi: 10.1016/j.ctrv.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musgrove EA, Lee CS, Buckley MF, Sutherland RL. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci U S A. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brustmann H. Poly(adenosine diphosphate-ribose) polymerase expression in serous ovarian carcinoma: correlation with p53, MIB-1, and outcome. Int J Gynecol Pathol. 2007;26:147–153. doi: 10.1097/01.pgp.0000235064.93182.ec. [DOI] [PubMed] [Google Scholar]
- 31.Wesierska-Gadek J, Schmid G. Poly(ADP-ribose) polymerase-1 regulates the stability of the wild-type p53 protein. Cell Mol Biol Lett. 2001;6:117–140. [PubMed] [Google Scholar]
- 32.Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ. 2013;20:1465–1474. doi: 10.1038/cdd.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treinin M, Gillo B, Liebman L, Chalfie M. Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc Natl Acad Sci U S A. 1998;95:15492–15495. doi: 10.1073/pnas.95.26.15492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yassin L, Gillo B, Kahan T, Halevi S, Eshel M, Treinin M. Characterization of the deg-3/des-2 receptor: a nicotinic acetylcholine receptor that mutates to cause neuronal degeneration. Mol Cell Neurosci. 2001;17:589–599. doi: 10.1006/mcne.2000.0944. [DOI] [PubMed] [Google Scholar]
- 35.Donnelly-Roberts DL, Lentz TL. Binding sites for alpha-bungarotoxin and the noncompetitive inhibitor phencyclidine on a synthetic peptide comprising residues 172-227 of the alpha-subunit of the nicotinic acetylcholine receptor. Biochemistry. 1991;30:7484–7491. doi: 10.1021/bi00244a017. [DOI] [PubMed] [Google Scholar]