

Abstract
We have recently shown that the novel anthelmintic drug monepantel (MPL) inhibits growth, proliferation and colony formation, arrests the cell cycle and induces cleavage of PARP-1 in ovarian cancer cell lines. Here we report on the mechanism behind the anticancer properties of MPL. The cytotoxic effect of MPL on ovarian cancer cells (OVCAR-3 and A2780) was investigated employing a panel of tests used for the detection of apoptosis and autophagy. Apoptosis and autophagy were defined by caspase activity, DNA-laddering, Annexin-V and acridine orange (AO) staining. Autophagy markers such as LC3B, SQSTM1/p62 and mammalian target of rapamycin (mTOR) pathway related proteins were assessed by western blotting and ELISA techniques. MPL did not activate caspases 3 or 8, nor did it alter the percentage of Annexin V positive stained cells. Failure to cause DNA laddering and the inability of z-VAD-fmk to block the MPL antiproliferative effects led to the ruling out of apoptosis as the mechanism behind MPL-induced cell death. On the other hand, accumulation of acidic vacuoles with distinct chromatin morphology and an increase in punctuate localization of green fluorescent protein-LC3B, and MPL-induced changes in the expression of SQSTM1/p62 were all indicative of MPL-induced autophagy. Consistent with this, we found inhibition of mTOR phosphorylation leading to suppression of the mTOR/p70S6K signalling pathway. Our findings provide the first evidence to show that MPL triggers autophagy through the deactivation of mTOR/p70S6K signalling pathway.
Keywords: Monepantel, ovarian cancer, apoptosis, PARP, autophagy, mTOR, p70S6K
Introduction
Monepantel [(1S)-1-Cyano-2-(5-cyano-2-trifluoromethyl-phenoxy)-1-methyl-ethyl]-4-trifluoromethylsulfanyl-benzamide] (MPL) was recently introduced in veterinary practice for the treatment of nematode infections in farm animals [1,2]. As a nematicide, MPL affects ligand-gated ion channels thus interfering with signal transduction at neuromuscular synapse and causing dysregulation in muscle contraction and eventual paralysis [1,3]. Interestingly, results from the toxicology studies conducted in various rodents and farm animals have shown that the drug is well tolerated in these animal species [1]. The level of safety seen in farm animals has added weight to the theory that MPL may have a completely novel mode of action involving a specific type of nicotinic acetylcholine receptor (nAChR) that is exclusively expressed in certain nematodes. So far, three nAChR related genes have been identified as the primary targets of MPL [4]. Interestingly, all these three genes encode for the proteins representing the DEG-3 subfamily of nAChR subunits that are currently thought to be only present in MPL-sensitive nematodes [5,6]. MPL has been shown to modulate DEG-3/DES-2 and ACR-23 channels, encoding nAChR α-subunit with resemblance to that of α7 subunit in second transmembrane domain [3]. We have recently shown for the first time that MPL exerts anticancer effects in a wide variety of cancer cells (Submitted article). In particular we found that, under cell culture conditions, MPL inhibits growth, proliferation and colony formation of ovarian cancer cell lines in time and concentration-dependent manner. Further studies revealed that, MPL interferes with the cell cycle progression and arrests cells in the G1 phase of the cell cycle. In line with this, we observed activation of PARP-1 by MPL, suggesting that the MPL-induced cell death may be an apoptotic mediated process. Additionally, we found that the anticancer effects of MPL are not nAChR dependent thus suggesting the involvement of other molecular pathways. These observations prompted the design of the current study where we looked at the mechanisms behind the anticancer activity of MPL. Interestingly, we found that MPL does not induce apoptosis but rather causes persistent autophagy through the deactivation of the mTOR/p70S6K signalling pathway.
Materials and methods
Chemicals and antibodies
Unless otherwise stated, all drugs and chemicals used in this study were obtained from Sigma-Aldrich (Australian subsidiary, Sydney). Monepantel was kindly gifted by Novartis, Basel, Switzerland or purchased from a contract manufacturer. MPL was initially made up in 100% ethanol and then diluted with media to provide the desired final concentration and final ethanol concentration of 1% (V/V). The primary antibodies were throughout this study: rabbit polyclonal or monoclonal antibodies specific for caspase-3 (#9662S), caspase-8 (#4790S), LC3B (#2775S), SQSTM1/p62 (D5E2) (#8025S), P-mTOR (Ser2448) (#2971S), mTOR (7C10) (#2983S), P-Raptor (Ser792) (#2083S), P-p70S6K (Thr389) (#9205S), p70S6K (49D7) (#2708S), P-4E-BP1 (Thr37/46) (236B4) (#2855S), 4E-BP1 (53H11) (#9644S), and secondary antibody; anti-rabbit IgG, HRP-linked (#7074) (dilution: 1/1000) (These were purchased from Cell signalling Technology, Sydney, Australia), whereas mouse Monoclonal anti-GAPDH (dilution: 1/35,000-1/70,000) was obtained from Sigma-Aldrich, Sydney, Australia, the secondary antibody was goat anti mouse IgG conjugated with horseradish peroxidise (dilution: 1/500-1/5000) (Santa Cruz Biotechnology, Sydney, Australia).
Cell lines and media
The human ovarian cancer cell lines OVCAR-3 (PIK3CA gene status; wild [7]) and A2780 (PIK3CA gene status; mutant [8]) used in this study, were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI 1640 medium with 2 mM l-glutamine, 2 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, 1 mM sodium pyruvate (Invitrogen, Sydney, Australia) supplemented with 10% heat inactivated fetal bovine serum (FBS) and penicillin-streptomycin (50 U/ml) at 37°C in a humidified atmosphere containing 5% CO2. Together with human ovarian surface epithelial (HOSEpiC) in OEpiCM (SienCell, CA, USA). The culture media used were all supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin mixture (Invitrogen, CA, USA).
Western blot analysis
Treated cells were washed in ice-cold PBS and extracted for 30 min with a RIPA buffer containing 10% phosphatase inhibitor and 10% protease inhibitor cocktail (Sigma, St. Louis, MO). Lysates were cleared by centrifugation at 13,000 × g for 30 min and protein concentrations were determined using Bio-red protein assay. Equivalent amounts of whole cell extracts were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore Corporation, MA, USA). The membranes were then probed with specific antibodies. Immune-complexes were detected using horseradish peroxidase conjugated with either anti-mouse or anti-rabbit followed by chemiluminescence detection (Perkin Elmer Cetus, Foster City, CA, USA). To demonstrate equal protein loading, blots were stripped and re-probed with a specific antibody recognizing GAPDH.
Caspase activity assay
Caspase-3 and -8 colorimetric assay kits were used according to the manufacturer’s instructions (Bio scientific, R&D Systems, Gymea, NSW, Australia). Briefly, after treatment the cells with indicated concentration of MPL (0, 10 and 25 µM) for 48 and 72 h, cells were harvested, centrifuged at 250 g for 10 min. The cell pellet lysed by the addition of the lyses buffer, then incubated on ice for 10 min followed by centrifugation at 10,000 g for 5 min. The supernatant was used to start the enzymatic reaction in 96 well plates based on manufacturer protocol. Each concentration was tested in replications of 4 and each experiment was repeated twice.
Cell proliferation assay
The effect of MPL with or without other agents on cell proliferation was assessed using the sulforhodamine B (SRB) assay [9]. Briefly, cells were seeded in 96-well plates (2500 cells/well) overnight followed by treatment with desired concentrations of MPL. After 72 h cells were fixed with 200 µl of 0.1% TCA, washed with tap water and stained with 100 µl of 0.4% (w/v) SRB dissolved in 1% acetic acid. Unbound dye was removed by five washes with 1% acetic acid before air drying. Bound SRB was solubilized with 100 µl of 10 mM Tris base (pH 10.5) and the absorbance read at 570 nm. Each concentration was tested in replications of 8 and each experiment was repeated twice. Data represent mean ± SEM from two independent experiments combined.
Annexin V/7-AAD staining
To determine the type of cell death induced by MPL, at indicated time points (24, 48 and 72 h) cells treated with MPL (0, 10 and 25 µM), washed twice in phosphate-buffered saline and then stained with Annexin V-FITC and 7-AAD (7-Aminoactinomycin D), according to the manufacturer’s instructions (BD Biosciences, Sydney, Australia). Then, samples were analysed by fluorescence-activated cell scanner (FACScan) flow cytometer (BD FACSCanto II). Data represent mean ± SEM from three independent experiments combined.
Analysis of inter-nucleosomal DNA fragmentation (DNA ladder)
Genomic DNA was isolated from cells using apoptotic DNA ladder kit (Millipore), subjected to electrophoresis through a 1.5% agarose gel in Tris-borate EDTA buffer, and stained with SYBR® Safe DNA gel stain (Invitrogen, Life technology, Sydney, Australia). Briefly, 2 × 106 cells seeded in 75 ml, medium size flasks and allowed to adhere overnight. Following treatment with MPL for 48 h, cells were trypsinized and counted. The pellet of 5-10 × 105 cells was processed according to the manufacturer protocol. A representative from three independent experiments has been shown.
Morphology
To find out the effect of MPL on cell morphology, cells treated with MPL (0, 10 and 25 µM) stained with giemsa. Briefly, cells (104 cells/well) seeded in six-well plates allowed to adhere overnight on cover sleeps followed by treatment with MPL. Cells fixed with methanol for 10 min. then stained with giemsa (10% in PBS) for 15 min. following by washing with tap water. Cell images captured using Zeiss, AxioCam, AxioSkop microscope, (West Germany) with 40x and 100x oil immersion lenses. A representative image from two independent experiments has been shown.
Quantification of acidic vesicular organelles (AVO) by acridine orange (AO) staining
AO is a fluorescent molecule used either to identify apoptotic cell death or autophagy. It can interact with DNA emitting green fluorescence or accumulate in acidic organelles in which it becomes protonated forming aggregates that emit bright red fluorescence [10]. Briefly, cells were treated with indicated concentrations of MPL (0, 5, 10 and 25 µM) for indicated time points followed by staining with 1 μM acridine orange for 15 min. Cells were then washed, re-suspended in PBS and subjected to FACS analysis. The green (510-530 nm, FL-1) and red (650 nm, FL-3) fluorescence of AO with blue (488 nm) excitation, was determined over 10,000 events and measured on a FACScan cytofluorimeter using the Cell Quest software (Becton Dickinson, San Jose`, CA, USA). Presented results are as the mean ± SEM of duplicate samples from two experiments (P<0.05).
Immuno-fluorescence, confocal scanning microscopy
After incubation with indicated concentration of MPL (0, 10 and 25 µM), cells were fixed in 4% paraformaldehyde prepared in phosphate-buffered saline (PBS), Permeabilized with ice-cold 100% methanol, and immuno-stained with indicated antibodies and related secondary. Fluorescence images were obtained using OlympusIX71 laser scanning microscopy with 60x oil immersion lenses and Zeiss, Vert.A1, AxioCam, MRm with 40x lenses. A microscopic image has been represented from two independent experiments.
Enzyme-linked immunosorbent assay (ELISA) for p70S6K and phosphorylated form
To quantify the endogenous levels of p70S6K and its phosphorylated form, PathScan® intracellular p70S6 Kinase (#7038) and P-p70S6 Kinase (Thr389) (#7063) sandwich ELISA kits was used according to the manufacturer’s instructions (Cell Signaling Technology, Sydney, Australia). Briefly, following treatment with 10 µM MPL, cells were harvested, washed with ice-cold PBS, lysed, and then incubated on ice for 5 min. Lysed cells were scrapped and sonicated on ice followed by 10 min microcentrifuge at 4°C. The lysate was added to well in quadruplicate and incubated for 2 h at room temperature. Following washing with “wash buffer 1X”, the detection antibody cocktail was added to each well and incubated for 1 h at room temperature. The HRP-linked streptavidin was then added to each well for another 30 min at room temperature. Following last washes, TMB substrate was added to each well for 10 min and the process terminated by adding stop solution. Absorbance was read at 450 nm. Data represent mean ± SEM from three independent experiments combined (P<0.05).
Statistical analysis
All statistical analyses were done with Graph Pad Prism software version 6.0. Data are presented as mean ± SEM. The student t-test was used to compare two independent groups’ means. Statistical significance was established at the P<0.05.
Results
MPL inhibits apoptosis
We have previously shown the inhibitory effect of MPL on different human tumour cell lines and observed that ovarian cancer cell lines are highly sensitive to MPL with IC50 values of 7.2 ± 0.2 µM (OVCAR-3) and 10.5 ± 0.4 μM (A2780) [30]. Our previous results showed MPL susceptibility was associated with induction of cleaved PARP-1, a molecule that is vital in DNA repair, genomic integrity, and cell death (apoptosis or necroptosis) [11]. We therefore looked for the presence of MPL-induced active caspase-3 and caspase-8, which are apoptosis indicators. Western blot analysis was used to assess the levels of cleavage of procaspase-3 and procaspase-8 to active forms. MPL treated cell lines failed to show active caspase-3 and -8 (Figure 1A). Quantitative analysis of those caspases activities by colorimetric assay confirmed that MPL treatment has led to significant reduction of both caspases in OVCAR-3 cell line. The percentage of reduction of caspase-3 was 23.1 ± 0.4 (P=0.005) and 31.42 ± 1.68 (P=0.018) after 72 h treatment with 10 and 25 µM MPL, respectively. Similarly, the percentage reduction of caspase-8 after 72 h treatment with 10 µM MPL was 12.9 ± 4.9 and with 25 µM MPL was 41 ± 2.6, P=0.017 (Figure 1B).
Figure 1.
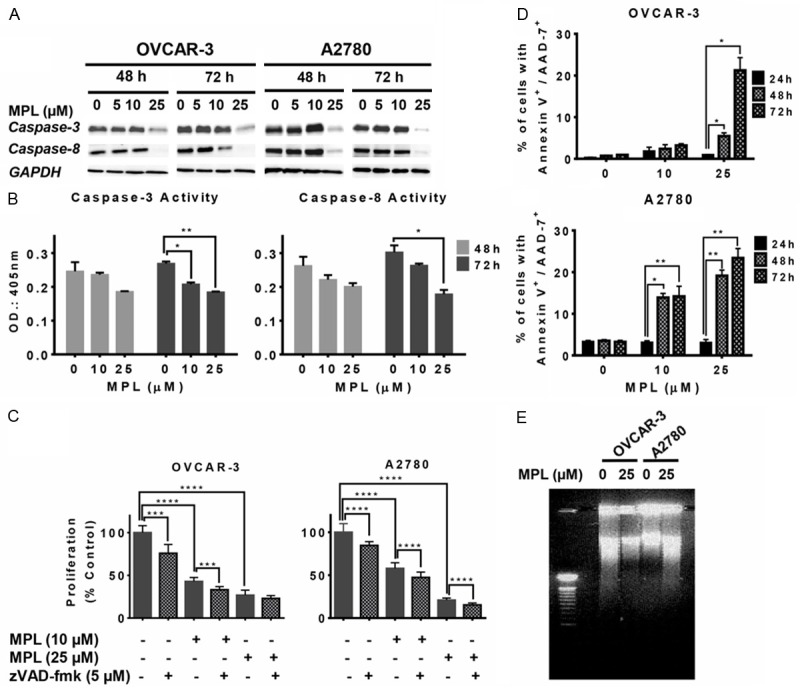
MPL inhibits apoptosis through down regulating caspases. A. Western blot of lysates prepared from OVCAR-3 and A2780 cells treated with MPL (0, 5, 10, 25 μM for 48 and 72 h) were analysed for the expression of caspase-3 and caspase-8 proteins. The house-keeping gene (GAPDH) was used to confirm similar protein loading and blot transfer. A representative from three independent experiments has been shown. B. The impact of MPL (0, 10 and 25 μM) on caspase-3 and -8 activities was assessed in MPL-treated OVCAR-3 cells for the indicated time points (48-72 h). Each concentration was tested in replications of 4 and each experiment was repeated twice. For statistical comparisons, each drug treated group was compared with the control group using Student’s t-test. C. Cell viability estimated by SRB assay in OVCAR-3 and A2780 cell lines after treatment with MPL (0, 10, 25 μM) 30 min. after pan-caspase inhibitor z-VAD-fmk (5 μM) up to 72 h. Each concentration was tested in replications of 8 and each experiment was repeated twice. Data represent mean ± SEM from two independent experiments combined. D. Cells treated with MPL (0, 10 and 25 μM) for the indicated time points (24, 48 and 72 h), stained with Annexin-V/7-AAD and cell death rates analysed by FACS. Experiment repeated twice for each concentration and time points. Data represent mean ± SEM from two independent experiments combined. E. DNA ladders were detected on 1.5% of agarose gel after OVCAR-3 and A2780 cells were treated with 25 μM MPL for 48 h. A representative from three independent experiments is shown.
For further proof, cells were pre-treated with the pan-caspase inhibitor z-VAD-fmk and the effect on cell death induced by MPL (0, 10 and 25 µM) was assessed, using SRB assay. As seen in Figure 1C, addition of z-VAD-fmk did not block the antiproliferative effect of MPL, indicating that the MPL-induced cell death is not caspase-dependent.
Interestingly, we observed a concentration-dependent increase in the extent of Annexin-V+/7-AAD+ labelling (Late apoptosis or necrosis) after 48 and 72 h of treatment with 10 and 25 µM MPL. After 72 h treatment with 25 µM MPL the percentage of dead cells (Annexin-V+/7-AAD+) compared with control increased from 0.95 ± 0.05 to 21.25 ± 2.15, P=0.0136 (22 fold increase) in OVCAR-3. 10 µM MPL had no significant changes. In A2780 cells, compared with control 10 µM MPL led to a 4.5 fold increase in the percentage of dead cells (from 3.1 ± 0.3% to 14.2 ± 1.7%, P=0.0068) and with higher concentration of MPL (≥ 25 µM) peaked to 7.6 fold after 72 h treatment (From 3.05 ± 0.55% to 23.4 ± 1.6%, P=0.0046) (Figure 1D). The percentage increase of Annexin-V+ alone (indicator of early apoptosis) in both cell lines was not significant (Data not shown). Treatment with MPL also failed to cause DNA fragmentation (Figure 1E). Put together, these data suggest that MPL-induced cell death mediated through inhibition of caspases activation and a non-apoptotic process.
MPL induces accumulation of acidic vacuoles
To determine the anticancer effect of MPL, we next examined other cellular responses associated with cell death under MPL treatment and in particular autophagy. Figure 2A shows representative examples of both treated and untreated cell lines. After 72 h treatment with 10 and 25 µM MPL, significant elevation in the number of visible vacuoles in malignant cells was observed, while the normal cells (HOSE) were minimally affected. Compared with the control group, the ultrastructure of giemsa stained OVCAR-3 and A2780 cells treated with MPL up to 72 h showed morphological changes in whole cytoplasm and membrane, including loss of plasma membrane integrity and obvious vacuole formation. The vacuolization was much less pronounced in MPL-treated normal human ovarian surface epithelial cells (HOSE) with no sign of changes in membrane integrity or cell death. This dramatic vacuolization of the cytoplasm without apparent loss of nuclear material is consistent with the described macrostructure of autophagy [12]. Having seen that MPL induces vacuole formation, we next performed FACS analysis of AO stained acidic vacuoles on MPL treated cells. Based on the work of Paglin [10], we used the red to green ratio as an indicator of acidic vacuolar organelle (AVO) accumulation and therefore autophagic progression. Quantification of AVO’s showed a concentration-dependent increase of AVO’s in both OVCAR-3 and A2780 cells (Figure 2B). Treatment with 25 µM MPL up to 72 h, led to 12 fold increase vs. control and the percentage of the bright red fluorescence intensity (y-axis) increased from 0.15 ± 0.05 to 1.8 ± 0.3, P=0.32 in OVCAR-3 cells. In A2780 the percentage of increase was from 0.3 ± 0.1 to 13.55 ± 0.26, P=0.0004 (45 fold increase). These data suggest that treatment with indicated concentration of MPL leads to enlargement of the endosomal/lysosomal system and induction of AVOs in ovarian cancer cells.
Figure 2.
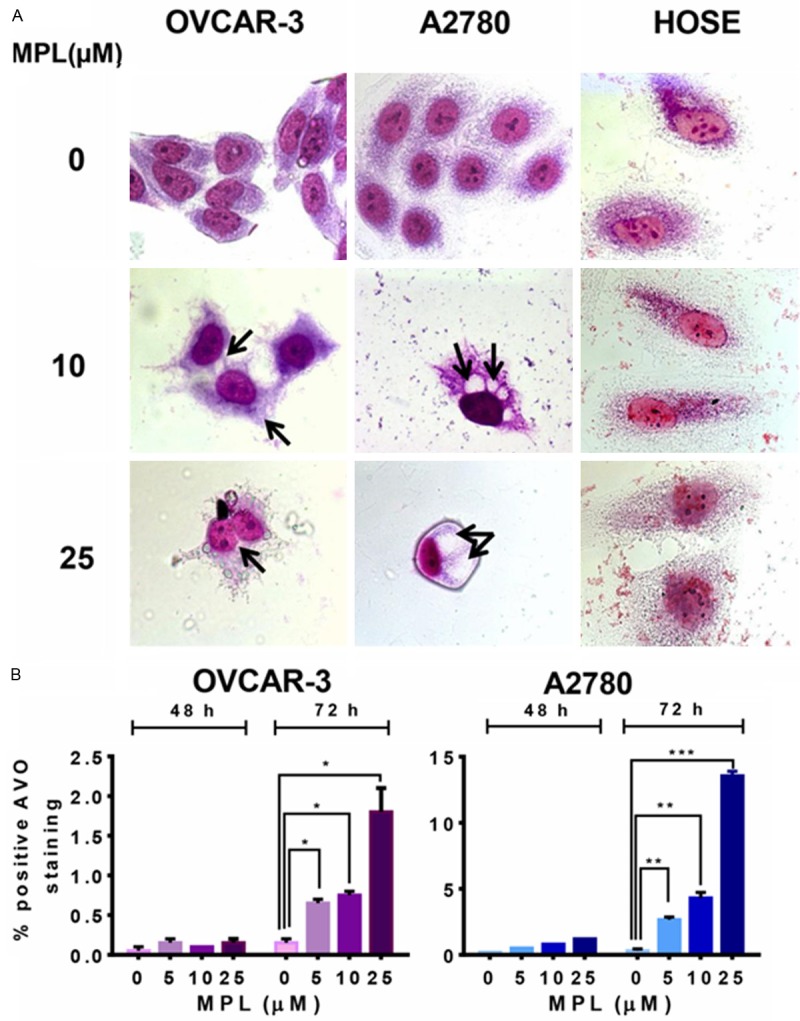
MPL induces accumulation of acidic vacuoles. A. Human ovarian cancer cell lines (OVCAR-3 and A2780) and Normal human ovarian surface epithelial (HOSE) were grown in six-well tissue culture plates under standard cell culture conditions in the presence of MPL (0, 10, 25 μM) for 72 h. Cells were then stained with giemsa, washed and photographed under Leica DM IRB light microscopy (magnification 100x) with oil immersion lenses. A representative from two independent experiments has been shown. B. The percentage of bright red fluorescence (FL3)-positive cells was evaluated by AO staining and FACS analysis in OVCAR-3 and A2780 cells untreated or treated with MPL (0, 5, 10, 25 μM) at different time points (48 and 72 h). Results are the mean ± SEM of duplicate samples from one experiment and repeated three with similar results experiments.
MPL induces autophagy
To determine the possible autophagic nature of the MPL-induced compartments, we next evaluated the effect of MPL on the intracellular localization of microtubule-associated protein 1 light chain 3 beta (LC3B), a specific marker of autophagosomes [13]. Autophagy is associated with LC3B-I being modified to a membrane-bound form, LC3B-II, which is relocated to the autophagosomal membranes during the process of autophagy [14,15]. As shown in Figure 3A, representative fluorescence micrographs show a punctate pattern of LC3B-II. After 48 h treatment with MPL (0, 10 and 25 µM), a marked elevation in the number of cells with visibly increased punctate fluorescence, particularly in the peri-nuclear region of the cytoplasm was observed. This suggests that MPL induces autophagosome formation in these cancer cell lines.
Figure 3.
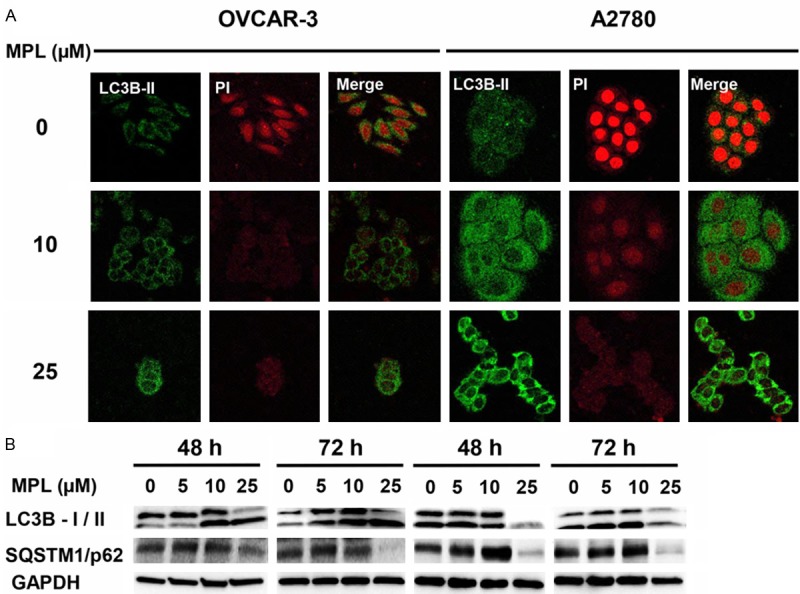
MPL induces autophagy. A. Human ovarian cancer cell lines (OVCAR-3 and A2780) were grown in six-well tissue culture plates under standard cell culture conditions in the presence of MPL (0, 10, 25 μM) for 48 h. Cells were washed, fixed and incubated with primary antibodies in 1% BSA followed by related secondary antibodies in 1% BSA. Fluorescence images were observed and collected under Olympus IX71 laser scanning microscopy, magnification 60x with oil immersion lenses. A representative from two independent experiments has been shown. B. Western blot of lysates prepared from OVCAR-3 and A2780 cells treated with MPL (0, 5, 10 and 25 μM) for the indicated period of time (48-72 h), were analysed for detection of autophagy markers (LC3B and SQSTM1/p62). The house-keeping gene (GAPDH) was used to confirm similar protein loading and blot transfer. A representative from three independent experiments has been shown.
To further verify that MPL does induce autophagy, we assessed the effect of MPL on the conversion of the cytoplasmic form of LC3B-I protein (18 kDa) to the pre-autophagosomal and autophagosomal membrane-bound form of LC3B-II (16 kDa). As shown in Figure 3B, increasing concentrations of MPL induced accumulation of both the unprocessed LC3B-I, and the processed form (LC3B-II). Immunoblot analysis revealed that MPL causes a time and concentration-dependent increase in the levels of LC3B-II proteins. Of note, in A2780 cells after 48 and 72 h treatment with 10 µM MPL, there was a clear increase in both forms of LC3B expression but the LC3B-I was significantly decreased with higher concentration of MPL (≥ 25 µM). Furthermore, MPL-induced autophagy was accompanied by enhanced accumulation of SQSTM1/p62 (Figure 3B).
These results imply that MPL-induced autophagy in OVCAR-3 and A2780 cell lines is SQSTM1/p62 dependent.
Inhibition of MPL-induced autophagy enhanced cell death
Our results so far suggest that MPL induces autophagy. The induction of autophagy in response to metabolic and therapeutic stresses can have a pro-death or a pro-survival role, which contributes to the anticancer efficacy. To evaluate whether MPL-induced autophagy promotes cell death or survival, we applied pharmacologic inhibition of autophagy. 3-methyladenine (3-MA) and wortmannin are known autophagy blockers at an early stage based on their inhibitory effect on class III PI3K activity [16]. While, 3-MA has transient and short period of time effect, wortmannin another early stage autophagy inhibitor, has persistent effects [17].
Figure 4A shows that in A2780 pre-treatment with 3-MA potentiated MPL-induced cell death compared with MPL or 3-MA treatment alone (cell proliferation dropped from 66.4 ± 2.26% to 49.6 ± 3.9% (P=0.001) with 10 µM MPL and from 28 ± 3.5% to 24 ± 3.3% observed with higher concentration, 25 µM MPL). These data are consistent with the morphological changes. Figure 4B presents pre-treatment with 3-MA resulted in amplified accumulation of autophagic marker (vacuoles) and dead bodies.
Figure 4.
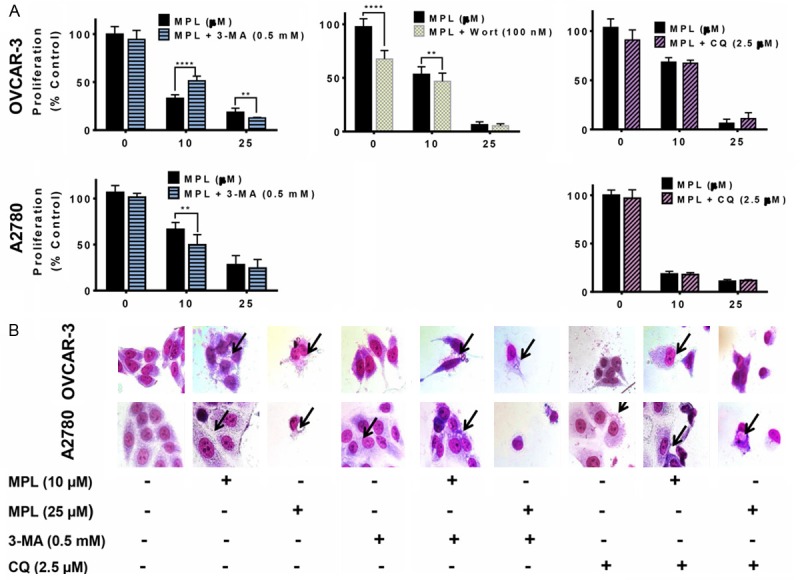
Inhibition of MPL-induced autophagy enhanced cell death. A. OVCAR-3 and A2780 cells pre-treated with 3- methyl adenine (3-MA, class III PI3K inhibitor, 0.5 mM), Wortmannin (selective and irreversible inhibitor of PI3K, 100 nM) and chloroquine (CQ, lysosomal inhibitors, 2.5 μM), 30 min. prior to treatment with indicated concentrations of MPL under the cell culture conditions. Proliferation was assessed using SRB assay. Each concentration was tested in replications of 8 and each experiment was repeated 3 times. Data represent mean ± SEM from all three experiments combined. B. Cells were grown in six-well tissue culture plates under standard cell culture conditions, pre-treated (30 min) with 3-MA (0.5 mM) and CQ (2.5 μM) followed by addition of MPL (0, 10, 25 μM) for 72 h. Cells were then stained with Giemsa (10% in PBS), washed and photographed under Leica DM IRB light microscope (magnification 40x). Black arrows show vacuoles. A representative from two independent experiments has been shown.
3-MA in combination with 10 µM MPL led to an increase in OVCAR-3 cell survival, from 33 ± 1.3% to 51 ± 1.9% (P<0.0001). However, in the presence of 3-MA higher concentration of MPL (25 µM) cell death increased (percentage of cell proliferation decreased from 18 ± 1.4 to 12.7 ± 0.2, P=0.0048). Pre-treatment with wortmannin has no additional effect on 25 µM MPL, but reduced the percentage of cell proliferation from 55.41 ± 2.01 to 46.73 ± 2.12, P=0.0086, with 10 µM MPL in OVCAR-3 (Figure 4A).
To distinguish whether the accumulation of autophagosomes are caused by increased production or alternatively through reduced clearance due to disruption of the autophagic flux, we examined MPL-stimulated autophagosome formation in the presence and absence of chloroquine (CQ). CQ is a specific inhibitor of the lysosomal proton pump that disrupts the autophagic flux by preventing the fusion of autophagosomes with lysosomes [18]. Pre-treatment with CQ did not influence MPL-induced cytotoxicity, suggesting that MPL-induced accumulation of autophagosomes were probably consequence of increased production rather than clearance (Figure 4A and 4B).
mTOR inhibitors sensitize ovarian cancer cells to MPL-induced cell death
To investigate whether promotion of the autophagic pathway has an influence on MPL induced cell death, rapamycin and everolimus, potent inhibitors of mTOR which has been shown to enhance autophagy [19], were used in our study. Co-treatment with rapamycin and everolimus suppressed A2780 cell proliferation further (the percentage of suppression in combination with rapamycin went down from 61.66 ± 1.3 to 33.72 ± 2.8 (P<0.0001) and with everolimus, from 73.50 ± 7.1 to 52.21 ± 3.5, P=0.044). These data are in the same line as morphological evidence of accumulation of big vacuoles as shown in Figure 5B. However, in OVCAR-3 the combination of MPL with mTOR inhibitors had no suppressive effect on cell proliferation compared with either treatment alone (Figure 5A).
Figure 5.
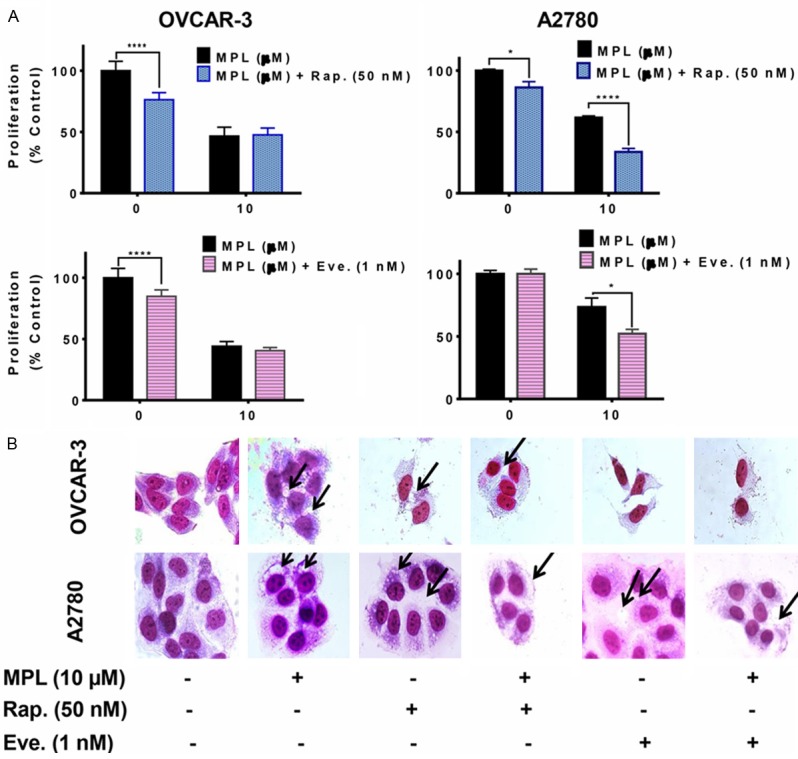
mTOR inhibitors sensitize ovarian cells to MPL-induced cell death. A. Cells pre-treated with 50 nM rapamycin (Rap. mTOR inhibitor, autophagy inducer) and 1 nM everolimus (Eve., ROD001, mTOR inhibitor, autophagy inducer), 30 min. prior to treatment with MPL (10 μM) under the cell culture conditions. Each concentration was tested in replications of 8 and each experiment was repeated 3 times. Data represent mean ± SEM from three independent experiments combined. B. Cells were grown in six-well tissue culture plates under standard cell culture conditions, pre-treated (30 min) with rapamycin and everolimus followed by MPL (0 and 10 μM) for 72 h. Cells were then stained with giemsa (10% in PBS), washed and photographed under Leica DM IRB light microscope (magnification 40x). Black arrows show vacuoles. A representative from three independent experiments has been shown.
MPL suppresses the mTOR/p70S6K signalling pathway
The mTOR/p70S6K signalling pathway is one of the well-established pathways negatively involves in the regulation of autophagy. mTOR associates with tumorigenesis and often activates in numerous types of tumors [20]. Therefore, we examined the effect of MPL on this pathway. To investigate whether mTOR was affected by MPL, we determined its cellular staining pattern using immuno-fluorescence confocal microscopy. Treated cells with MPL (0, 10 and 25 µM) after 4 h exhibited a markedly reduced level of punctate staining indicating the suppression of phosphorylated mTOR at Ser2448, which is consistent with western blot results (Figure 6A and 6B). MPL also decreased the expression of phosphorylated Raptor at Ser792, which is one of the mTORC1 complex members.
Figure 6.
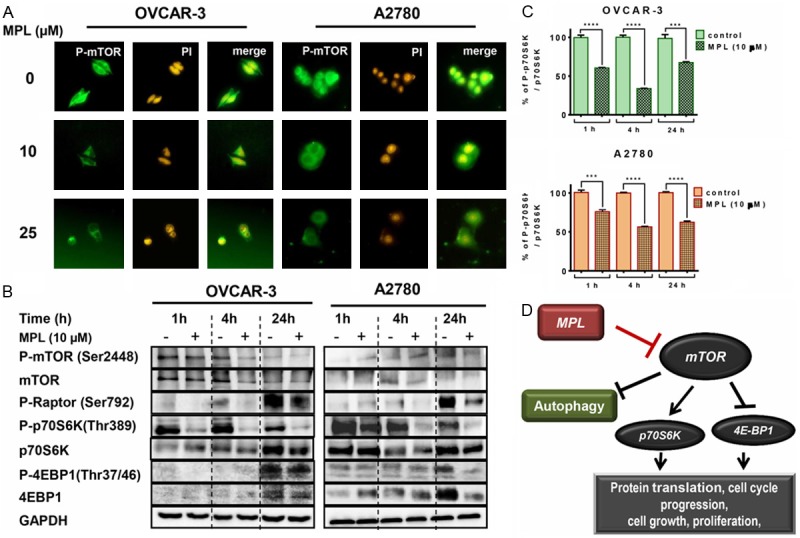
MPL induces autophagy through suppression of the mTOR/p70S6K signalling pathway. A. OVCAR-3 and A2780 were grown in six-well tissue culture plates under standard cell culture conditions in the presence of MPL (0, 10 and 25 μM) for 4 h. Cells were washed, fixed and incubated with primary antibodies in 1% BSA followed by related secondary antibodies in 1% BSA. Fluorescence images were observed and collected under Zeiss, Vert.A1, AxioCam, MRm (magnification 40x). A representative from two independent experiments has been shown. B. Western blot of lysates prepared from OVCAR-3 and A2780 cells treated with 10 μM MPL for the indicated period of time (1, 4 and 24 h), were analysed for detection of mTOR, P70S6K, 4EBP1 and related phosphorylated proteins and p-Raptor. The house-keeping gene (GAPDH) was used to confirm similar protein loading and blot transfer. A representative from three independent experiments has been shown. C. ELISA technique was performed to quantify the level of Phospho-p70S6K at Thr389 and p70S6K in OVCAR-3 and A2780 cells after treatment (1, 4 and 24 h) with 10 μM MPL. Each concentration was tested in quadruplicate and each experiment was repeated 2 times. Data represent mean ± SEM from 2 independent experiments combined. D. The proposed mechanism of MPL-induced autophagy through inhibition of mTOR/p70S6K signalling pathway.
If the mTOR is inhibited in these cell lines, then down-stream signalling molecules, p70S6K and 4E-BP1 are also likely to be affected. We therefore examined the expression of p70S6K and 4E-BP1 after treatment with MPL. As shown in Figure 6B, the expression of mTOR target proteins, 4E-BP1 and p70S6K, were highly reduced in a time-dependent manner. Quantification of P-p70S6K revealed that in both cell lines inhibitory effects started after 1 h treatment with MPL. Percentage of inhibition in OVCAR-3 was 60.6 ± 0.54, P<0.0001 and in A2780, 56.38 ± 0.73, P=0.0007. Maximum inhibition was after 4 h of treatment. Compare to control, phosphorylation of p70S6K suppressed by 33.87 ± 0.5%, P<0.0001 and 56.38 ± 0.73%, P<0.0001 in OVCAR-3 and A2780, respectively (Figure 6C). MPL-treatment for up to 24 h resulted in complete inhibition of phosphorylated 4E-BP1 Thr37/46 in A2780, but it was partial inhibition in OVCAR-3 cells (Figure 6B).
These data confirm that suppression of the mTOR/p70S6K signalling pathway is involved in MPL-induced autophagy.
Discussion
This study provides the first experimental evidence for the induction of autophagy by MPL in human ovarian cancer cells. Here, we have found that MPL-induced toxicity is manifested with features of autophagy (presented with deformation of cytoplasmic content and extensive vacuole formation) exerted through inhibition of mTOR/p70S6K signalling pathway. Several independent pieces of evidence support this conclusion.
Consequent to our recent described data on MPL-induced cleavage of PARP-1 and cell death, and the association of this marker with apoptosis (Submitted article), we anticipated that MPL may act as an apoptogenic agent. Published literature is divided as to whether PARP-1 cleavage is an event that precedes apoptotic cell death or is a marker of another distinct mechanism of cell death [21]. Our results however, did not show caspase-3 or caspase-8 activation in MPL-treated cells. Apoptotic features such as morphological changes in favour of apoptosis, increased level of Annexin-V+ or DNA fragmentation, were not detected. Additionally, MPL-induced antiproliferative effect was not prevented by pre-treatment with the pan-caspase inhibitor z-VAD-fmk, thus further confirming the induction of cell death is independent of the caspase-mediated apoptotic pathways. These results collectively suggest that cell death induced by MPL in ovarian cancer cell lines is not an apoptotic mediated event.
Instead, we conclusively found that cells treated with MPL undergo autophagy. Through LC3B localization studies, we were able to find that MPL treated cells present with typical autophagic morphology and biochemical signature. The autophagic effect of MPL was evident through drug induced expression of SQSTM1/p62 together with the conversion of LC3B-I to LC3B-II in a time and concentration dependent manner. SQSTM1/p62 protein interacts with LC3B-II [14,22] and is degraded in autophagolysosomes. Therefore, its reduction indicates increased autophagic degradation, whereas an increase of SQSTM1/p62 indicates incomplete autophagy [23]. On this line of thought, through accumulation of SQSTM1/p62 and LC3B-II, 10 µM MPL induces incomplete and non-productive autophagy while, higher concentration of MPL (25 µM) triggers active and complete autophagy (elevated LC3B-II along with degradation of other marker). The concept of “autophagic cell death” is commonly accepted based on the presence of autophagic features in dying cells and cell survival via suppression of autophagy [24,25]. Additionally, because autophagy may play a role as a cell survival pathway in response to therapeutic agents the induction of autophagy may work in favour of cancer cells [26]. Cell rescue experiments in which OVCAR-3 and A2780 cells were pre-treated with autophagy inhibitors like 3-MA resulted in decreasing cell viability compared with cells treated with MPL alone. Similar results were obtained in OVCAR-3 from experiments using wortmannin, an agent that inhibits the autophagic process at early stage [17]. The potentiated cell death through pre-treatment with autophagy inhibitors in cells treated with MPL indicates that other mechanisms of cell death may also be involved in MPL-induced cytotoxicity. Moreover, the specific end stage autophagy inhibitor, CQ targeting lysosomal proton pump did not influence the antiproliferative effect of MPL or the drug-induced morphologically changes.
The role of the mTOR, an atypical protein kinase as a positive regulator of protein synthesis, cell growth and proliferation has been well established. mTOR/p70S6K signalling is also known for its ability to halt and regulate cellular catabolic processes such as autophagy and it is frequently dysregulated in cancer and metabolic disorders [27,28]. Consistent with this, here we have described the negative effect of MPL on the mTOR/p70S6K pathway. Combined use of mTOR inhibitors (rapamycin and everolimus) and MPL resulted in further suppression cell viability, suggesting that the inhibition of mTOR signalling may play a role in MPL-induced autophagy. We found that MPL markedly inhibits mTOR phosphorylation at Ser2448, which then prevents activation of the mTOR signalling. On this basis, inhibition of p70S6K and 4E-BP1 may be directly resulting from their upstream, inhibition of mTOR by MPL. Thus, the pharmacologic significance of MPL-induced inhibition of mTOR leads to vacuole formation and autophagy. This is justified by the solid evidence showing inhibition of apoptosis followed by the induction of autophagy. Additionally and in line with that, our results have shown that MPL induced inhibition of protein synthesis and the arrest of cell cycle. Together these observations explain inhibition of growth and proliferation seen in MPL-treated cells.
It is well established that induction of autophagy can provide therapeutic benefits and the modulators of autophagy may provide novel therapeutic tools and may ultimately lead to new therapeutic strategies in cancer. Current data suggest that depending on the context, there are various types of antineoplastic therapies which act through induction of autophagy. Anticancer agents (doxorubicin, temozolomide, etoposide), histone deacetylase inhibitors, imatinib and anti-estrogen hormonal therapy have been shown to induce autophagy [29].
In conclusion, our studies provide the general mechanism behind the MPL-induced autophagy in ovarian cancer cells. Drug-induced autophagy is increasingly becoming a therapeutic strategy in the management of cancer patients. We provide proof-of-concept that MPL induces autophagy by suppressing mTOR/p70S6K pathway (Figure 6D). Studies to further explore the anticancer properties of MPL are currently in progress.
Acknowledgements
We thank Samina Badar for technical support and Novartis Animal Health Inc. (Basel, Switzerland), for providing us with monepantel.
Disclosure of conflict of interest
FB & AHM: No competing interest exist. LR: is an employee of Novartis, who participated in the development of monepantel as a nematicide. DLM & MHP: Own stocks in PharmAust and were the inventors of the use of MPL in cancer.
References
- 1.Kaminsky R, Ducray P, Jung M, Clover R, Rufener L, Bouvier J, Weber SS, Wenger A, Wieland-Berghausen S, Goebel T, Gauvry N, Pautrat F, Skripsky T, Froelich O, Komoin-Oka C, Westlund B, Sluder A, Maser P. A new class of anthelmintics effective against drug-resistant nematodes. Nature. 2008;452:176–180. doi: 10.1038/nature06722. [DOI] [PubMed] [Google Scholar]
- 2.Rufener L, Keiser J, Kaminsky R, Maser P, Nilsson D. Phylogenomics of ligand-gated ion channels predicts monepantel effect. PLoS Pathog. 2010;6:e1001091. doi: 10.1371/journal.ppat.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rufener L, Baur R, Kaminsky R, Maser P, Sigel E. Monepantel allosterically activates DEG-3/DES-2 channels of the gastrointestinal nematode Haemonchus contortus. Mol Pharmacol. 2010;78:895–902. doi: 10.1124/mol.110.066498. [DOI] [PubMed] [Google Scholar]
- 4.Rufener L, Maser P, Roditi I, Kaminsky R. Haemonchus contortus acetylcholine receptors of the DEG-3 subfamily and their role in sensitivity to monepantel. PLoS Pathog. 2009;5:e1000380. doi: 10.1371/journal.ppat.1000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mongan NP, Jones AK, Smith GR, Sansom MS, Sattelle DB. Novel alpha7-like nicotinic acetylcholine receptor subunits in the nematode Caenorhabditis elegans. Protein Sci. 2002;11:1162–1171. doi: 10.1110/ps.3040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufener L, Keiser J, Kaminsky R, Maser P, Nilsson D. Phylogenomics of ligand-gated ion channels predicts monepantel effect. PLoS Pathog. 2010;6:e1001091. doi: 10.1371/journal.ppat.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzenaki N, Andreou M, Stratigi K, Vergetaki A, Makrigiannakis A, Vanhaesebroeck B, Papakonstanti EA. High levels of p110delta PI3K expression in solid tumor cells suppress PTEN activity, generating cellular sensitivity to p110delta inhibitors through PTEN activation. FASEB J. 2012;26:2498–2508. doi: 10.1096/fj.11-198192. [DOI] [PubMed] [Google Scholar]
- 8.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 10.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 11.Chaabane W, User SD, El-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, Los MJ. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (Warsz) 2013;61:43–58. doi: 10.1007/s00005-012-0205-y. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G Nomenclature Committee on Cell D. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009;452:199–213. doi: 10.1016/S0076-6879(08)03613-6. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 15.Barth S, Glick D, KF M. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci. 2010;117:152–162. doi: 10.1093/toxsci/kfq170. [DOI] [PubMed] [Google Scholar]
- 17.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase. J Biol Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moretti L, Yang ES, Kim KW, Lu B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist Updat. 2007;10:135–143. doi: 10.1016/j.drup.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
- 23.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 24.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 25.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16:12–20. doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- 26.Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med. 2009;9:417–424. doi: 10.2174/156652409788167131. [DOI] [PubMed] [Google Scholar]
- 27.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 28.Sridharan S, Jain K, Basu A. Regulation of autophagy by kinases. Cancers (Basel) 2011;3:2630–2654. doi: 10.3390/cancers3022630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bahrami F, Morris DL, Rufener L, Pourgholami MH. Anticancer properties of novel aminoacetonitrile derivative monepantel (ADD 1566) in pre-clinical models of human ovarian cancer. Am J Cancer Res. 2014;4:545–557. [PMC free article] [PubMed] [Google Scholar]