

Interferon-β mediates the activation of ISGF3 and induction of interferon-stimulated genes, by lipopolysaccharide in macrophages.
Keywords: endotoxin, TLR4, ISGF3, NF-κB, STAT
Abstract
TLR agonists such as LPS and poly(I:C) induce expression of type I IFNs, such as IFN-α and -β, by macrophages. To examine the role of IFN-β in the induction of ISGs by LPS, we compared the ability of LPS to induce ISGF3 activity and ISG expression in bone marrow–derived macrophages from WT and Ifnb1−/− mice. We found that LPS treatment activated ISGF3 and induced expression of ISGs such as Oas1, Mx1, Ddx58 (RIG-I), and Ifih1 (MDA5) in WT macrophages, but not in macrophages derived from Ifnb1−/− mice or Ifnar1−/− mice. The inability of LPS to induce activation of ISGF3 and ISG expression in Ifnb1−/− macrophages correlated with the failure of LPS to induce activation of STAT1 and -2 in these cells. Consistent with these findings, LPS treatment also failed to induce ISG expression in bone marrow–derived macrophages from Stat2 KO mice. Although activation of ISGF3 and induction of ISG expression by LPS was abrogated in Ifnb1−/− and Ifnar1−/− macrophages, activation of NF-κB and induction of NF-κB-responsive genes, such as Tnf (TNF-α) and Il1b (IL-1β), were not affected by deletion of either the IFN-β or IFN-αR1 genes. These findings demonstrate that induction of ISGF3 activity and ISG expression by LPS is critically dependent on intermediate production of IFN-β and autocrine signaling through type I IFN receptors.
Introduction
TLRs mediate responsiveness to a wide variety of infectious agents and facilitate induction of many proinflammatory genes. LPS, or endotoxin, is a major outer membrane component of Gram-negative bacteria and induces expression of a wide variety of genes that constitute the innate immune response to Gram-negative bacterial infections. LPS signals through TLR4 on the cell surface of many cell types, especially macrophages and neutrophils. It binds to MD2, a TLR4-associated protein, to facilitate the dimerization of TLR4 that is necessary for productive signaling [1]. Signaling through TLR4 induces rapid activation of 2 distinct intracellular signaling pathways that mediate activation of specific transcription factors, including NF-κB via the MyD88-dependent pathway and IRF-3 via the MyD88-independent or TRIF-dependent signaling pathway [2, 3]. Activation of these signaling pathways results in expression of distinct gene signatures. For example, activation of NF-κB via the MyD88-dependent signaling pathway induces expression of multiple proinflammatory cytokines, such as TNF-α, IL-1β, and IL-12. These cytokines in turn signal through cytokine-specific receptors to induce a cascade of secondary biological responses.
A second major group of cytokines that are produced by LPS-activated macrophages is the type I IFNs [4]. The murine genome contains 14 highly related IFN-α genes and single versions of the more distantly related IFN-β, -ε, and -κ genes [5, 6]. Activation of IRF-3 by viral infection or exposure to TLR agonists such as LPS results in coexpression of several type I IFNs, including IFN-α4, and -β [7–9]. Type I IFNs mediate their biological activities by binding and signaling through type I IFN receptor complexes. IFN-α/β receptor complexes are composed of 2 noncovalently associated transmembrane proteins, IFN-αR1 and -2, which are encoded by the corresponding Ifnar1 and Ifnar2 genes [10]. Signaling through type I IFN receptors induces activation and nuclear translocation of ISGF3 transcription factor complexes that are composed of STAT1, STAT2, and IRF9. ISGF3 complexes subsequently bind to ISREs in the promoters of various ISGs, to induce transcription of these genes [11]. Signaling through type I IFN receptors can also induce formation of STAT1 homodimers that bind to GAS elements in the promoters of a distinct subset of IFN-responsive genes. Studies in Ifnar1−/− mice showed that deletion of this gene abolishes responsiveness to all type I IFNs and markedly increases susceptibility to viral infections [12, 13].
The ability of LPS to induce IFN production by macrophages was first reported many years ago [14, 15], and the importance of NF-κB activation in the induction of proinflammatory gene expression by TLR agonists is well established. However, the role of type I IFNs, particularly IFN-β, in the induction of macrophage gene expression is not as well defined. In this study, we used bone marrow–derived macrophages from Ifnb1−/− mice and Ifnar1−/− mice to examine the role of IFN-β and its receptor in the induction of ISG expression by LPS in macrophages. We found that LPS activates ISGF3 and induces expression of ISGs in WT macrophages, but the ability of LPS to induce activation of ISGF3 and ISG expression is abrogated in IFN-β-null macrophages, as well as in IFN-α/β receptor-null macrophages. These findings demonstrate a critical requirement for endogenous IFN-β expression in the induction of ISG expression by LPS in macrophages.
MATERIALS AND METHODS
Culture medium
The complete medium used for culturing macrophages consisted of RPMI-1640 medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% FBS (HyClone, Logan, UT, USA), 2 mM l-glutamine, and 50 μg/mL gentamicin.
Reagents
Ultrapure LPS from the Escherichia coli 0111:B4 strain was obtained from InvivoGen (San Diego, CA, USA). poly(I:C), MALP-2, and recombinant FliC were obtained from Imgenex Corp. (San Diego, CA, USA). Recombinant murine IFN-β was obtained from R&D Systems, Inc. (Minneapolis, MN, USA).
Cells
Bone marrow–derived macrophage cultures were generated as described elsewhere [16] from bone marrow aspirates extracted from the femurs of WT BALB/c or C57BL/6 mice obtained from Jackson Laboratories (Bar Harbor, ME, USA) or from specific gene KO mice, including Ifnb1−/− mice [17], Ifnar1−/− mice [12], and Stat2−/− mice [18]. The cells were cultured at 1 × 106 cells/mL in complete RPMI-1640 medium containing recombinant murine M-CSF (50 ng/mL) for 7–10 days at 37°C, after which the M-CSF was washed off the cultures, and the cells were treated with LPS or recombinant murine IFN-β, as indicated in the text.
EMSA
Nuclear protein extracts were prepared from cells after treatment with LPS or IFN-β, according to a modification [19] of the original method described by Dignam et al. [20]. A double-stranded oligonucleotide, based on a DNA sequence in the promoter of the human ISG15 gene, was used as a probe for ISGF3 in the gel shift assays [21]. This oligonucleotide contains an ISRE that has a high affinity for ISGF3 complexes. The NF-κB probe, obtained from Promega Corp. (Madison, WI, USA; catalog no. E3292), was a consensus oligonucleotide based on an NF-κB binding site in the Igκ gene [22]. These probes were end-labeled with [γ-32P]-ATP using T4 polynucleotide kinase, and binding reactions were performed [23]. A portion of each binding-reaction mixture (8 μL per sample) was electrophoresed on nondenaturing, 6% polyacrylamide gels (Invitrogen, Carlsbad, CA, USA), using 0.25× Tris-borate-EDTA buffer (22 mM Tris-HCl [pH 8.0], 22 mM borate, and 0.5 mM EDTA). The gels were subsequently dried and visualized by autoradiography.
Antibodies
The mouse monoclonal anti-phospho-Y701-STAT1 Ab was obtained from Invitrogen Corp., and the rabbit polyclonal anti-phospho-Y689-STAT2 Ab was obtained from Millipore Corp. (Temecula, CA, USA). The mouse monoclonal anti-STAT1 Ab (sc-417) and the rabbit polyclonal anti-STAT2 Ab (sc-839) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Western blot analysis
The levels of tyrosine-phosphorylated STAT1 and -2 were measured by Western blot analysis, as described previously [24]. After treatment with LPS or recombinant IFN-β, the cells were washed 3 times with Dulbecco's PBS, and whole-cell lysates were prepared. Proteins were resolved by electrophoresis on 8% SDS-PAGE gels (Invitrogen), and then transferred to polyvinylidene difluoride membranes. The levels of tyrosine-phosphorylated STAT1 and tyrosine-phosphorylated STAT2 were visualized by ECL with mouse monoclonal anti-phospho-Y701-STAT1 Ab or rabbit polyclonal anti-phospho-Y689-STAT2 Ab, respectively.
Northern blot analysis
Total RNA was isolated from macrophages with RNAzol B (Tel-Test, Friendswood, TX, USA) by the acid/guanidinium thiocyanate/phenol/chloroform extraction method [25]. Equivalent amounts of RNA (10 μg/lane) were size fractionated by electrophoresis in 1% agarose gels containing 0.66 M formaldehyde. The RNA was then transferred onto Nytran membranes (GE Life Sciences, Pittsburgh, PA, USA) and cross-linked by exposure to UV light. The membranes were hybridized and washed according to standard procedures [26]. The cDNA probes used to detect ISG expression have been described previously. Gel-purified insert DNA was radiolabeled by the random-primer method of Feinberg and Vogelstein [27].
RPA
RPAs were performed with the RiboQuant Multiprobe RNase Protection Assay System (BD Biosciences, San Jose, CA, USA) [16]. The assays were performed with a custom-designed template set containing probes for the following genes: Tnf, Il1b, Il10, Socs1, and Socs3 and the housekeeping genes Rpl32 and Gapdh. The probes were transcribed in the presence of [α-33P]-UTP (ICN Biomedicals, Irvine, CA, USA; catalog no. 58103). Protected RNA fragments were separated by electrophoresis through 6% polyacrylamide-7 M urea sequencing gels (CastAway Precast Sequencing System; Stratagene, La Jolla, CA, USA) and visualized by autoradiography.
Real-time qPCR
Changes in gene expression identified by Northern blot analysis were verified by qRT-PCR analyses of individual ISGs [28]. Total cellular RNA was isolated and treated with DNase, to remove any residual genomic DNA, by using RT2 qPCR-grade RNA isolation kits from Qiagen, Inc. (Valencia, CA, USA). One microgram of purified RNA from each treatment group was used as a template for synthesis of first-strand cDNAs with ReactionReady First Strand cDNA synthesis kits (Qiagen). Specific primer assays for selected ISGs were obtained from Qiagen and were analyzed on the Mx3000P system (Agilent Technologies, Santa Clara, CA, USA). PCR amplification was performed by thermal cycling with SuperArray RT2 Real-Time SYBR Green PCR Master Mix with real-time detection by SYBR Green and 5-carboxy-X-rhodamine dyes on the Mx3000P instrument, per the manufacturer's instructions. Changes in gene expression levels were analyzed by using MxPro software v.4.10 (Agilent), and the results are expressed as the mean fold increase relative to the control levels after normalization to the housekeeping gene, GAPDH. Changes in the relative levels of gene expression in treated vs. nontreated control cells were calculated by using the 2−ΔΔCT method [29]. Graphing and statistical analysis of qPCR results were performed with Prism 5.0 (Graph Pad Software, San Diego, CA, USA). Values represent the mean ± sd of triplicate determinations. Each of the experiments in this study was performed at least 3 times (n=3) with 3 separate sets of bone marrow–derived macrophages from WT, Ifnb1−/−, and Ifnar1−/− mice. The Northern blots and confirmatory qPCR analyses were each performed 3 times with similar results.
RESULTS
Induction of ISGF3 activity by TLR agonists in macrophages is IFN-β dependent
The activation of macrophages by signaling through TLRs induces expression of many genes, including proinflammatory cytokines, such as TNF-α, IL-1, and IL-12 and IFNs such as IFN-α and -β. IFN-α and -β can, in turn, induce autocrine expression of various ISGs in macrophages. To examine the role of IFN-β in induction of ISGF3 activity by various TLR agonists, we prepared cultures of bone marrow–derived macrophages from WT and Ifnb1−/− mice [17] and treated the cells with MALP-2 (100 ng/mL), poly(I:C) (10 μg/mL), LPS (100 ng/mL), or FliC (100 ng/mL) for 4 h at 37°C. At the end of the incubation period, nuclear protein extracts were prepared and assayed for ISRE DNA-binding activity by EMSA. As shown in Fig. 1A, treatment with the TLR3 agonist poly(I:C), TLR4 agonist LPS, or TLR5 agonist FliC, but not the TLR2 agonist MALP-2, induced ISGF3 activity in the WT macrophages. In contrast, none of these agents induced ISGF3 activity in the Ifnb1−/− macrophages. Although none of the TLR agonists (MALP-2, poly(I:C), LPS, or FliC) induced ISGF3 activity in the IFN-β-null macrophages, all 4 of these agents induced activation of NF-κB in both WT (Ifnb1+/+) and Ifnb1−/− macrophages (Fig. 1B), indicating that the cells responded to all 4 of these TLR agonists.
Figure 1. Induction of ISGF3 activity by TLR agonists in macrophages is IFN-β dependent.

Cultures of bone marrow–derived macrophages from WT or Ifnb1−/− mice were treated with MALP-2 (100 ng/mL), poly(I:C) (10 μg/mL), LPS (100 ng/mL), or flagellin (FliC) (100 ng/mL) for 4 h at 37°C. At the end of the incubation period, nuclear protein extracts were prepared and assayed by EMSA for (A) ISGF3 activity with an ISRE probe based on the promoter of the human ISG15 gene and (B) NF-κB activity with a consensus probe based on the promoter of the Igκ gene.
Induction of ISGF3 activity by LPS in macrophages requires expression of type I IFN receptors
IFN-α and -β induce ISG expression by signaling through type I IFN receptor complexes that are composed of the IFN-α receptor-1 and -2 polypeptide chains encoded by the Ifnar1 and Ifnar2 genes, respectively [10]. To determine whether type I IFN receptor expression is necessary for induction of ISGF3 activity by LPS, cultures of bone marrow–derived macrophages were prepared from WT, Ifnb1−/−, and Ifnar1−/− mice, and treated with LPS (100 ng/mL) or recombinant IFN-β (10 ng/mL) for 1 or 4 h at 37°C. Nuclear protein extracts were then prepared and assayed for ISGF3 activity by EMSA. Fig. 2A shows that LPS treatment induced delayed activation of ISGF3 in the WT macrophages, but this was not observed in the IFN-β-null macrophages or in the IFN-α/β receptor–null macrophages. In contrast, recombinant IFN-β induced rapid activation of ISGF3 in the WT macrophages and in the IFN-β-null macrophages, but not in the IFN-α/β receptor–null macrophages.
Figure 2. Induction of ISGF3 activity by LPS in macrophages requires expression of type I IFN receptors.
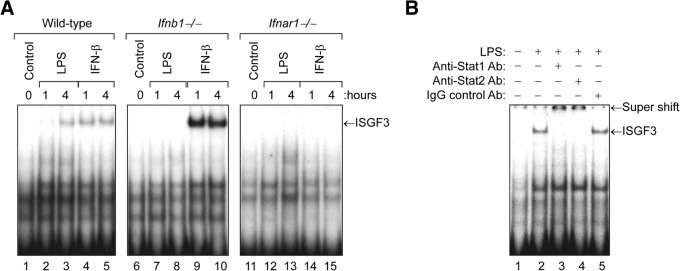
Cultures of bone marrow–derived macrophages from WT, Ifnb1−/−, or Ifnar1−/− mice were treated with LPS (100 ng/mL) or recombinant murine IFN-β (10 ng/mL) for 1 or 4 h at 37°C. (A) At the end of the incubation period, nuclear protein extracts were prepared and assayed for ISGF3 activity by EMSA with the ISRE probe. (B) To confirm that the ISGF3 complexes contained both STAT1 and -2, we preincubated the nuclear protein extract derived from LPS-stimulated WT macrophages (4 h) with rabbit anti-STAT1 or -2 Ab for 15 min before addition of the radiolabeled ISRE probe. Pretreatment with either the anti-STAT1 or -2 Ab, but not a control rabbit IgG Ab, resulted in retardation of the ISGF3 complexes.
The magnitude of ISGF3 activity induced by LPS in the WT macrophages was much greater at the 4 h time point than at the 1 h time point, because it takes several hours for the cells to produce endogenous IFN-β protein to induce autocrine activation of ISGF3 [30, 31]. In contrast, induction of ISGF3 activity by recombinant IFN-β in the WT macrophages was maximal at the 1-hour time point, because no lag period is necessary for production of IFN-β protein. Although Ifnb1−/− macrophages cannot produce IFN-β, they are fully responsive to exogenous IFN-β. In contrast, the Ifnar1−/− macrophages do not express type-I IFN receptors and therefore cannot respond to endogenous IFN-β produced in response to LPS stimulation or to treatment with exogenous IFN-β.
To confirm that the ISGF3 complexes detected by EMSA contained both STAT1 and -2, we preincubated the nuclear protein extract derived from LPS-stimulated WT macrophages (Fig. 2A, lane 3) with rabbit anti-STAT1 or -2 Ab for 15 min before addition of the radiolabeled ISRE probe. As shown in Fig. 2B, pretreatment with either the anti-STAT1 or -2 Ab, but not a control rabbit IgG Ab, caused a supershift of the ISGF3 complexes, thereby confirming that these complexes contain both STAT1 and -2.
The inability of LPS to induce ISGF3 activity in IFN-β-null macrophages correlates with failure to induce tyrosine phosphorylation of STAT1 and -2
ISGF3 complexes are heterotrimers composed of STAT1, STAT2, and IRF-9, and formation of ISGF3 transcription factor complexes is dependent on tyrosine phosphorylation of STAT1 and -2. To examine the role of STAT1 and -2 in the activation of ISGF3 by LPS, we treated bone marrow–derived macrophages from WT and Ifnb1−/− mice with LPS (100 ng/mL) at 37°C for 0 to 24 h. At the specified time points, nuclear protein extracts or whole-cell lysates were prepared and assayed for ISGF3 activity by EMSA and for activation of STAT1 and -2 by Western blot analysis with anti-phospho-STAT1 and anti-phospho-STAT2 Abs. Fig. 3A shows that LPS treatment induced time-dependent activation of ISGF3 in the WT macrophages but not in the Ifnb1−/− macrophages, confirming the data in Fig. 2A. The time-dependent activation of ISGF3 induced by LPS in the WT macrophages correlated well with the activation of STAT1 and -2, as measured by Western blot analysis with anti-phospho-STAT1 and anti-phospho-STAT2 Abs (Fig. 3B). In contrast, LPS treatment did not induce phosphorylation of STAT1 or -2 in IFN-β-null macrophages. These findings indicate that other type I IFNs, such as IFN-α, do not play a significant role in the activation of STAT1 and -2 or the generation of ISGF3 complexes by LPS-stimulated macrophages.
Figure 3. The inability of LPS to induce ISGF3 activity in IFN-β-null macrophages correlates with failure to induce tyrosine phosphorylation of STAT1 and -2.
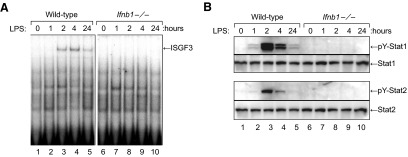
Cultures of bone marrow–derived macrophages from WT or Ifnb1−/− mice were treated with LPS (100 ng/mL) at 37°C for 0 to 24 h. (A) At each time point, nuclear protein extracts were prepared and assayed for ISGF3 activity by EMSA. (B) A matching set of whole-cell lysates was also prepared and used to measure activation of STAT1 and -2 by Western blot analysis with anti-phospho-STAT1 and anti-phospho-STAT2 Abs.
The MyD88-dependent pathway leading to proinflammatory cytokine expression is mediated via activation of NF-κB [1, 32]. To determine whether the inability of LPS to induce ISGF3 activity in IFN-β-null macrophages correlates with a similar inability to induce activation of NF-κB, we examined the ability of LPS to induce NF-κB activity in WT vs. Ifnb1−/− macrophages. As shown in Fig. 4A, LPS treatment induced equivalent activation of NF-κB in both the WT and the IFN-β-null macrophages. In addition, the TLR3 agonist poly(I:C), which also induced activation of ISGF3 in the WT but not the Ifnb1−/− macrophages (Fig. 1A), induced equivalent levels of NF-κB activity in WT and Ifnb1−/− macrophages (Fig. 4B). Therefore, in contrast to the activation of ISGF3, activation of NF-κB by TLR agonists such as LPS and poly(I:C) is not dependent on IFN-β.
Figure 4. Activation of NF-κB by LPS or poly(I:C) is not suppressed in IFN-β-null macrophages.
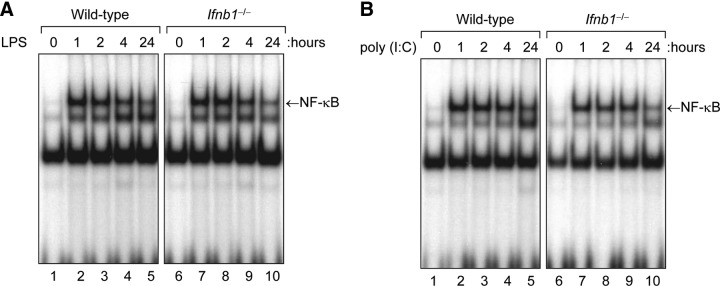
. Cultures of bone marrow–derived macrophages from WT or Ifnb1−/− mice were treated with (A) LPS (100 ng/mL) or (B) poly(I:C) (10 μg/mL) at 37°C for 0 to 24 h. At the specified time points, nuclear protein extracts were prepared and assayed for NF-κB activity by EMSA.
IFN-β expression is necessary for induction of ISG expression by LPS in macrophages
Activated ISGF3 catalyzes expression of multiple ISGs. To examine the role of IFN-β in the induction of ISG expression by LPS, we prepared bone marrow–derived macrophages from WT, Ifnb1−/−, and Ifnar1−/− mice and treated the cells with LPS (100 ng/mL) or IFN-β (10 ng/mL) for 1 or 5 h at 37°C. Total RNA was then extracted and analyzed by Northern blot analysis for expression of several ISGs, including Oas1, Mx1, Ddx58 (RIG-I), and Ifih1 (MDA5). As shown in Fig. 5A, both LPS and IFN-β induced expression of all 4 ISGs in the WT macrophages; however, only IFN-β, but not LPS induced expression of these genes in the Ifnb1−/− macrophages. It is noteworthy that although LPS failed to induce expression of the Oas1, Mx1, or Ddx58 genes in the Ifnb1−/− macrophages, we observed weak induction of the Ifih1 gene that encodes MDA5 in the Ifnb1−/− macrophages, suggesting that expression of this gene is also positively regulated by cytokines other than IFN-β. As expected, neither LPS nor IFN-β induced ISG expression in the Ifnar1−/− macrophages, because these cells do not express type I IFN receptors.
Figure 5. IFN-β expression is required for induction of ISG expression by LPS or poly(I:C) in macrophages.

Cultures of bone marrow–derived macrophages from WT, Ifnb1−/−, and Ifnar1−/− mice were treated with LPS (100 ng/mL) or IFN-β (10 ng/mL) for 1 or 5 h at 37°C. (A) At the specified time points, total RNA extracts were prepared and analyzed by Northern blot analysis for expression of several ISGs, including Oas1, Mx1, Ddx58 (RIG-I), and Ifih1 (MDA5). The blot was also probed to measure TNF-α mRNA expression levels. (B) Another set of bone marrow–derived macrophages from WT or Ifnb1−/− mice were treated with LPS or poly(I:C) for 4 or 24 h at 37°C. At the specified time points, RNA extracts were prepared and assayed by qPCR for expression of several ISGs, including Mx1, Isg20, and Irf7.
In a related set of experiments, we compared the ability of LPS and poly(I:C) to induce ISG expression in WT vs. Ifnb1−/− macrophages. The cells were treated with LPS or poly(I:C) for 4 or 24 h at 37°C, and then RNA extracts were prepared and assayed by qPCR for expression of several ISGs, including Mx1, Isg20, and Irf7. As shown in Fig. 5B, treatment with LPS or poly(I:C) induced significant expression of these ISGs in the WT macrophages, but the levels of expression were markedly decreased in the IFN-β-null macrophages. Therefore, although LPS and poly(I:C) signal via distinct TLRs in macrophages, they share a common requirement for IFN-β expression to induce downstream ISG expression.
To determine whether the induction of proinflammatory genes, such as Tnf and Il1b, by LPS is also dependent on IFN signaling, we compared the ability of LPS to induce expression of these genes in macrophages derived from WT and Ifnar1−/− mice. Macrophage cultures were treated with LPS (100 ng/mL) or IFN-β (10 ng/mL) for 0, 2, 4, or 6 h at 37°C. At each time point, RNA was extracted and analyzed by RPA for expression of the following genes: Tnf (TNF-α), Il1b (IL-1β), Il10 (IL-10), Socs1, and Socs3. As shown in Fig. 6, IFN-β treatment induced detectable expression of Il10 and Socs1 mRNA in the WT macrophages, but not in Ifnar1−/− macrophages. In contrast, LPS induced strong expression of TNF-α, IL-1β, and Socs3 mRNA in both the WT and the Ifnar1−/− macrophages. LPS treatment also induced delayed expression (t=4 h) of the IL-10 gene in the WT macrophages but not in the Ifnar1−/− macrophages. Therefore, the Tnf, Il1b, and Socs3 genes are inducible by LPS, but do not require IFN-α/β receptor signaling for induction; whereas, LPS induction of other genes, such as Il10 and Socs1, is mediated via production of endogenous IFN-β and signaling through cognate IFN-α/β receptors.
Figure 6. The ability of LPS to induce expression of pro-inflammatory cytokine genes, such as TNF-α and IL-1, does not require signaling through type I IFN receptors.
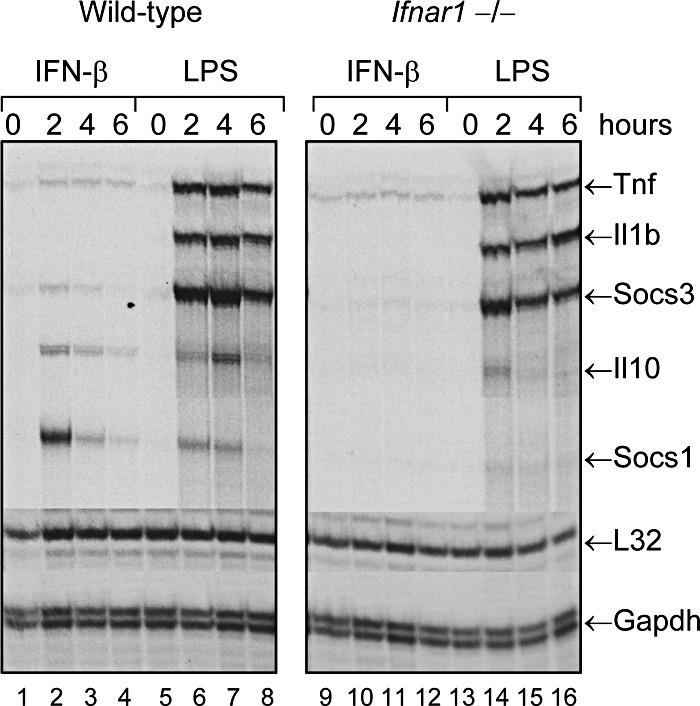
Bone marrow–derived macrophages from WT or Ifnar1−/− mice were treated with LPS (100 ng/mL) at 37°C for 0, 2, 4, or 6 h. At the specified time points, RNA extracts were prepared and analyzed by RPA for expression of several genes, including Tnf (TNF-α), Il1b (IL-1β), Il10 (IL-10), Socs1, and Socs3.
For most of the experiments described in this report, the cells were treated with LPS at a concentration of 100 ng/mL. To determine whether higher concentrations of LPS (e.g., 1 or 10 μg/mL) would overcome the failure of LPS at 100 ng/mL to induce activation of ISGF3 and ISG expression in IFN-β-null macrophages, we examined a full range of LPS concentrations (10, 1, 0.1, 0.01 and 0.001 μg/mL) for their ability to induce activation of STAT1 and -2 and ISG expression in WT vs. Ifnb1−/− macrophages. As shown in Fig. 7A, treatment with LPS at higher concentrations (i.e., 1 or 10 μg/mL) induced activation of STAT1 and -2 in the WT macrophages, but not in the IFN-β-null macrophages. In addition, treatment with LPS at 1 or 10 μg/mL induced ISG expression (Irf7, Isg20, Mx1, Cxcl10, Oas1g, and Ddx58) in the WT macrophages, but not in the IFN-β-null macrophages (Fig. 7B). These findings demonstrate that higher concentrations of LPS (1 or 10 μg/mL) do not overcome its inability to induce STAT activation and ISG expression in IFN-β-null macrophages.
Figure 7. The inability of LPS to induce STAT activation and ISG expression in IFN-β-null macrophages cannot be overcome with higher concentrations of LPS.

Cultures of bone marrow–derived macrophages from WT and Ifnb1−/− mice were treated with LPS (10, 1, 0.1, 0.01, or 0.001 μg/mL) at 37°C for 4 h. (A) Protein extracts were then prepared and used to measure activation of STAT1 and -2 by Western blot analysis with anti-phospho(Y)-STAT1 and -2 Abs. A companion set of bone marrow–derived macrophages from WT and Ifnb1 KO mice were treated with LPS at the specified concentrations for 24 h. (B) RNA extracts were then prepared and assayed by qPCR to measure expression levels of several ISGs, including Irf7, Isg20, Mx1, Cxcl10, Oas1g, and Ddx58.
STAT2 is necessary for induction of ISG expression by LPS in macrophages
Several studies have shown that activation of ISGF3 and induction of ISGF3-responsive genes by LPS necessitates activation and nuclear translocation of STAT1 [7, 30, 31]. However, the role of STAT2 in the induction of macrophage ISG expression by LPS has not been defined. To examine the role for STAT2 in the induction of ISGF3-responsive genes by LPS, we compared the ability of LPS and IFN-β to induce expression of several ISGs in bone marrow–derived macrophages from WT vs. Stat2−/− mice. As shown in Fig. 8A, LPS treatment induced expression of all 4 of the ISGs that we examined, including Irf7, Mx1, Oas1, and Ifih1 (MDA5), in WT macrophages but not in Stat2−/− macrophages. IFN-β treatment also induced expression of these ISGs in the WT macrophages but not in the Stat2−/− macrophages.
Figure 8. STAT2 is necessary for induction of ISG expression by LPS in macrophages.
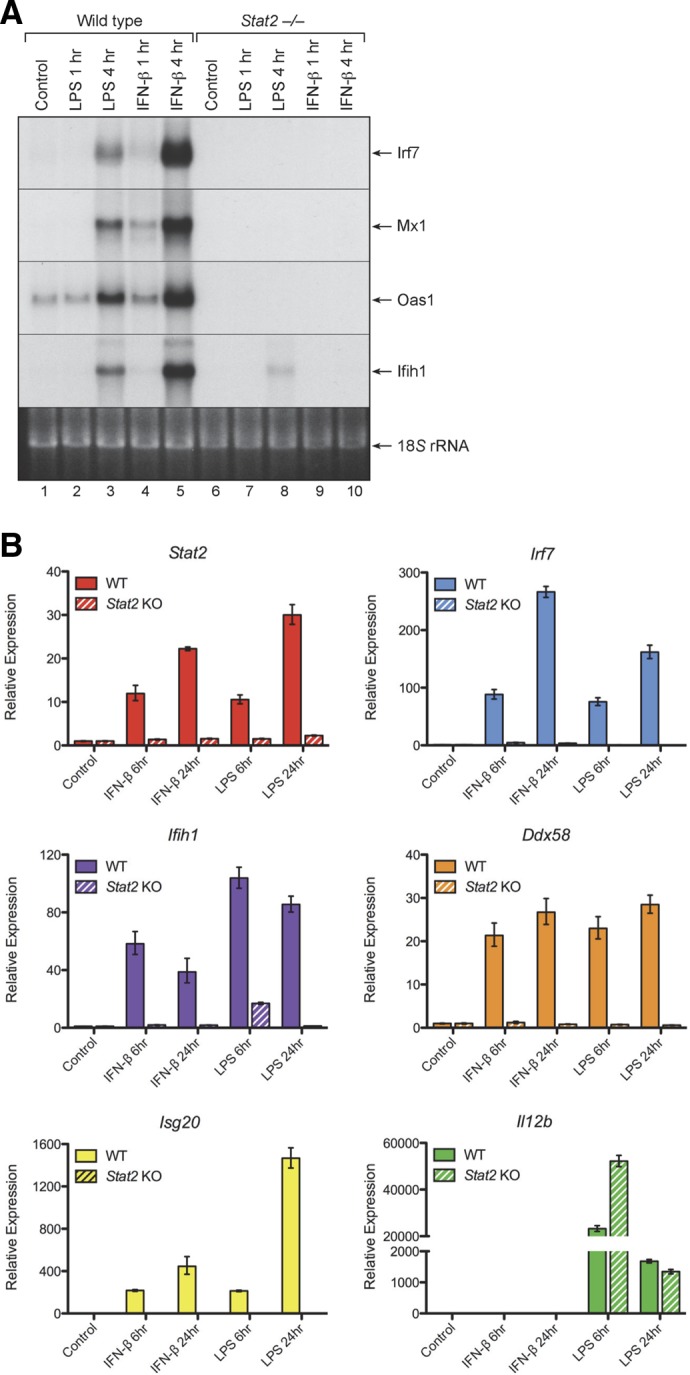
Cultures of bone marrow–derived macrophages from WT or Stat2−/− mice were treated with LPS (100 ng/mL) or IFN-β (10 ng/mL) for 1 or 4 h at 37°C. (A) At the specified time points, RNA extracts were prepared and analyzed by Northern blot analysis for expression of several ISGs, including Irf7, Mx1, Oas1, and Ifih1 (MDA5). (B) Another set of bone marrow–derived macrophages from WT and Stat2−/− mice were treated with recombinant IFN-β (10 ng/mL) or LPS (100 ng/mL) for 6 or 24 h at 37°C. At the specified time points, RNA extracts were prepared and assayed by qPCR for expression of several genes, including Stat2, Irf7, Ifih1 (MDA5), Ddx58 (Rig-I), Isg20, and Il12b.
A companion set of bone marrow–derived macrophages from WT and Stat2−/− mice were treated with recombinant IFN-β (10 ng/mL) or LPS (100 ng/mL) for 6 or 24 h at 37°C. At the specified time points, RNA extracts were prepared and assayed by qPCR for expression of several genes, including Stat2, Irf7, Ifih1 (MDA5), Ddx58 (Rig-I), Isg20, and Il12b. As shown in Fig. 8B, both IFN-β and LPS markedly up-regulated ISG expression levels in the WT macrophages but not in the Stat2−/− macrophages. In addition, the Stat2 gene itself was up-regulated by treatment with IFN-β or LPS in the WT macrophages, but not in the Stat2−/− macrophages. Therefore, STAT2 is essential for the induction of ISG expression by both IFN-β and LPS in macrophages.
DISCUSSION
Our studies showed that LPS activates ISGF3 in WT macrophages but not in IFN-β-null macrophages. In contrast, the ability of LPS to induce activation of NF-κB and expression of NF-κB-responsive genes, such as TNF-α and IL-1β, was not diminished in IFN-β-null macrophages. Although the ability of LPS to induce ISGF3 activity was abrogated in Ifnb1-deficient macrophages, ISGF3 activity could still be induced in these cells by treatment with exogenous IFN-β. These findings demonstrate that although Ifnb1−/− macrophages cannot produce IFN-β in response to LPS, they remain fully responsive to exogenous IFN-β. Moreover, our findings show that IFN-β is the predominant type I IFN produced by WT macrophages when stimulated by LPS. In contrast, both LPS and recombinant IFN-β failed to induce activation of ISGF3 in macrophages derived from Ifnar1−/− mice, thus demonstrating an essential role for type I IFN receptor signaling in the activation of ISGF3 by both LPS and IFN-β in macrophages.
The murine genome contains 14 distinct but highly homologous IFN-α genes, but only a single IFN-β gene [5, 6]. It is remarkable that deletion of the IFN-β gene alone was sufficient to abrogate the ability of LPS to induce activation of ISGF3 and ISG expression in macrophages. We were surprised to find that there is no significant role for the IFN-α genes in these responses. Furthermore, a study by others showed that treatment with neutralizing anti-IFN-β but not anti-IFN-α Abs blocks the activation of STAT1 and induction of ISG expression by LPS in macrophages [31]. These findings provide additional evidence to support our conclusion that the activation of ISGF3 and induction of ISG expression by LPS in macrophages is largely mediated by IFN-β, with little or no role for IFN-α.
Formation of ISGF3 complexes necessitates activation and assembly of heterotrimers consisting of STAT1, STAT2, and IRF9 [11]. We found that induction of ISGF3 activity by LPS in macrophages correlated temporally with tyrosine phosphorylation of STAT1 and -2. The ability of LPS to induce ISGF3 activity also correlated well with its ability to induce expression of ISGs such as Mx1, Oas1, Ddx58 (RIG-I), and Ifih1 (MDA5). LPS treatment markedly up-regulated expression of these genes in WT macrophages, but not in Ifnb1−/− macrophages or in macrophages derived from Ifnar1−/− mice. Several other ISGF3-inducible genes, including Irf7 and Isg20, were also markedly up-regulated by LPS or the TLR3 ligand poly(I:C) in the WT macrophages, but not in the Ifnb1−/− macrophages. In contrast, induction of NF-κB-responsive genes such as Tnf (TNF-α), Il1b (IL-1β), and Socs3 by LPS did not require intermediate production of IFN-β or expression of IFN-α/β receptors.
We found that LPS stimulation differentially regulates expression of the Socs1 and Socs3 genes in macrophages. The ability of LPS to induce expression of Socs3 was IFN independent; whereas, the ability of LPS to induce expression of Socs1 was IFN dependent (Fig. 6). These findings are consistent with those in other studies that also suggest a role for IFN-β production in the induction of Socs1 gene expression by TLR agonists such as LPS and CpG [33–35]. Like the Socs1 gene, the IL-10 gene (Il10) was also inducible by LPS in WT macrophages but not in Ifnar1-deficient macrophages. This observation is consistent with a previous report that implicated IFN-β in the induction of macrophage IL-10 expression by LPS [36].
Other studies have shown that induction of genes such as Ccl12 (MCP-5), Cxcl10 (IP-10), and Nos2 (iNOS) by LPS is mediated in a STAT1-dependent manner via intermediate expression of IFN-β [30, 31]. In contrast, induction of other LPS-responsive genes such as Tnf (TNF-α), Il1b (IL-1β), and Cxcl1 (KC) is not dependent on activation of STAT1 or expression of IFN-β. A subsequent study by Thomas et al. [37] using macrophages derived from Ifnb1−/− mice showed that IFN-β is essential for induction of STAT1-dependent LPS-responsive genes, such as Ccl12 (MCP-5), Cxcl10 (IP-10), and Nos2 (iNOS), but not for induction of STAT1-independent genes, such as Tnf (TNF-α), Il1b (IL-1β), and Cxcl1 (KC). Our findings extend these earlier reports by demonstrating that LPS-mediated induction of classic ISGF3-reponsive genes, such as Oas1, Mx1, Ddx58 (RIG-I), and Irf7, is critically dependent on the expression and activity of endogenous IFN-β.
The original analyses of IFN-mediated ISG expression in macrophages derived from Stat1−/− and Stat2−/− mice showed that induction of classic ISGs, such as Mx1, Oas1, Eif2ak2 (PKR), and Irf7, is STAT1 and -2 dependent [18, 38, 39]. Transcription of these ISGs is driven, in large part, by the binding of ISGF3 complexes to ISRE elements in the promoters of these genes. In contrast, a distinct subset of ISGs can be induced in a STAT1-dependent, but STAT2- and ISGF3-independent, manner. This subset includes ISGs, such as Irf1, Cxcl9 (Mig), Cxcl10 (IP-10), Cxcl11 (I-Tac), and Gbp, whose transcription is preferentially induced by the binding of activated STAT1 homodimers to GAS elements in the promoters of these genes [40]. Like the ISGF3-responsive genes, induction of these genes by LPS is dependent on IFN-β signaling; however, transcription of these genes is predominantly mediated by the binding of STAT1 homodimers to GAS elements in their promoters, rather than the binding of ISGF3 complexes to ISREs.
We found that LPS treatment induces activation (i.e., tyrosine phosphorylation) of STAT1 and -2 and expression of classical ISGs, such as Mx1, Oas1, and Irf7, in WT macrophages, but not in macrophages derived from Stat2−/− mice. Stat1−/− mice exhibit modest resistance to endotoxin challenge [41]; whereas, Stat2−/− mice display heightened sensitivity to LPS [42]. The molecular basis for the increased sensitivity of Stat2−/− mice to endotoxin challenge is not clear, because although LPS-induced expression of STAT2-dependent ISGs, such as Mx1, Oas1, and Irf7, is largely abolished in these animals, expression of proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, and IL-12, is also significantly decreased.
A recent report showed that IFN-β can bind and signal via the IFN-αR1 chain alone without recruiting the IFN-αR2 chain [43]. Activation of this IFN-αR2-independent signaling pathway by IFN-β results in expression of a unique subset of ISGs, including Irg1, Prok2, Trem1, and Tgm2. Ifnar1−/− mice are protected from challenge with high-dose LPS; whereas, Ifnar2−/− mice are as susceptible as WT mice. These findings suggest that the subset of genes induced by signaling through the IFN-αR1 chain alone, independent of the IFN-αR2 chain, contribute more substantially to the pathogenesis of Gram-negative endotoxemia than the classical ISGs induced by signaling through conventional IFN-αR1/IFN-αR2 heterodimeric complexes. Furthermore, related studies by others showed that treatment with a neutralizing anti-IFN-αR1 mAb protects mice against development of endotoxin-induced lethality in 2 distinct murine models of Gram-negative septicemia [44]. Together, these studies suggest that blocking signaling through the IFN-αR1 chain, but not the IFN-αR2 chain, provides a useful therapeutic approach to the treatment of bacterial endotoxemia.
The role of IFNs in regulating host responses to viral infections has been explored extensively. However, the role of IFNs in regulating host responses to bacterial infections has received less attention. Nevertheless, several studies have been undertaken to address this subject, and they showed that type I IFNs can play either a positive or negative role in regulating the host response to bacterial infection [45]. Mancuso et al. [46] demonstrated that type I IFN signaling is essential for host resistance to infectious challenge by several distinct bacterial species, including group B streptococci, Streptococcus pneumonia, and E. coli. After an infectious challenge with these bacteria, IFN-β KO mice and IFN-α/β receptor KO mice died from unrestrained bacteremia; whereas, the WT mice survived. In a related study, Kaplan et al. [47] reported that most Gram-positive and -negative bacteria induce production of IFN-β by macrophages. However, Staphylococcus aureus induced very little IFN-β expression in macrophages, resulting in failure to protect against a cutaneous challenge with live bacteria. These in vivo studies underscore the essential role for IFN-β and type I IFN receptor signaling in host responses to bacterial infection.
The precise role of IFN-β in the pathogenesis and resolution of bacterial endotoxemia is not yet fully defined. However, several studies have shown that IFN-β KO mice are more resistant to high-dose LPS challenge than are WT mice [37, 41]. These findings indicate that IFN-β may contribute substantially to the pathogenesis of endotoxemia induced by infection with Gram-negative bacteria and suggest that selectively antagonizing the activity of IFN-β could have therapeutic value in the treatment of Gram-negative septicemia.
ACKNOWLEDGMENTS
This study was supported in part by the Intramural Research Program of the U.S. Food and Drug Administration, and in part by U.S. National Institutes of Health (NIH) grant R01 CA140499 to A.M.G. and NIH grant R01 AI18797 to S.N.V.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
- FliC
- flagellin
- GAS
- (IFN) gamma-activated sequence
- IFN-αR
- IFN-α receptor
- Ifnar1−/−
- IFN-α/β receptor gene KO mouse
- Ifnb1−/−
- IFN-β gene KO mouse
- IRF
- IFN regulatory factor
- ISG
- IFN-stimulated gene
- ISGF3
- interferon-stimulated gene factor 3
- ISRE
- IFN-stimulated response element
- KO
- knockout
- MALP-2
- macrophage-activating lipopeptide-2 kDa
- MyD88
- myeloid differentiation primary response gene 88
- Poly(I:C)
- polyinosinic:polycytidylic acid
- qPCR
- quantitative PCR
- RPA
- RNase protection assay
- STAT
- signal transducer and activator of transcription
- Stat1−/− (or Stat2−/−)
- STAT1 (or STAT2) gene KO mouse
- TRIF
- TIR domain adapter protein inducing IFN-β
- WT
- wild-type
AUTHORSHIP
F.S., H.D., and R.P.D. designed and performed the experiments, analyzed the data, and composed the manuscript. A.M.G. and S.N.V. provided critical research materials, assisted with analysis of the results, and edited the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Takeuchi O., Fujita T., Inoue J., Mühlradt P. F., Sato S., Hoshino K., Akira S. (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894. [DOI] [PubMed] [Google Scholar]
- 3. Doyle S., Vaidya S., O'Connell R., Dadgostar H., Dempsey P., Wu T., Rao G., Sun R., Haberland M., Modlin R., Cheng G. (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263. [DOI] [PubMed] [Google Scholar]
- 4. Noppert S. J., Fitzgerald K. A., Hertzog P. J. (2007) The role of type I interferons in TLR responses. Immunol. Cell Biol. 85, 446–457. [DOI] [PubMed] [Google Scholar]
- 5. Hardy M. P., Owczarek C. M., Jermiin L. S., Ejdebäck M., Hertzog P. J. (2004) Characterization of the type I interferon locus and identification of novel genes. Genomics 84, 331–345. [DOI] [PubMed] [Google Scholar]
- 6. van Pesch V., Lanaya H., Renauld J. C., Michiels T. (2004) Characterization of the murine alpha interferon gene family. J. Virol. 78, 8219–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao J. J., Filla M. B., Fultz M. J., Vogel S. N., Russell S. W., Murphy W. J. (1998) Autocrine/paracrine IFN-alpha/beta mediates the lipopolysaccharide-induced activation of transcription factor Stat1-alpha in mouse macrophages: pivotal role of Stat1-alpha in induction of the inducible nitric oxide synthase gene. J. Immunol. 161, 4803–4810. [PubMed] [Google Scholar]
- 8. Marié I, Durbin J. E., Levy D. E. (1998) Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17, 6660–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakaguchi S., Negishi H., Asagiri M., Nakajima C., Mizutani T., Takaoka A., Honda K., Taniguchi T. (2003) Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 306, 860–866. [DOI] [PubMed] [Google Scholar]
- 10. de Weerd N. A., Samarajiwa S. A., Hertzog P. J. (2007) Type I interferon receptors: biochemistry and biological functions. J. Biol. Chem. 282, 20053–20057. [DOI] [PubMed] [Google Scholar]
- 11. Stark G. R., Kerr I. M., Williams B. R., Silverman R. H., Schreiber R. D. (1998) How cells respond to interferons. Annu. Rev. Biochem. 67, 227–264. [DOI] [PubMed] [Google Scholar]
- 12. Müller U., Steinhoff U., Reis L. F., Hemmi S., Pavlovic J., Zinkernagel R. M., Aguet M. (1994) Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 13. Hwang S. Y., Hertzog P. J., Holland K. A., Sumarsono S. H., Tymms M. J., Hamilton J. A., Whitty G., Bertoncello I., Kola I. (1995) A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. U. S. A. 92, 11284–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vogel S. N., Fertsch D. (1984) Endogenous interferon production by endotoxin-responsive macrophages provides an autostimulatory differentiation signal. Infect. Immun. 45, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gessani S., Belardelli F., Pecorelli A., Puddu P., Baglioni C. (1989) Bacterial lipopolysaccharide and gamma interferon induce transcription of beta interferon mRNA and interferon secretion in murine macrophages. J. Virol. 63, 2785–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dickensheets H., Vazquez N., Sheikh F., Gingras S., Murray P. J., Ryan J. J., Donnelly R. P. (2007) Suppressor of cytokine signaling (SOCS)-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun. 8, 21–27. [DOI] [PubMed] [Google Scholar]
- 17. Deonarain R., Alcamí A, Alexiou M., Dallman M. J., Gewert D. R., Porter A. C. (2000) Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J. Virol. 74, 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park C., Li S., Cha E., Schindler C. (2000) Immune response in Stat2 knockout mice. Immunity 13, 795–804. [DOI] [PubMed] [Google Scholar]
- 19. Schreiber E., Matthias P., Müller M. M., Schaffner W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reich N., Evans B., Levy D., Fahey D., Knight E, Jr., Darnell JE., Jr. (1987) Interferon-induced transcription of a gene encoding a 15-kDa protein depends on an upstream enhancer element. Proc. Natl. Acad. Sci. U. S. A. 84, 6394–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lenardo M. J., Fan C. M., Maniatis T., Baltimore D. (1989) The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell 57, 287–294. [DOI] [PubMed] [Google Scholar]
- 23. Kotenko S. V., Gallagher G., Baurin V. V., Lewis-Antes A., Shen M., Shah N. K., Langer J. A., Sheikh F., Dickensheets H., Donnelly R. P. (2003) IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77. [DOI] [PubMed] [Google Scholar]
- 24. Romero-Weaver A. L., Wang H. W., Steen H. C., Scarzello A. J., Hall V. L., Sheikh F., Donnelly R. P., Gamero A. M. (2010) Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol. Cancer Res. 8, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chomczynski P., Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 26. Sheikh F., Baurin V. V., Lewis-Antes A., Shah N. K., Smirnov S. V., Anantha S., Dickensheets H., Dumoutier L., Renauld J. C., Zdanov A., Donnelly R. P., Kotenko S. V. (2004) Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J. Immunol. 172, 2006–2010. [DOI] [PubMed] [Google Scholar]
- 27. Feinberg A. P., Vogelstein B. (1983) A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132, 6–13. [DOI] [PubMed] [Google Scholar]
- 28. Dickensheets H., Sheikh F., Park O., Gao B., Donnelly R. P. (2013) Interferon-lambda (IFN-λ) induces signal transduction and gene expression in human hepatocytes, but not in lymphocytes or monocytes. J. Leukoc. Biol. 93, 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 30. Ohmori Y., Hamilton T. A. (2001) Requirement for STAT1 in LPS-induced gene expression in macrophages. J. Leukoc. Biol. 69, 598–604. [PubMed] [Google Scholar]
- 31. Toshchakov V., Jones B. W., Perera P. Y., Thomas K., Cody M. J., Zhang S., Williams B. R., Major J., Hamilton T. A., Fenton M. J., Vogel S. N. (2002) TLR4, but not TLR2, mediates IFN-beta-induced STAT1 alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3, 392–398. [DOI] [PubMed] [Google Scholar]
- 32. Hirotani T., Yamamoto M., Kumagai Y., Uematsu S., Kawase I., Takeuchi O., Akira S. (2005) Regulation of lipopolysaccharide-inducible genes by MyD88 and Toll/IL-1 domain containing adaptor inducing IFN-beta. Biochem. Biophys. Res. Commun. 328, 383–392. [DOI] [PubMed] [Google Scholar]
- 33. Crespo A., Filla M. B., Russell S. W., Murphy W. J. (2000) Indirect induction of suppressor of cytokine signalling-1 in macrophages stimulated with bacterial lipopolysaccharide: partial role of autocrine/paracrine interferon-alpha/beta. Biochem. J. 349, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gingras S., Parganas E., de Pauw A., Ihle J. N., Murray P. J. (2004) Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J. Biol. Chem. 279, 54702–54707. [DOI] [PubMed] [Google Scholar]
- 35. Baetz A., Frey M., Heeg K., Dalpke A. H. (2004) Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J. Biol. Chem. 279, 54708–54715. [DOI] [PubMed] [Google Scholar]
- 36. Chang EY, Guo B, Doyle SE, Cheng G. (2007) Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J. Immunol. 178, 6705–6709. [DOI] [PubMed] [Google Scholar]
- 37. Thomas K. E., Galligan C. L., Newman R. D., Fish E. N., Vogel S. N. (2006) Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J. Biol. Chem. 281, 31119–31130. [DOI] [PubMed] [Google Scholar]
- 38. Meraz M. A., White J. M., Sheehan K. C., Bach E. A., Rodig S. J., Dighe A. S., Kaplan D. H., Riley J. K., Greenlund A. C., Campbell D., Carver-Moore K., DuBois R. N., Clark R., Aguet M., Schreiber R. D. (1996) Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84, 431–442. [DOI] [PubMed] [Google Scholar]
- 39. Durbin J. E., Hackenmiller R., Simon M. C., Levy D. E. (1996) Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84, 443–450. [DOI] [PubMed] [Google Scholar]
- 40. Decker T., Kovarik P., Meinke A. (1997) GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 17, 121–134. [DOI] [PubMed] [Google Scholar]
- 41. Karaghiosoff M., Steinborn R., Kovarik P., Kriegshäuser G., Baccarini M., Donabauer B., Reichart U., Kolbe T., Bogdan C., Leanderson T., Levy D., Decker T., Müller M. (2003) Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 4, 471–477. [DOI] [PubMed] [Google Scholar]
- 42. Alazawi W., Heath H., Waters J. A., Woodfin A., O'Brien A. J., Scarzello A. J., Ma B., Lopez-Otalora Y., Jacobs M., Petts G., Goldin R. D., Nourshargh S., Gamero A. M., Foster G. R. (2013) Stat2 loss leads to cytokine-independent, cell-mediated lethality in LPS-induced sepsis. Proc. Natl. Acad. Sci. U. S. A. 110, 8656–8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Weerd N. A., Vivian J. P., Nguyen T. K., Mangan N. E., Gould J. A., Braniff S. J., Zaker-Tabrizi L., Fung K. Y., Forster S. C., Beddoe T., Reid H. H., Rossjohn J., Hertzog P. J. (2013) Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat. Immunol. 14, 901–907. [DOI] [PubMed] [Google Scholar]
- 44. Dejager L., Vandevyver S., Ballegeer M., Van Wonterghem E., An L. L., Riggs J., Kolbeck R., Libert C. (2014) Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J. Infect. Dis. 209, 960–970. [DOI] [PubMed] [Google Scholar]
- 45. Decker T., Müller M., Stockinger S. (2005) The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5, 675–687. [DOI] [PubMed] [Google Scholar]
- 46. Mancuso G., Midiri A., Biondo C., Beninati C., Zummo S., Galbo R., Tomasello F., Gambuzza M., Macrì G, Ruggeri A., Leanderson T., Teti G. (2007) Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178, 3126–3133. [DOI] [PubMed] [Google Scholar]
- 47. Kaplan A., Ma J., Kyme P., Wolf A. J., Becker C. A., Tseng C. W., Liu G. Y., Underhill D. M. (2012) Failure to induce IFN-β production during Staphylococcus aureus infection contributes to pathogenicity. J. Immunol. 189, 4537–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]