

ABSTRACT
Emerin is a conserved membrane component of nuclear lamina structure. Here, we report an advance in understanding the molecular basis of emerin function: intermolecular emerin–emerin association. There were two modes: one mediated by association of residues 170–220 in one emerin molecule to residues 170–220 in another, and the second involving residues 170–220 and 1–132. Deletion analysis showed residues 187–220 contain a positive element essential for intermolecular association in cells. By contrast, deletion of residues 168–186 inactivated a proposed negative element, required to limit or control association. Association of GFP–emerin with nuclear BAF in cells required the LEM domain (residues 1–47) and the positive element. Emerin peptide arrays revealed direct binding of residues 170–220 to residues 206–225 (the proposed positive element), residues 147–174 (particularly P153MYGRDSAYQSITHYRP169) and the LEM domain. Emerin residues 1–132 and 159–220 were each sufficient to bind lamin A or B1 tails in vitro, identifying two independent regions of molecular contact with lamins. These results, and predicted emerin intrinsic disorder, support the hypothesis that there are multiple ‘backbone’ and LEM-domain configurations in a proposed intermolecular emerin network at the nuclear envelope.
KEY WORDS: Barrier to autointegration factor, Emerin, Emery-Dreifuss muscular dystrophy, Lamin, LEM domain, Nestor-Guillermo progeria, Nuclear envelope, Nucleoskeleton, Nuclear lamina
INTRODUCTION
Emerin is a conserved nuclear membrane LEM domain protein that binds lamins and barrier-to-autointegration factor (BAF, also known as BANF1) as part of the nuclear ‘lamina’ structure (Burke and Stewart, 2013; Margalit et al., 2007; Simon and Wilson, 2011; Wagner and Krohne, 2007). Mutations in nuclear lamina proteins cause cardiomyopathy, muscular dystrophy, metabolic syndrome, ‘accelerated aging’ and other conditions (Méndez-López and Worman, 2012; Schreiber and Kennedy, 2013), through unknown mechanisms.
Emerin, a 254-residue protein, is ubiquitous in human tissues. However, its functions are masked by overlap or collaboration with other nuclear membrane proteins. For example, loss of emerin primarily affects striated muscle (Emery–Dreifuss muscular dystrophy) (Bione et al., 1994; Emery and Dreifuss, 1966). In myoblasts and muscle, emerin functionally overlaps with other LEM-domain-containing proteins: LEM2 (also known as LEMD2) and MAN1 (also known as LEMD3) in mice (Huber et al., 2009), and LEM-2 in C. elegans (Barkan et al., 2012). Emerin function overlaps with MAN1 during muscle and eye development in Xenopus (Reil and Dabauvalle, 2013). In muscle, emerin is also specifically ‘backed up’ by a nuclear membrane partner named lamina associated polypeptide 1 (LAP1, also known as TOR1AIP1) (Foisner and Gerace, 1993; Shin et al., 2013). In C. elegans, emerin and LEM-2 are co-essential for mitotic chromosome segregation, nuclear assembly and embryogenesis (Liu et al., 2003), and are required during meiosis for centrosome attachment to the nuclear envelope (Meyerzon et al., 2009) and double-strand DNA break repair (Dittrich et al., 2012). Human emerin binds proteins that are crucial for centrosome positioning and nuclear envelope structure (SUN1, SUN2, and nesprins) (Haque et al., 2010; Salpingidou et al., 2007) and influences actin cytoskeletal dynamics (Chang et al., 2013; Ho et al., 2013; Holaska et al., 2004). Emerin activates HDAC3, inhibits β-catenin and LMO7 (Berk et al., 2013b; Demmerle et al., 2012; Holaska et al., 2006; Markiewicz et al., 2006), helps ‘tether’ silent chromatin (Amendola and van Steensel, 2014), and is required to activate force-responsive genes (‘mechano-transduction signaling’) (Lammerding et al., 2005).
The nature and mechanisms of emerin function have been obscured by collaboration and its apparent lack of structure between the LEM-domain fold (residues 1–47; sufficient to bind BAF; Cai et al., 2007) and the C-terminal transmembrane domain. We report biochemical and cellular evidence that emerin intermolecular association is fundamental to its localization and contact with nuclear BAF. This evidence predicts that emerin–emerin networks act as adaptable molecular frameworks for contact with lamins and BAF at the nuclear envelope.
RESULTS
An in vitro binding assay revealed direct intermolecular binding between polypeptides each comprising the full nucleoplasmic domain of human emerin (residues 1–220). We used immobilized N-terminally His-tagged emerin residues 1–221 (His–emerin-1–221; 1 µM) incubated with different concentrations (31.3–500 nM) of recombinant GST or GST–emerin-1–222 from urea-solubilized bacterial lysates. Beads were washed and bound proteins were eluted with SDS, resolved (100%) by SDS-PAGE, Ponceau-stained to verify input ‘bait’ His–emerin-1–221 (Fig. 1A, Ponceau) and immunoblotted for GST (Fig. 1A, WB: GST). Control lanes show 5% of unbound 500 nM input GST (∼28 kDa) or GST–emerin-1–222 (∼55 kDa; Fig. 1A). His–emerin-1–221 bound GST–emerin-1–222 specifically at all concentrations tested (Fig. 1A, arrowhead; n = 2). Another bait, C-terminally His-tagged emerin 1–220 (Emerin-1–220–His), gave the same results (data not shown; n = 3). We concluded emerin polypeptides can associate in vitro through the nucleoplasmic domain.
Fig. 1.
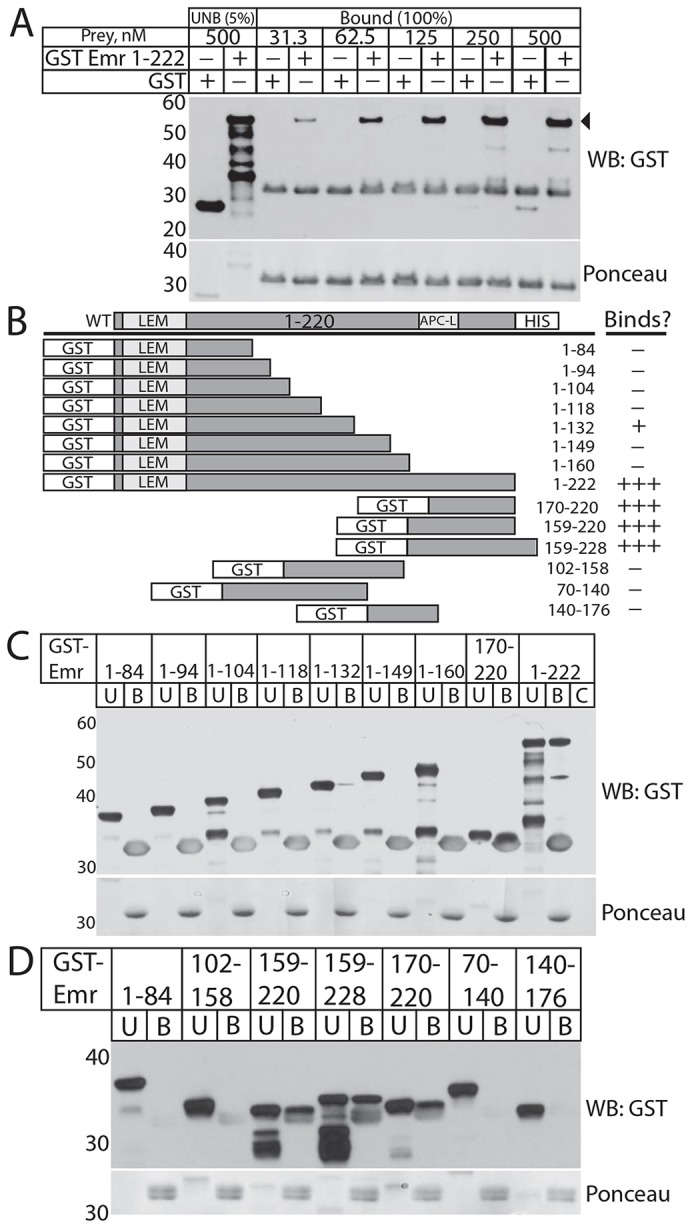
Direct emerin–emerin binding in vitro requires residues 170–220. (A) Immunoblot of His–emerin-1–221 (1 µM) binding to 31.3–500 nM ‘prey’ GST or GST–emerin-1–222 (GST Emr 1-222; 100% of bead-bound eluates shown), probed with anti-GST antibodies (WB: GST), with 5% of each 500 nM unbound sample as markers. Ponceau staining (contrast enhanced) verified similar levels of emerin bait. GST antibodies cross-reacted weakly with His–emerin (∼33 kDa). The blot shown is representative of five independent experiments [two with His–emerin-1–221 bait (shown); three with emerin-1–220–His bait). (B) Schematic summary of GST–emerin polypeptides tested in C and D for binding to emerin-1–220–His. The number of ‘+’ symbols indicates the degree of binding; ‘–’ indicates no binding. (C,D) Immunoblots and corresponding (contrast enhanced) Ponceau-stain showing bound (B label, 100%) versus unbound (U label, 5%) ‘prey’ GST–emerin incubated with emerin-1–220–His (1 µM) ‘bait’, probed for GST. GST–emerin-1–222 ‘prey’ control was incubated with beads alone (C label, 100% of bound). Blots in C and D are representative of four (n = 4) and six independent experiments [n = 6, three with His–emerin-1–221 bait (shown), and three with emerin-1–220–His], respectively.
To locate regions involved in emerin–emerin association, several recombinant GST–emerin fragments (Fig. 1B) were tested for binding to Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose-immobilized emerin-1–220–His at protein concentrations of 1 µM. Six GST–emerin constructs (containing residues 1–84, 1–94, 1–104, 1–118, 1–149 or 1–160) showed little or no detectable binding (Fig. 1C; n = 4). Two constructs (1–222 and 170–220) showed consistent binding to emerin-1–220–His; consistent but weaker signals were detected for GST–emerin-1–132 (Fig. 1C; n = 4) as summarized in Fig. 1B. Thus, two regions were each sufficient to bind emerin-1–220–His; one strongly (170–220) and one weakly (1–132; Fig. 1C). Negative results for 1–149 and 1–160 could be due to recombinant protein artifacts, or interference (e.g. auto-inhibition) by residues 133–160. Three internal polypeptides (70–140, 102–158 and 140–176) also failed to bind the His–emerin-1–221 bait (Fig. 1D, n = 3) and emerin-1–220–His bait (n = 3; unpublished observations). However, GST–emerin-170–220 and two related polypeptides, 159–220 and 159–228, showed strong binding to His–emerin-1–221 (Fig. 1D; n = 6). Thus, residues 170–220 were sufficient to bind the nucleoplasmic domain of another emerin polypeptide(s).
Two modes of emerin–emerin intermolecular binding in vitro: between residues 170–220 and 170–220, and residues 170–220 and 1–132
To locate region(s) bound by residues 170–220, we used N-terminally S-tagged and C-terminally His-tagged emerin 170–220 (S–emerin-170–220–His) as bait for several GST–emerin fragments (Fig. 2A). Residues 170–220 bound each other directly (Fig. 2B), defining this region as sufficient for one mode of intermolecular association in vitro. Residues 170–220 also consistently bound GST–emerin-1–132 and (with lower signals) GST–emerin-1–149 (Fig. 2B). This result revealed a second mode of binding that involved residues 1–132, and hinted that this mode of binding might be facilitated by residues 119–132 and weakened by residues 133–160.
Fig. 2.

Residues 170–220 are sufficient for two modes of emerin–emerin intermolecular association. (A) Schematic summary of recombinant prey GST–emerin polypeptides tested for binding to S–emerin-170–220–His bait (1 µM each). The number of ‘+’ symbols indicates the degree of binding; ‘−’ indicates no binding. (B) Immunoblot and corresponding (contrast enhanced) Ponceau staining of bound (B label, 100% loaded) versus unbound (U label, 5%) fractions of each GST–emerin prey, or control GST–emerin-1–222 incubated with beads alone (C label; 100% of bound), probed for GST (WB: GST; n = 4). (C) Intrinsic disorder in emerin predicted by PONDR-VLXT (black line; Romero et al., 2001) or PONDR-FIT (blue line; gray shadows indicate prediction error; Xue et al., 2010).
Predicted intrinsic disorder in emerin
Approximately 30% of eukaryotic proteins (70% of signaling proteins) are characterized by a lack of stable structure, defining a spectrum of intrinsically disordered proteins (IDPs) (Iakoucheva et al., 2002). IDPs have conformational plasticity; they can undergo disorder-to-order transitions when bound to partners, form stable intertwined structures or promote the formation of supra-molecular complexes (Cozzetto and Jones, 2013; Uversky, 2013). IDPs are strongly influenced by post-translational modifications and are uniquely suited for roles in signaling (Uversky et al., 2008). Many kinases and the essential enzyme O-GlcNAc transferase (OGT), which O-GlcNAc-modifies Ser/Thr residues, favor flexible extended substrates (Lazarus et al., 2011; Trinidad et al., 2012). Emerin shares many IDP features including compositional bias (e.g. 50% Ala, Arg, Gly, Gln, Ser, Glu and Pro residues), and its most fundamental role – binding to BAF – is controlled by O-GlcNAc versus phosphate modifications at Ser173 in vivo (Berk et al., 2013a). We analyzed potential disorder in emerin using the PONDR-VLXT algorithm (primary sequence complexity; Romero et al., 2001) and PONDR-Fit (multiple criteria; Fig. 2C; gray indicates prediction error; Xue et al., 2010). PONDR-Fit correctly recognized the LEM-domain residues ∼14–45 as ‘not-disordered’ (Fig. 2C); predicted disorder in LEM-domain residues ∼1–13 is of unknown significance. Both algorithms predicted disorder in emerin residues ∼45–62, ∼105–118 and ∼140–215 (Fig. 2C), suggesting potential disorder in the emerin self-associating residues 170–220. Considering emerin in this new light, as a potential IDP, we realized previously studied emerin mutations might have disrupted its functions directly (by perturbing partner contact sites) or by multiple indirect mechanisms, e.g. perturbing association-dependent ‘platforms’ for partners, biochemical freedom to switch conformation(s), or post-translational control of conformations or switching in cells. This motivated us to locate the regions of emerin that directly contact lamins.
Two regions of emerin contact lamin tails
Previous work has shown lamin A binding is disrupted by centrally located mutations in emerin (residues 70–170) (Berk et al., 2013b; Lee et al., 2001). We challenged this result by identifying regions of emerin sufficient to bind lamin tails in vitro. GST–emerin fragments were incubated with purified recombinant lamin tail domains, starting with NTA-agarose-immobilized N-terminally T7-tagged and C-terminally His-tagged mature lamin A tail residues 385–646 (T7–mLmA; Fig. 3A). GST–emerin-1–84, 1–94, 1–104 and 1–118 gave no detectable signals, whereas GST–emerin-1–132, 1–149 and 1–160 bound lamin A consistently above background (Fig. 3A; n = 3). These positive fragments were further tested against the lamin B1 tail (residues 395–586) with mature lamin A tails as positive control, or incubated without lamins as the negative control (Fig. 3B). All three emerin fragments bound lamin A, as expected, and two (1–132 and 1–149) also bound the lamin B1 tail with lower signals (Fig. 3B; n = 3). Thus, emerin residues 1–132 were sufficient to bind lamin A or B1 tails; in this context, residues 118–132 were important or essential (Fig. 3D). In previous studies, emerin binding to full-length lamin A was not reduced by LEM-domain or S54F mutations, but was abolished by mutations m70 (D70ADMY74 to A70AAMA74), m76 (L76PKKEDAL83 to A76PAKADAA83), Δ95–99 (an in-frame EDMD-causing deletion) and m112 (G112PSRAVRQSVT122 to A112ASRAVAAAVA122) (Lee et al., 2001). These considerations conservatively predict emerin residues 55–132 as sufficient for molecular contact with lamin tails (Fig. 3E). Mutation m141 (S141SSEEECKDR150 to A141ASAEECKAA150) also abolished lamin binding (Lee et al., 2001); whether residues 133–150 are sufficient to bind lamins was untested.
Fig. 3.

Lamin tails contact two regions of emerin. (A–C) Immunoblots of bound (B label, 25% loaded) vs unbound (U label; 4% loaded) fractions of each recombinant GST–emerin polypeptide (1.0 µM) incubated with NTA-agarose-immobilized recombinant lamin A or B1 tail baits, probed for GST (WB: GST), then stripped and reprobed for the lamin T7 tag (WB: T7). (A) Lamin bait: N-terminally T7-tagged and C-terminally His-tagged mature lamin A tail residues 385–646 (‘T7-mLmA’; n = 3). (B,C) Two baits were tested: mature lamin A tail (‘mA’; see A) or lamin B1 tail residues 395–586 (‘B1’; n = 3 each). (D) Schematic summary of results in A–C. The number of ‘+’ symbols indicates the degree of binding; ‘–’ indicates no binding; ‘+/–’ indicates inconsistent, weak binding. *Binding to lamin B1 not detected in two independent trials (our unpublished observations). (E) Emerin schematic indicating the LEM domain (LEM; sufficient to bind BAF), residues S159AYQ162, region ‘A’ (residues 168–186), region ‘R’ (residues 187–220), the transmembrane domain (TM) and two regions (residues 1–132 and 159–220) each sufficient to bind lamin A or B1 tails in vitro.
We hypothesized that it was also possible that lamin tails also contacted the membrane-proximal region of emerin. To test this idea we incubated GST–emerin residues 159–220 or 170–220 with lamin A or B1 tails, or neither as control (Fig. 3C). Lamin A tails bound GST–emerin-159–220 strongly, but gave low or no signals with GST–emerin-170–220 (Fig. 3C; n = 3). Similarly lamin B1 tails bound GST–emerin-159–220, but not 170–220 (Fig. 3C; n = 3). This established a new second site of molecular contact with lamin A and B1 tails, comprising emerin residues 159–220, wherein residues 159–169 were important or essential (Fig. 3D). Among six previous mutations in this region (m164, m179, P183H, m196, m207 and m214) only one – m164 (I164THYRPV170 to A164AHARPA170) – was shown to disrupt the interaction with lamin A (Lee et al., 2001), implicating membrane-proximal emerin residues 159–178 as the second independent site of molecular contact with lamin tails (Fig. 3E). Mutation m164 also disrupted binding to F-actin (six other partners were unaffected) (Berk et al., 2013b; Ho et al., 2013).
Emerin–emerin association validated in cells
To test the biological relevance of emerin–emerin association, we made five deletions in Flag–emerin and GFP–emerin (Fig. 4A). Deletion ‘ΔL’ removed residues 1–47 (the LEM domain) as the negative control for binding to BAF. Deletion ‘ΔM’ removed ‘middle’ residues 67–108; this region is required to bind full-length lamin A in vitro (Lee et al., 2001), but ΔM still localizes at the nuclear envelope in cells (Tsuchiya et al., 1999) and was the positive control for binding to BAF. Deletion ‘ΔA’ removed residues 168–186 (included ‘APC-like’ residues) from the domain that meditates the first mode of emerin–emerin binding [i.e. that between two 170–220 domains] (Cartegni et al., 1997; Markiewicz et al., 2006), ‘ΔR’ removed its right portion (residues 187–220), and ‘ΔAR’ removed the entire association domain (residues 170–220; Fig. 4A). We first tested intermolecular association in HeLa cells that co-expressed each GFP–emerin construct (or GFP, as control), plus the same deletion in Flag–emerin, for 24 h. Equal protein concentrations of lysates were immunoprecipitated with GFP antibodies; eluates were resolved by SDS-PAGE (5 or 10%) with control input lysates (1%), immunoblotted with anti-Flag antibodies, then stripped and reprobed with anti-GFP antibodies. The GFP control did not precipitate wild-type Flag–emerin (Fig. 4B; n = 2). Wild-type GFP–emerin and wild-type Flag–emerin co-precipitated (Fig. 4B; n = 2), validating emerin–emerin association in HeLa cells. Association was undetectable when both constructs lacked either AR (ΔAR/ΔAR; n = 1), or R (ΔR/ΔR; Fig. 4B; n = 2). This suggested R might be essential for emerin intermolecular association in HeLa cells.
Fig. 4.

Emerin intermolecular association in cells – deletions reveal positive and negative elements. (A) Schematic of GFP–emerin polypeptides studied in vivo. LEM, LEM domain; APC-L, APC-like homology domain; TM, predicted transmembrane domain. (B) GFP–emerin and Flag–emerin associate in HeLa cells. Sonicated (total) whole cell lysates (WCLT) from HeLa cells 24 h post-co-transfection with GFP–emerin (or GFP) and Flag–emerin were immunoprecipitated (IP) using GFP antibodies, resolved by SDS-PAGE (5% or 10%) alongside input lysates (1%), probed with Flag antibodies, then stripped and reprobed for GFP. Wild-type (WT) GFP–emerin specifically co-precipitated wild-type Flag–emerin (n = 2). Association was undetectable when both constructs lacked AR (ΔAR/ΔAR; n = 1) or R (ΔR/ΔR; n = 2). (C–F) GFP–emerin and Flag–emerin associate in HEK293T cells. (C,D) Sonicated ‘total’ whole cell lysates (WCLT) from HEK293T cells 24 h after co-transfection with wild-type Flag–emerin plus GFP or GFP–emerin (WT, ΔL, ΔA, ΔR or ΔAR) were immunoprecipitated using anti-GFP antibodies (IP: GFP). Precipitates (2–2.5%) and input control lysates (1%) from cells co-expressing both WT constructs were resolved by SDS-PAGE, immunoblotted for Flag (WB: Flag), stripped and reprobed for GFP (WB: GFP). Results (C) were quantified by densitometry and presented as a graph (D) as the amount of wild-type Flag–emerin precipitated per GFP–emerin mutant, relative to wild-type GFP–emerin. *P<0.05, **P<0.005 (n = 3). Results are mean±s.e.m. (E,F) Residues 67–108 (ΔM) are not essential for emerin–emerin association in HEK293T cells. Equal concentrations of whole cell lysates from HEK293T cells 24 h post co-transfection with GFP–emerin and Flag–emerin (both wild-type or both ΔM) were immunoprecipitated with anti-GFP antibodies, resolved by SDS-PAGE (2.5%) with input lysate controls (1%), immunoblotted for Flag or GFP (E), and quantified (F) as the Flag-to-GFP ratio for ΔM/ΔM, relative to WT/WT. Results are mean±s.e.m. (n = 2). (G,H) Immunoprecipitation from HEK293T cells that co-expressed two deletion constructs. Precipitates (2.5%; IP: GFP) and corresponding input lysates (1%; Inputs) were resolved separately, immunoblotted for Flag (WB: Flag), stripped and reprobed for GFP (WB: GFP). Black lines separate sections from same blot. Results (G) were quantified (H) as the ratio of Flag–emerin for each GFP–emerin construct, relative to cells expressing WT/WT. Results are mean±s.e.m. ΔAR/ΔR and ΔAR/ΔA were each tested twice (n = 2); others three times (n = 3). *P<0.05; **P<0.005.
Co-expression of ΔL, ΔA, ΔR and ΔAR constructs in cis and in trans
To further dissect the contributions of regions A and R, we co-expressed pairs of GFP- versus Flag-fused emerin constructs for 24 h in HEK293T cells and used anti-GFP antibodies to immunoprecipitate from equal concentrations of sonicated (‘total’) whole cell lysates (WCLT) (Berk et al., 2013a). Immunoprecipitates (2–2.5%) and control input lysates (1%) were resolved by SDS-PAGE, immunoblotted with anti-Flag antibodies, then stripped and reprobed with anti-GFP antibodies (Fig. 4C,D). We first co-expressed GFP–emerin (WT, ΔL, ΔA, ΔR, ΔAR) with wild-type Flag–emerin (Fig. 4C). The control pair of wild-type constructs co-immunoprecipitated specifically (no Flag signal with GFP alone; Fig. 4C), independently confirming intermolecular emerin–emerin association in HEK293T cells. The effects of each deletion on binding to wild-type Flag-emerin were quantified by densitometry as the Flag-to-GFP ratio for each deletion, relative to the WT-to-WT ratio (Fig. 4D). Association with wild-type Flag–emerin was increased slightly but insignificantly by ΔL (Fig. 4D; P<0.06; n = 3). Interestingly ΔA and ΔR had opposite effects in cells: ΔR significantly decreased association with wild-type Flag–emerin (by 32%; Fig. 4D; P<0.002, n = 3) suggesting R has a positive role, whereas ΔA increased association threefold (Fig. 4D; P<0.02, n = 3). This suggested that the A element is not essential for the emerin–emerin association between two AR (170–220) domains, and that ΔA inactivated a negative element required to limit or control association. Loss of both elements (ΔAR) decreased association with wild-type Flag–emerin by 56% (Fig. 4D; P<0.03, n = 3); this reduction was statistically indistinguishable from that caused by ΔR alone (Fig. 4D, P<0.2), suggesting R contained a positive element crucial for emerin–emerin association in cells.
To determine whether the R element alone was responsible for the emerin–emerin association between two AR domains, we co-expressed combinations of deletions in HEK293T cells (Fig. 4E–H). Compared to control pairs of wild-type constructs, emerin association in HEK293T cells was not significantly altered when both constructs lacked residues 67–108 (ΔM/ΔM; Fig. 4E,F; n = 2), but increased significantly (twofold) in cells when both constructs lacked the LEM domain (ΔL/ΔL; Fig. 4G,H; P<0.02; n = 4). This suggested that the LEM domain normally limits the extent of emerin–emerin association in cells, either through association with BAF (hence, chromatin) or through a novel role for the LEM domain. Association increased further (fivefold) when both constructs lacked A (ΔA/ΔA; Fig. 4G,H; P<0.02; n = 4), supporting the idea that A is required to inhibit emerin–emerin association. Strongly supporting the hypothesis that R is a positive element, emerin–emerin association dropped significantly when both constructs lacked R alone (ΔR/ΔR; reduced by 82%; Fig. 4G,H; P<0.0003; n = 4) or AR (ΔAR/ΔAR; reduced 90%; Fig. 4G,H; P<0.0003; n = 5).
To determine which element (A or R) was required for residues 170–220 to associate with emerin residues 1–132 (Fig. 1B), we tested the functionality of single A or R regions by co-expressing ΔR and ΔA either in trans (on different molecules), or in combination with GFP–emerin-ΔAR (Fig. 4G,H). Reciprocal ΔA versus ΔR experiments showed that A on one molecule, and R on the other, associated at wild-type levels (ΔR/ΔA and ΔA/ΔR; Fig. 4G,H; n = 3 each; statistically indistinguishable from wild type). This result supported at least two interpretations: A might directly bind R (i.e. complementation through antiparallel association), or R might be sufficient to bind the other polypeptide through residues 1–132. Supporting the latter model, the R element on Flag–emerin was sufficient to associate with GFP–emerin-ΔAR at levels indistinguishable from wild type (ΔAR/ΔA; Fig. 4G,H; n = 2), whereas the A element on Flag–emerin was insufficient: emerin–emerin association was reduced by 92% (ΔAR/ΔR; Fig. 4G,H; n = 2).
Localizations and phenotypes in cells
To further characterize the positive and negative emerin–emerin association elements, we localized deletion constructs by indirect immunofluorescence in HEK293T cells, either 5 or 24 h post-transfection. All Flag–emerin constructs localized at the nuclear envelope and endoplasmic reticulum (ER) at 5 h post-transfection, with occasional perinuclear ‘puncta’ (a potential overexpression artifact; Fig. 5A). At 24 h we saw interesting phenotypes and differences in emerin localization, which were scored by a single-blinded observer (K.L.W.; n = 3; >300 Flag-positive cells counted per construct).
Fig. 5.

Flag–emerin localization and overexpression phenotypes in HEK293T cells. (A) Cells were fixed 5 h (n = 1) or 24 h (n = 3) after transfection with each Flag–emerin construct, stained with anti-Flag antibodies and visualized by indirect immunofluorescence. DNA was DAPI-stained. Scale bars: 10 µm. Arrowheads indicate wild-type (WT) Flag–emerin accumulation in nuclear envelope ‘patches/caps’. (B–G) Fluorescence images were scored ‘blind’, then quantified and are presented as the percentage of Flag-positive cells showing each characteristic. (B) Cells showing the SCC phenotype [small nuclei, ‘condensed’ chromatin, irregular nuclear envelope (NE)]. ΔL, *P<0.018; ΔA, **P<0.004; compared with wild-type Flag–emerin. (C) Cells showing accumulation in nuclear envelope patches and or caps. (D) Cells showing nuclear envelope accumulation (scored negative if this was less than the ER signal). (E) Cells showing ER accumulation. (F) Cells showing aggregation outside nucleus. (G) Cells showing enrichment at or near plasma membrane. *P<0.05; **P<0.005; ***P<0.0005. Results are mean±s.e.m.
At 5 h post-transfection, overexpressed wild-type Flag–emerin localized mainly at the nuclear envelope, with an even distribution (Fig. 5A, WT, 5 h). By 24 h, most wild-type Flag–emerin was concentrated at discrete regions of the nuclear envelope, forming ‘patches’ or concave ‘caps’ in 80% of cells (arrowheads, Fig. 5A, WT; quantified in Fig. 5C), consistent with intermolecular association of overexpressed wild-type Flag–emerin. At these high overexpression levels, most cells (nearly 70%; Fig. 5B) also showed an unexpectedly dominant chromatin phenotype at 24 h, termed small nuclei with potentially condensed chromatin and irregular nuclear envelope (‘SCC’ phenotype; Fig. 5A, 24 h; see also supplementary material Figs S1–S3). Cells with the SCC phenotype were not apoptotic (supplementary material Fig. S4A). Chromatin was condensed in only 7.8% of mock-transfected control cells, essentially all of which were mitotic (mitotic index 7.7%; n = 3; 863 cells counted). Based on fixed cell images, we speculate that ‘curved’ structures formed by wild-type Flag-emerin might enlarge and disconnect from chromatin as rounded structures (supplementary material Fig. S1, WT). Given that emerin can bind tubulin (Salpingidou et al., 2007), we tested potential colocalization of these round structures with the microtubule-organizing center (MTOC), using antibodies against CEP192 (centriolar protein 192, also known as Spd2), but found no correlation (arrows, supplementary material Fig. S4B). Cells expressing wild-type Flag–emerin at moderate levels typically showed a milder misshaped-nucleus phenotype (Fig. 5A; supplementary material Fig. S1). We attribute this unexpected dominant SCC phenotype to the very high exogenous Flag–emerin overexpression permitted in HEK293T cells. The SCC phenotype was also seen in a minority of transfected HeLa cells (highest expressers; J.M.B. and K.L.W., unpublished observations) and at 48 h, but not 24 h, post-transfection in the independent ‘Hek293FA’ cell line with reduced A-type lamins (Takeshi Shimi, Northwestern University, Chicago, IL, personal communication). The percentage of SCC-positive cells was reduced significantly (∼3–4 fold; Fig. 5B) by any deletion that weakened (ΔL, ΔR, ΔAR) or disrupted (ΔA) Flag–emerin localization at the nuclear envelope (discussed below). ΔM localized at the nuclear envelope (statistically insignificant ER localization; Fig. 5D,E) and retained some penetrance for the SCC phenotype (∼34% of cells were SCC positive; twofold reduction; Fig. 5B; P<0.05). The mechanisms and timing (e.g. interphase versus postmitotic nuclear assembly) of emerin-overexpression-induced chromatin ‘condensation’ will be characterized in future work.
Region A was essential for Flag–emerin steady-state localization: at 24 h post transfection, the vast majority of ΔA was absent from the nuclear envelope and ER (Fig. 5D,E), having formed perinuclear aggregates in 97% of cells (Fig. 5F; P<0.0005), consistent with the fivefold increase in intermolecular association in vivo (Fig. 4H). We conclude that residues 168–186 are essential to inhibit or control emerin intermolecular association in vivo.
The LEM domain was required for efficient emerin retention at the nuclear envelope: Flag–emerin-ΔL showed significant ER localization in 98% of expressing cells (Fig. 5E; P<0.003). This result, and the inefficient nuclear envelope retention of native emerin in BAF-deficient Nestor-Guillermo progeria patient cells (Puente et al., 2011), support a BAF-dependent mechanism for emerin retention at the nuclear envelope, but do not rule out additional role(s) for the LEM domain.
Positive element R was important for Flag–emerin localization at the nuclear envelope: at 24 h, ΔR and ΔAR were detectable at the nuclear envelope (Fig. 5A,D) but showed significantly higher ER localization (Fig. 5E) and a tendency to aggregate (especially ΔAR; Fig. 5F). ΔR and especially ΔAR were significantly less able to form patches and/or caps at the nuclear envelope (Fig. 5C), consistent with their ‘dilution’ in the ER and reduced intermolecular association (Fig. 4H). Interestingly, ΔAR was the only construct that localized substantially at or near the plasma membrane, as seen in 52% of Flag–ΔAR-positive cells (∼25-fold increase over wild-type; Fig. 5G, P<0.005), including potential sites of cell–cell contact (Fig. 5A; lower plane of focus, supplementary material Fig. S2). Thus, the positive and negative elements together – hence, properly controlled emerin intermolecular association – might contribute to emerin nuclear envelope localization by blocking an alternative trafficking fate(s). The juxta-plasma-membrane localization of ΔAR, in particular, under these highly overexpressed conditions, suggests that cells might target native emerin to new locations, including cardiomyocyte intercalated disks (Berk et al., 2013b; Cartegni et al., 1997; Wheeler et al., 2010), by post-translationally inhibiting emerin–emerin association.
Emerin residues 170–220 are required to bind BAF in cells
Compared to Flag-tagged constructs, the GFP-tagged constructs expressed at lower levels, and GFP–ΔR and GFP–ΔAR were more enriched at the nuclear envelope; GFP–ΔR in particular also showed some ability to ‘condense’ chromatin (24 h; Fig. 6A), suggesting this phenotype is influenced by different tags (GFP is 34 kDa; Flag, 2 kDa). To test association with native nuclear lamina partners, we expressed each GFP–emerin construct in HEK293T cells for 24 h and then isolated nuclear pellets, prepared sonicated (total) nuclear (‘NT’) lysates and immunoprecipitated from equal protein concentrations using bead-conjugated llama anti-GFP antibodies, or beads alone as control. Immunoprecipitates (20%) and control input lysates (1%) were resolved by SDS-PAGE and probed with antibodies specific for emerin, A- or B-type lamins or BAF, then stripped and reprobed with anti-GFP antibodies (Fig. 6B). Results were quantified as the partner-to-GFP ratio for each deletion, normalized to that of wild-type GFP–emerin. All six GFP–emerin deletions specifically co-precipitated endogenous emerin, A-type lamins and B-type lamins at levels statistically indistinguishable from wild-type GFP–emerin; controls verified GFP alone or beads alone (in lysates containing wild-type GFP–emerin) did not precipitate lamina proteins (Fig. 6B; quantified in supplementary material Fig. S4C–E). There was one exception: nuclear ΔL co-precipitated 25% less endogenous emerin (supplementary material Fig. S4C), suggesting the LEM-domain contributes to emerin–emerin association. Other potential changes might not have been detected, e.g. due to construct overexpression, or because emerin and lamins are relatively stable components of nuclear structure. Note that each construct still possessed at least one region sufficient to bind native emerin (e.g. through a different domain) and lamins (through emerin residues 1–132 or 159–220). The ΔL and ΔM controls behaved as expected: BAF association was eliminated by ΔL, and unaffected by ΔM (Fig. 6B,C; n = 3; P<0.0002 for ΔL). This result for ΔM, which shortened emerin to the same extent as ΔAR, eliminated a strict ‘molecular length’ requirement for emerin to bind BAF in vivo. Nuclear BAF did not associate with ΔA (Fig. 6C; n = 3; P<0.00005), as expected (Fig. 5). Intriguingly, nuclear BAF association with nuclear GFP–emerin was reduced significantly by ΔR and ΔAR (by 93% and 87%, respectively; Fig. 6B,C; n = 3; ΔR P<0.0004; ΔAR P<0.004). Why was this region critical for contact with BAF in cells?
Fig. 6.

GFP–emerin localizations and interactions in HEK293T cells: residues 170–220 are required to bind BAF in vivo. (A) Localization of GFP–emerin polypeptides in HEK293T cells either fixed 5 h post-transfection and stained by indirect immunofluorescence using GFP antibodies (n = 2), or fixed 24 h post-transfection for direct GFP fluorescence detection (n = 3). (B,C) Sonicated (total) nuclear lysates (NT fraction) from HEK293T cells at 24 h post-transfection with GFP–emerin polypeptides were immunoprecipitated with anti-GFP antibodies (IP: GFP), resolved by SDS-PAGE and immunoblotted for A-type lamins (LmA/C), B-type lamins (LmB), GFP, emerin (Emr; the endogenous ∼34 kDa band is shown) or BAF. The vertical line divides separate blots. Results were quantified by densitometry in C as the endogenous BAF-to-GFP–emerin signal ratio, relative to that of wild-type GFP–emerin (n = 3). **P<0.004. Results are mean±s.e.m.
Peptide array analysis
We next wanted to investigate why the AR region was crucial for emerin contact with BAF in cells. To specifically locate the regions contacted by AR (residues 170–220), we used recombinant GST–emerin-170–220 to probe arrayed 20-mer rat emerin peptides, spotted with three-residue offsets on cellulose membranes (Wanichawan et al., 2011). This probe bound two singletons (the ‘R-peptide’ and the ‘LEM-peptide’) and four overlapping peptides (the ‘SAYQ-region’; Fig. 7A; n = 2). To evaluate potential conservation of these peptides, we manually aligned the sequences of human (‘H’) and rat (‘R’) emerin with two more-distant species: nine-banded armadillo (‘A’) and zebrafish (‘Z’; Fig. 7B).
Fig. 7.

Peptide mapping and modeling. (A) Rat emerin 20-mer peptide array probed with (+) or without (−) GST–emerin-170–220; positive peptide sequences are shown (n = 2). (B) Amino acid sequences of human (H., UniProt P50402), rat (R., UniProt Q63190), nine-banded armadillo (A., GenBank XP_004482931.1) and zebrafish (Z., GenBank XP_001924046.1) emerin, numbered according to the human sequence. The LEM domain, transmembrane domain (TMD), positive element R and other functional elements are boxed (see text). Solid lines summarize peptide array results. Stippled line indicates ‘APC-like’ homology (Cartegni et al., 1997).
The R-peptide and two SAYQ-region peptides showed the qualitatively strongest signals. Rat R-peptide IRPEKQAPTAALGQDRQVPL corresponded to human residues 206–225 in region R, plus most of a Val-Pro-aliphatic-Trp (‘VPaW’) motif abutting the transmembrane domain (Fig. 7B). We propose region R (potentially the R-peptide itself) mediates emerin homotypic association (‘R-to-R’ model; Fig. 8A).
Fig. 8.

Models of the proposed emerin intermolecular association. Emerin–emerin ‘R-to-R’ (A) and ‘R-to-S’ (B) ‘backbone’ configurations, with hypothetical ‘up’ (backbone-associated) versus ‘down’ (BAF- or chromatin-associated) positions of the LEM domain (L). INM, inner nuclear membrane.
The ‘SAYQ-region’ peptides (Fig. 7A) identified a strong heterotypic partner for the AR probe, corresponding to human residues ∼147–174 (Fig. 7B). The two best signals (rat peptides RERPIYGRDSAYQSIAEYRP and PIYGRDSAYQSIAEYRPISN) shared the sequence PIYGRDSAYQSIAEYRP (Fig. 7A). Cross-species comparison suggests this peptide has conserved features, underlined in human residues P153MYGRDSAYQSITHYRP169 (Fig. 7B). This peptide might have multiple roles: it includes S159AYQSITHYRP169 (important or essential to bind lamins; Fig. 3) and was ‘clipped’ (RP169 removed) by ΔA. We conclude this SAYQ-region peptide (‘S’) is a strong binding partner for AR. Because R was essential for both modes of domain association in vivo (Fig. 4H), we favor ‘S-to-R’ models of intermolecular binding (Fig. 8B) but cannot rule out potential involvement of region A.
Rat LEM-peptide YEKKIFEYETQRRRLSPPSS identified a second binding partner for the 170–220 region, corresponding to human LEM-domain α-helical residues 34–47 (Cai et al., 2007) plus adjacent residues 48–53 (Fig. 7B). This peptide identified the LEM-domain itself as a binding partner for residues 170–220 (‘L-to-AR’ models; Fig. 8).
DISCUSSION
This biochemical and cellular evidence fits models for emerin in which positive elements (region R) associate with each other (proposed ‘R-to-R’ backbone, Fig. 8A) and with different domains [proposed ‘R-to-SAYQ-region (‘R-to-S’) backbone, Fig. 8B] giving rise to a strong potential to form emerin intermolecular networks. Emerin–emerin association in cells was supported by the time-dependent concentration of overexpressed emerin at discrete nuclear envelope regions, and GFP–emerin association with endogenous emerin. The emerin network hypothesis and underlying intrinsic disorder are new concepts in nuclear envelope organization and signaling. Direct LEM-peptide binding to AR, and cellular evidence that BAF association requires positive element R, suggest functional ‘backbone’ coupling to the LEM-domain. ‘Backbones’ might include or exclude the LEM domain (‘up’ or ‘down’ models, respectively; Fig. 8) as mechanisms to regulate or respond to contact with BAF and chromatin. These models, and multipartite emerin–emerin association per se, predict emerin ‘network’ re-organization in response to mechanical force as the basis for mechano-transduction signaling relevant to Emery–Dreifuss muscular dystrophy (Guilluy et al., 2014; Ho et al., 2013; Holaska et al., 2004; Lammerding et al., 2005).
We do not know which molecular framework(s) or LEM-domain conformation(s) actually bind BAF or chromatin (modeled ‘down’ for simplicity in Fig. 8). Major biophysical unknowns include the stoichiometry, orientation and affinities of emerin intermolecular association. However, intermolecular association is conserved in C. elegans emerin, which has a shorter nucleoplasmic domain (125 versus 228 residues) and self-associates with an equilibrium affinity of 14 nM, comparable to its range of affinities for lamin (4–27 nM) and stronger than its affinity for BAF (57 nM) (Bar et al., 2014). Future characterization of emerin intrinsic disorder is also warranted. This concept fits all current evidence for emerin, including its signaling roles, post-translational ‘hotspots’ (Berk et al., 2013b) and participation in diverse multi-protein complexes, some of which lack BAF or lamins (Berk et al., 2013a; Holaska and Wilson, 2007). Our models satisfy this biological requirement for flexibility, with high predictive value.
Positive element and juxta-membrane elements
The positive element (residues 187–220) removed by ΔR has two parts. ‘Left’ residues 187–206 were Ser-rich with conservation in mammals (human residues S199WLxRRAI206) but not zebrafish (Fig. 7B). By contrast residues 207–220 showed conservation in zebrafish (e.g. human R207PxxxA212; Fig. 7B) and corresponded to residues 2–14 of the R-peptide. Thus, the peptide array and in vivo ΔR results together implicate human residues 207–220, at least, as direct mediators of emerin–emerin association. The last four residues of the R-peptide, R221QVPL225, though nonessential for the association between the two 170–200 domains in vitro (Fig. 1D), revealed a conserved juxta-membrane ‘VPaW’ motif with flanking Arg and Gly residues (Fig. 7B). Pro (‘kinks’ peptide) and Gly (potential ‘swivel’) residue conservation suggests that they have roles in emerin conformation near the membrane. Conserved C-terminal residues A248EEGNPF254 (Fig. 7B) likely contact lumenal partners.
Three positive element mutations – m196 (S196S to AA), m207 (R207P to AA) and m214 (G214AGL to AAGA) – were previously tested for binding to eight or more partners. None affected BAF, lamin A or F-actin binding; however, all three reduced or eliminated binding to transcription-regulating partners including LMO7 (Berk et al., 2013b; Mull et al., 2014; Wozniak et al., 2013). Whether such partners contact the positive element, or require a specific type of emerin ‘backbone’ are open questions.
Mechanisms and implications of the negative element
Residues 168–186 were non-essential for emerin–emerin association in vivo, and their absence (ΔA) increased emerin–emerin association fivefold in vivo. Thus ΔA inactivated or removed an element required to control or limit emerin intermolecular association. This negative element might bind partner(s) that competitively inhibit intermolecular association, or structural partner(s) that mechanically counterbalance emerin–emerin association. One candidate ‘inhibitor’, F-actin, is sensitive to negative element mutations m164 (I164THYRPV170 to A164AHARPA170), m175 (S175SL to A175AA) and m179 (L179S to A179A) (Berk et al., 2013b), and is also abundant in the cytoplasm. Other potential ‘inhibitors’ include cell adhesion and focal adhesion signaling partners; LMO7 (Mull et al., 2014; Wozniak et al., 2013) is sensitive to emerin mutations m175 and m179 (Holaska et al., 2006) and β-catenin is sensitive to ΔA (Markiewicz et al., 2006). As ‘counterbalancers’ we suggest lamins, which might directly contact the negative element: emerin residues 159–178 were found to be sufficient to bind lamin tails in vitro, and this region was disrupted by ΔA. We also suggest counterbalancing by chromatin (through BAF), because emerin–emerin association in HEK293T cells increased twofold when the LEM domain was missing (ΔL). We speculate ‘counterbalancing’ might specifically involve silent chromatin, because mutations m179 (L179S to A179A) and P183H in the negative element disrupt binding to HDAC3 (Demmerle et al., 2012).
The negative element is also likely crucial for post-translational control of emerin–emerin association. This element and the adjacent SAYQ-region have nine known phosphorylation sites, with competing O-GlcNAc modifications at Ser171 and Ser173 (Berk et al., 2013a; Berk et al., 2013b). These modifications might promote, block or bias conformational options, allowing signaling pathways to compete or engage in ‘crosstalk’ by direct manipulation of emerin–emerin association. We speculate inhibitory modifications might promote native emerin localization near the cell surface, as seen in cardiomyocyte intercalated disks (Berk et al., 2013b; Cartegni et al., 1997; Wheeler et al., 2010).
Post-translational control and auto-inhibition of LEM-domain ‘tethering’?
We unexpectedly discovered direct binding of the emerin ‘backbone’ (AR; residues 170–220) to the LEM-domain, specifically LEM-domain α-helical residues 34–47 plus residues L48SPPSS53. Three ‘LEM-peptide’ residues (Tyr34, Lys36, Lys37) are highly conserved and directly interface with BAF (Cai et al., 2007), supporting the possibility that ‘backbone-associated’ LEM domains cannot contact BAF (Fig. 8). Our conclusion that distal regions of emerin influence the LEM domain is consistent with a previous study: Xenopus LAP2 isoforms with identical LEM domains but distinct C-terminal domains varied ninefold in their affinity for BAF (Shumaker et al., 2001), through unknown mechanisms. For emerin, we predict LEM-domain association with the emerin ‘backbone’ is controlled by post-translational modifications of the LEM-domain and the neighboring residues L48SPPSS53 (Fig. 7B), which include major sites of mitotic phosphorylation (Ser49) and O-GlcNAc regulation (Ser53) (Berk et al., 2013a).
The peptide arrays predict residues 170–220 (AR) should have bound any recombinant polypeptide in Fig. 2 that included the LEM-peptide (e.g. residues 1–84 through 1–118, and 1–160). Its failure to bind, suggested a potential recombinant protein artifact. However, another possibility is that, enabled by molecular flexibility, there is auto-inhibition of the LEM domain in cis: residues 48–118 might ‘hide’ the LEM peptide, with ‘counter-inhibition’ by residues 119–132 in cis. Similarly, residues 1–160, containing the LEM peptide and half of region ‘S’, also failed to bind AR. These negative results might be biologically significant because this hypothetical auto-inhibition of the LEM-peptide in cis involves residues 48–118 and 133–160 – the same two regions implicated in contact with lamins (Fig. 3C). Residues 133–160, which includes part of ‘S’, also contribute to the proposed S-to-R ‘backbone’. These correlations suggest auto-inhibition in cis as a mechanism to coordinate emerin intermolecular association with nuclear lamina structure.
LEM-domain association with the emerin ‘backbone’ suggests other LEM-domain proteins might behave similarly, and provides a context to understand why mutations in the SAYQ peptide (Y161F) or negative element (S173D, S175A) interfere with LEM-domain binding to BAF in vivo (Berk et al., 2013a; Tifft et al., 2009). Ser173 in particular is a ‘battleground’ for OGT (permissive) versus kinase (inhibitory) control of emerin–BAF association in cells (Berk et al., 2013a). These models for emerin associations have broad implications for nuclear lamina structure, nuclear envelope signaling, chromatin organization and postmitotic nuclear assembly (Amendola and van Steensel, 2014; Demmerle et al., 2012).
MATERIALS AND METHODS
Cell culture
HEK293T cells (from Susan Michaelis, Johns Hopkins School of Medicine) and HeLa cells (American Type Culture Collection, Manassas, VA) were cultured in complete medium (high glucose Dulbecco's modified Eagle's medium (DMEM) plus 10% (v/v) fetal bovine serum (FBS; Life Technologies, Grand Island, NY) with 1% (v/v) penicillin-streptomycin (Quality Biological, Gaithersburg, MD). For transfection, cells were plated at ∼30–40% confluence, incubated for 24 h, then supplemented with 100 µl Optimem (Life Technologies, Grand Island, NY), 2.5 µl Trans-IT LT1 (Mirus, Madison, WI) and 0.5 µg DNA/ml of complete medium. Cells were incubated for 5 or 24 h, then fixed or harvested.
Immunoprecipitation
Cell pellets were lysed with ice-cold lysis buffer (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 0.3% v/v Triton-X100, 50 mM GlcNAc, 1 mM DTT, 100 µM PMSF, 1 µg/ml Pepstatin A, 1× Roche Protease Cocktail Inhibitor, 1× Roche PhoStop Phosphatase Inhibitor), sonicated and processed as described previously (Berk et al., 2013a) to obtain ‘total’ whole-cell lysates. For Fig. 6, fractionated (17,000 g) nuclear-enriched pellets were sonicated as described previously (Berk et al., 2013a) to obtain ‘total’ nuclear lysates. Protein concentrations (Quick Start Bradford Protein assay; Bio-Rad, Hercules, CA) were adjusted to identical levels between replicates. To co-immunoprecipitate GFP–emerin and Flag–emerin polypeptides, whole-cell lysate protein concentrations were adjusted to 1.5–2.0 mg/ml with lysis buffer. To co-immunoprecipitate endogenous partners with GFP–emerin, total nuclear lysate protein concentrations were adjusted to 0.75–1.0 mg/ml; for each reaction 1 ml lysate was incubated overnight (rotating, 4°C) with 10 µl pre-washed GFP-specific agarose-conjugated llama antibodies, or agarose alone as control (gta-200 and bab-20, respectively; Chromotek, Martinsried, Germany). Agarose was washed three times (50 mM Tris-HCl pH 7.4, 0.3 M NaCl, 0.3% v/v Triton X-100), and proteins were eluted in SDS sample buffer. For GFP–emerin and Flag–emerin co-immunoprecipitations, 2–2.5% of each eluate was resolved on NuPage Novex 4–12% Bis-Tris gradient gels in 1× NuPage MES SDS Running Buffer (Life Technologies) along with 1% input lysate from each reaction on the same or parallel gels. For GFP–emerin and endogenous partner co-immunoprecipitations, we resolved 20% of each eluate and 1% of input, transferred onto nitrocellulose (Bio-Rad) and immunoblotted as described below.
Immunoblotting
After transfer, membranes were stained with Ponceau (Sigma), scanned (Perfection 2450 Photo; Epson, Long Beach, CA) if needed, and blocked for 1 h (22–25°C) in PBST (PBS, 0.1% v/v Tween-20) containing 3% (w/v) BSA (Sigma), and incubated overnight in PBST with 3% (w/v) BSA with either mouse anti-GST (1∶2000–3000; SC-138, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-BAF serum 3273 (1∶1000–2000; Haraguchi et al., 2001), rabbit anti-lamin A/C (1∶2000; SC-20681, Santa Cruz Biotechnology), rabbit anti-lamin B serum NC-7 (1∶2000, gift from Nilabh Chaudhary), rabbit anti-emerin serum 2999 (1∶2000–10000; Lee et al., 2001), rabbit anti-histone H3 (1∶200,000; 1791, Abcam, Cambridge, MA), mouse anti-Flag (1∶10,000; F3165, Sigma), mouse anti-GFP (1∶200,000; SC-9996, Santa Cruz Biotechnology), rabbit anti-GFP (1∶20,000; A6455, Life Technologies) or mouse anti-T7-tag (69048-3, Novagen; 1∶10,000) antibodies. Blots were incubated for 1 h (22–25°C) with horseradish-peroxidase-conjugated mouse or rabbit secondary antibodies (1∶10,000; NA931V and NA934V, respectively; GE Healthcare Biosciences) in PBST with 3% (w/v) BSA and visualized by chemiluminescence (HyGlo; Denville) on autoradiography film (HyBlot CL, Denville, South Plainfield, NJ). Films were scanned (Perfection 2450 Photo; Epson), quantified (Quantity One, v. 4.5.2; Bio-Rad) and processed using Adobe Photoshop CS4 and Illustrator CS4 (Adobe; San Jose, CA).
Fluorescence microscopy
HEK293T cells plated on sterile, acid-washed, poly-lysine-coated coverslips were transfected with GFP, GFP–emerin or Flag–emerin, or were mock transfected, and then incubated for 5 or 24 h. Coverslips were rinsed three times in PBS, fixed in PBS with 3.7% (w/v) formaldehyde (VWR International, Radnor, PA) for 15 min, washed three times (PBS), permeabilized 18 min in PBS with 0.2% (v/v) Triton X-100 (Sigma), washed three times (PBS) and blocked 1 h in PBS with 2% BSA (w/v), all at 22–25°C. Where indicated coverslips were incubated with 5% (w/v) BSA in KCM (120 mM KCl, 20 mM NaCl, 10 mM Tris-HCl pH 7.7, 0.1% v/v Triton-X 100) with rabbit anti-GFP (1∶500; A6455, Life Technologies) or mouse anti-Flag (1∶200; F3165, Sigma) antibodies overnight at 4°C. Coverslips were washed three times in PBS, then incubated (1 h, 22–25°C) in KCM with 5% (w/v) BSA containing either FITC-conjugated goat anti-rabbit or Cy3-conjugated goat anti-mouse antibodies (Jackson ImmunoResearch, West Grove, PA; each 1∶1000), rinsed three times in PBS, briefly incubated with 25 µg/ml 4′,6-diamidino-2-phenylindole (DAPI) and mounted on 1.0-mm glass slides (Vectashield mounting media; Vector Laboratories, Burlingame CA). Coverslips were visualized with a 60× oil immersion DIC H objective with 1.4 numerical aperture mounted on an Eclipse E600 wide-field fluorescent microscope (Nikon, Melville, NY), captured using a Retiga Exi charge-coupled device camera (Q Imaging, British Columbia, Canada) and iVision-MacTM image capturing software (BioVision Technologies, Exton, PA). Images were adjusted with Photoshop CS4 (Adobe; San Jose, CA).
Recombinant protein purification
Protein expression was induced in transformed bacteria (BL21 DE3 competent E. coli) at an optical density at 600 nm (OD600) of 0.6–0.7 using 0.4 or 1.0 mM isopropyl-β-D-thio-galactoside (IPTG) for 4 h (37°C). GST–emerin-1–222 polypeptides were purified and refolded as described previously (Tifft et al., 2009). Other GST-tagged polypeptides were purified as described previously (Berk et al., 2013a). Recombinant His-tagged emerin polypeptides (pET29b Emerin-His 1–220; pET29b S-Emerin-His 170–220; pET29b S-Emerin-His 1–176; pET15b His-Emerin 1–118; pET15b His-emerin 1–132; pET15b His-emerin 1–221) were purified under denaturing conditions as described previously (Berk et al., 2013a).
Recombinant lamin tail peptides were expressed in BL21 DE3 competent E. coli, induced using 1.0 mM isopropyl-β-D-thio-galactoside (IPTG; 3 h, 37°C), purified on Ni2+-NTA-agarose as described previously (Simon et al., 2010) and stored in 50 mM NaHPO4 pH 8.0, 300 mM NaCl, 100 mM imidazole, 0.5 mM PMSF at −80°C.
In vitro binding reactions
Purified His–emerin-1–220 and S–emerin-170–220–His polypeptides in 4 M urea buffer (see above) were bound to Ni2+-NTA agarose in 4 M urea storage buffer (pH 7.4) for 1 h (4°C), and then washed with 80 mM imidazole in protein binding buffer (PBB, 20 mM HEPES-NaCl, pH 7.9, 20% v/v glycerol, 0.5 mM EDTA, 60 mM NaCl, 6 mM MgCl2, 0.1% v/v NP-40, 1 mM PMSF, 1∶1000 β-ME; adapted from Takemaru and Moon, 2000). Bead-bound ‘bait’ polypeptides were then resuspended (final concentration 1.0 µM) in 200 µl PBB with 80 mM imidazole containing either purified GST or GST–emerin polypeptides (final ‘prey’ concentration: ∼0.03–1.0 µM), rotated for 1 h (4°C), and washed three times in PBB with 80 mM imidazole. Bead-bound proteins were eluted using SDS-sample buffer. Binding reactions with His–emerin-1–220 as bait were resolved on NuPage Novex 4–12% Bis-Tris gradient gels in 1× NuPage MES SDS Running Buffer (Invitrogen, Grand Island, NY). Binding reactions with His–emerin-170–220 as bait were resolved on 10% Bis-Tris gels in 1× NuPage MOPS SDS Running Buffer (Invitrogen). Resolved proteins were transferred onto nitrocellulose, Ponceau stained and immunoblotted as described below, or Coomassie stained with GelCode Blue Safe Protein Stain (Thermo Fisher Scientific, Waltham, MA).
Purified N-terminally-T7-tagged and C-terminally-His-tagged lamin tail peptides were dialyzed overnight to remove imidazole, bound to Ni2+-NTA agarose (Qiagen) in storage buffer (50 mM NaHPO4 pH 8.0, 300 mM NaCl) for 1 h (4°C), then washed with 20 mM imidazole in PBB (detailed above). Bead-bound lamin ‘bait’ polypeptides were resuspended in 200 µl PBB with 20 mM imidazole (1.0 µM final concentration) and 1.0 µM GST–emerin, rotated (1 h, 4°C), washed three times in PBB with 20 mM imidazole and eluted using SDS-sample buffer. Binding reactions were resolved on NuPage Novex 4–12% Bis-Tris gradient gels in 1× NuPage MES SDS Running Buffer (Invitrogen, Grand Island, NY), transferred onto nitrocellulose, Ponceau-stained and immunoblotted as described above.
Plasmids
Bacterial expression plasmids (pET29b) expressing His-tagged emerin residues 1–220 or S- and His-tagged residues 170–220, and pGEX plasmids expressing GST-tagged emerin residues 1–220, 70–140, 140–176 or 170–220 were kind gifts from Juliet Ellis (King's College, London, UK) (Wheeler et al., 2007). pGST-emerin-1–222 was described previously (Tifft et al., 2009). GST-tagged emerin polypeptides (1–84, 1–94, 1–104, 1–118, 1–132, 1–149, 1–160) were made using site-directed mutagenesis to insert stop codons in pGEX 4T3 GST-emerin 1–220 (Quikchange, Agilent Technologies, Clara, CA), and verified by DNA sequencing. To create GST-fused emerin residues 102–158, 159–220 or 159–228, we PCR-amplified from emerin cDNA using primers flanked with 5′BamHI and 3′SalI restriction sites and subcloned into pGEM-T Easy (Promega, Madison, WI). Each insert was excised using appropriate restriction enzymes, then ligated (T4 ligase; New England Biolabs, Ipswich, MA) into BamHI- and SalI-digested pGEX-4T3.
The pET15b His-emerin 1–221 was made by amplifying full-length emerin cDNA from pEGFP-C1 emerin (Tsuchiya et al., 1999) using primers with flanking 5′NdeI and 3′BamHI restriction sites, then subcloned into pGEM-T Easy (Promega, Madison, WI). The amplicon was excised and ligated (T4 ligase) into NdeI- and BamHI-cut pET15b, and truncated by site-directed mutagenesis as described above. Human lamin tail constructs for mature lamin A (residues 385–646) and lamin B1 (residues 395–586) in pET23b (Novagen) which places a T7 tag at the N-terminus and a His6-tag at the C-terminus, were described (Simon et al., 2010).
For expression in mammalian cells, we used plasmids encoding N-terminally GFP-tagged (pEGFP-C1) or Flag-tagged (pCMV Tag2b; kindly provided by Juliet Ellis; Wheeler et al., 2010) full-length wild-type human emerin (residues 1–254) to generate the ΔL (residues 1–47 deleted), ΔM (67–108 deleted), ΔA (168–186 deleted), ΔR (187–220 deleted) and ΔAR (170–220 deleted) constructs. For ΔL, the emerin cDNA encoding residues 48–254 was amplified using primers flanked by 5′BglII and 3′SalI sites, subcloned into pGEM-T Easy, then excised and ligated into either BglII-and-SalI-cut pEGFP-C1 or BamHI-and-SalI-cut pCMV Tag2b. GFP–emerin-ΔM was constructed as described previously (Tsuchiya et al., 1999). GFP–emerin-ΔR was constructed by site-directed mutagenesis (Quikchange, Agilent Technologies). Flag–emerin-ΔM and Flag–emerin-ΔR were constructed by using 5′BamHI- and 3′SalI-site flanked primers to amplify the ΔM and ΔR cDNAs from pEGFP-C1, then subcloning into pGEM-T Easy, excision and ligation into BamHI-and-SalI-cut pCMV Tag2b. Both ΔA plasmids were constructed as described previously (Markiewicz et al., 2006). The ΔAR deletions were made by site-directed mutagenesis. All plasmids were verified by DNA sequencing. PCR primer sequences are available upon request.
Peptide arrays
Rat emerin 20-mer peptides synthesized with three-residue offsets were spotted on cellulose membranes, incubated (18 h, 4°C) with or without recombinant GST–emerin-170–220, washed and visualized using anti-GST–HRP and chemiluminescence as described previously (Wanichawan et al., 2011).
Statistical analysis
Statistical significance was determined using a two-tailed Student's t-test.
Supplementary Material
Acknowledgments
We gratefully acknowledge Takeshi Shimi and Robert Goldman (Northwestern University, Chicago, IL) for communicating results, Juliet Ellis (King's College, London, UK) for plasmids, Keith Dunker (Indiana University, Indianapolis, IN) and Bin Xue (University of South Florida, Tampa, FL) for help with intrinsic disorder predictions, and our colleagues from Johns Hopkins University School of Medicine, Carolyn Machamer and Andrew Holland, for antibodies and insightful discussions.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
Experiments shown in Figs 1–6 were designed and analyzed by J.M.B. and K.L.W., performed by J.M.B., D.N.S. and J.W.W. with scoring by K.L.W. and modeling by J.M.B. and K.L.W. Peptide arrays were designed by C.R.C., L.G.W. and K.L.W., and performed by C.R.C. and L.G.W. Supplementary experiments were designed and analyzed by J.M.B., C.R.J.-H. and K.L.W., and performed by J.M.B. and C.R.J.-H. The manuscript was written by J.M.B. and K.L.W.
Funding
We acknowledge funding from the Research Council of Norway (to C.R.C.); the National Institutes of Health [grant number GM048646 to K.L.W.]; the Institute for Basic Biomedical Sciences at Johns Hopkins School of Medicine [grant number 80031412; and the Mills Fund to K.L.W. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.148247/-/DC1
References
- Amendola M., van Steensel B. (2014). Mechanisms and dynamics of nuclear lamina-genome interactions. Curr. Opin. Cell Biol. 28, 61–68 10.1016/j.ceb.2014.03.003 [DOI] [PubMed] [Google Scholar]
- Bar D. Z., Davidovich M., Lamm A. T., Zer H., Wilson K. L., Gruenbaum Y. (2014). BAF-1 mobility is regulated by environmental stresses. Mol. Biol. Cell 25, 1127–1136 10.1091/mbc.E13-08-0477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan R., Zahand A. J., Sharabi K., Lamm A. T., Feinstein N., Haithcock E., Wilson K. L., Liu J., Gruenbaum Y. (2012). Ce-emerin and LEM-2: essential roles in Caenorhabditis elegans development, muscle function, and mitosis. Mol. Biol. Cell 23, 543–552 10.1091/mbc.E11-06-0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk J. M., Maitra S., Dawdy A. W., Shabanowitz J., Hunt D. F., Wilson K. L. (2013a). O-Linked β-N-acetylglucosamine (O-GlcNAc) regulates emerin binding to barrier to autointegration factor (BAF) in a chromatin- and lamin B-enriched “niche” J. Biol. Chem. 288, 30192–30209 10.1074/jbc.M113.503060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk J. M., Tifft K. E., Wilson K. L. (2013b). The nuclear envelope LEM-domain protein emerin. Nucleus 4, 298–314 10.4161/nucl.25751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bione S., Maestrini E., Rivella S., Mancini M., Regis S., Romeo G., Toniolo D. (1994). Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 8, 323–327 10.1038/ng1294-323 [DOI] [PubMed] [Google Scholar]
- Burke B., Stewart C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13–24 10.1038/nrm3488 [DOI] [PubMed] [Google Scholar]
- Cai M., Huang Y., Suh J. Y., Louis J. M., Ghirlando R., Craigie R., Clore G. M. (2007). Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J. Biol. Chem. 282, 14525–14535 10.1074/jbc.M700576200 [DOI] [PubMed] [Google Scholar]
- Cartegni L., di Barletta M. R., Barresi R., Squarzoni S., Sabatelli P., Maraldi N., Mora M., Di Blasi C., Cornelio F., Merlini L. et al. (1997). Heart-specific localization of emerin: new insights into Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 6, 2257–2264 10.1093/hmg/6.13.2257 [DOI] [PubMed] [Google Scholar]
- Chang W., Folker E. S., Worman H. J., Gundersen G. G. (2013). Emerin organizes actin flow for nuclear movement and centrosome orientation in migrating fibroblasts. Mol. Biol. Cell 24, 3869–3880 10.1091/mbc.E13-06-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzetto D., Jones D. T. (2013). The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr. Opin. Struct. Biol. 23, 467–472 10.1016/j.sbi.2013.02.001 [DOI] [PubMed] [Google Scholar]
- Demmerle J., Koch A. J., Holaska J. M. (2012). The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J. Biol. Chem. 287, 22080–22088 10.1074/jbc.M111.325308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich C. M., Kratz K., Sendoel A., Gruenbaum Y., Jiricny J., Hengartner M. O. (2012). LEM-3 - A LEM domain containing nuclease involved in the DNA damage response in C. elegans. PLoS ONE 7, e24555 10.1371/journal.pone.0024555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery A. E., Dreifuss F. E. (1966). Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 29, 338–342 10.1136/jnnp.29.4.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R., Gerace L. (1993). Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73, 1267–1279 10.1016/0092-8674(93)90355-T [DOI] [PubMed] [Google Scholar]
- Guilluy C., Osborne L. D., Van Landeghem L., Sharek L., Superfine R., Garcia-Mata R., Burridge K. (2014). Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381 10.1038/ncb2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque F., Mazzeo D., Patel J. T., Smallwood D. T., Ellis J. A., Shanahan C. M., Shackleton S. (2010). Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 285, 3487–3498 10.1074/jbc.M109.071910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T., Koujin T., Segura-Totten M., Lee K. K., Matsuoka Y., Yoneda Y., Wilson K. L., Hiraoka Y. (2001). BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 114, 4575–4585 [DOI] [PubMed] [Google Scholar]
- Ho C. Y., Jaalouk D. E., Vartiainen M. K., Lammerding J. (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511 10.1038/nature12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska J. M., Wilson K. L. (2007). An emerin “proteome”: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 46, 8897–8908 10.1021/bi602636m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska J. M., Kowalski A. K., Wilson K. L. (2004). Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2, e231 10.1371/journal.pbio.0020231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska J. M., Rais-Bahrami S., Wilson K. L. (2006). Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 15, 3459–3472 10.1093/hmg/ddl423 [DOI] [PubMed] [Google Scholar]
- Huber M. D., Guan T., Gerace L. (2009). Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell. Biol. 29, 5718–5728 10.1128/MCB.00270-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L. M., Brown C. J., Lawson J. D., Obradović Z., Dunker A. K. (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584 10.1016/S0022-2836(02)00969-5 [DOI] [PubMed] [Google Scholar]
- Lammerding J., Hsiao J., Schulze P. C., Kozlov S., Stewart C. L., Lee R. T. (2005). Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 170, 781–791 10.1083/jcb.200502148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M. B., Nam Y., Jiang J., Sliz P., Walker S. (2011). Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567 10.1038/nature09638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. K., Haraguchi T., Lee R. S., Koujin T., Hiraoka Y., Wilson K. L. (2001). Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 114, 4567–4573 [DOI] [PubMed] [Google Scholar]
- Liu J., Lee K. K., Segura-Totten M., Neufeld E., Wilson K. L., Gruenbaum Y. (2003). MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 100, 4598–4603 10.1073/pnas.0730821100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalit A., Brachner A., Gotzmann J., Foisner R., Gruenbaum Y. (2007). Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol. 17, 202–208 10.1016/j.tcb.2007.02.004 [DOI] [PubMed] [Google Scholar]
- Markiewicz E., Tilgner K., Barker N., van de Wetering M., Clevers H., Dorobek M., Hausmanowa-Petrusewicz I., Ramaekers F. C., Broers J. L., Blankesteijn W. M. et al. (2006). The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 25, 3275–3285 10.1038/sj.emboj.7601230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-López I., Worman H. J. (2012). Inner nuclear membrane proteins: impact on human disease. Chromosoma 121, 153–167 10.1007/s00412-012-0360-2 [DOI] [PubMed] [Google Scholar]
- Meyerzon M., Fridolfsson H. N., Ly N., McNally F. J., Starr D. A. (2009). UNC-83 is a nuclear-specific cargo adaptor for kinesin-1-mediated nuclear migration. Development 136, 2725–2733 10.1242/dev.038596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mull A., Kim G., Holaska J. M. (2014). Lmo7-null mice exhibit phenotypes consistent with Emery-Dreifuss muscular dystrophy. Muscle Nerve [Epub ahead of print] doi:10.1002/mus.24286 [DOI] [PubMed] [Google Scholar]
- Puente X. S., Quesada V., Osorio F. G., Cabanillas R., Cadiñanos J., Fraile J. M., Ordóñez G. R., Puente D. A., Gutiérrez-Fernández A., Fanjul-Fernández M. et al. (2011). Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 88, 650–656 10.1016/j.ajhg.2011.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reil M., Dabauvalle M. C. (2013). Essential roles of LEM-domain protein MAN1 during organogenesis in Xenopus laevis and overlapping functions of emerin. Eur. J. Cell Biol. 92, 280–294 10.1016/j.ejcb.2013.10.008 [DOI] [PubMed] [Google Scholar]
- Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001). Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- Salpingidou G., Smertenko A., Hausmanowa-Petrucewicz I., Hussey P. J., Hutchison C. J. (2007). A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 178, 897–904 10.1083/jcb.200702026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber K. H., Kennedy B. K. (2013). When lamins go bad: nuclear structure and disease. Cell 152, 1365–1375 10.1016/j.cell.2013.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. Y., Méndez-López I., Wang Y., Hays A. P., Tanji K., Lefkowitch J. H., Schulze P. C., Worman H. J., Dauer W. T. (2013). Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell 26, 591–603 10.1016/j.devcel.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker D. K., Lee K. K., Tanhehco Y. C., Craigie R., Wilson K. L. (2001). LAP2 binds to BAF.DNA complexes: requirement for the LEM domain and modulation by variable regions. EMBO J. 20, 1754–1764 10.1093/emboj/20.7.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D. N., Wilson K. L. (2011). The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat. Rev. Mol. Cell Biol. 12, 695–708 10.1038/nrm3207 [DOI] [PubMed] [Google Scholar]
- Simon D. N., Zastrow M. S., Wilson K. L. (2010). Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 1, 264–272 10.4161/nucl.1.3.11799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru K. I., Moon R. T. (2000). The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 149, 249–254 10.1083/jcb.149.2.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tifft K. E., Bradbury K. A., Wilson K. L. (2009). Tyrosine phosphorylation of nuclear-membrane protein emerin by Src, Abl and other kinases. J. Cell Sci. 122, 3780–3790 10.1242/jcs.048397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad J. C., Barkan D. T., Gulledge B. F., Thalhammer A., Sali A., Schoepfer R., Burlingame A. L. (2012). Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteomics 11, 215–229 10.1074/mcp.O112.018366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y., Hase A., Ogawa M., Yorifuji H., Arahata K. (1999). Distinct regions specify the nuclear membrane targeting of emerin, the responsible protein for Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 259, 859–865 10.1046/j.1432-1327.1999.00112.x [DOI] [PubMed] [Google Scholar]
- Uversky V. N. (2013). A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 22, 693–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N., Oldfield C. J., Dunker A. K. (2008). Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 10.1146/annurev.biophys.37.032807.125924 [DOI] [PubMed] [Google Scholar]
- Wagner N., Krohne G. (2007). LEM-Domain proteins: new insights into lamin-interacting proteins. Int. Rev. Cytol. 261, 1–46 10.1016/S0074-7696(07)61001-8 [DOI] [PubMed] [Google Scholar]
- Wanichawan P., Louch W. E., Hortemo K. H., Austbø B., Lunde P. K., Scott J. D., Sejersted O. M., Carlson C. R. (2011). Full-length cardiac Na+/Ca2+ exchanger 1 protein is not phosphorylated by protein kinase A. Am. J. Physiol. 300, C989–C997 10.1152/ajpcell.00196.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler M. A., Davies J. D., Zhang Q., Emerson L. J., Hunt J., Shanahan C. M., Ellis J. A. (2007). Distinct functional domains in nesprin-1alpha and nesprin-2beta bind directly to emerin and both interactions are disrupted in X-linked Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 313, 2845–2857 10.1016/j.yexcr.2007.03.025 [DOI] [PubMed] [Google Scholar]
- Wheeler M. A., Warley A., Roberts R. G., Ehler E., Ellis J. A. (2010). Identification of an emerin-beta-catenin complex in the heart important for intercalated disc architecture and beta-catenin localisation. Cell. Mol. Life Sci. 67, 781–796 10.1007/s00018-009-0219-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M. A., Baker B. M., Chen C. S., Wilson K. L. (2013). The emerin-binding transcription factor Lmo7 is regulated by association with p130Cas at focal adhesions. PeerJ. 1, e134 10.7717/peerj.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B., Dunbrack R. L., Williams R. W., Dunker A. K., Uversky V. N. (2010). PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 10.1016/j.bbapap.2010.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.