

Abstract
Background
PALB2 has emerged as a breast cancer susceptibility gene. Mutations in PALB2 have been identified in almost all breast cancer populations studied to date, but the rarity of these mutations and lack of information regarding their penetrance makes genetic counseling for these families challenging. We studied BRCA1/2 -negative breast and/or ovarian cancer families to a) assess the contribution of PALB2 mutations in this series and b) identify clinical, pathological and family history characteristics that might make PALB2 screening more efficient.
Methods
The coding region of the PALB2 gene was analyzed in 175 probands with family histories of breast and/or ovarian cancer ascertained from a single Canadian institution in Eastern Ontario.
Results
We identified 2 probands with PALB2 mutations that are known or strongly considered to be pathogenic and 3 probands with missense mutations that are possibly pathogenic. One of the identified truncating mutations [c.3113G > A (p.Gly1000_Trp1038del – major product)], has been previously described while the other four mutations [c.3507_3508delTC (p.H1170Ffs*19), c.1846G > C (p.D616H), c.3418 T > G (p.W1140G), c.3287A > G (p.N1096S)] have not been previously reported. Loss of heterozygosity was detected in two breast tumors from one c.3507_3508delTC mutation carrier but not in other available tumors from that family or in tumors from carriers of other mutations.
Conclusions
PALB2 mutation screening identifies a small, but significant number of mutations in BRCA1/2 -negative breast and/or ovarian cancer families. We show that mutations are more likely to be found in families with three or more breast cancers as well as other BRCA2-related cancers. In our cohort, both clearly pathogenic mutations were identified in premenopausal breast cancer cases (2/77, 2.6%). Testing should be preferentially offered to affected women from such families.
Keywords: PALB2, Hereditary breast cancer, Pancreatic cancer, Melanoma, BRCA1 and BRCA2 mutation-negative
Introduction
Since first being identified as a BRCA2-interacting protein, Partner and Localizer of BRCA2 (PALB2) has been shown to also interact with BRCA1, effectively bridging these two well-known high-risk breast cancer susceptibility genes and aiding to regulate their function in DNA damage response and homologous recombination [1,2]. Not surprisingly, PALB2 has emerged in the last few years as an important breast cancer susceptibility gene in its own right (reviewed in [3]).
Germline mutations in PALB2 have been identified worldwide (reviewed in [4]), albeit rarely (1–4% of breast cancer families negative for BRCA1/BRCA2 mutations), and these mutations are associated with an increased risk of breast cancer that varies from approximately 2.3 to as high as ~6.0 depending on the mutations being studied and the populations under investigation [5-7]. While the degree of breast cancer susceptibility is still unclear, some studies examining recurrent PALB2 mutations tested in patients unselected for family history have demonstrated a risk and penetrance as high as those arising from BRCA2 mutations [6,7].
Similar to mutations in BRCA1 and BRCA2, the risks associated with monoallelic mutations in PALB2 seem to extend beyond breast cancer. To date, the spectrum of malignancies associated with PALB2 mutations remains unclear, however mutations confer increased risks for pancreatic cancer [8] and possibly ovarian cancer [9].
With the advent and increasing use of multiplex panels that test PALB2 alongside the BRCA genes [10], the greatest barrier for the implementation of PALB2 analysis into the clinic is no longer its testing efficiency but instead the lack of clear information and recurrence risks with which to counsel patients should a mutation be identified. Determining PALB2 mutation status is important, however, as it may allow female relatives of mutation positive patients the opportunity to make informed decisions about options to mitigate their elevated risk for disease. Also, new effective targeted therapeutic options are becoming available (PARP inhibitors) that have shown promising results in in vitro studies with PALB2 deficient cells exhibiting a defect in homologous repair [11].
Given that in order to determine clear criteria for genetic testing, we must first identify the likelihood of finding mutations in different populations, here we report our analysis of PALB2 in 175 breast and ovarian cancer pedigrees from a clinical cohort in Eastern Ontario, Canada, in which BRCA1 and BRCA2 testing failed to identify any causal variants.
Materials and methods
Cases and case selections
Participants were accrued from May 2009 to July 2012 at the Eastern Ontario Regional Genetics Program at the Children’s Hospital of Eastern Ontario. For this investigation, we selected participants affected with breast or ovarian cancer who had been previously screened for BRCA1 and BRCA2 mutations and excluded individuals with pathogenic mutations. Most individuals (169/175) were tested for BRCA1 and BRCA2 mutations by denaturing high-performance liquid chromatography, enhanced mismatch mutation analysis or sequencing. In six of eight individuals of Ashkenazi Jewish (AJ) descent, testing was limited to screening for the three common AJ mutations accounting for 98% of mutations in that population. Two individuals of AJ decent underwent full gene analysis, and 163/175 individuals were screened by MLPA for large insertions/deletions. Two separate cohorts of patients were specified: 1) those recruited May 2009 to September 2010, who were affected with breast or ovarian cancer, met Ontario provincial criteria for BRCA1 and BRCA2 genetic testing, and had a minimum BRCAPRO score of 0.10; 2) those recruited September 2010 to July 2012, who met select Ontario provincial criteria, were affected with breast or ovarian cancer, and had no BRCAPRO requirement (see Supplemental Materials for detailed list of Ontario Criteria, Additional file 1: Table S1). The criteria for study entry was changed to make sure we did not miss persons with a family history of pancreas cancer that might not otherwise be included on the basis of the BRCAPRO score.
Enrollment of probands, ascertainment of families and PALB2 gene analysis proceeded identically for each participant. Their respective geneticist or genetic counselor initially identified potential participants, and informed consent was obtained for those meeting criteria and interested in enrolling. In total, 195 patients were originally accrued. Of these, 13 participants showed initial interest yet failed to submit a blood sample, 4 were found to not meet selection criteria following enrollment. Of the 178 participants tested, in three cases the DNA sample was of insufficient quality. Therefore, of the 195 participants originally enrolled, 175 were eligible and able to be tested. Characteristics of the study participants are given in Table 1. Characteristics of breast tumor specimens are given in Additional file 1: Table S2.
Table 1.
Characteristics of 175 probands with clinical diagnoses of female unilateral breast cancer, female multiple primary breast cancer, male breast cancer or ovarian cancer
| Female unilateral | Female multiple primary | Male breast cancer | Ovarian cancer | |||||
|---|---|---|---|---|---|---|---|---|
|
Number of probands: |
126 |
24 |
12 |
13* |
||||
|
Age at Diagnosis |
(N) |
(%) |
(N) |
(%) |
(N) |
(%) |
(N) |
(%) |
|
20-29 |
7 |
5.6 |
0 |
0 |
0 |
0 |
1 |
7.7 |
|
30-39 |
26 |
20.6 |
5 |
20.8 |
0 |
0 |
1 |
7.7 |
|
40-49 |
44 |
34.9 |
13 |
54.2 |
3 |
25.0 |
5 |
38.5 |
|
50-59 |
30 |
23.8 |
6 |
25.0 |
2 |
16.7 |
0 |
0 |
|
60-69 |
16 |
12.7 |
0 |
0 |
3 |
25.0 |
5 |
38.5 |
|
70 and over |
3 |
2.3 |
0 |
0 |
4 |
33.3 |
1 |
7.7 |
|
Mean (years) |
47.1 |
43.4 |
60.8 |
51.3 |
||||
|
Range (years) |
23-78 |
31-58 |
43-76 |
23-76 |
||||
|
Mean time from diagnosis (years) |
4.7 |
12.4 |
1.8 |
10.3 |
||||
|
Ethnicity** |
(N) |
(%) |
(N) |
(%) |
(N) |
(%) |
(N) |
(%) |
|
British Isles |
47 |
37.3 |
8 |
33.3 |
6 |
50.0 |
5 |
38.5 |
|
French Canadian |
27 |
21.4 |
5 |
20.8 |
1 |
8.3 |
5 |
38.5 |
|
Other European |
19 |
15.1 |
7 |
29.2 |
2 |
16.6 |
2 |
15 |
|
Ashkenazi Jewish |
8 |
6.3 |
1 |
4.2 |
0 |
0 |
0 |
0 |
|
Asian |
4 |
3.2 |
0 |
0 |
1 |
8.3 |
0 |
0 |
|
Mixed Ethnicity |
16 |
12.7 |
3 |
12.5 |
1 |
8.3 |
0 |
0 |
| Other or unknown | 5 | 4.0 | 0 | 0 | 1 | 8.3 | 1 | 7.7 |
*Includes 4 probands affected with both breast and ovarian cancer.
**All self-reported ethnicities from the side of the family eligible for BRCA1 and BRCA2 sequencing based on Ontario guidelines.
Of the 13 patients who were diagnosed with ovarian cancer, four of whom were also diagnosed with breast cancer, over half (7/13) of the ovarian cancers were papillary serous or serous morphology. Two probands had ovarian cancers with mucinous morphology. The other 4 participants were affected with one case each of clear cell carcinoma, carcinosarcoma, mixed mucinous and endometrioid cystadenocarcinoma, and borderline papillary serous tumor of low malignant potential.
Of the 24 cases of female multiple primary breast cancer, 16 cases were bilateral metachronous, 5 cases were bilateral synchronous, and 3 were synchronous and in the same breast.
Three hundred and eleven index cases from suspected hereditary breast and ovarian cancer families from Northern Portugal, all Caucasian, were screened for the N1096S variant only. Cases included 256 female breast cancer patients with an average age at diagnosis of 42.6 yrs, 24 male breast cancer cases and 31 index cases with other (or no) cancer.
Molecular methods
a. PALB2 sequencing and MLPA
We screened the 13 coding exons of PALB2 (NCBI reference sequences NG_007406.1 and NM_024675.3), by high-resolution melting (HRM) analysis using the LightScanner instrument (Idaho Technologies Inc., Utah, USA). The PCR reactions for HRM were performed in 96 well plates from Bio Rad (Ontario, Canada) using the mastermix and the LCGreen Plus from Transition Technologies (Ontario, Canada). Following amplification, the plates were then transferred to the LightScanner instrument and the melting curves were analysed with the software provided by Idaho Technologies. This technique was used as a presequencing selection for amplicons harboring variants. Samples having a different melting curve than the majority were analysed by Sanger sequencing. The primer sequences and the protocol for amplification have been previously published [12]. Samples were also screened for large insertions/deletions overlapping coding regions that would be missed using the PCR-based method above using a commercially available Multiplex Ligation-dependent Probe Amplification mix (MRC-Holland, Amsterdam, Netherlands) using the supplier’s recommended protocol.
b. c.3287A > G (p.N1096S) screening
Screening for the c.3287A > G (N1096S) variant in 311 Portuguese index cases was performed using KASPar® SNP genotyping on demand by KBiosciences on a Roche LightCycler® 480 Real-Time PCR System.
c. LOH testing in tumor samples
DNA extracted from formalin-fixed, paraffin-embedded (FFPE) tumors that were macro-dissected to enrich for tumor cells was used to perform loss of heterozygosity (LOH) analysis in mutation carriers. The region surrounding each variant was amplified by PCR using variant-specific primers (Additional file 1: Table S3) that were designed using the Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/). Amplicons were analysed by Sanger sequencing.
d. Expression analysis of c.3507_3508delTC (p.H1170Ffs*19)
Lymphoblastoid cell lines (LCLs) harboring the c.3507_3508delTC (p.H1170Ffs*19) mutation were grown in Dulbecco’s Modified Iscove’s Media (Life Technologies Burlington, Ontario, Canada). RNA was extracted from 10 million cells using Trizol reagent (Life Technologies Burlington, Ontario, Canada) following the manufacturer’s protocol. 1 ug of RNA was used for reverse transcription (Qiagen California, USA) following the manufacture’s protocol, followed by cDNA amplification of PALB2. The primers used were: forward primer, 5’-GCAGGCTGGCAGGTTCCT-3’ (covers the junction of exon 12 and 13 of PALB2 in the cDNA); and reverse primer, 5’-CATCCAAGATCAGTGGTGCTA-3’ (in the 3’UTR).
Results and discussion
Mutation analysis of PALB2 identified two monoallelic deleterious mutations in 2/126 (1.6%) women with diagnoses of unilateral breast cancer. Of the two mutations, c.3113G > A has been previously reported by several groups and is thought to be a founder mutation from the United Kingdom [5,7,12-15]. It has been shown to produce three distinct transcripts resulting in p.Gly1000_Trp1038del (56%), p.Thr1029Ilefs*2 (40%) and p.Trp1038* (4%) [12] (Additional file 1: Figure S1). The frameshift mutation c.3507_3508delTC (p.H1170Ffs*19) has not been previously reported.
The index case with the PALB2 c.3113G > A mutation (Figure 1A) was of British descent and had been diagnosed with invasive ductal (ER+, PR+, HER2-) breast cancer at 36 years of age. Evaluation of her mother revealed that the mutation was most likely paternally inherited. Her paternal family history is significant for an aunt diagnosed with pancreatic cancer at 60 and two first cousins with breast cancer diagnosed in their 40s.
Figure 1.

Pedigree and LOH Testing in Tumors of c.3113G > A Mutation Family. (A) Pedigree of the family carrying the c.3113G > A mutation. Circles = females; Squares = males. Filled symbols = affected with cancer. Slashed symbols = deceased. +/+ = wild-type; +/- = heterozygous carrier; d. = death, numbers denote age, the arrow denotes the proband. (B) Loss of heterozygosity analysis of the proband’s breast cancer shows both alleles are still present in the tumor. (C) Hematoxylin-eosin stain of the tumor tissue analyzed in panel B.
The index case with the PALB2 c.3507_3508delTC (p.H1170Ffs*19) (Figure 2A) mutation was of British descent and had been diagnosed with invasive ductal (ER+, PR+, HER2+) breast cancer at the age of 44. We evaluated the presence of the mutation in samples from four of the proband’s relatives, from whom genetic material was available; two individuals (the proband’s mother and sister) affected with breast cancer were carriers of the family mutation. The proband’s unaffected sister, who has a daughter with a diagnosis of malignant melanoma at age 11, was also found to carry the familial mutation. An unaffected uncle was found to not carry the familial mutation.
Figure 2.
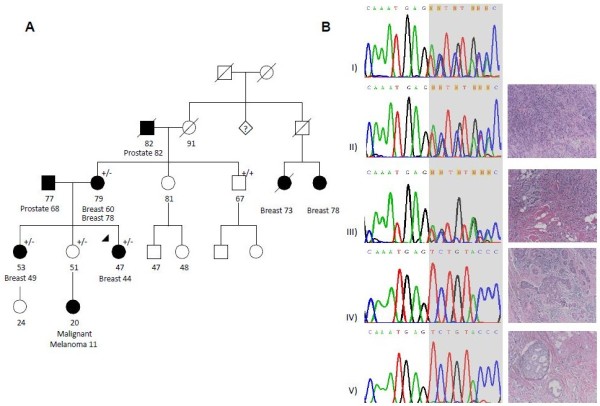
Pedigree and LOH Testing in Tumors of c.3507_3508delTC Mutation Family. (A) Pedigree of the family carrying the c.3507_3508delTC mutation. Circles = females; Squares = males. Filled symbols = affected with cancer. Slashed symbols = deceased. +/+ = wild-type; +/- = heterozygous carrier; numbers denote age, the arrow denotes the proband. (B) Loss of heterozygosity (LOH) analysis in tumor tissue from mutation carriers. The reverse sequence is shown. A hematoxylin-eosin stain of the tumor tissue analyzed is shown to the right of each chromatogram. I) Normal tissue showing the heterozygous mutation; II) Tumor tissue from the proband, (no LOH); III) Tumor tissue from the proband’s sister (no LOH); IV) Tumor tissue from the left breast of the proband’s mother (loss of the wild-type allele); V) Tumor tissue from the right breast of the proband’s mother (loss of the wild-type allele).
Three additional missense variants predicted to be pathogenic by Sorting Intolerant From Tolerant (SIFT) [16], Polymorphism Phenotyping (Polyphen) [17], MutationTaster [18] and Consensus Deleteriousness (Condel) [19] were identified (Table 2). The first, c.1846G > C (p.D616H) was observed in a proband diagnosed with unilateral breast cancer from a French Canadian family (Additional file 1: Figure S2A). The second, c.3418 T > G (p.W1140G), was observed in a proband diagnosed with unilateral breast cancer from a French Canadian and British family (Additional file 1: Figure S2B). Finally, the third variant, c.3287A > G (p.N1096S), was identified in a proband diagnosed with bilateral breast cancer from a Portuguese family (Additional file 1: Figure S2C). Given the previously reported existence of a Portuguese founder mutation in BRCA2[20], we considered the possibility that this novel PALB2 variant was a founder mutation and subsequently analyzed for its presence in a larger Portuguese series. The c.3287A > G (p.N1096S) variant was not observed in an additional 311 Caucasian index cases primarily from Northern Portugal who were negative for BRCA1 and BRCA2 mutations. While these variants are possibly pathogenic functionally, c.1846G > C (p.D616H) does not appear to segregate with the disease, and DNA from additional affected individuals was not available to perform informative segregation analysis of the other two variants. In total, 86 participants, including the 5 with known/predicted pathogenic mutations, were identified to carry a PALB2 variant (Additional file 1: Table S4).
Table 2.
Summary of identified variants and tumor characteristics of the PALB2 carriers
| |
|
|
Tumor characteristics |
|
In silico analysis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Exon | Age | Type | Stage | ER | PR | Her2 | Previously reported? | SIFT | Polyphen | |
| Truncating |
c.3113G > A, |
10 |
36 |
IDC |
1 |
Pos |
Pos |
Neg |
yes |
|
|
| p.Gly1000_Trp1038del | |||||||||||
| p.Thr1029Ilefs*2 | |||||||||||
| p.W1038* | |||||||||||
| |
c.3507_3508delTC |
12 |
44 |
IDC |
1 |
Pos |
Pos |
Pos |
no |
|
|
| p.H1170Ffs*19 | |||||||||||
| Missense |
c.1846G > C |
4 |
25 |
IDC |
1 |
U |
U |
U |
no |
0.02 |
1.00 |
| p.D616H | |||||||||||
| |
c.3418 T > G |
12 |
32 |
IDC |
1 |
Pos |
Pos |
Pos |
no |
0.00 |
1.00 |
| p.W1140G | |||||||||||
| |
c.3287A > G |
10 |
50/58 |
DCIS/IDC |
0/2 |
NP/Pos |
NP/Pos |
NP/U |
no |
0.00 |
1.00 |
| p.N1096S | |||||||||||
ER = estrogen receptor; PR = progesterone receptor; HER2 = human epidermal growth factor receptor 2; IDC = infiltrating ductal carcinoma; DCIS = ductal carcinoma in situ; U = unknown; Pos = positive; Neg = negative; NP = not performed.
We performed loss of heterozygosity (LOH) analysis on available breast tumors from the five families. Tumor tissue was not available for testing for any c.3287A > G (p.N1096S) and c.1846G > C (p.D616H) carriers. LOH was observed in both the left and right breast tumors from the mother carrying the c.3507_3508delTC (p.H1170Ffs*19) mutation, but none of the other tumors showed loss of the wild-type allele [Figures 1B and 2B, data not shown for the tumor from the proband with c.3418 T > G (p.W1140G)]. These results are consistent with previous observations that LOH is an inconsistent feature of PALB2 tumors, even among tumors carrying identical predisposing mutations [13,14,21,22].
In 175 probands tested, we identified PALB2 mutations that can alter the length of the PALB2 protein. The first, c.3113G > A, was previously identified in multiple unrelated families from Australia, the United States and the UK and is associated with an estimated 91% (95% CI = 44–100) cumulative risk of breast cancer to age 70 [7,23]. As our family with c.3113G > A is of British descent, our results support the hypothesis that c.3113G > A is a founder mutation in the British population. Most recently, Teo et al.[23] identified this particular mutation in 8 of 871 (0.92%) probands from “high-risk” breast and/or ovarian cancer families evaluated in familial cancer clinics in Australia and New Zealand.
The c.3507_3508delTC (p.H1170Ffs*19) mutation is located in the final exon (exon 13) of PALB2. Sequencing of RNA from cultured LCLs showed expression of the mutant allele (Additional file 1: Figure S3), indicating that mRNA derived from the mutated allele is not subject to nonsense-mediated decay. The frameshift introduces 18 new amino acids that are not seen in the native protein (Additional file 1: Figure S1B), resulting in a mutant protein of 1187 amino acids, compared to 1186 amino acids in the wild-type protein. The predicted structure of PALB2 includes an amino-terminal coiled-coil structure and a carboxy-terminal WD40-repeats motif that has the characteristics of a seven-bladed beta-propeller, a domain commonly involved in protein-protein interactions. The WD40-repeats domain on amino acids 836–1186 provides a binding site for the N-terminus of BRCA2 [24]. Mutations in this region, even if they do not result in nonsense-mediated decay, may lead to alteration of the structure of WD40-repeats (Additional file 1: Figure S1) [5], and many are known to be pathogenic. Thus, independent of the genetic data we present here (segregation and LOH), there is fairly strong a priori evidence that this novel mutation c.3507_3508delTC (p.H1170Ffs*19) we describe here is deleterious, probably as a result of disruption of the BRCA1-PALB2-BRCA2 interaction, resulting in defective homologous repair of double-stranded breaks.
In terms of immunohistochemical features seen in the tumors of our participants with pathogenic or suspected pathogenic PALB2 mutations, although earlier studies performed in Finnish, French Canadian and other European populations suggested a possible overrepresentation of triple negative (basal-like) tumors in PALB2-related breast cancers [4,25,26], our overall results support more recent studies of Australasian breast cancer families that have not found any association between PALB2 mutations and hormone-receptor negative breast tumors [27,28]. Although the number of probands with multiple breast primaries in our series is small (24 cases) it is interesting that, of the tumors that are available for hormone receptor and HER2 analysis in this specific subgroup of patients, an increased proportion (24%) had triple negative phenotype (Additional file 1: Table S2). Further studies in larger cohorts will be necessary to determine whether the triple negative phenotype is overrepresented in PALB2 carriers with specific (founder) mutations or specific clinical presentations (early onset or bilateral disease).
Pathogenic or likely pathogenic PALB2 mutations were observed in 2 of 89 families (2.2%) with 3 or more cases of breast cancer (Table 3A). Although not statistically significant, these numbers suggest a greater than 1% chance of identifying mutations in PALB2 in families with 3 or more breast cancer cases where BRCA1/2 mutations have been excluded.
Table 3.
Total patient population screened
|
(A)Number of Breast Cancer Cases in Families | |||||||
|---|---|---|---|---|---|---|---|
| Total | 5 BC in family | 4 BC in family | 3 BC in family | 2 BC in family | 1 BC in family | 0 BC in family | |
|
Female Unilateral Breast |
126 |
19 |
25 |
31 |
39 |
12 |
0 |
|
Female Multiple Primary Breast |
24 |
1 |
4 |
2 |
12 |
5 |
0 |
|
Male Breast |
12 |
0 |
0 |
1 |
4 |
7 |
0 |
|
Breast and Ovarian |
4 |
1 |
1 |
0 |
1 |
1 |
0 |
|
Ovarian |
9 |
0 |
2 |
2 |
2 |
1 |
2 |
|
Total screened |
175 |
21 |
32 |
36 |
58 |
26 |
2 |
|
PALB2
mutations* |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
|
B)Proximity to Breast Cancer Cases in Families | |||||||
| |
Total |
FDR with BC |
SDR with BC |
TDR with BC |
No relatives with BC |
||
|
Female Unilateral Breast |
126 |
79 |
28 |
7 |
12 |
||
|
Female Multiple Primary Breast |
24 |
11 |
5 |
3 |
5 |
||
|
Male Breast |
12 |
2 |
1 |
2 |
7 |
||
|
Breast and Ovarian |
4 |
2 |
0 |
1 |
1 |
||
|
Ovarian |
9 |
4 |
2 |
1 |
2 |
||
|
Total screened |
175 |
98 |
36 |
14 |
27 |
||
| PALB2 mutations* | 2 | 1 | 0 | 1 | 0 | ||
*Only the two definitely deleterious mutations were included in these tables. BC = breast cancer; FDR = first degree relative; SDR = second degree relative; TDR = third degree relative.
The majority of probands with a pathogenic or suspected pathogenic PALB2 mutation had a first or second-degree relative with breast cancer (Table 3B), and if they did not, had a first degree or second-degree relative with a tumor suspected to be associated with a BRCA2 or PALB2 mutation (Additional file 1: Figure S2). Thus it does not appear to be worthwhile to screen families for PALB2 mutations if the proband does not have close relatives who are affected by breast cancer or a BRCA2/PALB2 related cancer.
Similar to the observations of Casadei et al.[13], the cancer profiles of the two PALB2 mutation families seemed similar to cancer profiles of BRCA2 families; including relatives affected with pancreatic cancer and melanoma. The frequencies of these cancers in the family histories of our cohort are described in Additional file 1: Table S5A and Additional file 1: Table S5B, respectively. Multiple studies have identified PALB2 mutations in familial pancreatic cancer probands, several of which were found to have family histories including cases of breast cancer [8,29-31]. It should be noted that these mutations occur at relatively low frequencies (0–4%), which depend on the population being studied [8,32].
Whether or not PALB2 mutations are associated with an increased risk of melanoma remains unclear and further studies are needed. Rahman et al.[5] described a proband with a truncating PALB2 mutation (p.Y1183X) and diagnoses of melanoma at 47 years of age and breast cancer at 56 years. To the best of our knowledge, this is the only case in the literature describing melanoma in PALB2 mutation carriers. In addition, Sabbaghian et al.[33] did not find a single PALB2 mutation in 53 cases of familial melanoma. While we do not know PALB2 status in the niece of our c.3507_3508delTC proband (given her young age she was not yet offered testing), the extremely young age of malignant melanoma diagnosis (at 11 years) is intriguing.
The contribution of PALB2 mutations to ovarian cancer risk also remains uncertain with previous studies being conflicting (reviewed in 3,4). Some groups have observed an increased likelihood for PALB2 mutation carriers to have relatives affected with ovarian cancer, albeit this was not a statistically significant finding [13]. One study which used massively parallel sequencing to examine the burden of loss of function mutations in tumor suppressor genes in women diagnosed with primary ovarian, peritoneal, or fallopian tube carcinoma identified 2 PALB2 mutations in 360 probands tested [9]. It should be noted that the majority (139/175) of the families we screened did not contain any reported cases of ovarian cancer (Additional file 1: Table S5C).
Both the truncating pathogenic and the non-truncating frameshifting (and likely pathogenic) mutation in PALB2 were identified amongst the 77 women diagnosed with premenopausal (<50 years old) unilateral breast cancer. Therefore, 2/77 (2.6%) mutations were identified in this particular group of women. Data from other reported series also suggests that PALB2 mutations may be preferentially identified in premenopausal women, but not all studies report on the number of individuals in the series with diagnoses under the age of 50. Rahman et al.[5] noted that the median age of diagnosis for those with PALB2 mutations in their study was 46 years, while those individuals without a PALB2 mutation had a median age at diagnosis of 49 years. In their cohort, 8/10 probands with pathogenic mutations had their breast cancer diagnosed at or less than 50 years of age. In the case series reported by Southey et al., 4/5 had breast cancer diagnosed at or less than 50 years of age [7]. In the study of cases with bilateral breast cancer reported by Tischkowitz [12], the median age at diagnosis of first breast cancer in the 5 women identified to carry a pathogenic mutation was 46 years. Notably in their series, none of the probands had an additional young onset case in the family. Taken together, these data suggest that it may be reasonable to preferentially offer PALB2 clinical testing to BRCA1/2-negative breast and/or ovarian cancer families with multiple cases of breast cancer, including at least one case of premenopausal breast cancer.
Finally, although the sample size in our study is small, the presence of PALB2 mutations in our cohort confirms that they are present at low frequency in familial breast cancer cases. It is also possible that deep intronic mutations may exist which would have escaped detection with our screening methodology.
Conclusion
In 175 probands from a clinic based series of breast and/or ovarian cancer families with no detectable BRCA1/BRCA2 mutations, we have identified two mutations in PALB2: one, c.3113G > A, is known to be deleterious and the other, c.3507_3508delTC (p.H1170Ffs*19), is likely to be deleterious. Both probands had family histories in which three or more individuals had been diagnosed with breast cancer. Both of these two families also had a family member with a diagnosis of another BRCA2-related cancer. PALB2 mutations are not as frequent in the population as BRCA1 and BRCA2 mutations; however, the role of PALB2 in breast cancer susceptibility is still significant, some studies suggesting that particular PALB2 mutations may predispose individuals to breast cancer to a similar extent as BRCA2. It therefore warrants knowing who should be tested.
This study supports the notion that PALB2 mutations are present at a relatively low frequency in hereditary breast cancer cases and that in women with breast cancer, who have a family history including 3 or more cases of breast cancer, PALB2 testing may identify a mutation in >2% of cases and restricting testing to affected premenopausal women in such families may capture the vast majority of these mutations. Clinical testing could also be considered in the context of another BRCA2-related malignancy, and when at least one family member is diagnosed with premenopausal breast cancer.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors made substantial contributions to the acquisition and interpretation of data and critical revision of the manuscript. MT, ET and WDF made substantial contributions to the conception and design of the study. TH, RSS, ES, VH, NH and ET were involved in patient care/coordination of the study. TH, NH, ET, WDF wrote the manuscript. LC, NS, PP performed the molecular genetic studies. OA provided pathology expertise for the study. NH and LC performed the LOH studies on tumor tissue. All authors read and approved the final manuscript.
Supplementary Material
Predicted effect of truncating mutations on PALB2 protein. Figure S2. Pedigrees of the PALB2 missense mutation-carrier families. Figure S3. The c.3507_3508delTC (p.H1170Ffs*19) variant is expressed in lymphoblastoid cells from the patient. Table S1. Ontario guidelines for molecular analysis of BRCA1 and BRCA2. Table S2. Breast Tumor Characteristics. Table S3. Primers. Table S4. Variants found (n = 176) 89 individuals had no variants and 86 had 1 or more variants. Table S5. Total patient population screened. (A) Number of Pancreatic Cancer Cases in Families. (B) Number of Melanoma Cancer Cases in Families. (C) Number of Ovarian Cancer Cases in Families.
Contributor Information
Taila Hartley, Email: thartley@cheo.on.ca.
Luca Cavallone, Email: lucacavallone@gmail.com.
Nelly Sabbaghian, Email: nelly.sabbaghian@mail.mcgill.ca.
Rachel Silva-Smith, Email: rachel.silva-smith@mail.mcgill.ca.
Nancy Hamel, Email: nancy.hamel@mail.mcgill.ca.
Olga Aleynikova, Email: oaleynikova@jgh.mcgill.ca.
Erika Smith, Email: esmith@cheo.on.ca.
Valerie Hastings, Email: vhastings@cheo.on.ca.
Pedro Pinto, Email: pedropinto18@hotmail.com.
Marc Tischkowitz, Email: mdt33@medschl.cam.ac.uk.
Eva Tomiak, Email: etomiak@cheo.on.ca.
William D Foulkes, Email: william.foulkes@mcgill.ca.
Acknowledgements
This research was supported by a grant to WDF from Susan G Komen © and to MDT and WDF from the Cancer Research Society and the Ministère du Développement Économique, de l'Innovation et de l’Exportation (Québec). Marc Tischkowitz is funded by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n.310018. We would like to thank Professor Manuel Teixeira, (Portuguese Oncology Institute-Porto, Porto, Portugal) for overseeing the analysis of the N1096S variant we found in our study patient and in his collection of Portuguese breast cancer patients. We thank Ms. Kelly Anderson for all of her help with this project. We would also like to thank Mr. James Mattina for technical help with mutation testing of samples.
References
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poumpouridou N, Kroupis C. Hereditary breast cancer: beyond BRCA genetic analysis; PALB2 emerges. Clin Chem Lab Med. 2011;50:423–434. doi: 10.1515/cclm-2011-0840. [DOI] [PubMed] [Google Scholar]
- Tischkowitz M, Xia B. PALB2/FANCN: Recombining cancer and Fanconi Anemia. Cancer Res. 2010;70:7353–7359. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D. Breast Cancer Suscetibility Collaboration (UK), Easton DF, Stratton MR. PALB2 which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkko H, Dowty JG, Nikkila J, Syrjakoski K, Mannermaa A, Pylkas K, Southey MC, Holli K, Kallioniemi A, Jukkola-Vuorinen A, Kataja V, Kosma VM, Xia B, Livingston DM, Winqvist R, Hopper JL. Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res. 2008;14:4667–4671. doi: 10.1158/1078-0432.CCR-08-0210. [DOI] [PubMed] [Google Scholar]
- Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, Sabbaghian N, Apicella C, Byrnes GB, Winship I, Baglietto L, Giles GG, Goldgar DE, Foulkes WD, Hopper JL. kConFab for the Breast Cancer Family Registry. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010;12:R109. doi: 10.1186/bcr2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzier KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, King MC, Swisher EM. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek SM, Bradbury A. Multiplex Genetic Testing for Cancer Susceptibility: Out on the High Wire Without a Net? J Clin Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–1255. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallee MP, Tavtigian SV, Concannon P, Foulkes WD, Bernstein L. WECARE Study Collaborative Group, Bernstein JL, Begg CB. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. 2012;33:674–680. doi: 10.1002/humu.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, Stamatoyannopoulos JA, King MC. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–2229. doi: 10.1158/0008-5472.CAN-10-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MJ, Fernandez V, Osorio A, Barroso A, Llort G, Lazaro C, Blanco I, Caldes T, de la Hoya M, Ramon Y, Cajal T, Alonso C, Tejada MI, San Roman C, Robles-Diaz L, Urioste M, Benitez J. Analysis of FANCB and FANCN/PALB2 Fanconi Anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009;113:545–551. doi: 10.1007/s10549-008-9945-0. [DOI] [PubMed] [Google Scholar]
- Wong MW, Nordfors C, Mossman D, Pecenpetelovska G, Avery-Kiejda KA, Talseth-Palmer B, Bowden NA, Scott RJ. BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res Treat. 2011;127:853–859. doi: 10.1007/s10549-011-1443-0. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Adzubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashev AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- González-Pérez A, López-Bigas N. Improving the Assessment of the Outcome of Nonsynonymous SNVs with a Consensus Deleteriousness Score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto A, Santos C, Pinheiro M, Pinto P, Soares MJ, Rocha P, Gusmão L, Amorim A, van der Hout A, Gerdes AM, Thomassen M, Kruse TA, Cruger D, Sunde L, Bignon YJ, Uhrhammer N, Cornil L, Rouleau E, Lidereau R, Yannoukakos D, Pertesi M, Narod S, Royer R, Costa MM, Lazaro C, Feliubadaló L, Graña B, Blanco I, de la Hoya M, Caldés T, Maillet P. et al. International distribution and age estimation of the Portuguese BRCA2 c.156_157insAlu founder mutation. Breast Cancer Res Treat. 2011;127:671–679. doi: 10.1007/s10549-010-1036-3. [DOI] [PubMed] [Google Scholar]
- Balia C, Sensi E, Lombardi G, Roncella M, Bevilacqua G, Caligo MA. PALB2: a novel inactivating mutation in a Italian breast cancer family. Fam Cancer. 2010;9:531–536. doi: 10.1007/s10689-010-9382-1. [DOI] [PubMed] [Google Scholar]
- Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, Omeroglu A, Poll A, Lepage P, Wong N, Nederlof PM, Ashworth A, Tonin PN, Narod SA, Livingston DM, Foulkes WD. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci U S A. 2007;104:6788–6793. doi: 10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo ZL, Sawyer SD, James PA, Mitchell G, Trainer AH, Lindeman GJ, Shackleton K, Cicciarelli L, Southey MC. The incidence of PALB2 c.3113G > A in women with a strong family history of breast and ovarian cancer attending famlial cancer centres in Australia. Fam Canc. 2013;12:587–595. doi: 10.1007/s10689-013-9620-4. [DOI] [PubMed] [Google Scholar]
- Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009;10:990–996. doi: 10.1038/embor.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, Derlatka P, Cendrowski K, Kupryjanczyk J. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;2:11–20. doi: 10.1186/1471-2350-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, Blomqvist C, Nevanlinna H. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15:3214–3212. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- Teo ZL, Park DJ, Provenzano E, Chatfield CA, Odefrey FA, Nguyen-Dumont T, kConFab, Dowty JG, Hopper JL, Winship I, Goldgar DE, Southey MC. Prevalence of PALB2 mutations in Australasian multiple-case breast cancer families. Breast Canc Res. 2013;15:R17. doi: 10.1186/bcr3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo ZL, Provenzano E, Dite GS, Park DJ, Apicella C, Sawyer SD, James PA, Mitchell G, Trainer AH, Lindeman GJ, Shackleton K, Cicciarelli L, kConFab, Buys SS, Andrulis IL, Mulligan AM, Glendon G, John EM, Terry MB, Daly M, Odefrey FA, Nguyen-Dumont T, Giles GG, Dowty JG, Winship I, Goldgar DE, Hopper JL, Southey MC. Tumour morphology predicts PALB2 germline mutations status. Br J Cancer. 2013;109:154–163. doi: 10.1038/bjc.2013.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstatter EW, Domchek SM, Miron A, Garber J, Wang M, Componeschi K, Boghossian L, Miron PL, Nathanson KL, Tung N. PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer. 2011;10:225–231. doi: 10.1007/s10689-011-9426-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, Srivastava A, Holter S, Rothenmund H, Ghadirian P, Foulkes WD, Gallinger S. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137:1183–1186. doi: 10.1053/j.gastro.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler ZK, Salo-Mullen E, Sabbaghian N, Simon JA, Zhang L, Olson SH, Kurtz R, Offit K, Foulkes WD, Robson ME, Tischkowitz M. Germline PALB2 mutation analysis in breast-pancreas families. J Med Genet. 2011;48:523–525. doi: 10.1136/jmg.2010.087379. [DOI] [PubMed] [Google Scholar]
- Harinck F, Kluijt I, van Mil SE, Waisfisz Q, van Os TA, Aalfs CM, Wagner A, Olderode-Berends M, Sijmons RH, Kuipers EJ, Poley JW, Fockens P, Bruno MJ. Routine testing for PALB2 mutations in familial pancreatic cancer families and breast cancer families with pancreatic cancer is not indicated. Eur J Hum Genet. 2012;20:577–579. doi: 10.1038/ejhg.2011.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbaghian N, Kyle R, Hao A, Hogg D, Tischkowitz M. Mutation analysis of the PALB2 cancer predisposition gene in familial melanoma. Fam Cancer. 2011;10:315–317. doi: 10.1007/s10689-010-9405-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Predicted effect of truncating mutations on PALB2 protein. Figure S2. Pedigrees of the PALB2 missense mutation-carrier families. Figure S3. The c.3507_3508delTC (p.H1170Ffs*19) variant is expressed in lymphoblastoid cells from the patient. Table S1. Ontario guidelines for molecular analysis of BRCA1 and BRCA2. Table S2. Breast Tumor Characteristics. Table S3. Primers. Table S4. Variants found (n = 176) 89 individuals had no variants and 86 had 1 or more variants. Table S5. Total patient population screened. (A) Number of Pancreatic Cancer Cases in Families. (B) Number of Melanoma Cancer Cases in Families. (C) Number of Ovarian Cancer Cases in Families.