

Abstract
Cardiovascular diseases are affected by multiple factors like genetic as well as environmental hence they reveal factorial nature. The evidences that genetic factors are susceptible for developing cardiovascular diseases come from twin studies and familial aggregation. Different ethnic populations reveal differences in the prevalence coronary artery disease (CAD) pointing towards the genetic susceptibility. With progression in molecular techniques different developments have been made to comprehend the disease physiology. Molecular markers have also assisted to recognize genes that may provide evidences to evaluate the role of genetic factors in causation of susceptibility towards CAD. Numerous studies suggest the contribution of specific “candidate genes”, which correlate with various roles/pathways that are involved in the coronary heart disease. Different studies have revealed that there are large numbers of genes which are involved towards the predisposition of CAD. However, these reports are not consistent. One of the reasons could be weak contribution of genetic susceptibility of these genes. Genome wide associations show different chromosomal locations which dock, earlier unknown, genes which may attribute to CAD. In the present review different ApoAI-CIII-AIV gene clusters have been discussed.
Keywords: ApoAI-CIII-AIV gene cluster, Haplotype analysis, Single nucleotide polymorphism, Candidate gene study, Genome wide association studies
Core tip: Cardiovascular disease analysis requires holistic approach using genomic, epigenomic and exposomic techniques to improve the quality of life of patients and contribution towards personalised medicine.
INTRODUCTION
Coronary artery disease (CAD), is mostly fatal if remain untreated result into atherosclerosis in the epicardial coronary arteries[1]. Atherosclerotic plaques progressively narrow the coronary artery lumen and impair antegrade myocardial blood flow. This reduction in coronary artery flow may lead to a myocardial infarction.
Cardiovascular disease is a multifarious disorder showing large diversity of phenotypes. The accurate, and analogous phenotypic evidences are crucial for detailed understanding of the affiliation between disease and genes, as well as understanding the role of various extrinsic factors on different component of various genotypes. This complexity also contributes to difficulties in diagnosis and prognosis of the disease. Diagnostic difficulties also hamper the optimal and personalised treatment for patients. In recent years the role of genetic variability on the development of CAD has been extensively been studied[1,2] which is impacting upon our understanding of phenotypic outcomes and clinical complications. New developments in genomics, epigenomics and exposomics (environmental risk factors across the life span) would result into the improved understanding of the different phenotypes observed in CAD and would help in the better regimen of treatment. In the last century, there has been rapid increases in the global prevalence of CAD, which has become the important cause of cardiovascular mortality all over the world, is > 4.5 million deaths in the developing countries. By 2020, it is predictable that CAD will be the major source of disease burden universally[2]. The prevalence of CAD varies in different ethnic groups which may show higher/lower genetic and environmental susceptibilities. India has also witnessed consistent increases in the prevalence of CAD over the past few decades and could become the number one killer if appropriate interventions are not planned and implemented. In Table 1 the incidence of CAD is shown in different parts of India.
Table 1.
Prevalence of coronary artery disease in different Indian surveys
| City | Prevalence | Ref. |
| Urban population | ||
| Chandigarh | (6.60%) | Sarvotham et al[49] |
| Rohtak | (3.80%) | Gupta et al[50] |
| Jaipur | (7.60%) | Gupta et al[51] |
| Delhi | (9.70%) | Chada et al[52] |
| Rural population | ||
| Jaipur | (3.50%) | Gupta et al[53] |
| Ludhiana | (3.08%) | Wander et al[54] |
| South Indians | ||
| Tamil Nadu | (14.30%) | Ramachandran et al[55] |
| Tamil Nadu | (11.00%) | Mohan et al[56] |
| Migrant Indians | ||
| London, United Kingdom | (17.00%) | Bahl et al[57] |
| Illinois, United States | (10.00%) | Enas et al[3] |
It has been reported that CAD is increasing in a linear fashion as it has increased from 4% in 1960 to 11% in 2001 i.e., almost every 25th individual in 1960 was having CAD, while in 2001 every 9th individual was having CAD. The CAD is declining internationally among Indians settled abroad, whereas, these rates are growing in the Indian subcontinent. Presently, 10%-12% of metropolitan Indians have CAD compared to 3% of the United States population. Many studies document that Asian Indians are at 3-4 times greater risk of CAD than white Americans/Europeans, 6 times higher than Chinese, and almost 20 times higher than Japanese[3-7]. CAD prevalence has increased from 3.5% in the 1960s to 9.5% in the 1990s in urban populations of India[8]. Current studies recognized occurrence of CAD to be 13.9% in the urban south Indians, 9.6% in urban north Indians[9-11]. In 1990s, 33% of cardiovascular deaths have been reported from India[12] and it is likely that deaths from non-communicable diseases such as CAD will increase two times higher i.e., 4.5 million in 1998 to 8 million in 2020 in India[13].
As CAD has a multifactorial nature and the occurrence of the familial clustering in CAD led investigators to start searching for susceptibility genes. In Figure 1 different risk factors involved in the causation of CAD are summarized.
Figure 1.
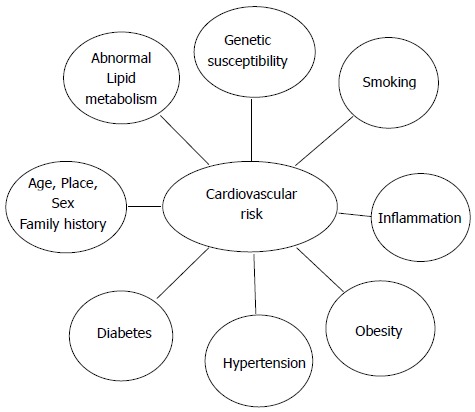
Cardiovascular risk factors.
It is vital to keep in mind that certain genes may show population specific effects. There are hundreds of genes known to have functional allelic variations that may be important for determining an individual’s vulnerability to CAD. There is much argument on results of published epidemiological studies until now. The differences may be due to differences in the techniques used or the population used to calculate the incidence, prevalence and other risks. It has been proposed that if multiple markers are used for assigning the risk, the results would be more conclusive clinically. Most important reason of concern in developing countries like India is the incomplete detection, treatment and control of CAD risk factors. The benefits of addressing the root cause of CAD, such as inflammation, smoking and cholesterol, together with preventive methodology will be useful in improving quality of life and saving lives. This in turn may be translated into preventive approaches to help reduce the risk of CAD using genetic and epigenetic approaches. Although CAD mortality in the Indians is highest than other populations[14,15], the reason for increased risk, which has been recorded in both the Asian immigrants and among Indians in urban India; are not yet clear hence more systematic and comprehensive studies are required to understand the spectrum of genetic and epigenetic influences on CAD.
GENETIC BASIS OF CAD
Atherosclerosis involves multiple factors, hence understanding the genetic and environmental basis of this complex disease requires holistic approaches[16-18]. A range of candidate genes (e.g., APOE, APOB, LPL, iNOS, ACE, COX2, CD14, P-Selectin, E-Selectin, MTHFR, PON1, TNFα) have been investigated in relation to initiation, development and progression of CAD[16-18]. A large number of studies using of candidate genes and genome-wide association analyses have shown some promising signals, but only a few have been confirmed to some extent which may be playing a role in CAD.
There are very few examples where single genes have played a role in causing atherosclerosis[19,20]. Mostly, CAD is caused by the environmental factors however the risk increases when some risk associated genes are also present. Research on identical twins consistently shows significant genetic effect in the development of CAD or its risk factors (Table 2). Heritability for CHD vary from 40% to 60%[21,22], suggesting a strong role of genes in the development of the disease. A detailed analysis of the many known CAD susceptibility genes and studies is beyond the scope of this overview. This overview will focus on selected candidate genes in the ApoAI-CIII-AIV gene region.
Table 2.
Genetic and environmental risk factors for coronary heart disease
| Risk factors with a significant genetic component (heritability) |
| Elevated LDL and VLDL cholesterol (40%-60%) |
| Low HDL cholesterol (45%-75%) |
| Elevated triglycerides (40%-80%) |
| Increased body mass index (25%-60%) |
| Elevated systolic blood pressure (50%-70%) |
| Elevated diastolic blood pressure (50%-65%) |
| Elevated lipoprotein(a) levels (90%) |
| Elevated homocysteine levels (45%) |
| Type 2 diabetes mellitus (40%-80%) |
| Elevated fibrinogen (20%-50%) |
| Elevated C-reactive protein (40%) |
| Elevated homocysteine levels (45%) |
| Gender |
| Age |
| Family history |
| Environmental risk factors |
| Smoking |
| Diet |
| Exercise |
| Infection |
| Foetal environment |
| Air pollution (particulates) |
Risk factors for coronary heart disease can be subdivided into those that are determined significantly by genetic differences and those that are largely environmental (Based on Lusis et al[58] 2004). VLDL: Very low density lipoprotein; LDL: Low density lipoprotein; HDL: High density lipoprotein.
SINGLE GENE DISORDERS AND CAD
Familial hypercholesterolemia
Familial hyper cholesterolemia (FH) is a classic genetic disease in which increased cholesterol, tendon xanthomas, and early heart disease segregates together. Joseph Goldstein and Michael Brown showed that FH results from mutations in the low-density lipoprotein (LDL) receptor, which leads to impaired binding, internalization and degradation of LDL. Dose dependent relationship was observed, homozygotes patients had higher levels of cholesterol (> 600 mg/dL), whereas heterozygotes had levels of approximately 400 mg/dL. This variable penetrance is modified by genes and other risk factors such as diet, smoking, and physical activity level[23]. Heterozygote frequency for this disease relatively high, approximately 1 in 500[24] in most populations, however DNA screening and effective treatments are available now[25,26].
Familial defective apolipoprotein B causing hypercholesterolemia
This comparatively common hypercholesterolemia (approximately 1 in 800), results from mutations in the major protein of LDL called Apolipoprotein B (ApoB). The mutations in ApoB prevent LDL binding to the LDL receptor. The majority of patients of this disorder carry a dominant mutation (codon 3500) and have lower cholesterol levels compared to FH patients. Other single-gene CHD/CAD traits are rare and of lower clinical/population significance[20].
CANDIDATE GENES AND CAD
During last 30 years, there have been many advancements in molecular genetic technology, development of sophisticated statistical tools and analyses which have contributed to improvements in human genetic research. One of the early developments was positional cloning technique, which allowed genetic mapping of many Mendelian diseases and traits. However for complex diseases, which involve many genes and environmental influences, this technique did not provide any major insights into genetic basis. Majority of our understanding of the genetic basis of CAD/CHD has been gained from studies of “candidate genes,” and more recently genome wide association (GWA) studies. These population based studies have provided further insights into genetic susceptibilities/contributions to complex diseases. Some examples of these are given below.
APOLIPOPROTEIN E AND APOAI-CIII-AIV-AV GENE CLUSTER
Apolipoprotein E (ApoE) is one of the extensively studied genetic locus as it plays a pivotal role in lipid metabolism and mediates the uptake of chylomicron and very low-density lipoprotein (VLDL) remnants. Utermann and colleagues[27] identified genetic polymorphism at ApoE locus and its association with cholesterol levels and type III hyperlipidemia. The polymorphism and its CAD associations have been replicated in many global populations. E3 allele is the most common (approximately 60%) followed by E4 allele (approximately 30%) and E2 (approximately 10) in world populations. E4 allele carriers have increased plasma cholesterol levels compared to E3 allele carriers while E2 carriers have decreased plasma cholesterols. The allelic variation at ApoE locus explains approximately 5% of the variation in cholesterol levels[28]. Type III hyperlipidemia, a relatively rare phenotype, are homozygous for the E2 allele, but not all E2 homozygous individuals have this disorder[29]. Therefore, genotype-phenotype relationships may require contribution of other genetic or environmental factors.
In addition to ApoE, there is now strong evidence that mutations in hepatic lipase influence the levels of high-density lipoprotein (HDL)[30], and the ApoAI-CIII-AIV-AV locus contributes to plasma triglyceride levels[31]. Many studies have shown that Lp(a) levels are strongly influenced by Apo(a) gene[32] In addition, both hepatic lipase and the ApoAI-CIII-AIV-AV cluster influence LDL particle size, which significantly contributes to CHD risk[33]. However, taken together, these genetic differences only explain a small amount of variation in plasma lipids and CHD/CAD phenotypes.
Dyslipidemia, a metabolic disorder, caused due to the defects in the synthesis, processing and catabolism of lipoprotein particles. Increased total cholesterol (TC)[34], triglyceride (TG)[35], LDL cholesterol (LDL-C)[36], and apolipoprotein (Apo)B[37], together with lower levels of ApoA1[37] and HDL cholesterol (HDL-C)[38] have been found to increase coronary artery disease (CAD) risk. Epidemiological and clinical studies have documented that above genetic factors/polymorphisms play a significant role in dyslipidemia[39] susceptibilities along with environmental factors. Twin and family studies suggest there are considerable genetic contributions in the inter-individual variation in plasma lipid phenotypes with the heritability estimates ranging from 40%-60%[40]. It has been suggested that understanding variation at these loci along with other newer genetic loci will provide a better understanding of the disease processes and contribution to personalized medicine.
ApoA1, is the main protein component of HDL-C, it functions in the activation of lecithin: cholesterol acyltransferase, and facilitates the reverse cholesterol transport from peripheral tissues[41]. ApoC3, is a 79-amino-acid protein formed mainly in the liver, is one of the major component of chylomicrons and VLDL and a minor component of HDL. ApoC3 prevents lipoprotein lipase and plays a key role in the catabolism of TG-rich lipoproteins. ApoA5 is detectable in very low-density lipoprotein, HDL, and chylomicrons and its concentrations are low compared to other apolipoproteins. Human ApoA1/C3/A5 genes resides in the ApoA1/C3/A4/A5 gene cluster on chromosome 11q23-q24[42-45]. The ApoA1/C3/A4/A5 gene cluster has emerged as a significant risk factor for hypertriglyceridemia and atherosclerosis[41,42]. A number of studies have shown significant associations between single nucleotide polymorphisms (SNPs) in the ApoA1/C3/A4/A5 gene cluster and raised plasma or serum lipid levels in humans, while others have reported negative or inconsistent results[42-46]. In addition there are many other SNPs involved in the inflammation and cell signalling with CAD and/or MI, some of these are summarized in Table 3.
Table 3.
Example of association studies of factors involved in inflammation and cell signalling with coronary artery disease and/or myocardial infarction
| Gene | Polymorphism | Ref. | Suggested results |
| CRP | 1059G/C | Zee et al[59] | No significant association with non-fatal MI, stroke or cardiovascular death |
| ICAM-1 | Lys-469-glu | Jiang et al[60] | Association with MI and CAD |
| E-selectin | Ser-128-Arg, Leu-554-phe, G98T | Wenzel et al[61] | Associated with angiographic proof of severe CAD in patients < 50 yr |
| Ser-128-Arg, G98T | Herrmann et al[62] | No association with MI | |
| Zheng et al[63] | T allele more common in younger patients with angiographic CAD | ||
| Ser-128-Arg | Ye et al[64] | Association with early-onset CAD | |
| P-selectin | Pro715 | Herrmann et al[65], Kee et al[66] | Possibly has a protective role from MI |
| S290N, N562D, V599L, T715P, T741T | Tregouet et al[67] | Protective effect of the P715; S290N and N562D associated with MI, when carried by certain haplotype | |
| C-2123G, A-1969G, Thr715Pro | Barbaux et al[68] | Polymorphisms associated with P-selectin levels but not with MI | |
| TNF-α and β | -863C/A, -308G/A (TNF-α), 252G/A (TNF-β) | Koch et al[69] | No association of TNF or IL-10 polymorphisms with MI or CAD |
| TNF-α | Five polymorphisms | Herrmann et al[65] | No association to MI or CAD |
| TNF-α and β | TNF- α 308 G/A , | Padovani et al[70] | No association to MI |
| TNF- β 252 A/G | |||
| TNF-α and β | TNF- β 308 G/A , TNF- β 252 A/G | Keso et al[71] | No association to old MI by autopsy or CAD |
| TNF-α | 308 G/A | Francis et al[72] | No association to angiographic CAD |
| IL-1 cluster | IL-1α (-889), IL-1b (-511), | Francis et al[72] | No association to angiographic CAD |
| IL-1β (+3953), IL-1RA intron 2 VNTR | IL-1RA VNTR allele 2 associated with single-vessel CAD | ||
| IL-1-RA | IL-1RA intron 2 VNTR | Manzoli et al[73] | No clear-cut association to CAD or MI |
| IL-1 cluster | IL-1β 511 C/T, IL-1RA intron 2 VNTR | Vohnout et al[74] | No association to angiographic CAD with either polymorphisms |
| IL-1-RA | IL 1RN-VNTR | Zee et al[75] | No association with risk for future MI |
| IL-1 β, IL-RA | IL-1β 511 C/T, IL-1RA intron 2 VNTR | Momiyama et al[76] | IL-1 β (-511)C/C and IL-1Ra (intron 2)2-or 3- repeat allele both associated with CAD, association with MI only in patient who are seropositive for Chlamydia pneumoniae |
| IL-6 | IL-6 G (-174)C promoter polymorphism | Nauck et al[77] | No association with the risk for CAD or MI |
| -174 (G/C), -572 (G/C), -596 (G/A), +528 I/D | Georges et al[78] | -174 C associated with MI (OR = 1.34)-174 C more frequent in patients with two or fewer stenosed vessels than in patients with three vessel lesions | |
| IL-10 | 3 IL-10 promotor polymorphisms (1082G/A, - 819C/T and -592C/A) | Koch et al[69] | No association with MI or CAD |
| 7 polymorphisms | Donger et al[79] | No association with risk for MI | |
| TGF- β1 | 29 T/C | Yokota et al[80] | T allele is a risk factor for MI in middle-aged Japanese men |
| -509T | Wang et al[81] | No association with CAD | |
| 7 polymorphisms | Cambien et al[82] | No association with degree of angiographic CAD, Pro25 allele associated with MI in some regions. | |
| Stromelysin (MMP-3) | 5 polymorphisms | Syrris et al[83] | No association of either polymorphisms with CAD |
| 5A-117/6A promoter polymorphism (5A/6A) | Schwarz et al[84] | No association with the risk for MI, 6A allele marker for progression of CAD | |
| 5A/6A | Terashima et al[85] | 5A allele associated with risk for MI | |
| 5A/6A | Kim et al[86] | 5A allele associated with stable angina | |
| 5A/6A | Humphries et al[87] | 6A genotypes at greater risk for CAD related events in nonsmokers, 5A/5A genotypes amplifies risk in smokers | |
| 5A/6A | Ye et al[88] | Homozygosis for 6A associated with greater progression of angiographic CAD | |
| PECAM-1 (CD31) | Val 125Leu, Asn563Ser and Gly670Arg | Sasaoka et al[89] | 563Ser/Ser and 670Arg/Arg genotypes associated with MI |
| Val 125Leu, Asn563Ser | Wenzel et al[90] | 125 Val and 563Asn associated with early onset of CAD (< 50 yr) | |
| Leu 125Val, Ser563Asn | Song et al[91] | 125Val and 563Asn associated with CAD | |
| Val125Leu | Gardemann et al[92] | No association with MI; weak association of Val125 with CAD in low-risk patients without HTN or DM (OR = 1.54; 95%CI: 1.03-2.3) |
CRP: C-reactive protein; DM: Diabetes mellitus; HTN: Hypertension; ICAM: Intercellular adhesion molecule; IL: Interleukin; MI: Myocardial infarction; MMP: Matrix metalloproteinase; PECAM: Platelet endothelial cell adhesion molecule; TGF: Transforming growth factor; TNF: Tumor necrosis factor; VNTR: Variable number of tandem repeats.
One of the limitations of case control studies is that many false positive or false negative associations may emerge between different genetic markers and complex diseases like CAD. The reason for such results are: (1) controls are not properly selected; (2) sample size of both controls and cases because of which accurate power of the study is not generated and replication of results is not possible; and (3) position of single-nucleotide polymorphisms (SNPs) in terms of their effect on transcription of gene or protein expression. In general, results of small sample size studies (200-300 patients and control subjects) should be interpreted with caution and should be replicated with larger sample sizes. It is important to confirm that genotype distributions are not skewed, especially in the control group. Large deviations from the Hardy-Weinberg equilibrium, may suggest that the control group is not necessarily the representative of healthy and randomly sampled individuals. This departure may also highlight issues with genotype scoring. Recent genome-wide sequencing research has revealed extensive level of variation and heterogeneity between individuals and populations, which should be considered when choosing SNPs and interpreting SNP data. Some of the early SNP association studies failed to include the effect of the polymorphism on gene expression or protein function and genotype-phenotype correlations. This information could reveal if an SNP is the actual cause or solely a marker which may be in linkage disequilibrium another causal variant. These analyses could provide significant clues for understanding the pathophysiologic mechanisms behind clinical outcomes. It is important to correct/control for the age, gender, ethnicity, and other confounders in heart disease genetic association studies. There should be a holistic approach to understand the role of genes, environment and life style factors in CAD susceptibilities and progression.
Recently, genetic analyses have expanded to whole genome sequence analysis and genome-wide association studies (GWAS) as these analyses eliminates biases in the selection of the candidate genes. A number of GWAS studies have identified new loci in previously unsuspected genomic regions. These analyses have shown, novel biological pathways involved in the disease states and development of novel therapies. Many recent studies have shown only limited evidences may exist where the genetic variants may be associated with MI or only with CAD. A care has to be taken in interpreting the GWAS data as large number of variant alleles may be found but one should consider only elegant systems genetics approach to Plaisier et al[47] used similar approach and found that FADS3 is a causal gene for familial combined hyperlipidemia (FCHL) and elevated triglycerides in Mexicans. The authors used network gene co-expression analysis and SNP data to assign a function to the genetic variants rs3737787 (1q21-q23) in USF1 gene, which was previously identified to be associated with FCHL. It is envisaged that new methods like Network medicine[48] will play an important role in these analyses and the advancement of our understanding of pathophysiological mechanisms of diseases like CAD and MI.
CONCLUSION
This overview has highlighted some of the important challenges regarding the use of genetic approaches to investigate complex diseases. The recent research using genomic, epigenomics and exposomic approaches is providing a range of patient centric tools which will help better classification of phenotypes and personalised medicine for CAD patients. The mechanisms underlying the association of these loci to CAD/MI remain largely unknown and the effects are relatively small. Hence the future challenges are (1) discovering new genetic variants through large-scale meta-analyses, using pathway-based approaches, and high throughput sequencing; (2) illustrating the mechanisms for the identified loci to CAD; and (3) translating the findings from CAD- GWASs and epigenetic analyses to novel and optimized therapeutic strategies.
Footnotes
P- Reviewer: Iacoviello M, Ong HT S- Editor: Wen LL L- Editor: A E- Editor: Wu HL
References
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL. Measuring the Global Burden of Disease and Risk Factors, 1990–2001. In: Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL, et al., editors. Washington (DC): World Bank; 2006. p. Chapter 1. [Google Scholar]
- 2.World Health Organization. Facts and figures (The World Health report 2003–shaping the future) Available from: http://www.who.int/whr/2003/en/Facts_and_Figures-en.pdf.
- 3.Enas EA, Garg A, Davidson MA, Nair VM, Huet BA, Yusuf S. Coronary heart disease and its risk factors in first-generation immigrant Asian Indians to the United States of America. Indian Heart J. 1996;48:343–353. [PubMed] [Google Scholar]
- 4.Gupta R. Epidemiological evolution and rise of coronary heart disease in India. South Asian J Prev Cardiol. 1997;1:14–20. [Google Scholar]
- 5.Enas EA, Yusuf S. Third Meeting of the International Working Group on Coronary Artery Disease in South Asians. 29 March 1998, Atlanta, USA. Indian Heart J. 1999;51:99–103. [PubMed] [Google Scholar]
- 6.Ghaffar A, Reddy KS, Singhi M. Burden of non-communicable diseases in South Asia. BMJ. 2004;328:807–810. doi: 10.1136/bmj.328.7443.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanaya AM, Kandula N, Herrington D, Budoff MJ, Hulley S, Vittinghoff E, Liu K. Mediators of Atherosclerosis in South Asians Living in America (MASALA) Study: Objectives, Methods, and Cohort Description. Clin Cardiol. 2013;36:713–720. doi: 10.1002/clc.22219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta R, Goyle A, Kashyap S, Agarwal M, Consul R, Jain BK. Prevalence of atherosclerosis risk factors in adolescent school children. Indian Heart J. 1999;50:511–515. [PubMed] [Google Scholar]
- 9.Gupta R, Misra A, Pais P, Rastogi P, Gupta VP. Correlation of regional cardiovascular disease mortality in India with lifestyle and nutritional factors. Int J Cardiol. 2006;108:291–300. doi: 10.1016/j.ijcard.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 10.Gupta R, Gupta KD. Coronary heart disease in low socioeconomic status subjects in India: “an evolving epidemic”. Indian Heart J. 2009;61:358–367. [PubMed] [Google Scholar]
- 11.Gupta R, Gupta HP, Keswani P, Sharma S, Gupta VP, Gupta KD. Coronary heart disease and coronary risk factor prevalence in rural Rajasthan. J Assoc Physicians India. 1994;42:24–26. [PubMed] [Google Scholar]
- 12.Reddy KS. Rising burden of cardiovascular disease in India. In Sethi KK (ed). Coronary artery disease in Indians: a global perspective. Mumbai: Cardiological Society of India; 1998. p. 63–72. [Google Scholar]
- 13. Available from: http://www.who.int/whr/1998/en/
- 14.Enas EA, Singh V, Munjal YP, Bhandari S, Yadave RD, Manchanda SC. Reducing the burden of coronary artery disease in India: challenges and opportunities. Indian Heart J. 2008;60:161–175. [PubMed] [Google Scholar]
- 15.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 16.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 18.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 19.Milewicz DM, Seidman CE. Genetics of cardiovascular disease. Circulation. 2000;102:IV103–IV111. doi: 10.1161/01.cir.102.suppl_4.iv-103. [DOI] [PubMed] [Google Scholar]
- 20.Nabel EG. Cardiovascular disease. N Engl J Med. 2003;349:60–72. doi: 10.1056/NEJMra035098. [DOI] [PubMed] [Google Scholar]
- 21.Lusis AJ, Weinreb A, Drake TA. Genetics of atherosclerosis. In: Topol EJ, Califf RM, Isner J, eds , et al., editors. Textbook of Cardiovascular Medicine. Philadelphia, Pa: Lippincott Williams & Wilkins; 1998. p. 2389–2413. [Google Scholar]
- 22.Motulsky AG, Brunzell JD. Genetics of coronary artery disease. In: King RA, Rotter JI, Motulsky AG, eds , et al., editors. The Genetic Basis of Common Disease. 2nd ed. New York, NY: Oxford University Press; 2002. p. 105–126. [Google Scholar]
- 23.Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ. Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree mortality study. BMJ. 2001;322:1019–1023. doi: 10.1136/bmj.322.7293.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 25.Thorsson B, Sigurdsson G, Gudnason V. Systematic family screening for familial hypercholesterolemia in Iceland. Arterioscler Thromb Vasc Biol. 2003;23:335–338. doi: 10.1161/01.atv.0000051874.51341.8c. [DOI] [PubMed] [Google Scholar]
- 26.Heath KE, Humphries SE, Middleton-Price H, Boxer M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet. 2001;9:244–252. doi: 10.1038/sj.ejhg.5200633. [DOI] [PubMed] [Google Scholar]
- 27.Utermann G, Pruin N, Steinmetz A. Polymorphism of apolipoprotein E. III. Effect of a single polymorphic gene locus on plasma lipid levels in man. Clin Genet. 1979;15:63–72. [PubMed] [Google Scholar]
- 28.Sing CF, Davignon J. Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am J Hum Genet. 1985;37:268–285. [PMC free article] [PubMed] [Google Scholar]
- 29.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 30.Cohen JC, Vega GL, Grundy SM. Hepatic lipase: new insights from genetic and metabolic studies. Curr Opin Lipidol. 1999;10:259–267. doi: 10.1097/00041433-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Talmud PJ, Hawe E, Martin S, Olivier M, Miller GJ, Rubin EM, Pennacchio LA, Humphries SE. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet. 2002;11:3039–3046. doi: 10.1093/hmg/11.24.3039. [DOI] [PubMed] [Google Scholar]
- 32.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90:52–60. doi: 10.1172/JCI115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lusis AJ, Ivandic B, Castellani LW. Lipoprotein and lipid metabolism. In: Rimoin DL, Conner JM, Pyeritz RE, eds , et al., editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. 4th ed. London, England: Churchill Livingstone; 2002. p. 2500–2537. [Google Scholar]
- 34.Satko SG, Freedman BI, Moossavi S. Genetic factors in end-stage renal disease. Kidney Int Suppl. 2005;(94):S46–S49. doi: 10.1111/j.1523-1755.2005.09411.x. [DOI] [PubMed] [Google Scholar]
- 35.Wattanakit K, Coresh J, Muntner P, Marsh J, Folsom AR. Cardiovascular risk among adults with chronic kidney disease, with or without prior myocardial infarction. J Am Coll Cardiol. 2006;48:1183–1189. doi: 10.1016/j.jacc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 36.Hsu CC, Kao WH, Coresh J, Pankow JS, Marsh-Manzi J, Boerwinkle E, Bray MS. Apolipoprotein E and progression of chronic kidney disease. JAMA. 2005;293:2892–2899. doi: 10.1001/jama.293.23.2892. [DOI] [PubMed] [Google Scholar]
- 37.Fredman D, White SJ, Potter S, Eichler EE, Den Dunnen JT, Brookes AJ. Complex SNP-related sequence variation in segmental genome duplications. Nat Genet. 2004;36:861–866. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- 38.Kilis´-Pstrusin’ska K. Genetic factors in the development and progression of chronic kidney disease. Postepy Hig Med Dosw. 2010;64:50–57. [PubMed] [Google Scholar]
- 39.Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–731. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- 40.Ioannidis JP. Prediction of cardiovascular disease outcomes and established cardiovascular risk factors by genome-wide association markers. Circ Cardiovasc Genet. 2009;2:7–15. doi: 10.1161/CIRCGENETICS.108.833392. [DOI] [PubMed] [Google Scholar]
- 41.Fullerton SM, Buchanan AV, Sonpar VA, Taylor SL, Smith JD, Carlson CS, Salomaa V, Stengård JH, Boerwinkle E, Clark AG, et al. The effects of scale: variation in the APOA1/C3/A4/A5 gene cluster. Hum Genet. 2004;115:36–56. doi: 10.1007/s00439-004-1106-x. [DOI] [PubMed] [Google Scholar]
- 42.Eichenbaum-Voline S, Olivier M, Jones EL, Naoumova RP, Jones B, Gau B, Patel HN, Seed M, Betteridge DJ, Galton DJ, et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2004;24:167–174. doi: 10.1161/01.ATV.0000099881.83261.D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chien KL, Chen MF, Hsu HC, Su TC, Chang WT, Lee CM, Lee YT. Genetic association study of APOA1/C3/A4/A5 gene cluster and haplotypes on triglyceride and HDL cholesterol in a community-based population. Clin Chim Acta. 2008;388:78–83. doi: 10.1016/j.cca.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Delgado-Lista J, Perez-Jimenez F, Ruano J, Perez-Martinez P, Fuentes F, Criado-Garcia J, Parnell LD, Garcia-Rios A, Ordovas JM, Lopez-Miranda J. Effects of variations in the APOA1/C3/A4/A5 gene cluster on different parameters of postprandial lipid metabolism in healthy young men. J Lipid Res. 2010;51:63–73. doi: 10.1194/jlr.M800527-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin RX, Li YY, Lai CQ. Apolipoprotein A1/C3/A5 haplotypes and serum lipid levels. Lipids Health Dis. 2011;10:140. doi: 10.1186/1476-511X-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JY, Lee BS, Shin DJ, Woo Park K, Shin YA, Joong Kim K, Heo L, Young Lee J, Kyoung Kim Y, Jin Kim Y, et al. A genome-wide association study of a coronary artery disease risk variant. J Hum Genet. 2013;58:120–126. doi: 10.1038/jhg.2012.124. [DOI] [PubMed] [Google Scholar]
- 47.Plaisier CL, Horvath S, Huertas-Vazquez A, Cruz-Bautista I, Herrera MF, Tusie-Luna T, Aguilar-Salinas C, Pajukanta P. A systems genetics approach implicates USF1, FADS3, and other causal candidate genes for familial combined hyperlipidemia. PLoS Genet. 2009;5:e1000642. doi: 10.1371/journal.pgen.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarvotham SG, Berry JN. Prevalence of coronary heart disease in an urban population in northern India. Circulation. 1968;37:939–953. doi: 10.1161/01.cir.37.6.939. [DOI] [PubMed] [Google Scholar]
- 50.Gupta SP, Malhotra KC. Urban--rural trends in the epidemiology of coronary heart disease. J Assoc Physicians India. 1975;23:885–892. [PubMed] [Google Scholar]
- 51.Gupta R, Prakash H, Majumdar S, Sharma S, Gupta VP. Prevalence of coronary heart disease and coronary risk factors in an urban population of Rajasthan. Indian Heart J. 1995;47:331–338. [PubMed] [Google Scholar]
- 52.Chadha SL, Gopinath N, Shekhawat S. Urban-rural differences in the prevalence of coronary heart disease and its risk factors in Delhi. Bull World Health Organ. 1997;75:31–38. [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta R, Gupta VP, Ahluwalia NS. Educational status, coronary heart disease, and coronary risk factor prevalence in a rural population of India. BMJ. 1994;309:1332–1336. doi: 10.1136/bmj.309.6965.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wander GS, Khurana SB, Gulati R, Sachar RK, Gupta RK, Khurana S, Anand IS. Epidemiology of coronary heart disease in a rural Punjab population--prevalence and correlation with various risk factors. Indian Heart J. 1994;46:319–323. [PubMed] [Google Scholar]
- 55.Ramachandran A, Snehalatha C, Latha E, Satyavani K, Vijay V. Clustering of cardiovascular risk factors in urban Asian Indians. Diabetes Care. 1998;21:967–971. doi: 10.2337/diacare.21.6.967. [DOI] [PubMed] [Google Scholar]
- 56.Mohan V, Deepa R, Rani SS, Premalatha G. Prevalence of coronary artery disease and its relationship to lipids in a selected population in South India: The Chennai Urban Population Study (CUPS No. 5) J Am Coll Cardiol. 2001;38:682–687. doi: 10.1016/s0735-1097(01)01415-2. [DOI] [PubMed] [Google Scholar]
- 57.Bahl VK, Prabhakaran D, Karthikeyan G. Coronary artery disease in Indians. Indian Heart J. 2001;53:707–713. [PubMed] [Google Scholar]
- 58.Lusis AJ, Fogelman AM, Fonarow GC. Genetic basis of atherosclerosis: part I: new genes and pathways. Circulation. 2004;110:1868–1873. doi: 10.1161/01.CIR.0000143041.58692.CC. [DOI] [PubMed] [Google Scholar]
- 59.Zee RY, Ridker PM. Polymorphism in the human C-reactive protein (CRP) gene, plasma concentrations of CRP, and the risk of future arterial thrombosis. Atherosclerosis. 2002;162:217–219. doi: 10.1016/s0021-9150(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 60.Jiang H, Klein RM, Niederacher D, Du M, Marx R, Horlitz M, Boerrigter G, Lapp H, Scheffold T, Krakau I, et al. C/T polymorphism of the intercellular adhesion molecule-1 gene (exon 6, codon 469). A risk factor for coronary heart disease and myocardial infarction. Int J Cardiol. 2002;84:171–177. doi: 10.1016/s0167-5273(02)00138-9. [DOI] [PubMed] [Google Scholar]
- 61.Wenzel K, Ernst M, Rohde K, Baumann G, Speer A. DNA polymorphisms in adhesion molecule genes--a new risk factor for early atherosclerosis. Hum Genet. 1996;97:15–20. doi: 10.1007/BF00218826. [DOI] [PubMed] [Google Scholar]
- 62.Herrmann SM, Ricard S, Nicaud V, Mallet C, Evans A, Ruidavets JB, Arveiler D, Luc G, Cambien F. The P-selectin gene is highly polymorphic: reduced frequency of the Pro715 allele carriers in patients with myocardial infarction. Hum Mol Genet. 1998;7:1277–1284. doi: 10.1093/hmg/7.8.1277. [DOI] [PubMed] [Google Scholar]
- 63.Zheng F, Chevalier JA, Zhang LQ, Virgil D, Ye SQ, Kwiterovich PO. An HphI polymorphism in the E-selectin gene is associated with premature coronary artery disease. Clin Genet. 2001;59:58–64. doi: 10.1034/j.1399-0004.2001.590110.x. [DOI] [PubMed] [Google Scholar]
- 64.Ye SQ, Usher D, Virgil D, Zhang LQ, Yochim SE, Gupta R. A PstI polymorphism detects the mutation of serine128 to arginine in CD 62E gene - a risk factor for coronary artery disease. J Biomed Sci. 1999;6:18–21. doi: 10.1007/BF02256419. [DOI] [PubMed] [Google Scholar]
- 65.Herrmann SM, Ricard S, Nicaud V, Mallet C, Arveiler D, Evans A, Ruidavets JB, Luc G, Bara L, Parra HJ, et al. Polymorphisms of the tumour necrosis factor-alpha gene, coronary heart disease and obesity. Eur J Clin Invest. 1998;28:59–66. doi: 10.1046/j.1365-2362.1998.00244.x. [DOI] [PubMed] [Google Scholar]
- 66.Kee F, Morrison C, Evans AE, McCrum E, McMaster D, Dallongeville J, Nicaud V, Poirier O, Cambien F. Polymorphisms of the P-selectin gene and risk of myocardial infarction in men and women in the ECTIM extension study. Etude cas-temoin de l’infarctus myocarde. Heart. 2000;84:548–552. doi: 10.1136/heart.84.5.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tregouet DA, Barbaux S, Escolano S, Tahri N, Golmard JL, Tiret L, Cambien F. Specific haplotypes of the P-selectin gene are associated with myocardial infarction. Hum Mol Genet. 2002;11:2015–2023. doi: 10.1093/hmg/11.17.2015. [DOI] [PubMed] [Google Scholar]
- 68.Barbaux SC, Blankenberg S, Rupprecht HJ, Francomme C, Bickel C, Hafner G, Nicaud V, Meyer J, Cambien F, Tiret L. Association between P-selectin gene polymorphisms and soluble P-selectin levels and their relation to coronary artery disease. Arterioscler Thromb Vasc Biol. 2001;21:1668–1673. doi: 10.1161/hq1001.097022. [DOI] [PubMed] [Google Scholar]
- 69.Koch W, Kastrati A, Böttiger C, Mehilli J, von Beckerath N, Schömig A. Interleukin-10 and tumor necrosis factor gene polymorphisms and risk of coronary artery disease and myocardial infarction. Atherosclerosis. 2001;159:137–144. doi: 10.1016/s0021-9150(01)00467-1. [DOI] [PubMed] [Google Scholar]
- 70.Padovani JC, Pazin-Filho A, Simões MV, Marin-Neto JA, Zago MA, Franco RF. Gene polymorphisms in the TNF locus and the risk of myocardial infarction. Thromb Res. 2000;100:263–269. doi: 10.1016/s0049-3848(00)00315-7. [DOI] [PubMed] [Google Scholar]
- 71.Keso T, Perola M, Laippala P, Ilveskoski E, Kunnas TA, Mikkelsson J, Penttilä A, Hurme M, Karhunen PJ. Polymorphisms within the tumor necrosis factor locus and prevalence of coronary artery disease in middle-aged men. Atherosclerosis. 2001;154:691–697. doi: 10.1016/s0021-9150(00)00602-x. [DOI] [PubMed] [Google Scholar]
- 72.Francis SE, Camp NJ, Dewberry RM, Gunn J, Syrris P, Carter ND, Jeffery S, Kaski JC, Cumberland DC, Duff GW, et al. Interleukin-1 receptor antagonist gene polymorphism and coronary artery disease. Circulation. 1999;99:861–866. doi: 10.1161/01.cir.99.7.861. [DOI] [PubMed] [Google Scholar]
- 73.Manzoli A, Andreotti F, Varlotta C, Mollichelli N, Verde M, van de Greef W, Sperti G, Maseri A. Allelic polymorphism of the interleukin-1 receptor antagonist gene in patients with acute or stable presentation of ischemic heart disease. Cardiologia. 1999;44:825–830. [PubMed] [Google Scholar]
- 74.Vohnout B, Di Castelnuovo A, Trotta R, D’Orazi A, Panniteri G, Montali A, Donati MB, Arca M, Iacoviello L. Interleukin-1 gene cluster polymorphisms and risk of coronary artery disease. Haematologica. 2003;88:54–60. [PubMed] [Google Scholar]
- 75.Zee RY, Lunze K, Lindpaintner K, Ridker PM. A prospective evaluation of the interleukin-1 receptor antagonist intron 2 gene polymorphism and the risk of myocardial infarction. Thromb Haemost. 2001;86:1141–1143. [PubMed] [Google Scholar]
- 76.Momiyama Y, Hirano R, Taniguchi H, Nakamura H, Ohsuzu F. Effects of interleukin-1 gene polymorphisms on the development of coronary artery disease associated with Chlamydia pneumoniae infection. J Am Coll Cardiol. 2001;38:712–717. doi: 10.1016/s0735-1097(01)01438-3. [DOI] [PubMed] [Google Scholar]
- 77.Nauck M, Winkelmann BR, Hoffmann MM, Böhm BO, Wieland H, März W. The interleukin-6 G(-174)C promoter polymorphism in the LURIC cohort: no association with plasma interleukin-6, coronary artery disease, and myocardial infarction. J Mol Med (Berl) 2002;80:507–513. doi: 10.1007/s00109-002-0354-2. [DOI] [PubMed] [Google Scholar]
- 78.Georges JL, Loukaci V, Poirier O, Evans A, Luc G, Arveiler D, Ruidavets JB, Cambien F, Tiret L. Interleukin-6 gene polymorphisms and susceptibility to myocardial infarction: the ECTIM study. Etude Cas-Témoin de l’Infarctus du Myocarde. J Mol Med (Berl) 2001;79:300–305. doi: 10.1007/s001090100209. [DOI] [PubMed] [Google Scholar]
- 79.Donger C, Georges JL, Nicaud V, Morrison C, Evans A, Kee F, Arveiler D, Tiret L, Cambien F. New polymorphisms in the interleukin-10 gene--relationships to myocardial infarction. Eur J Clin Invest. 2001;31:9–14. doi: 10.1046/j.1365-2362.2001.00754.x. [DOI] [PubMed] [Google Scholar]
- 80.Yokota M, Ichihara S, Lin TL, Nakashima N, Yamada Y. Association of a T29--& gt; C polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to myocardial infarction in Japanese. Circulation. 2000;101:2783–2787. doi: 10.1161/01.cir.101.24.2783. [DOI] [PubMed] [Google Scholar]
- 81.Wang XL, Sim AS, Wilcken DE. A common polymorphism of the transforming growth factor-beta1 gene and coronary artery disease. Clin Sci (Lond) 1998;95:745–746. [PubMed] [Google Scholar]
- 82.Cambien F, Ricard S, Troesch A, Mallet C, Générénaz L, Evans A, Arveiler D, Luc G, Ruidavets JB, Poirier O. Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Témoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension. 1996;28:881–887. doi: 10.1161/01.hyp.28.5.881. [DOI] [PubMed] [Google Scholar]
- 83.Syrris P, Carter ND, Metcalfe JC, Kemp PR, Grainger DJ, Kaski JC, Crossman DC, Francis SE, Gunn J, Jeffery S, et al. Transforming growth factor-beta1 gene polymorphisms and coronary artery disease. Clin Sci (Lond) 1998;95:659–667. doi: 10.1042/cs0950659. [DOI] [PubMed] [Google Scholar]
- 84.Schwarz A, Haberbosch W, Tillmanns H, Gardemann A. The stromelysin-1 5A/6A promoter polymorphism is a disease marker for the extent of coronary heart disease. Dis Markers. 2002;18:121–128. doi: 10.1155/2002/418383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Terashima M, Akita H, Kanazawa K, Inoue N, Yamada S, Ito K, Matsuda Y, Takai E, Iwai C, Kurogane H, et al. Stromelysin promoter 5A/6A polymorphism is associated with acute myocardial infarction. Circulation. 1999;99:2717–2719. doi: 10.1161/01.cir.99.21.2717. [DOI] [PubMed] [Google Scholar]
- 86.Kim JS, Park HY, Kwon JH, Im EK, Choi DH, Jang YS, Cho SY. The roles of stromelysin-1 and the gelatinase B gene polymorphism in stable angina. Yonsei Med J. 2002;43:473–481. doi: 10.3349/ymj.2002.43.4.473. [DOI] [PubMed] [Google Scholar]
- 87.Humphries SE, Martin S, Cooper J, Miller G. Interaction between smoking and the stromelysin-1 (MMP3) gene 5A/6A promoter polymorphism and risk of coronary heart disease in healthy men. Ann Hum Genet. 2002;66:343–352. doi: 10.1017/S0003480002001264. [DOI] [PubMed] [Google Scholar]
- 88.Ye S, Watts GF, Mandalia S, Humphries SE, Henney AM. Preliminary report: genetic variation in the human stromelysin promoter is associated with progression of coronary atherosclerosis. Br Heart J. 1995;73:209–215. doi: 10.1136/hrt.73.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sasaoka T, Kimura A, Hohta SA, Fukuda N, Kurosawa T, Izumi T. Polymorphisms in the platelet-endothelial cell adhesion molecule-1 (PECAM-1) gene, Asn563Ser and Gly670Arg, associated with myocardial infarction in the Japanese. Ann N Y Acad Sci. 2001;947:259–269; discussion 269-270. doi: 10.1111/j.1749-6632.2001.tb03948.x. [DOI] [PubMed] [Google Scholar]
- 90.Wenzel K, Baumann G, Felix SB. The homozygous combination of Leu125Val and Ser563Asn polymorphisms in the PECAM1 (CD31) gene is associated with early severe coronary heart disease. Hum Mutat. 1999;14:545. doi: 10.1002/(SICI)1098-1004(199912)14:6<545::AID-HUMU20>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 91.Song FC, Chen AH, Tang XM, Zhang WX, Qian XX, Li JQ, Lu Q. Association of platelet endothelial cell adhesion molecule-1 gene polymorphism with coronary heart disease. Diyi Junyi Daxue Xuebao. 2003;23:156–158. [PubMed] [Google Scholar]
- 92.Gardemann A, Knapp A, Katz N, Tillmanns H, Haberbosch W. No evidence for the CD31 C/G gene polymorphism as an independent risk factor of coronary heart disease. Thromb Haemost. 2000;83:629. [PubMed] [Google Scholar]