

Summary
Thrombotic thrombocytopenic purpura (TTP) and atypical haemolytic uraemic syndrome (aHUS) are acute, rare life‐threatening thrombotic microangiopathies that require rapid diagnosis and treatment. They are defined by microangiopathic haemolytic anaemia and thrombocytopenia, with renal involvement primarily in aHUS and neurological and cardiological sequelae in TTP. Prompt treatment for most cases of both conditions is with plasma exchange initially and monoclonal therapy (rituximab in TTP and eculizumab in aHUS) as the mainstay of therapy. Here we discuss the diagnosis and therapy for both disorders.
Keywords: thrombotic thrombocytopenic purpura, atypical haemolytic uraemic syndrome, treatment, diagnosis, complement
The last 10 years has been associated with a paradigm shift in our understanding, diagnosis, treatment and continuing clinical and basic research into Thrombotic thrombocytopenic purpura (TTP) and atypical haemolytic uraemic syndrome (aHUS) and other thrombotic microangiopathies (TMAs). Here we will concentrate specifically on TTP and aHUS, which, for many years were considered to be the same disease, but it is now clear that their pathophysiology and therapies are quite distinct. TTP results from a deficiency of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), a serine metalloprotease required for the cleavage of von Willebrand factor (VWF). In the majority of cases, the low ADAMTS13 levels are due to Anti‐ADAMTS13 autoantibodies and an underlying disturbance in immune function. aHUS is the consequence of dysregulation of the complement system associated with gain‐ or loss‐of‐function mutations causing excessive complement activation.
However, in both disorders, further environmental and possible genetic triggers will be needed for disease presentation.
Diagnosis of TTP and aHUS
The underlying pathophysiology of TTP is the presence of widespread microthrombi that are platelet‐ and VWF‐rich. aHUS is associated with platelet fibrin thrombi, but other TMAs have varying amounts of lymphocytic or neutrophil infiltration as part of, or related to, the microthrombi (Hosler et al, 2003).
Previously, TTP was diagnosed as a pentad (thrombocytopenia, microangiopathic haemolytic anaemia (MAHA), neurological features, fever and renal involvement) and HUS was classified by the presence or absence of bloody diarrhoea. Indeed, the relevance of ‘diarrhoea negative’ HUS was unclear. This has now changed, and central to the diagnosis of TMAs is thrombocytopenia, MAHA and the clinical or histological effects of microvascular thrombosis. The degree of thrombocytopenia at initial presentation of TTP and aHUS is, broadly, <50 × 109/l for TTP and usually >50 × 109/l for aHUS. However, this is only a general rule and TTP cases with platelet counts >50 × 109/l and aHUS cases with platelet counts <50 × 109/l can be recalled and are not anecdotal. Therefore, a clinical suspicion and full diagnostic work up is required. Presentation may not be ‘classic’. In patients presenting with neurological, cardiac or bleeding symptoms (such as haematuria) as an emergency, and is associated with thrombocytopenia, TTP should be considered. The presence of neurological and cardiological involvement signifies severe disease and should be considered, especially in young patients. At least 10% of presenting TTP patients require intubation and ventilation. Identification of subgroups of TTP, such as human immunodeficiency virus (HIV)‐associated, pregnancyand drug‐related cases, require associated treatment of the underlying condition as well as plasma exchange (PEX).
Atypical haemolytic uraemic syndrome (aHUS) is a renal thrombotic microangiopathy characterized by excessive complement activation on the surface of the renal microvasculature. It can be both sporadic and familial. Approximately 60% of individuals affected by aHUS also have an inherited and/or acquired abnormality affecting components of the complement pathway. These include mutations of genes encoding both complement regulators (factor H, factor I, CD46 and thrombomodulin) and complement activators (C3 and factor B) (Kavanagh & Goodship, 2011). In the United Kingdom, criteria for the diagnosis of aHUS have been established by the aHUS Rare Disease Group (RDG) (http://rarerenal.org). The criteria agreed for the diagnosis of aHUS are shown in Table 1. When a patient presents with clinical features compatible with a diagnosis of aHUS, all other causes of a renal thrombotic microangiopathy are excluded with the investigations listed in Table 2. Blood samples to be sent for measurement ofcomplement levels (C3, C4, factor H and factor I), mutation screening (CFH, CFI, CD46, C3, CFB, THBD and DGKE) and detection of factor H autoantibodies should be undertaken.
Table 1. aHUS diagnostic criteria.
| Exclusion |
| Shiga toxin‐associated HUS |
| Secondary causes |
| Drugs |
| Infection (HIV, Streptococcus pneumonia) |
| Transplantation (bone marrow, liver, lung, cardiac but not de‐novo renal) |
| Cobalamin deficiency |
| Systemic lupus erythematosus |
| Antiphospholipid syndrome |
| Syndrome |
| Scleroderma |
| ADAMTS13 antibodies or deficiency |
| Inclusion |
| Renal biopsy showing a TMA |
| And/or |
| The classic triad of microangiopathic haemolytic anaemia, thrombocytopenia, renal failure |
(a)HUS, (atypical) haemaolytic uraemic syndrome; HIV, human immunodeficiency virus; ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; TMA, thrombotic microangiopathy.
Table 2. Investigations to exclude other causes of a renal thrombotic microangiopathy.
| TTP | ADAMTS13 activity |
| STEC HUS | Stool culture |
| E. coli endotoxin antibodies (IgM) | |
| APL Antibody syndrome | APL antibody |
| SLE | DsDNA |
| HIV | HIV test |
| Scleroderma | ANA |
| Anticentromere antibodies | |
| Anti‐ACL‐70 | |
| Cobalamin C disease | Plasma homocysteine levels |
| Plasma and urine methylmalonic acid levels |
TTP, thrombotic thrombocytopenic purpura; ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; STEC‐HUS, Shiga‐like toxin‐producing E. coli haemaolytic uraemic syndrome; APL, Antiphospholipid; SLE, Systemic lupus erythematosus; HIV, human immunodeficiency virus; ANA, anti‐nuclear antibodies; ACL, anti‐cardiolipin antibody.
Presentation of thrombocytopenia, MAHA and acute anuric/oliguric renal failure requiring dialysis, with no obvious underlying precipitant, would be in keeping with aHUS and requires urgent PEX. The caveat to this is late onset congenital TTP, which has previously been undiagnosed, but may present with renal failure as a result of recurrent, probably sub‐acute TTP episodes. Therefore, an ADAMTS13 activity measurement is required, and a sample taken pre‐PEX for exclusion (Fig 1).
Figure 1.
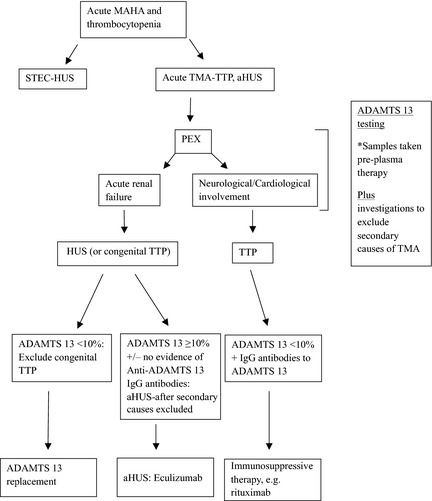
Summary in the diagnosis and treatment of TTP and aHUS. TTP, thrombotic thrombocytopenic purpura; (a)HUS (atypical) haemaolytic uraemic syndrome; STES‐HUS, Shiga‐like toxin‐producing E. coli haemaolytic uraemic syndrome; TMA, thrombotic microangiopathy; PEX, plasma exchange; ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; IgG, immunoglobulin G.
The presence of diarrhoea may not always help differentiate these conditions and further investigations to exclude verotoxin, include stool culture, serology and/or polymerase chain reaction.
The importance of diagnosis and prompt treatment, specifically with PEX in the first instance, is to help preserve renal function in aHUS and in TTP and increase platelet counts by delivering high volumes of ADAMTS13 present in plasma, preventing organ sequelae.
Mortality in acute TTP remains at 10–20% and is up to 25% for aHUS, dependent on the mutational trigger. Therefore, on consideration of these conditions, swift referral and therapy must be instituted.
The role of ADAMTS13 measurement in TMAs
There remains considerable debate on the role of ADAMTS13 measurements in TTP. These are fuelled by the lack of reproducibility between the various assays using VWF multimers or collagen binding, which provide an indirect measure of ADAMTS13 (Studt et al, 2003). They could identify very low ADAMTS13 levels but their reproducibility was poor, with very high intra‐assay coefficients. More recently, fluorescence resonance energy transfer (FRETS) assay has become the main methodology of choice for many institutions undertaking ADAMTS13 assays, but other assay methods are used, such as mass spectrometry and enzyme‐linked immunosorbent assay, e.g., technozyme (Technoclone, Vienna, Austria) (Tripodi et al, 2004; Peyvandi et al, 2010).
Atypical HUS, and other TMAs by definition, will not have low (<10%) ADAMTS13 activity or the presence of significant Anti‐ADAMTS13 immunoglobulin G (IgG) autoantibodies. In contrast, idiopathic TTP will have low ADAMTS13 activity (<10%) and, in the majority of cases (excluding congenital TTP), antibody can be detected. It has been suggested that there are cases of normal ADAMTS13 activity in TTP, but in our experience, these have turned out to be other diagnosis such as aHUS, mainly associated with CD46 mutations. Assuming a sample has been taken pre‐PEX, a mid range/normal ADAMTS13 is not consistent with TTP. The only subgroup of TTP with ADAMTS13 activity that is not low (<10%) is pancreatitis‐associated TTP (McDonald et al, 2009).
Atypical haemolytic uraemic syndrome may have a normal ADAMTS13 at presentation or it can be reduced, usually to about 30–40%. Similar results can be seen in other TMAs, such as malignant hypertension, scleroderma, autoimmune‐mediated TMA and transplant‐associated TMA. The reason some secondary cases have a normal ADAMTS13 at presentation and some are reduced is unclear. It may be because of the raised VWF/thrombin levels, which result in consumption of ADAMTS13, but this does not explain the full picture.
Differentiating TTP from aHUS
It has been suggested that unless there is prompt availability of ADAMTS13 assays, a serum Creatinine level >150–200 μmol/l or a platelet count >30 × 109/l almost always eliminates a diagnosis of severe ADAMTS13 deficiency (Zuber et al, 2012a). While the overall Creatinine levels in TTP cases are not increased and platelet counts are more usually reduced, i.e., <30 × 109/l, in large cohorts, there is enough evidence in daily practice of patients with TTP who have renal dysfunction or a platelet count >30 × 109/l (Scully et al, 2008; McDonald et al, 2009; Coppo et al, 2010). However, we would suggest that samples are taken for ADAMTS13 testing to differentiate the disorders, before patients initiate PEX.
Treatment
When a diagnosis of an acute TMA has been made, suggesting TTP or aHUS, it is imperative that patients are transferred to specialist centres for treatment as soon as possible.
How I treat TTP
Thrombotic thrombocytopenic purpura is a haematological emergency. Referral to an apheresis unit, ensuring central venous line access and defrosting of plasma, may result in delays to initiation of treatment. However, patients should not be left overnight without PEX. Therefore, provision of centres with a 24/7 apheresis service is required. If there is a delay in transfer, pulsed methyl prednisolone and plasma infusion should be considered (Scully et al, 2012).
Plasma exchange is the most important treatment, as it replaces large volume of plasma containing the missing enzyme and in removes the associated anti‐ADAMTS13 IgG antibody, which is directly related to the pathogenesis of the disease. In conjunction, high dose steroids, either oral prednisolone or pulsed methylprednisolone, should be used. While determining the underlying precipitant, steroids should be considered.
Severe initial disease or progression of symptoms during admission may require that PEX is intensified, for example to twice daily, or increased from 1·0 to 1·5 plasma volumes. With an improvement in counts and clinical condition, PEX can be continued as a daily, single plasma volume. There is no indication to taper PEX, assuming close surveillance once PEX is stopped.
Further treatment modalities to improve response should then be instituted. For example, highly active anti‐retroviral therapy (HAART) should be commenced in patients for whom a diagnosis of HIV‐TTP is made. Therapy should be given after PEX and triple or quadruple therapy is preferable initially (Hart et al, 2011). As the majority of cases are idiopathic TTP, with no precipitating cause and antibody‐mediated, further immunosuppressive therapy should be considered. Ciclosporin is used, with satisfactory results. However, it is poorly tolerated by patients and currently rituximab is preferable.
Adjuvant therapy in the treatment of TTP
There is a widespread national and international experience with rituximab in idiopathic TTP. However, there remain a number of questions regarding the timing, dose and frequency of therapy and it has not been used in a randomized control trial setting.
Initial data showed, and confirmed in individual cases, small series and larger cohorts (relative to the disease incidence), the benefit of rituximab in relapsing and refractory TTP (Chemnitz et al, 2002; Gutterman et al, 2002; Zheng et al, 2003; Ahmad et al, 2004; Sallah et al, 2004; Fakhouri et al, 2005; Reddy et al, 2005; Scully et al, 2007; Jasti et al, 2008; Ling et al, 2009; de la Rubia et al, 2010). Indeed, funding restrictions are such that, in some areas, this remains the point at which rituximab is used. A phase II non‐randomized trial to demonstrate the efficacy and safety of rituximab compared to historical controls, showed the benefit of Rituximab administration in acute TTP. In Caucasian patients, there was a reduction in the number of PEX required to achieve remission and an overall (excluding cases treated in the Intensive Care Unit) reduction in inpatient days, which was also seen in the group overall compared to historical controls (Scully et al, 2011).
The standard rituximab dose of 375 mg/m2 has been used almost extensively in the literature to achieve B cell‐depletion in TTP. There is little evidence of low dose rituximab regimes in acute disease. Furthermore, despite B cell depletion measured by CD19 levels, they do not always correlate with ADAMTS13 autoantibody reduction acutely and remission and are not helpful in predicting relapse.
As per the lymphoma protocol, based on convenience with other chemotherapy infusions in this disease, rituximab was used weekly. However, confirmation of clearance of the anti‐CD20 therapy in PEX fluid and a review of peak and trough rituximab levels with PEX has resulted in a move to infusing rituximab every 3–4 d (McDonald et al, 2010).
Regardless of the timing of starting Rituximab in an acute TTP episode, the responses take a median of 10 d. Therefore, starting Rituximab earlier in an acute episode will result in reduced PEX, quicker over all responses and reduced inpatient admissions, with significant clinical and financial benefits (Westwood et al, 2013).
From the phase II trial, an unexpected finding was the reduction in relapse rates with acute TTP. Before rituximab, immunosuppressive therapies had not generally shown a consistent sustained benefit. This appears, from our historical control cases, to be as a result of failure to increase the ADAMTS13 activity by reducing antibody levels. In patients who do relapse after rituximab, the median time is 24 months, but far longer periods of remission are seen. This is in contrast to many having had multiple repeated episodes in the pre‐rituximab era and immunosuppressive therapy that had unacceptable side effects. The low relapse rate was considerable and confirmed in a further multicentre trial in relapsed/refractory acute TTP cases presented by the French collaborative group (Froissart et al, 2012). Previously documented relapse rates are between 30% and 50%. The Oklahoma registry had a 34% relapse rate in 60 TTP cases between 1989 and 2008, with ADAMTS13 activity <10% at diagnosis (Kremer Hovinga et al, 2010).
Monitoring patients following an episode of acute TTP
In the majority of acute idiopathic TTP cases, treatment is associated with a reduction in anti‐ADAMTS13 IgG levels and an increase in ADAMTS13 activity. The risk of relapse of TTP is associated with low ADAMTS activity and or the presence of antibody. Our practice is to monitor patients in clinical remission with normal ADAMTS13 activity. A reduction in enzyme (<10%) is a marker to consider elective treatment with rituximab, which results in normalization of ADAMTS13 activity and prevents an acute episode (Westwood et al, 2013).
The reduction in ADAMTS13 activity may occur some months before relapse, especially in Afro‐Caribbean patients, whereas in Caucasians, relapse typically occurs sooner if left untreated. ADAMTS13 is a useful surrogate marker of potential relapse, but the timing of relapse is variable and the use of prophylactic rituximab may be guided by the patient's previous history. Monitoring does not provide an absolute guarantee against relapse and patients need to be vigilant.
Within the elective therapy setting, we have used low dose rituximab (100 mg/m2), for which an increase in ADAMTS13 activity can be documented and relapse possibly prevented.
Failure to achieve normalization of ADAMTS13 activity despite remission
There are a small percentage (approximately 5%) of patients who may attain clinical remission, although this is typically associated with a longer acute admission, but the ADAMTS13 activity does not improve and remains low (<10%). These patients are very difficult to monitor regarding relapse and need to seek urgent medical attention for a blood count if unwell. However, a useful combination has been rituximab and mycophenolate, which has achieved normalization of levels in patients who previously have not demonstrated a rise in ADAMTS13 activity. However, these cases do still need regular monitoring and if ADAMTS13 activity falls (<10%), further rituximab therapy is given. Once again, although not exclusively, these are typically Afro‐Caribbean patients and a further understanding of the pathophysiology of these cases is required, together with the development of other therapies.
Other immunosuppressive treatments have been documented in the literature. Ciclosporin is effective (Cataland et al, 2007a,b), but we have not used it since the advent of monoclonal therapy for a number of reasons, including the risk of relapse during/on stopping treatment. Furthermore, patients do not tolerate side effects, particularly young females.
Congenital TTP (also known as Upshaw‐Schulman syndrome)
Despite being a rare subtype, this is increasingly being diagnosed, both in young children and late onset associated with pregnancy. Patients require monitoring of their platelet counts and therapy to ensure platelets remain >150 × 109/l. The frequency of treatment will vary with the age of presentation and the patient's genotype. Failure to achieve normal platelet counts will result in end organ damage, including renal failure, neurological or cardiac features. Normalization can be achieved with either plasma infusions (non‐UK sourced and virally inactivated‐Octaplas®; Octapharma, Vienna, Austria) or with an intermediated purity Factor VIII concentrate, which contains measurable ADAMTS13 (BioProducts Laboratory, Elstree, Herts, UK).
In women of child‐bearing age, pregnancy requires regular plasma infusion throughout to ensure improved outcomes for mother and baby.
Pregnancy‐associated TTP
Pregnancy may be a precipitant of TTP in a small proportion of cases and ADAMTS13 analysis is required to differentiate congenital from acquired TTP. TMAs in pregnancy can be difficult to differentiate, e.g., pre‐eclampsia (PET), HELLP syndrome (haemolysis, elevated liver enzyme levels, and low platelet counts) and TTP. In general, HUS occurs in the postpartum period. TTP may be associated with features suggestive of PET or HELLP. As a general guide, PEX should be started in patients with platelet counts <50 × 109/l and MAHA features or the presence of clinical features possibly suggestive of TTP at higher platelet counts.
How do I treat incident patients with aHUS?
Until recently it was recommended that all patients presenting with aHUS should be offered a trial of PEX and/or plasma infusions (Taylor et al, 2010). Despite this, the majority of patients need long term renal replacement therapy within 2 years of presentation (Noris et al, 2010). With the finding that excessive activation of the alternative pathway of complement underlies the pathogenesis of aHUS in most patients, it became clear that complement inhibition would be a logical therapy. Eculizumab is the only complement inhibitor currently licensed for any indication. It was first introduced for the treatment of paroxysmal nocturnal haemoglobinuria (Hillmen et al, 2006) and long term data has shown its efficacy in this disease (Kelly et al, 2011). Anecdotal reports of its efficacy in aHUS (Gruppo & Rother, 2009; Nurnberger et al, 2009) led to clinical trials, the results of which have recently been published (Legendre et al, 2013). These showed that eculizumab was a highly effective treatment for aHUS in patients who were either resistant to PEX or dependent on regular PEX to maintain remission. Based on these results, in 2011 both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved the use of eculizumab for the treatment of aHUS. In England an application was submitted to the Advisory Group for National Specialised Services (AGNSS) for a national service for aHUS, including funding for eculizumab. This was considered by AGNSS in June 2012. In January 2013 it was announced that. ‘they concluded that eculizumab would help save lives and improve the quality of life for children (among whom the condition is particularly prevalent) and adults with atypical haemolytic uraemic syndrome.’ (http://www.specialisedservices.nhs.uk/document/eculizumab-treatment-atypical-haemolytic-uraemic-syndrome/). Subsequently ‘Ministers agreed with AGNSS that there is evidence for the clinical effectiveness of eculizumab for the treatment of atypical haemolytic uraemic syndrome but wanted further advice on the affordability of the drug’. Ministers have asked NICE to consider eculizumab for the treatment of atypical haemolytic uraemic syndrome as part of its new Highly Specialised Technologies programme.’ The outcome of the National Institute for Health and Care Excellence (NICE) evaluation will be announced in 2014. In the meantime, National Health Service (NHS) England has published a Clinical Commissioning Policy Statement on the use of eculizumab for aHUS (http://www.england.nhs.uk/wp-content/uploads/2013/09/e03-hss-a.pdf). This states that NHS England will commission eculizumab for new patients with aHUS and for patients who are on dialysis and are suitable for a kidney transplant. In England, we are therefore now able to treat all new patients with aHUS. This service is delivered locally but coordinated through the expert centre in Newcastle. When a patient presents with features compatible with a diagnosis of aHUS we request that the investigations listed in Table 2 are undertaken. We try to initiate treatment with eculizumab as soon as possible because the clinical trials have suggested that the sooner that treatment is started, the greater is the chance of recovery of renal function. The only result that we request be available is the ADAMTS13 activity. If the ADAMTS13 activity is not immediately available we recommend that treatment with PEX is commenced and continued until the result is available. Provided that the ADAMTS13 activity is not <10% and that the clinical features are compatible with a diagnosis of aHUS we, with the approval of NHS England, authorize the onset of treatment with eculizumab. If subsequent investigations show that the thrombotic microangiopathy is due to another condition apart from aHUS, then eculizumab is withdrawn. The results of the genetic investigations are available within 8 weeks.
Two groups have recently published reports of mutations in the DGKE gene, encoding diacylglycerol kinase epsilon (DKGE) in young children with the clinical phenotype of aHUS (Lemaire et al, 2013; Ozaltin et al, 2013). DGKE is a lipid kinase that catalyses the conversion of diacylglycerol to phosphatidic acid. Failure of this leads to increased protein kinase C activity and it has been postulated that this is responsible for the development of a TMA (Quaggin, 2013). This form of aHUS presents usually in the first year of life and does not respond to eculizumab. The mutations in DGKE are either homozygous or compound heterozygous. If, on screening, we find such changes in DGKE in the absence of any mutations in complement genes, then we will consider withdrawing eculizumab.
The markers of an active TMA (thrombocytopenia, microangiopathic haemolytic anaemia, elevated lactate dehydrogenase, low haptoglobin) respond rapidly to treatment with eculizumab in virtually all patients with an underlying complement abnormality. In aHUS, whether renal function returns is dependent on the extent of irreversible renal damage sustained before the onset of treatment. In both anecdotal reports and the trials it has been found that some patients who are on dialysis at the onset of the treatment will recover sufficient renal function to stop dialysis. This can take several months. We, therefore, currently would not consider withdrawing eculizumab in a dialysis‐dependent patient until they had received at least 4 months treatment with eculizumab. If eculizumab is withdrawn in such a patient then we monitor closely the TMA markers. If there is any evidence of recurrent extra‐renal TMA then eculizumab is reinstituted. The other group of patients in whom we will consider withdrawing eculizumab is those in whom we subsequently find a mutation in CD46 in the absence of changes in any other gene. We counsel this patient that there is a risk of recurrent disease and withdrawal of the eculizumab is only undertaken if the patient agrees to this. If not then they continue on eculizumab indefinitely.
At present, in all other patientswe continue treatment with eculizumab indefinitely. The major risk associated with eculizumab treatment is the development of meningococcal disease. All patients are vaccinated with a tetravalent vaccine before the onset of treatment and, in addition, receive long‐term prophylactic antibiotics (either penicillin or erythromycin) because current vaccines do not cover Serogroup B strains. We will in the future be testing the response to vaccination after 12 weeks and re‐vaccinating if the response is suboptimal.
How do I treat prevalent aHUS patients?
The outcome of renal transplantation alone in aHUS patients on dialysis has been well documented to be poor, with a high rate of graft loss due to recurrent disease (Bresin et al, 2006; Le et al, 2013). Because of this, many patients known to have complement abnormalities associated with a high risk of recurrence have not been listed for a transplant. In those patients known to have mutations in the genes encoding complement proteins synthesized predominantly by the liver (such as factor H and factor I), combined liver‐kidney transplant is an alternative therapeutic strategy that can be successful (Saland et al, 2009). However, the 1‐year patient survival for this procedure is not as high as would be expected with a kidney transplant alone. Because of this and the advent of eculizumab, many patients do not opt for this procedure. Both anecdotal reports and trial data have shown that eculizumab can be used successfully to both treat and prevent recurrent disease post‐transplant (Zuber et al, 2012b). In England, aHUS patients on dialysis are now, therefore, being listed for a kidney transplant that is covered by eculizumab given immediately prior to transplantation and then long term to prevent recurrent disease. Whether in any of these patients it will be safe to withdraw eculizumab at a later stage not yet known.
There are a small number of aHUS patients who respond to PEX, but need long‐term therapy to maintain their remission. Most of these patients entered the clinical trials and are now receiving long‐term eculizumab. We recommend that those few who are still on PEX should be started on eculizumab and PEX withdrawn.
There are also a small number of patients who have responded to PEX and have stayed in remission without any prophylactic therapy. Screening in some of these patients has shown a pathogenic complement gene mutation. We believe that such patients are potentially at risk of recurrent disease in the future. Whether this warrants prophylactic treatment with eculizumab is uncertain. We currently discuss this with the individual patient.
Conclusion
The initial diagnosis and treatment of TMAs, specifically TTP and aHUS with PEX remains paramount, but is more justified as we understand different disease states. However, determining the underlying pathophysiology and diagnosis is relevant when further therapeutic modalities need to be introduced into the treatment pathway. The advent of eculizumab has dramatically changed the prognosis of aHUS. It is now possible that early treatment will prevent irreversible renal failure in patients who present for the first time and that recurrent disease can be prevented in those patients undergoing renal transplantation. Similarly, prompt diagnosis and initiation of PEX and monoclonal anti‐CD20 treatment in TTP has improved response and relapse rates.
Author contributions
MS and TG equally contributed to the writing of the paper.
References
- Ahmad, A., Aggarwal, A., Sharma, D., Dave, H.P., Kinsella, V., Rick, M.E. & Schechter, G.P. (2004) Rituximab for treatment of refractory/relapsing thrombotic thrombocytopenic purpura (TTP). American Journal of Hematology, 77, 171–176 [DOI] [PubMed] [Google Scholar]
- Bresin, E., Daina, E., Noris, M., Castelletti, F., Stefanov, R., Hill, P., Goodship, T.H. & Remuzzi, G. (2006) Outcome of renal transplantation in patients with non‐Shiga toxin‐associated hemolytic uremic syndrome: prognostic significance of genetic background. Clinical Journal of the American Society of Nephrology, 1, 88–99 [DOI] [PubMed] [Google Scholar]
- Cataland, S.R., Jin, M., Ferketich, A.K., Kennedy, M.S., Kraut, E.H., George, J.N. & Wu, H.M. (2007a) An evaluation of cyclosporin and corticosteroids individually as adjuncts to plasma exchange in the treatment of thrombotic thrombocytopenic purpura. British Journal of Haematology, 136, 146–149 [DOI] [PubMed] [Google Scholar]
- Cataland, S.R., Jin, M., Lin, S., Kennedy, M.S., Kraut, E.H., George, J.N. & Wu, H.M. (2007b) Cyclosporin and plasma exchange in thrombotic thrombocytopenic purpura: long‐term follow‐up with serial analysis of ADAMTS13 activity. British Journal of Haematology, 139, 486–493 [DOI] [PubMed] [Google Scholar]
- Chemnitz, J., Draube, A., Scheid, C., Staib, P., Schulz, A., Diehl, V. & Sohngen, D. (2002) Successful treatment of severe thrombotic thrombocytopenic purpura with the monoclonal antibody rituximab. American Journal of Hematology, 71, 105–108 [DOI] [PubMed] [Google Scholar]
- Coppo, P., Schwarzinger, M., Buffet, M., Wynckel, A., Clabault, K., Presne, C., Poullin, P., Malot, S., Vanhille, P., Azoulay, E., Galicier, L., Lemiale, V., Mira, J.P., Ridel, C., Rondeau, E., Pourrat, J., Girault, S., Bordessoule, D., Saheb, S., Ramakers, M., Hamidou, M., Vernant, J.P., Guidet, B., Wolf, M. & Veyradier, A. (2010) Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One, 5, e10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhouri, F., Vernant, J.P., Veyradier, A., Wolf, M., Kaplanski, G., Binaut, R., Rieger, M., Scheiflinger, F., Poullin, P., Deroure, B., Delarue, R., Lesavre, P., Vanhille, P., Hermine, O., Remuzzi, G. & Grunfeld, J.P. (2005) Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood, 106, 1932–1937 [DOI] [PubMed] [Google Scholar]
- Froissart, A., Buffet, M., Veyradier, A., Poullin, P., Provot, F., Malot, S., Schwarzinger, M., Galicier, L., Vanhille, P., Vernant, J., Bordessoule, D., Guidet, B., Azoulay, E., Rondeau, E., Mira, J., Wynckel, A., Clabault, K., Choukroun, G., Presne, C., Pourrat, J., Hamidou, M. & Coppo, P. (2012) Efficacy and safety of first‐line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Critical Care Medicine, 40, 104–111 [DOI] [PubMed] [Google Scholar]
- Gruppo, R.A. & Rother, R.P. (2009) Eculizumab for congenital atypical hemolytic‐uremic syndrome. New England Journal of Medicine, 360, 544–546 [DOI] [PubMed] [Google Scholar]
- Gutterman, L.A., Kloster, B. & Tsai, H.M. (2002) Rituximab therapy for refractory thrombotic thrombocytopenic purpura. Blood Cells, Molecules, & Diseases, 28, 385–391 [DOI] [PubMed] [Google Scholar]
- Hart, D., Sayer, R., Miller, R., Edwards, S., Kelly, A., Baglin, T., Hunt, B., Benjamin, S., Patel, R., Machin, S. & Scully, M. (2011) Human immunodeficiency virus associated thrombotic thrombocytopenic purpura – favourable outcome with plasma exchange and prompt initiation of highly active antiretroviral therapy. British Journal of Haematology, 153, 515–519 [DOI] [PubMed] [Google Scholar]
- Hillmen, P., Young, N.S., Schubert, J., Brodsky, R.A., Socie, G., Muus, P., Roth, A., Szer, J., Elebute, M.O., Nakamura, R., Browne, P., Risitano, A.M., Hill, A., Schrezenmeier, H., Fu, C.L., Maciejewski, J., Rollins, S.A., Mojcik, C.F., Rother, R.P. & Luzzatto, L. (2006) The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine, 355, 1233–1243 [DOI] [PubMed] [Google Scholar]
- Hosler, G.A., Cusumano, A.M. & Hutchins, G.M. (2003) Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Archives of Pathology and Laboratory Medicine, 127, 834–839 [DOI] [PubMed] [Google Scholar]
- Jasti, S., Coyle, T., Gentile, T., Rosales, L. & Poiesz, B. (2008) Rituximab as an adjunct to plasma exchange in TTP: a report of 12 cases and review of literature. Journal of Clinical Apheresis, 23, 151–156 [DOI] [PubMed] [Google Scholar]
- Kavanagh, D. & Goodship, T.H. (2011) Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology/The Education Program of the American Society of Hematology, 2011, 15–20 [DOI] [PubMed] [Google Scholar]
- Kelly, R.J., Hill, A., Arnold, L.M., Brooksbank, G.L., Richards, S.J., Cullen, M., Mitchell, L.D., Cohen, D.R., Gregory, W.M. & Hillmen, P. (2011) Long‐term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood, 117, 6786–6792 [DOI] [PubMed] [Google Scholar]
- Kremer Hovinga, J.A., Vesely, S.K., Terrell, D.R., Lammle, B. & George, J.N. (2010) Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood, 115, 1500–1511 [DOI] [PubMed] [Google Scholar]
- Le, Q.M., Zuber, J., Moulin, B., Kamar, N., Jablonski, M., Lionet, A., Chatelet, V., Mousson, C., Mourad, G., Bridoux, F., Cassuto, E., Loirat, C., Rondeau, E., Delahousse, M. & Fremeaux‐Bacchi, V. (2013) Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. American Journal of Transplantation, 13, 663–675 [DOI] [PubMed] [Google Scholar]
- Legendre, C.M., Licht, C., Muus, P., Greenbaum, L.A., Babu, S., Bedrosian, C., Bingham, C., Cohen, D.J., Delmas, Y., Douglas, K., Eitner, F., Feldkamp, T., Fouque, D., Furman, R.R., Gaber, O., Herthelius, M., Hourmant, M., Karpman, D., Lebranchu, Y., Mariat, C., Menne, J., Moulin, B., Nurnberger, J., Ogawa, M., Remuzzi, G., Richard, T., Sberro‐Soussan, R., Severino, B., Sheerin, N.S., Trivelli, A., Zimmerhackl, L.B., Goodship, T. & Loirat, C. (2013) Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. New England Journal of Medicine, 368, 2169–2181 [DOI] [PubMed] [Google Scholar]
- Lemaire, M., Fremeaux‐Bacchi, V., Schaefer, F., Choi, M., Tang, W.H., Le, Q.M., Fakhouri, F., Taque, S., Nobili, F., Martinez, F., Ji, W., Overton, J.D., Mane, S.M., Nurnberg, G., Altmuller, J., Thiele, H., Morin, D., Deschenes, G., Baudouin, V., Llanas, B., Collard, L., Majid, M.A., Simkova, E., Nurnberg, P., Rioux‐Leclerc, N., Moeckel, G.W., Gubler, M.C., Hwa, J., Loirat, C. & Lifton, R.P. (2013) Recessive mutations in DGKE cause atypical hemolytic‐uremic syndrome. Nature Genetics, 45, 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, H.T., Field, J.J. & Blinder, M.A. (2009) Sustained response with rituximab in patients with thrombotic thrombocytopenic purpura: a report of 13 cases and review of the literature. American Journal of Hematology, 84, 418–421 [DOI] [PubMed] [Google Scholar]
- McDonald, V., Laffan, M., Benjamin, S., Bevan, D., Machin, S. & Scully, M.A. (2009) Thrombotic thrombocytopenic purpura precipitated by acute pancreatitis: a report of seven cases from a regional UK TTP registry. British Journal of Haematology, 144, 430–433 [DOI] [PubMed] [Google Scholar]
- McDonald, V., Manns, K., MacKie, I.J., Machin, S.J. & Scully, M.A. (2010) Rituximab pharmacokinetics during the management of acute idiopathic thrombotic thrombocytopenic purpura. Journal of Thrombosis and Haemostasis, 8, 1201–1208 [DOI] [PubMed] [Google Scholar]
- Noris, M., Caprioli, J., Bresin, E., Mossali, C., Pianetti, G., Gamba, S., Daina, E., Fenili, C., Castelletti, F., Sorosina, A., Piras, R., Donadelli, R., Maranta, R., van der Meer, I., Conway, E.M., Zipfel, P.F., Goodship, T.H. & Remuzzi, G. (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clinical Journal of the American Society of Nephrology, 5, 1844–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberger, J., Philipp, T., Witzke, O., Opazo, S.A., Vester, U., Baba, H.A., Kribben, A., Zimmerhackl, L.B., Janecke, A.R., Nagel, M. & Kirschfink, M. (2009) Eculizumab for atypical hemolytic‐uremic syndrome. New England Journal of Medicine, 360, 542–544 [DOI] [PubMed] [Google Scholar]
- Ozaltin, F., Li, B., Rauhauser, A., An, S.W., Soylemezoglu, O., Gonul, I.I., Taskiran, E.Z., Ibsirlioglu, T., Korkmaz, E., Bilginer, Y., Duzova, A., Ozen, S., Topaloglu, R., Besbas, N., Ashraf, S., Du, Y., Liang, C., Chen, P., Lu, D., Vadnagara, K., Arbuckle, S., Lewis, D., Wakeland, B., Quigg, R.J., Ransom, R.F., Wakeland, E.K., Topham, M.K., Bazan, N.G., Mohan, C., Hildebrandt, F., Bakkaloglu, A., Huang, C.L. & Attanasio, M. (2013) DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. Journal of the American Society of Nephrology, 24, 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyvandi, F., Palla, R., Lotta, L.A., Mackie, I., Scully, M.A. & Machin, S.J. (2010) ADAMTS‐13 assays in thrombotic thrombocytopenic purpura. Journal of Thrombosis and Haemostasis, 8, 631–640 [DOI] [PubMed] [Google Scholar]
- Quaggin, S.E. (2013) DGKE and atypical HUS. Nature Genetics, 45, 475–476 [DOI] [PubMed] [Google Scholar]
- Reddy, P.S., Deauna‐Limayo, D., Cook, J.D., Ganguly, S.S., Blecke, C., Bodensteiner, D.C., Skikne, B.S. & Sahud, M.A. (2005) Rituximab in the treatment of relapsed thrombotic thrombocytopenic purpura. Annals of Hematology, 84, 232–235 [DOI] [PubMed] [Google Scholar]
- de la Rubia, J., Moscardo, F., Gomez, M.J., Guardia, R., Rodriguez, P., Sebrango, A., Zamora, C., Deben, G., Goterris, R., Lopez, R., Pena, F., Pujol, M., Vidaller, A., del Rio‐Garma, J. & Sanz, M.A. (2010) Efficacy and safety of rituximab in adult patients with idiopathic relapsing or refractory thrombotic thrombocytopenic purpura: results of a Spanish multicenter study. Transfusion and Apheresis Science, 43, 299–303 [DOI] [PubMed] [Google Scholar]
- Saland, J.M., Ruggenenti, P. & Remuzzi, G. (2009) Liver‐kidney transplantation to cure atypical hemolytic uremic syndrome. Journal of the American Society of Nephrology, 20, 940–949 [DOI] [PubMed] [Google Scholar]
- Sallah, S., Husain, A., Wan, J.Y. & Nguyen, N.P. (2004) Rituximab in patients with refractory thrombotic thrombocytopenic purpura. Journal of Thrombosis and Haemostasis, 2, 834–836 [DOI] [PubMed] [Google Scholar]
- Scully, M., Cohen, H., Cavenagh, J., Benjamin, S., Starke, R., Killick, S., Mackie, I. & Machin, S.J. (2007) Remission in acute refractory and relapsing thrombotic thrombocytopenic purpura following rituximab is associated with a reduction in IgG antibodies to ADAMTS‐13. British Journal of Haematology, 136, 451–461 [DOI] [PubMed] [Google Scholar]
- Scully, M., Yarranton, H., Liesner, R., Cavenagh, J., Hunt, B., Benjamin, S., Bevan, D., Mackie, I. & Machin, S. (2008) Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. British Journal of Haematology, 142, 819–826 [DOI] [PubMed] [Google Scholar]
- Scully, M., McDonald, V., Cavenagh, J., Hunt, B.J., Longair, I., Cohen, H. & Machin, S.J. (2011) A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood, 118, 1746–1753 [DOI] [PubMed] [Google Scholar]
- Scully, M., Hunt, B.J., Benjamin, S., Liesner, R., Rose, P., Peyvandi, F., Cheung, B. & Machin, S.J. (2012) Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. British Journal of Haematology, 158, 323–335 [DOI] [PubMed] [Google Scholar]
- Studt, J.D., Bohm, M., Budde, U., Girma, J.P., Varadi, K. & Lammle, B. (2003) Measurement of von Willebrand factor‐cleaving protease (ADAMTS‐13) activity in plasma: a multicenter comparison of different assay methods. Journal of Thrombosis and Haemostasis, 1, 1882–1887 [DOI] [PubMed] [Google Scholar]
- Taylor, C.M., Machin, S., Wigmore, S.J. & Goodship, T.H. (2010) Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. British Journal of Haematology, 148, 37–47 [DOI] [PubMed] [Google Scholar]
- Tripodi, A., Chantarangkul, V., Bohm, M., Budde, U., Dong, J.F., Friedman, K.D., Galbusera, M., Girma, J.P., Moake, J., Rick, M.E., Studt, J.D., Turecek, P.L. & Mannucci, P.M. (2004) Measurement of von Willebrand factor cleaving protease (ADAMTS‐13): results of an international collaborative study involving 11 methods testing the same set of coded plasmas. Journal of Thrombosis and Haemostasis, 2, 1601–1609 [DOI] [PubMed] [Google Scholar]
- Westwood, J.P., Webster, H., McGuckin, S., McDonald, V., Machin, S.J. & Scully, M. (2013) Rituximab for thrombotic thrombocytopenic purpura: benefit of early administration during acute episodes and use of prophylaxis to prevent relapse. Journal of Thrombosis and Haemostasis, 11, 481–490 [DOI] [PubMed] [Google Scholar]
- Zheng, X., Pallera, A.M., Goodnough, L.T., Sadler, J.E. & Blinder, M.A. (2003) Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Annals of Internal Medicine, 138, 105–108 [DOI] [PubMed] [Google Scholar]
- Zuber, J., Fakhouri, F., Roumenina, L.T., Loirat, C. & Fremeaux‐Bacchi, V. (2012a) Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nature Reviews Nephrology, 8, 643–657 [DOI] [PubMed] [Google Scholar]
- Zuber, J., Le, Q.M., Krid, S., Bertoye, C., Gueutin, V., Lahoche, A., Heyne, N., Ardissino, G., Chatelet, V., Noel, L.H., Hourmant, M., Niaudet, P., Fremeaux‐Bacchi, V., Rondeau, E., Legendre, C. & Loirat, C. (2012b) Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. American Journal of Transplantation, 12, 3337–3354 [DOI] [PubMed] [Google Scholar]