

Abstract
Intrahepatic fat deposition has been demonstrated in patients with nonalcoholic fatty liver disease (NAFLD). Genetic and environmental factors are important for the development of NAFLD. Diseases such as obesity, diabetes, and hypertension have been found to be closely associated with the incidence of NAFLD. Evidence suggests that obesity and insulin resistance are the major factors that contribute to the development of NAFLD. In comparing the factors that contribute to the buildup of excess calories in obesity, an imbalance of energy homeostasis can be considered as the basis. Among the peripheral signals that are generated to regulate the uptake of food, signals from adipose tissue are of major relevance and involve the maintenance of energy homeostasis through processes such as lipogenesis, lipolysis, and oxidation of fatty acids. Advances in research on adipose tissue suggest an integral role played by adipokines in NAFLD. Cytokines secreted by adipocytes, such as tumor necrosis factor-α, transforming growth factor-β, and interleukin-6, are implicated in NAFLD. Other adipokines, such as leptin and adiponectin and, to a lesser extent, resistin and retinol binding protein-4 are also involved. Leptin and adiponectin can augment the oxidation of fatty acid in liver by activating the nuclear receptor super-family of transcription factors, namely peroxisome proliferator-activated receptor (PPAR)-α. Recent studies have proposed downregulation of PPAR-α in cases of hepatic steatosis. This review discusses the role of adipokines and PPARs with regard to hepatic energy metabolism and progression of NAFLD.
Keywords: Nonalcoholic fatty liver disease, Adipose tissue, Energy homeostasis, Peroxisome proliferator-activated receptors, Adipokines
Core tip: Nonalcoholic fatty liver disease (NAFLD) is one of the principal causes for chronic liver disease. Recent reports suggested a positive association between cytokines secreted by the adipocytes, such as tumor necrosis factor-α, transforming growth factor-β, and interleukin (IL)-6 in NAFLD. Furthermore, hepatic natural killer T-cells produce IL-13 and IL-4; IL-13 may then activate hepatic stellate cells to produce pro-inflammatory cytokines and initiate oxidative stress, iron overload and fibrosis. Downregulation of peroxisome proliferator-activated receptors (PPAR), particularly PPAR-α in cases of hepatic steatosis, may facilitate the activity of hepatic proinflammatory cytokines. Hence, PPAR-γ and PPAR-α ligands have been considered for administration to prevent the initial inflammatory reactions and render protection to the liver cells.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a disease spectrum encompassing simple steatosis, steatohepatitis, fibrosis and, ultimately, liver cirrhosis[1]. It is reported to be one of the principal causes of chronic liver disease, and is predominant in developed countries. Results of several studies proposed NAFLD as the hepatic component of metabolic syndrome (MS), which is characterized by hyperinsulinemia, insulin resistance (IR), obesity, type II diabetes mellitus (TDM), dyslipidemia, and hypertension[2]. NAFLD has a wide histological spectrum ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), which may progress to cirrhosis. In simple NAFLD, the presence of macrovesicular fat droplets in hepatocytes, foci of lobular inflammation, mild portal inflammation, and lipogranulomas may be seen, while increased portal inflammation has been reported in untreated NAFLD patients[3]. In approximately 25% of cases, histological signs of fibrosis and necroinflammatory injury that define NASH are present[4]. Patients with NASH are at higher risk of developing fibrosis, cirrhosis and hepatocellular carcinoma[4]. The exact cause of NAFLD is still unknown. However, incidences of NAFLD have been associated with cases of IR in both diabetes and obesity, and with hypertension. It has been proposed that an imbalance in energy homeostasis may be the basis for obesity and thus NAFLD.
Complex physiologic processes, constant communication within and among tissues such as adipose tissue, liver, skeletal muscle, pancreas, and the central nervous system are required for energy homeostasis. The brain is referred to as the central regulator of energy homeostasis and thus the primary controller of body weight. Peripherally, the major organs participating in the regulation of food intake are the stomach, intestines, pancreas, and adipose tissue[5]. The imbalance in energy homeostasis stems etiologically from either excess food intake or insufficient energy expenditure. This may also be secondarily related to endocrinopathies such as hypothyroidism, Cushing’s syndrome, etc., that evolve into MS, which, in turn, is related to NAFLD[6,7].
Among various peripheral organs, the role played by adipose tissue in energy homeostasis remains central. Adipose tissue, far from being an inert depot of storage fat, is an active endocrine organ, as evidenced by the variety of hormones or adipokines it synthesizes, including leptin and adiponectin, among others[8]. Signals controlling energy intake originate from adipose tissue, mediated by leptin. The afferent and efferent signals fluctuate in a coordinated manner to maintain energy homeostasis. It is necessary to determine the role played by various adipokines and nuclear receptors such as peroxisome proliferators-activated receptors (PPARs), in the initiation and progression of NAFLD. Research in this field is evolving to explain the exact role of the adipokines in NAFLD. This review discusses the role of adipokines and PPARs in NAFLD.
ADIPOSE TISSUE IN ENERGY HOMEOSTASIS
Energy homeostasis is maintained by the integration of major metabolic functions such as lipogenesis, lipolysis, and fatty acid oxidation, and is mediated through adipose tissue[9]. Of the two types of adipose tissues, white (WAT) and brown (BAT), WAT is concerned with energy balance in adults and has been found to be increased in obesity[10,11]. Cells constituting WAT include preadipocytes, fibroblasts, endothelial cells, macrophages, and monocytes[12]. The functional adipocyte secretes several factors known as adipokines to communicate actively with the liver, muscle, and hypothalamus[13,14]. Such factors include pro-inflammatory cytokines produced by resident macrophages such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and monocyte chemoattractant protein (MCP)-1. The majority of adipokines, however, maintain equilibrium in the utilization of energy substrates between adipose and non adipose tissues in the intestine, liver, brain, and skeletal muscle[14]. Among such adipokines, leptin and adiponectin and, to a lesser extent, resistin and retinol binding protein-4 have a role in energy homeostasis.
ROLE OF LEPTIN, ADIPONECTIN, RESISTIN, AND RETINOL BINDING PROTEIN-4 IN LIPID METABOLISM
Leptin (16 kDa) is a peptide hormone synthesized by adipocytes and in inconsequential amounts by the liver and skeletal muscle[15]. Its gene expression (Lep/Ob gene) is regulated by food intake, energy status, hormones, and the overall inflammatory state[16]. Leptin signals are mediated through a membrane receptor and signaling pathways involve Janus activating kinase/signal transducer and activator of transcription, mitogen-activated protein kinase, phosphatidyl-inositol 3’ kinase, and AMP-dependent protein kinase (AMPK)[17,18]. Through such pathways, leptin acts on the hypothalamus to reduce appetite and thus function as an adipostat[16]. The stimulating pathways, in general, favor fatty acid oxidation and decrease lipogenesis. Leptin also tends to decrease the ectopic deposition of fat in liver and muscle. Furthermore, leptin can also act on the pancreas, inhibiting both insulin and glucagon secretion via short-term/non-genomic and long-term/genomic mechanisms, thereby promoting glucose homeostasis[19]. In excess calorie states, such as obesity, hyperleptinemia associated with leptin receptor inactivation/downregulation has been observed in the hypothalamus and liver of obese rats[20].
Adiponectin (30 kDa) is a protein hormone, which exists in several globular, trimeric, and high molecular weight forms in circulation. Adiponectin stimulates insulin sensitivity, decreases hepatic glucose production, increases glucose utilization in muscle and oxidation of fatty acid in muscle, liver, and peripheral tissues, thus down-regulating the secretion of pro-inflammatory cytokines IL-6, IL-8 and MCP-1[21,22]. Adiponectin receptors activate AMPK, p38 mitogen-activated protein kinase, and PPAR-α, which in turn regulate fatty acid metabolism[23]. Low levels of adiponectin are associated with low-grade inflammation, oxidative stress, and endothelial dysfunction[24]. Its receptors are largely decreased in obese animals and humans.
Both leptin and adiponectin stimulate AMPK in skeletal muscle, liver, and adipose tissue[25]. In skeletal muscle, AMPK activation promotes glucose transport, glycolysis, and fatty acid oxidation, and inhibits fatty acid synthesis (Figure 1). AMPK directly inhibits acetyl CoA carboxylase, preventing lipogenesis; consequentially, carnitine palmitoyl transferase-1 is stimulated leading to fatty acid oxidation[22]. AMPK is activated during exercise and is involved in glucose transport and fatty acid oxidation in skeletal muscle. Hepatic lipid turnover is regulated by transcription factors carbohydrate-responsive element-binding protein, sterol regulatory element binding protein 1c (SREBP-1c), CCAAT-enhancer-binding protein α, and PPARs. In liver, AMPK is a cellular metabolic sensor, inhibiting lipogenesis and cholesterol synthesis; its activation suppresses the SREBP1c[9]. Disequilibrium in lipid homeostasis causes triglycerides to accumulate in the liver.
Figure 1.
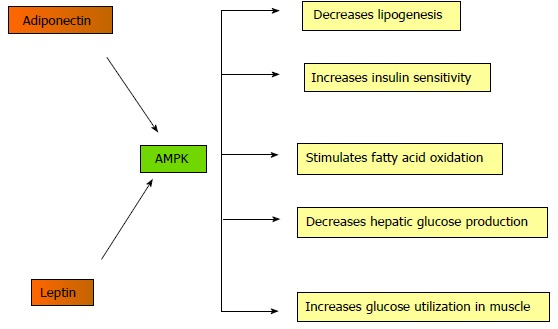
Mechanism of action of adenosine monophosphate-dependent protein kinase. AMP: Adenosine monophosphate; AMPK: AMP-dependent protein kinase.
Resistin (12.5 kDa) is a 108 amino acid protein; its circulatory form consists of a dimer united by a single disulfide bridge. Although its function in humans is still unclear, in mice it has been shown to increase blood glucose and insulin concentrations by means of promoting hepatic IR and gluconeogenesis[26]. Its exact role in IR and obesity has not yet been determined; however, studies have demonstrated that obese individuals display higher serum resistin values than lean subjects and a positive correlation may exist between BMI and resistin, although the latter remains under debate[15].
Correspondingly, retinol binding protein-4 (RBP4) (21 kDa), the transport protein for retinol in blood, has also been linked to cases of IR[27]. Although primarily hepatic in origin, high levels of RBP4 were detected in the adipose tissue of glucose transport protein 4 knockout mice[28]. The blood concentration of RBP4 was also found to be increased in obese and diabetic human individuals. Alternatively, RBP4 knockout mice displayed increased insulin sensitivity[27]. A more positive association, however, was distinguished between adipocyte inflammatory cytokine production in cases of IR and RBP4 levels[28].
ROLE OF LEPTIN, ADIPONECTIN, RESISTIN, AND RETINOL BINDING PROTEIN-4 IN NAFLD
NAFLD usually occurs concomitantly with obesity, TDM, and/or hyperlipidemia. The current explanation for the pathogenesis of NAFLD is two-fold; according to the “two-hit” theory, IR develops first. Excess free fatty acid flux occurs from adipose stores, and discrepancies in liver function lead to excessive fatty acid synthesis, insufficient fatty acid oxidation, and/or inadequate incorporation of fatty acids into very low density lipoproteins, eventually result in steatosis[12,26,29]. Secondly, oxidative stress paves the way for portal inflammation, lipid peroxidation, and ultimately fibrosis[12]. Assuming defective lipid metabolism as one of the underlying causes for intrahepatic fat deposition in NAFLD, serum adipokine levels are of certain interest. As such, leptin and adiponectin are of foremost importance in the formation of NAFLD.
Although the exact role of adipokines in the development and progression of NAFLD is unknown, Huang et al[29] proposed a positive association between blood leptin levels and incidences of NAFLD, while soluble leptin receptors were found to be significantly reduced in such cases. Other studies expounded on this claim and found that the elevated leptin levels cannot be tied definitively to IR in adults, but rather may be linked to the fibrogenesis noted in NAFLD[30]. It was found that hepatic stellate cells (HSCs) are responsible for the fibrotic changes that characterize NAFLD; HSCs produce leptin, suggesting that they may be implicated in fibrogenesis. According to Canbakan et al[30], leptin may increase the level of reactive oxygen species (ROS) in the liver, provoking Kupffer cells to produce TNF-α and other cytokines, thus enhancing collagen production and the progression of fibrosis (Figure 2). In children, hyperleptinemia was observed and was found to be correlated to the degree of liver impairment[31]. However, such studies performed in adults yielded inconclusive results, implying that hyperleptinemia may emerge in cases of early NAFLD, leading ultimately to IR and obesity. It is thought that the hyperleptin state only aggravates the inflammatory changes and fibrogenesis initiated by HCSs[31].
Figure 2.
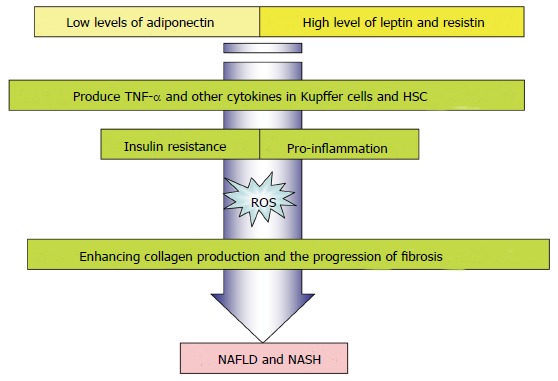
Role of adipokines in inflammation and fibrosis. NAFLD: Nonalcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis. TNF-α: Tumor necrosis factor alpha; HSCs: Hepatic stellate cells.
Adiponectin enhances glucose utilization and hepatic fatty acid oxidation via its receptors, AdipoR1 and AdipoR2. NAFLD patients displayed low adiponectin levels along with IR[12]. It was observed in mice that the hyperinsulinemic state in most liver disorders led to the downregulation of adiponectin receptors, leading to adiponectin resistance; studies have demonstrated, however, that the characteristically low adiponectin level observed in NAFLD is linked more to intrahepatic fat deposition rather than to the degree of liver damage[32]. High adiponectin levels in mice have succeeded in preventing intrahepatic fat deposition via inhibition of fatty acid synthesis. Bugianesi et al[32] identified increased serum activity of alanine transaminase (ALT) and γ-glutamyl transpeptidase (GGT) associated with hypoadiponectinemia in healthy individuals, highlighting the importance of adiponectin in preventing liver damage. Adiponectin plays a role in the inflammation observed in NAFLD; unlike leptin, however, high levels of adiponectin have been shown to inhibit the secretion of TNF-α by HSCs. One may surmise that there exists interplay between leptin-adiponectin levels and the inflammatory and fibrotic changes seen in NAFLD[12]. Adiponectin was reported to have antifibrogenic and antisteatogenic effects in the liver as opposed to leptin, which is involved in fibrogenesis. A retrospective study of serum adipokine levels in patients with NAFLD, who underwent liver biopsy due to elevated transaminase activities, found that a lower serum adiponectin/leptin ratio was useful as a non-invasive approach to discriminate NASH from simple steatosis[33]. The hepatic expression of AdipoR2 was found to be significantly higher in patients with NASH compared with controls and was related with necroinflammatory injury[33].
Adult NAFLD patients displayed increased serum resistin values. Although there is debate surrounding the exact relation between resistin and obesity and IR, recent studies seem to support the notion that increased resistin levels may be correlated with IR, BMI, and the severity of NAFLD[33]. Women were found to display a higher level of serum resistin than men, attributable to the disparity in body fat content. Murad et al[34] discovered that patients with moderate to severe steatosis displayed higher serum resistin values than those with mild steatosis. Such a finding may be related to the link between resistin and IR and later inflammation. Resistin may exacerbate the inflammation brought on by TNF-α and IL-6 secreted from HSCs[34]. Specifically, it is proposed that resistin can induce the secretion of TNF-α and IL-12 from macrophages via a nuclear factor-kappa B-dependent cascade to control the release of IL-6 and IL-1β[35-37]. There exists the likelihood that HSCs themselves may produce resistin, as they do leptin, contributing to the hyper-resistin state observed in NAFLD[34].
Serum RBP4 was found to be increased in cases with IR, including obesity, TDM, and impaired glucose tolerance, suggesting that RBP4 may play a similar role in NAFLD[38]. Earlier research indicated that RBP4 is raised in non-diabetic NAFLD patients, and selected studies claimed that there exists a positive association between RBP4 and liver enzymes, specifically ALT and GGT[38]. However, Alkhouri et al[39] reported an inverse relationship between the degree of fibrosis and RBP4 levels, such that patients with late fibrosis and cirrhosis displayed lower RBP4 values.
Additionally, there have been several studies investigating the association between polymorphisms in the genes for adipokines and the susceptibility to NAFLD within populations sharing common environmental and metabolic predisposing factors[40]. Of the many leptin polymorphisms reported, the LEPR G3057A variant was shown to be expressed by many NAFLD patients, implying that this polymorphism may be an independent risk factor for developing NAFLD in patients with TDM[41]. Adiponectin gene short nucleotide polymorphisms (SNPs) 45TG and 276GT were demonstrated in non-obese and non-diabetic NAFLD patients. Furthermore, these reported SNPs indicated the extent of liver injury in NAFLD[42]. A recent finding showed that a SNP in the Patatin-like phospholipase domain-containing 3 (PNPLA3), i.e., I148M PNPLA3 variant predicts the extent of steatosis in NAFLD[43]. The I148M PNPLA3 genotype may represent a genetic determinant of serum adiponectin levels. Therefore, in carriers of the 1148M PNPLA3 variant, modulation of serum adiponectin might be involved in mediating the susceptibility to steatosis[44].
FUNCTION OF PEROXISOME PROLIFERATOR-ACTIVATED RECEPTORS IN LIPID METABOLISM
The key element in the process of lipogenesis and lipolysis in adipose and non adipose tissues is mediated by PPARs. They are members of the steroid/retinoid nuclear receptor superfamily, and they act namely in two ways: transactivation and transrepression[45]. Three types of PPARs have been identified: alpha, gamma, and delta (beta)[46]. PPAR-α is expressed in the liver, kidney, heart, muscle, adipose tissue, and other organs. PPAR-β is expressed in many tissues, but markedly in the brain, adipose tissue, and skin[47]. Finally, PPAR-γ (γ2 and γ3) is expressed namely in adipose tissue.
PPAR-γ is activated by fatty acids and their derivatives. It plays a role in insulin sensitivity, adipogenesis, placental function, and transcription activation in concert with coactivators like steroid receptor coactivator-1. Coordination is required between PPAR-α and PPAR-γ activity for the maintenance of equilibrium between oxidation and synthesis of fatty acids. PPAR-α regulates the expression of genes involved in peroxisomal and mitochondrial beta-oxidation in liver and skeletal muscle; such genes encode for proteins such as fatty acid transport protein, fatty acid translocase, liver cytosolic fatty acid-binding protein, and uncoupling proteins-2 and 3. PPAR-γ2, on the other hand, is the central regulator of adipogenesis, favoring the deposition of excess calories in adipocytes. PPAR-γ2 is involved in growth arrest, clonal expansion, early and terminal differentiation, and anti-inflammatory effects in adipose tissue macrophages[48]. It modulates differentiation and cytokine production. Endogenous ligands of PPARs include FFA and eicosanoids[49].
Activation of PPAR-γ can promote secretion of anti-hyperglycemic adipokines like adiponectin, and shift the deposition of non-esterified fatty acids (NEFAs) to adipose tissue and away from liver and skeletal muscle, as it serves as an activator of lipoprotein lipase[50]. Moreover, adiponectin can increase PPAR-γ in adipose tissue. This process enhances its anti-inflammatory effects and thus insulin sensitivity in adipose tissue. The mechanism of activation of PPAR-γ is shown in Figure 3. It functions as a heterodimer with retinoic acid receptor (RXR), which binds direct repeat sequences AGGTCA in the promoter region of its target genes, enhancing their expression[51]. Histone deacetylase and RNA pol II activation are observed in response to PPAR-γ action in BAT and WAT[52]. Other ligands have been found to activate PPAR-γ and may be suitable as insulin-sensitizing agents to treat DM2.
Figure 3.
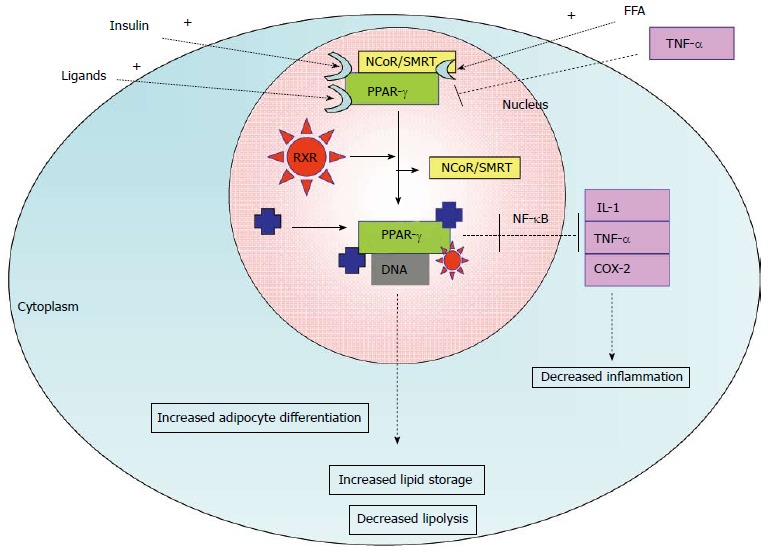
Activation of peroxisome proliferator-activated receptors α and associated biological response in adipose tissue. TNF-α: Tumor necrosis factor α; COX-2: Cyclooxygenase-2; IL-1: Interleukin-1; FFA: Free fatty acids; RXR: Retinoid receptor; SMRT: Silencing mediator of retinoid and thyroid hormone receptors; NCoR1: Nuclear receptor co-repressor 1; PPAR: Peroxisome proliferator-activated receptors.
ROLE OF PPARS IN NAFLD
Recent studies have proposed a downregulation of PPAR-α in cases of hepatic steatosis and obesity-related IR[53], thereby favoring lipogenesis over oxidation. This effect may be further aggravated by PPAR-γ upregulation, promoting overall lipogenesis. Adiponectin is responsible for the expression of PPAR-α in liver cells[54]. It has also been suggested that PPAR-α downregulation may facilitate the activity of hepatic pro-inflammatory cytokines, expediting the transition from steatosis to steatohepatitis; however, further research must be conducted to confirm such proposals[55]. Accordingly, PPAR-γ has been implicated in preventing pro-inflammatory cytokine gene expression via transrepression. As such, PPAR-γ ligands have been considered for administration in cases of inflammation, including NAFLD. In mice, PPAR-γ ligands successfully reversed the effects of cytokines produced by HSCs and managed to restore HSC quiescence. The thiazolidinedione class of antidiabetic drugs, which includes rosiglitazone and pioglitazone, acts as a PPAR-γ agonist in adipose tissue, reducing the release of NEFAs and enhancing hepatic insulin sensitivity. Although reversal of fibrosis was not observed in the 6-mo treatment with pioglitazone, improvement was noted in the 12-mo treatment of non-diabetic NASH subjects[45]. Treatment of NAFLD patients with n-3 polyunsaturated fatty acid, a known PPAR-α ligand, slightly alleviated steatosis and decreased transaminase activity[45].
Of the PPAR polymorphisms reported, a Leu162Val SNP for PPAR-α has been observed and severity of fibrosis in NAFLD[56]. Similarly, a Pro12Ala SNP has been documented for PPAR-γ. Though earlier studies have documented many NAFLD patients with the alanine variant of PPAR-γ, recent meta-analyses have failed to establish such an association between Pro12Ala SNP and NAFLD risk[50]. Further investigations are required to corroborate such findings.
ROLE OF PROINFLAMMATORY CYTOKINES IN NAFLD
Cytokines are soluble chemical mediators credited with a number of functions. They are renowned for their role in inflammatory diseases, including NAFLD. They are not normally secreted by the liver. Gradual hepatic lipid accumulation provokes HSCs and Kupffer cells to synthesize various cytokines, leading to portal inflammation, slow necrosis or apoptosis, and eventual fibrosis[57]. Of the cytokines implicated in NAFLD, TNF-α, TGF-β, and IL-6 will be discussed.
TNF-α is secreted by a number of body cells, but in particular by HSCs, Kupffer cells, and adipocytes. Selected studies have demonstrated a link between TNF-α expression and IR in steatohepatitis associated with NAFLD, indicating that adipocyte TNF-α has a key role in inflammation and IR by binding to the tyrosine kinase insulin receptor[57-59]. It has also been demonstrated that TNF-α induces swelling of hepatic mitochondria, resulting in eventual rupture and disruption of respiratory chain complexes, principally complexes I and III[60]. Reduced activity of mitochondrial complexes were reported in experimental animals followed by hepatotoxicity[61,62]. Although the results from rodent studies supported an association between enhanced TNF-α expression in inflammatory diseases, such as NAFLD and NASH, and IR, in human subjects, such an association is under debate. In both species, however, TNF-α upregulation was observed in obese subjects, and a positive association was observed between the severity of hepatic fibrosis and serum TNF-α levels. Additionally, in both mice and humans, treatment with either anti-TNF-α antibodies or a TNF-α inhibitor (pentoxifylline), respectively, served to alleviate inflammatory and fibrotic changes. In humans, the drug also succeeded in reducing serum aminotransferase activity. Excess TNF-α is also thought to enhance the expression of SREBP-1c, and promote lipogenesis[58].
Of the many isoforms of TGF-β, TGF-β1 is most predominant within the inflamed liver and has been suggested to induce the transformation of HSCs to myofibroblasts through the production of proteins like collagen 1[57]. The production of TGF-β1 leads to a cascade of irreversible events, including further synthesis of TGF-β1, connective tissue growth factors, and type I collagen; a reduction in cell turnover as a result of impeded DNA synthesis; inhibition of metalloprotease expression; and initiation of apoptosis via the phosphatidylinositol 3-kinase (PI3K)-AKT pathway and of fibrosis via increased production of fibronectin[63]. It was also suggested that elevated serum TGF-β1 may be considered an independent predictor of fibrosis[63].
IL-6 possesses a contradictory role; a study in a mouse model claimed that in addition to activating hepatocytes, stem cells, and osteoclasts, IL-6 acts as a protective shield for the liver, hindering mitochondrial dysfunction in cases of hepatic steatosis[57]. Recent reports suggested a positive association between IL-6 and IR through its inhibition of hepatic cytokine signaling[59]. Administration of anti-IL-6 antibodies in obese mice was observed to enhance insulin sensitivity, and NAFLD mice and human subjects have demonstrated elevated serum IL-6 and IL-6 receptor levels. Mahmoud et al[63] established IL-6 to be the most important biomarker for NAFLD in their study. In contrast, IL-10 has been suggested to inhibit inflammation, and prevent steatosis, while some research has also shown IL-10 serum levels to be inversely related to incidence of MS[57]. There is preliminary evidence that hepatic natural killer T-cells accumulate in liver diseases and produce IL-13 and IL-4; IL-13 may then activate HSCs to produce further pro-inflammatory cytokines, recruiting TGF-β and initiating fibrosis[58] .
Mild overload of iron is frequently observed in NAFLD. Significantly increased local as well as systemic TNF-α may favor the accumulation of iron in the liver. The progressive retention of iron can be ascribed to the impaired iron export from liver cells via cytokine-mediated downregulation of the iron exporter, ferroportin-1 (FP-1) in sinusoidal Kupffer cells. Iron accumulation may also result from an ineffective sensing of hepatic iron due to low hemojuvelin (HJV) expression. The increased hepatic and serum concentrations of TNF-α in NAFLD patients were inversely correlated with liver FP-1 and HJV mRNA levels whereas, it was positively associated with body mass index and hepatic hepcidin mRNA. Hepatic iron accumulation as well as increased level of TNF-α stimulates hepcidin formation, which decreases the duodenal and hepatic FP-1 expression and results in the blockage of duodenal iron uptake to compensate for liver iron overload. Nevertheless, decreased intestinal absorption of iron, and hepatic iron overload is found in NAFLD patients. Iron reduction therapy was found to be beneficial with regard to NAFLD disease activity and insulin sensitivity[64].
Mechanisms of NAFLD are closely linked to chronic inflammatory and oxidative stress responses. Increased intrahepatic levels of fatty acids as well as iron load provide a source of oxidative stress. Fatty liver is injured by ROS generated from microsomal, mitochondrial, and/or other hepatocellular pro-oxidant pathways when the antioxidant defenses are critically lowered. In the diminished antioxidant response, cells are susceptible to mitochondrial damage and cellular apoptotic injury. Ajith et al[65-67] reported reduced activity of antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase during acute and chronic liver injury in experimental animals. In NASH, FFAs induce lipoapoptosis in hepatocytes[68]. Cytotoxic products of lipid peroxidation (e.g., malondialdehyde, and 4-hydroxynonenal) may impair cellular functions including nucleotide and protein synthesis and may play a role in hepatic fibrogenesis. Increased ROS can release more TNF-α from hepatocytes, Kupffer cells, and adipose tissue[69], and can further upregulate pro-inflammatory pathways. Increased levels of nitric oxide and superoxide radical interact to form peroxynitrite, which is an important mediator of free radical toxicity. The role played by the toxic FFAs is depicted in Figure 4. TNF-α can induce mitochondrial ROS and thus exacerbate NAFLD by attenuating the anti-inflammatory effects of adiponectin and PPAR-γ[70], and results in secondary inflammation and fibrosis in NAFLD. Animal models of NAFLD suggest that an increased translocation of bacterial endotoxins lead to an activation of toll-like receptor-dependent signaling cascades and increased formation of ROS[71].
Figure 4.
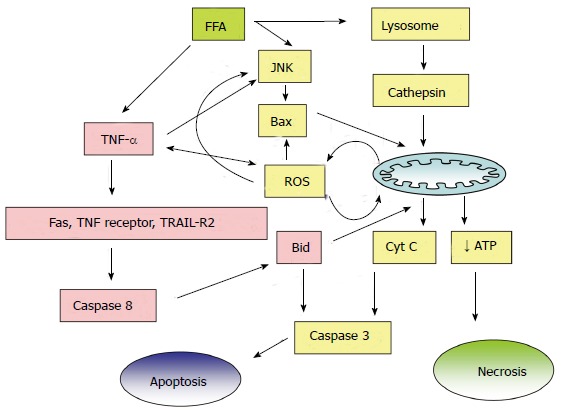
Cytotoxic effects of free fatty acids in nonalcoholic fatty liver disease. FFA: Free fatty acid; JNK: c-Jun NH(2)-terminal kinase; ROS: Reactive oxygen species; TRAIL-R2: Tumor necrosis factor-related apoptosis inducing ligand; ATP: Adenosine triphosphate.
CONCLUSION
NAFLD is one of the principal causes of chronic liver disease. Cytokines secreted by the adipocytes, such as TNF-α, TGF-β, and IL-6, are implicated in NAFLD. TNF-α, IL-6 and leptin have been shown to exert pro-inflammatory effects and adiponectin has been shown to exert anti-inflammatory effects at the liver level. Furthermore, preliminary evidence suggests that hepatic natural killer T-cells accumulate in liver diseases and produce IL-13 and IL-4; IL-13 may then activate HSCs to produce further pro-inflammatory cytokines, increase TGF-β and initiate fibrosis. Downregulation of PPAR-α in cases of hepatic steatosis favors lipogenesis over oxidation (Figure 5). PPAR-α downregulation may facilitate the activity of hepatic proinflammatory cytokines, expediting the transition from steatosis to steatohepatitis; however, further research must be conducted to confirm such proposals. PPAR-γ ligands have been considered for administration in cases of inflammation, including NAFLD; PPAR-γ ligands successfully reversed the effects of cytokines produced by HSCs and managed to restore HSC quiescence. Treatment of NAFLD patients with n-3 polyunsaturated fatty acid, a known PPAR-α ligand, slightly alleviated steatosis and decreased transaminase activity. Hence, ligands that activate these receptors may prevent the initial inflammatory reactions and render protection to the liver cells. Mechanisms of NAFLD are closely linked to chronic inflammatory and the oxidative stress response. However, there are insufficient data to support the use of antioxidant supplements for patients with NAFLD. Iron reduction therapy and lipid lowering drugs were found to be beneficial in NAFLD. Future research in these fields is required to design specific compounds to prevent fat deposition in liver cells.
Figure 5.
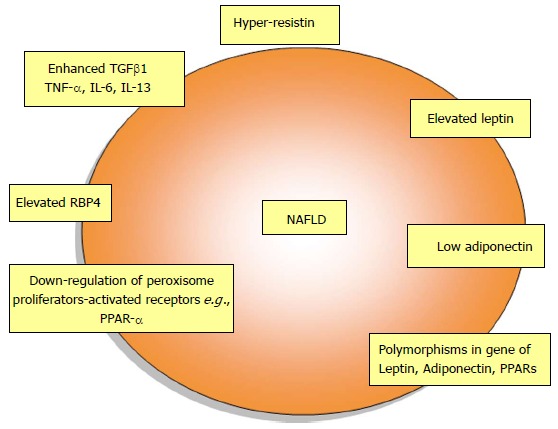
Changes of adipokines and peroxisome proliferator-activated receptors in nonalcoholic fatty liver disease. TGFβ1: Transforming growth factor beta 1; TNF-α: Tumor necrosis factor alpha; RBP4: Retinol binding protein 4. NAFLD: Nonalcoholic fatty liver disease; PPAR: Peroxisome proliferator-activated receptors.
Footnotes
P- Reviewer: Mascitelli L, Milic S, Navarro-Jarabo JM, Yoshiji H S- Editor: Wen LL L- Editor: Cant MR E- Editor: Wu HL
References
- 1.Méndez-Sánchez N, Arrese M, Zamora-Valdés D, Uribe M. Current concepts in the pathogenesis of nonalcoholic fatty liver disease. Liver Int. 2007;27:423–433. doi: 10.1111/j.1478-3231.2007.01483.x. [DOI] [PubMed] [Google Scholar]
- 2.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:5286–5296. doi: 10.3748/wjg.v16.i42.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 5.Friedman JM. Obesity in the new millennium. Nature. 2000;404:632–634. doi: 10.1038/35007504. [DOI] [PubMed] [Google Scholar]
- 6.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 7.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 8.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 9.Vázquez-Vela ME, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Arch Med Res. 2008;39:715–728. doi: 10.1016/j.arcmed.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Cannon B, Nedergaard J. Developmental biology: Neither fat nor flesh. Nature. 2008;454:947–948. doi: 10.1038/454947a. [DOI] [PubMed] [Google Scholar]
- 12.Mirza MS. Obesity, Visceral Fat, and NAFLD: Querying the Role of Adipokines in the Progression of Nonalcoholic Fatty Liver Disease. ISRN Gastroenterol. 2011;2011:592404. doi: 10.5402/2011/592404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahima RS, Qi Y, Singhal NS. Adipokines that link obesity and diabetes to the hypothalamus. Prog Brain Res. 2006;153:155–174. doi: 10.1016/S0079-6123(06)53009-2. [DOI] [PubMed] [Google Scholar]
- 14.Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology. 2007;132:2103–2115. doi: 10.1053/j.gastro.2007.03.058. [DOI] [PubMed] [Google Scholar]
- 15.Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem. 2004;50:1511–1525. doi: 10.1373/clinchem.2004.032482. [DOI] [PubMed] [Google Scholar]
- 16.Denver RJ, Bonett RM, Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. 2011;94:21–38. doi: 10.1159/000328435. [DOI] [PubMed] [Google Scholar]
- 17.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101:545–559. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 18.Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci (Landmark Ed) 2011;16:2771–2793. doi: 10.2741/3885. [DOI] [PubMed] [Google Scholar]
- 19.Marroquí L, Vieira E, Gonzalez A, Nadal A, Quesada I. Leptin downregulates expression of the gene encoding glucagon in alphaTC1-9 cells and mouse islets. Diabetologia. 2011;54:843–851. doi: 10.1007/s00125-010-2024-1. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes T, Ara I, Guadalupe-Grau A, Larsen S, Stallknecht B, Olmedillas H, Santana A, Helge JW, Calbet JA, Guerra B. Leptin receptor 170 kDa (OB-R170) protein expression is reduced in obese human skeletal muscle: a potential mechanism of leptin resistance. Exp Physiol. 2010;95:160–171. doi: 10.1113/expphysiol.2009.049270. [DOI] [PubMed] [Google Scholar]
- 21.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 22.Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:2562–2570. doi: 10.2337/db05-1322. [DOI] [PubMed] [Google Scholar]
- 23.Frqhbeck G, Salvador J. Role of adipocytokines in metabolism and disease. Nutrition Res. 2004;24:803–826. [Google Scholar]
- 24.Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010;2010:802078. doi: 10.1155/2010/802078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daval M, Foufelle F, Ferré P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol. 2006;574:55–62. doi: 10.1113/jphysiol.2006.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pagano C, Soardo G, Pilon C, Milocco C, Basan L, Milan G, Donnini D, Faggian D, Mussap M, Plebani M, et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J Clin Endocrinol Metab. 2006;91:1081–1086. doi: 10.1210/jc.2005-1056. [DOI] [PubMed] [Google Scholar]
- 27.Klöting N, Graham TE, Berndt J, Kralisch S, Kovacs P, Wason CJ, Fasshauer M, Schön MR, Stumvoll M, Blüher M, et al. Serum retinol-binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metab. 2007;6:79–87. doi: 10.1016/j.cmet.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Yao-Borengasser A, Varma V, Bodles AM, Rasouli N, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ, Rashidi AA, et al. Retinol binding protein 4 expression in humans: relationship to insulin resistance, inflammation, and response to pioglitazone. J Clin Endocrinol Metab. 2007;92:2590–2597. doi: 10.1210/jc.2006-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang XD, Fan Y, Zhang H, Wang P, Yuan JP, Li MJ, Zhan XY. Serum leptin and soluble leptin receptor in non-alcoholic fatty liver disease. World J Gastroenterol. 2008;14:2888–2893. doi: 10.3748/wjg.14.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canbakan B, Tahan V, Balci H, Hatemi I, Erer B, Ozbay G, Sut N, Hacibekiroglu M, Imeryuz N, Senturk H. Leptin in nonalcoholic fatty liver disease. Ann Hepatol. 2008;7:249–254. [PubMed] [Google Scholar]
- 31.Nobili V, Manco M, Ciampalini P, Diciommo V, Devito R, Piemonte F, Comparcola D, Guidi R, Marcellini M. Leptin, free leptin index, insulin resistance and liver fibrosis in children with non-alcoholic fatty liver disease. Eur J Endocrinol. 2006;155:735–743. doi: 10.1530/eje.1.02288. [DOI] [PubMed] [Google Scholar]
- 32.Bugianesi E, Pagotto U, Manini R, Vanni E, Gastaldelli A, de Iasio R, Gentilcore E, Natale S, Cassader M, Rizzetto M, et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J Clin Endocrinol Metab. 2005;90:3498–3504. doi: 10.1210/jc.2004-2240. [DOI] [PubMed] [Google Scholar]
- 33.Lemoine M, Ratziu V, Kim M, Maachi M, Wendum D, Paye F, Bastard JP, Poupon R, Housset C, Capeau J, et al. Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 2009;29:1431–1438. doi: 10.1111/j.1478-3231.2009.02022.x. [DOI] [PubMed] [Google Scholar]
- 34.Murad A, Hassan H, Husein H, Ayad A. Serum resistin levels in nonalcoholic fatty liver disease and their relationship to severity of liver disease. JEMDSA. 2010;15:53–56. [Google Scholar]
- 35.Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174:5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 36.Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun. 2005;334:1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- 37.Zhang LY, Jin YJ, Jin QS, Lin LY, Zhang DD, Kong LL. Association between resistin +299A/A genotype and nonalcoholic fatty liver disease in Chinese patients with type 2 diabetes mellitus. Gene. 2013;529:340–344. doi: 10.1016/j.gene.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Seo JA, Kim NH, Park SY, Kim HY, Ryu OH, Lee KW, Lee J, Kim DL, Choi KM, Baik SH, et al. Serum retinol-binding protein 4 levels are elevated in non-alcoholic fatty liver disease. Clin Endocrinol (Oxf) 2008;68:555–560. doi: 10.1111/j.1365-2265.2007.03072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alkhouri N, Lopez R, Berk M, Feldstein AE. Serum retinol-binding protein 4 levels in patients with nonalcoholic fatty liver disease. J Clin Gastroenterol. 2009;43:985–989. doi: 10.1097/MCG.0b013e3181a0998d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duvnjak M, Barsić N, Tomasić V, Lerotić I. Genetic polymorphisms in non-alcoholic fatty liver disease: clues to pathogenesis and disease progression. World J Gastroenterol. 2009;15:6023–6027. doi: 10.3748/wjg.15.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu H, Sun J, Sun L, Shu X, Xu Y, Xie D. Polymorphism of human leptin receptor gene is associated with type 2 diabetic patients complicated with non-alcoholic fatty liver disease in China. J Gastroenterol Hepatol. 2009;24:228–232. doi: 10.1111/j.1440-1746.2008.05544.x. [DOI] [PubMed] [Google Scholar]
- 42.Musso G, Gambino R, De Michieli F, Durazzo M, Pagano G, Cassader M. Adiponectin gene polymorphisms modulate acute adiponectin response to dietary fat: Possible pathogenetic role in NASH. Hepatology. 2008;47:1167–1177. doi: 10.1002/hep.22142. [DOI] [PubMed] [Google Scholar]
- 43.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valenti L, Rametta R, Ruscica M, Dongiovanni P, Steffani L, Motta BM, Canavesi E, Fracanzani AL, Mozzi E, Roviaro G, et al. The I148M PNPLA3 polymorphism influences serum adiponectin in patients with fatty liver and healthy controls. BMC Gastroenterol. 2012;12:111. doi: 10.1186/1471-230X-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Järvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 46.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, Rodan GA. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol Endocrinol. 1992;6:1634–1641. doi: 10.1210/mend.6.10.1333051. [DOI] [PubMed] [Google Scholar]
- 48.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 49.Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, Wahli W, Noy N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol Cell Biol. 2002;22:5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Guo X, Wu P, Song J, Ye C, Yu S, Zhang J, Dong W. Association between the Pro12Ala polymorphism of PPAR-γ gene and the non-alcoholic fatty liver disease: a meta-analysis. Gene. 2013;528:328–334. doi: 10.1016/j.gene.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 51.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2007;1771:936–951. doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Powell E, Kuhn P, Xu W. Nuclear Receptor Cofactors in PPARgamma-Mediated Adipogenesis and Adipocyte Energy Metabolism. PPAR Res. 2007;2007:53843. doi: 10.1155/2007/53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 55.Videla LA, Pettinelli P. Misregulation of PPAR Functioning and Its Pathogenic Consequences Associated with Nonalcoholic Fatty Liver Disease in Human Obesity. PPAR Res. 2012;2012:107434. doi: 10.1155/2012/107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Domenici FA, Brochado MJ, Martinelli Ade L, Zucoloto S, da Cunha SF, Vannucchi H. Peroxisome proliferator-activated receptors alpha and gamma2 polymorphisms in nonalcoholic fatty liver disease: a study in Brazilian patients. Gene. 2013;529:326–331. doi: 10.1016/j.gene.2013.06.091. [DOI] [PubMed] [Google Scholar]
- 57.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:727–735. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearce SG, Thosani NC, Pan JJ. Noninvasive biomarkers for the diagnosis of steatohepatitis and advanced fibrosis in NAFLD. Biomark Res. 2013;1:7. doi: 10.1186/2050-7771-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sudheesh NP, Ajith TA, Mathew J, Nima N, Janardhanan KK. Ganoderma lucidum protects liver mitochondrial oxidative stress and improves the activity of electron transport chain in carbon tetrachloride intoxicated rats. Hepatol Res. 2012;42:181–191. doi: 10.1111/j.1872-034X.2011.00906.x. [DOI] [PubMed] [Google Scholar]
- 62.Sudheesh NP, Ajith TA, Janardhanan KK. Hepatoprotective effects of DL-α-lipoic acid and α-Tocopherol through amelioration of the mitochondrial oxidative stress in acetaminophen challenged rats. Toxicol Mech Methods. 2013;23:368–376. doi: 10.3109/15376516.2013.769289. [DOI] [PubMed] [Google Scholar]
- 63.Mahmoud AA, Bakir AS, Shabana SS. Serum TGF-β, Serum MMP-1, and HOMA-IR as non-invasive predictors of fibrosis in Egyptian patients with NAFLD. Saudi J Gastroenterol. 2012;18:327–333. doi: 10.4103/1319-3767.101132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valenti L, Fracanzani AL, Dongiovanni P, Bugianesi E, Marchesini G, Manzini P, Vanni E, Fargion S. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case-control study. Am J Gastroenterol. 2007;102:1251–1258. doi: 10.1111/j.1572-0241.2007.01192.x. [DOI] [PubMed] [Google Scholar]
- 65.Ajith TA, Hema U, Aswathy MS. Zingiber officinale Roscoe prevents acetaminophen-induced acute hepatotoxicity by enhancing hepatic antioxidant status. Food Chem Toxicol. 2007;45:2267–2272. doi: 10.1016/j.fct.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 66.Ajith TA, Janardhanan KK. Chemopreventive activity of a macrofungus Phellinus rimosus against N-nitrosodiethylamine induced hepatocellular carcinoma in rat. J Exp Ther Oncol. 2006;5:309–321. [PubMed] [Google Scholar]
- 67.Ajith TA, Sheena N, Janardhanan KK. Phellinus rimosus protects carbon tetrachloride induced chronic hepatotoxicity in rat: Antioxidant defense mechanism. Pharm Biol. 2006;44:1–8. [Google Scholar]
- 68.Wree A, Kahraman A, Gerken G, Canbay A. Obesity affects the liver - the link between adipocytes and hepatocytes. Digestion. 2011;83:124–133. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 69.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95:2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moschen AR, Wieser V, Tilg H. Adiponectin: key player in the adipose tissue-liver crosstalk. Curr Med Chem. 2012;19:5467–5473. doi: 10.2174/092986712803833254. [DOI] [PubMed] [Google Scholar]
- 71.Kanuri G, Ladurner R, Skibovskaya J, Spruss A, Königsrainer A, Bischoff SC, Bergheim I. Expression of toll-like receptors 1-5 but not TLR 6-10 is elevated in livers of patients with non-alcoholic fatty liver disease. Liver Int. 2013:Dec 18; Epub ahead of print. doi: 10.1111/liv.12442. [DOI] [PubMed] [Google Scholar]