

Abstract
Human AKR1D1 (steroid 5β-reductase/aldo-keto reductase 1D1) catalyses the stereospecific reduction of double bonds in Δ4-3-oxosteroids, a unique reaction that introduces a 90° bend at the A/B ring fusion to yield 5β-dihydrosteroids. AKR1D1 is the only enzyme capable of steroid 5β-reduction in humans and plays critical physiological roles. In steroid hormone metabolism, AKR1D1 serves mainly to inactivate the major classes of steroid hormones. AKR1D1 also catalyses key steps of the biosynthetic pathway of bile acids, which regulate lipid emulsification and cholesterol homoeostasis. Interestingly, AKR1D1 displayed a 20-fold variation in the kcat values, with steroid hormone substrates (e.g. aldosterone, testosterone and cortisone) having significantly higher kcat values than steroids with longer side chains (e.g. 7α-hydroxycholestenone, a bile acid precursor). Transient kinetic analysis revealed striking variations up to two orders of magnitude in the rate of the chemistry step (kchem), which resulted in different rate determining steps for the fast and slow substrates. By contrast, similar Kd values were observed for representative fast and slow substrates, suggesting similar rates of release for different steroid products. The release of NADP+ was shown to control the overall turnover for fast substrates, but not for slow substrates. Despite having high kchem values with steroid hormones, the kinetic control of AKR1D1 is consistent with the enzyme catalysing the slowest step in the catabolic sequence of steroid hormone transformation in the liver. The inherent slowness of the conversion of the bile acid precursor by AKR1D1 is also indicative of a regulatory role in bile acid synthesis.
Keywords: bile acid deficiency, cholesterol metabolism, rate determining step, stopped-flow
INTRODUCTION
Human steroid 5β-reductase, which corresponds to AKR (aldo-keto reductase 1D1), catalyses the stereospecific reduction of double bonds in Δ4-3-ketosteroids to yield 5β-DHS (dihydrosteroids) [1,2]. The reaction is unique in that it introduces a 90° bend at the A/B ring fusion of the steroid. As a result, 5β-reduced steroids possess different properties from either the α,β-unsaturated or 5α-reduced steroids, which have a largely planar four-ring steroidal structure. With this activity, AKR1D1 is recognized for its important roles in steroid metabolism and bile acid biosynthesis.
AKR1D1 is most abundantly expressed in the liver, the primary site of steroid metabolism and bile acid synthesis. In the liver, all major classes of steroid hormones except oestrogens are metabolized to THS (tetrahydrosteroids) through consecutive Aring reduction steps [3,4]. THS are subsequently conjugated in phase 2 reactions leading to elimination. AKR1D1 is responsible for one of the two metabolic pathways of A-ring reduction (Figure 1A), and for some steroids such as cortisone the 5β-pathway is the predominant one. In addition to hepatic steroid catabolism, 5β-reduction generates an array of functionally diverse active 5β-reduced steroids that are neuroactive [5] and/or involved in erythropoiesis [6] and parturition [7].
Figure 1. Biological role of AKR1D1 (A and B) and chemical structures of the steroid substrates tested.
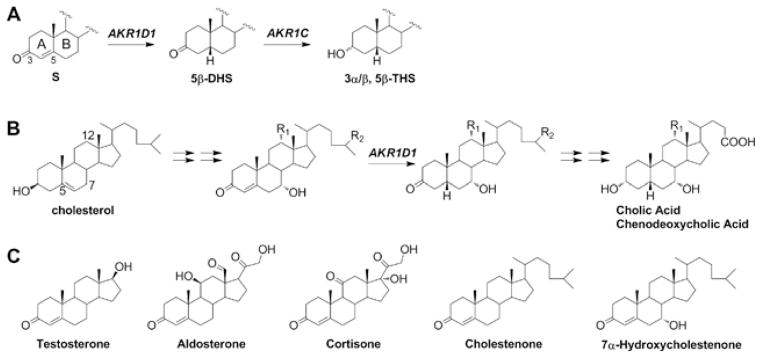
(A) Steroid hormones are metabolized by the concerted actions of AKR1D1 and AKR1C enzymes. S, steroid hormone. (B) AKR1D1 is responsible for the 5β-reduction in all pathways for the synthesis of primary bile acids cholic acid and chenodeoxycholic acid. R1, -H or -OH; R2, -CH3 or -COOH. (C) All substrates are similar in chemical structure and have an identical A-ring, where the reaction occurs.
The more appreciated biological role of AKR1D1 is in bile acid biosynthesis, where it catalyses the key step that introduces the 5β-configuration in primary bile acids [8,9] (Figure 1B). The 5β-configuration of bile acids gives these molecules detergent-like properties for proper emulsification of dietary cholesterol, fats and lipophilic vitamins. The conversion of cholesterol into bile acids also represents the main pathway of cholesterol degradation in the liver. In addition, bile acids are recognized as metabolically active signalling molecules regulating many crucial physiological events such as their own synthesis and lipid and glucose homoeostasis [10–13]. The role of AKR1D1 is underscored by the discovery of a growing number of genetic variants of AKR1D1 associated with bile acid deficiency, which is a fatal condition for neonates without bile-acid supplementation [14–18].
Previously, we have examined the specificity of AKR1D1 towards 11 steroid substrates of the C18, C19, C21 and C27 series, confirming the ability of AKR1D1 to mediate steroid metabolism and bile acid synthesis [19]. Considerable differences in steady-state kinetic parameters were observed despite the substrates having a similar chemical structure [19]. Specifically, the kcat values for the different steroids varied by 20-fold, and substrates with shorter side chains (e.g. steroid hormones) were generally turned over faster than substrates with longer side chains (e.g. bile acid precursors). The kinetic basis of this difference is unknown. AKR1D1 shares high sequence homology with other steroid transforming members of the AKR family. Crystal structures of AKR1D1 show that the enzyme harbours the (α/β)8-barrel core structure typical to the superfamily with the cofactor and the steroid substrate at the C-terminal end of the β-sheets [20] and the conserved catalytic tetrad (Tyr58, Lys87, Glu120 and Asp53) in the active site [21–23]. Like other members of the AKR family, the kinetics of AKR1D1 is believed to follow a sequential ordered bi-bi mechanism [24]. The macroscopic events in the minimal mechanism include: (i) binding of NADPH; (ii) binding of steroid substrate (SS); (iii) hydride transfer (rate constant defined as kchem); (iv) release of steroid product SP (rate constant defined as kr,Sp); and (v) release of NADP+ (rate constant defined as kr,NADP+) (Figure 2). The rate equations can be derived using the King–Altman method [25], where it is assumed that the conversion from enzyme–NADPH–SS into enzyme–NADP+–Sp is irreversible. Thus the apparent kcat of AKR1D1 is only related to the individual steps defined by eqn (1) [26,27]:
| (1) |
Figure 2. Macroscopic kinetic events in AKR1D1 catalysis.
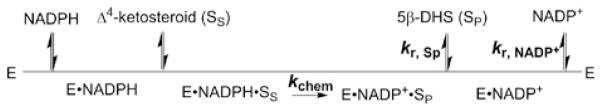
The turnover number (kcat) is defined by the rate constants of three steps: the chemistry step (kchem), the release of steroid product (kr,Sp) and the release of NADP+ (k r,NADP +). E, enzyme; SS, steroid substrate; SP, steroid product.
Different macroscopic events have been shown to be responsible for the determination of the overall rate of AKR catalysis [28–31]. However, the rate determining step for AKR1D1 catalysis is not known. In the present study, we have carried out detailed kinetic characterizations of AKR1D1 catalysis. We show that AKR1D1 exhibits differential kinetic behaviours in the reaction it catalyses, which may have important consequences for steroid hormone metabolism and bile acid synthesis.
EXPERIMENTAL
Materials
All steroids were obtained from Steraloids. Pyridine nucleotides were purchased from Roche Applied Science. All other reagents were purchased from Sigma–Aldrich and were of ACS (American Chemical Society) grade or better. Recombinant AKR1D1 was overexpressed and purified to homogeneity as described previously [22,32,33]. Under standard assay conditions, the specific activity of AKR1D1 was 80 nmol/min per mg for the NADPH-dependent reduction of 10 μM testosterone.
Unless otherwise noted, all experiments were carried out at 37°C in 100 mM potassium phosphate buffer (pH 6.0), containing 4 % acetonitrile as a co-solvent. All steroid stock solutions were prepared in acetonitrile except for finasteride, which was dissolved in DMSO.
Transient kinetics of substrate turnover
All stopped-flow experiments were performed on an Applied Photophysics SX.18 and SX.19 MV stopped-flow spectrophotometers. Data were collected and analysed using the Applied Photophysics software.
For stopped-flow turnover experiments, changes in the absorbance signal of NADPH at 340 nm or the fluorescence signal of NADPH (excitation at 340 nm, emission at 450 nm) were monitored upon rapid mixing of equal volumes of the premixed enzyme–cofactor solution from one syringe and the steroid solution from a second syringe.
For stopped-flow multiple turnover experiments, the enzyme–cofactor solution contained a fixed concentration of AKR1D1 and excess NADPH, and was mixed with excess steroid substrate. A typical reaction contained 2.5 μM AKR1D1, 18 μM NADPH and 25 μM steroid. For each steroid substrate, the averaged reaction traces from at least three replicates were fitted to either the ‘burst’ (eqn 2) or the linear equations (eqn 3) [34], where, kburst is the rate constant for the exponential burst phase, kss is the rate constant for the linear steady-state phase, y is the fluorescence signal, A is the amplitude, [E] is the enzyme concentration and t is time:
| (2) |
| (3) |
For cortisone and 7α-hydroxy-4-cholesten-3-one (7α-hydroxycholestenone), experiments were carried out over a complete range of steroid concentrations. Secondary plots of kburst and kss against [S] displayed saturation kinetics and were fitted to the hyperbolic equation (eqns 4a and 4b), where klimB is the limiting rate constant for the burst phase, klimL is the limiting rate constant for the steady-state linear phase, K1/2 is the apparent half-saturation constant and [S] is the concentration of the added steroid:
| (4a) |
| (4b) |
For the single-turnover experiments, the enzyme–cofactor solution contained a fixed concentration of AKR1D1 and a stoichiometric concentration of NADPH, and was mixed with steroid solution. A typical reaction contained 1.5 μM AKR1D1, 1.5 μM NADPH and 25 μM steroid. For each substrate, the averaged reaction traces from at least three replicates were fitted to single- or double-exponential equations (eqns 5a and 5b), where ksto is the single-turnover rate constant:
| (5a) |
| (5b) |
Dissociation constant for steroids
The dissociation constant (Kd) for the AKR1D1–NADPH–steroid ternary complex was determined by fluorescence titration using a Hitachi F-4500 fluorescence spectrophotometer (excitation at 295 nm and emission at 460 nm). Samples were maintained at 4 °C for the substrates (to prevent turnover) and at 4 °C or 37 °C for inhibitor titrations (to evaluate the effect of temperature). Each sample contained 0.14 μM AKR1D1 and 8 μM NADPH, to which small volumes of cortisone (final concentrations 0.3 μM– 32μM), 7α-hydroxycholestenone (final concentrations 1 μM–40 μM) or finasteride (final concentrations 1.5 μM–72 μM) were added incrementally. The total volume change from the addition of the steroid was less than 2 %. The quenching of the energy transfer fluorescence (peaked at 460 nm) of the AKR1D1–NADPH binary complex upon the addition of the steroid was monitored. Data were plotted as a percentage change in fluorescence at 460 nm against the steroid concentration and fitted to the Morrison equation (eqns 6a and 6b) using the program GraFit (Erithacus Software) to determine the dissociation constant, where ΔF is the difference in fluorescence emission in the absence and presence of steroid, ΔFmax is the maximum value of ΔF at saturating steroid concentration, [ECS] is the concentration of the enzyme–cofactor–steroid ternary complex, [EC] is the total concentration of the enzyme–cofactor binary complex and Kd is the apparent dissociation constant of the steroid:
| (6a) |
| (6b) |
Molecular docking of steroids to AKR1D1
In silico docking was performed using Autodock Vina following the protocol published in [35]. The crystal structure of AKR1D1 in complex with NADP+ and cortisone (PDB code 3CMF) minus solvent and steroid was used as the receptor. A docking box was defined to include the entire steroid-binding channel. Cortisone, testosterone and 7α-hydroxycholestenone were allowed to move freely inside the docking box. Tyr132 and Trp230, which are known to be involved in ligand binding in the crystal structures of AKR1D1, were allowed to have flexible side chains. The docking results were validated by demonstrating that the calculated lowest-energy conformation of cortisone adopted an almost identical conformation with that found in the crystal structure.
Transient kinetics of NADP+ release
The dissociation rate constant of NADP+ from AKR1D1 was measured by a competition method [36]. A pre-mixed enzyme–NADP+ solution (1 μM AKR1D1 and 5 μM NADP+) was mixed with a large excess of NADPH (50 μM) in the stopped-flow instrument. The formation of the AKR1D1–NADPH complex was monitored by the increase in the energy transfer band (fluorescence signal at 450 nm with 295 nm excitation). In the control experiments, the enzyme solution or the NADP+ solution was mixed with the NADPH solution in the stopped-flow instrument. For each condition, data from at least five replicates were averaged and fitted to either single- (eqn 7a) or double- (eqn 7b) exponential equations, where a is the intercept and kobs, kobs1 and kobs2 are the apparent rate constants:
| (7a) |
| (7b) |
RESULTS
The previous study in which steady-state kinetic parameters were determined for 11 different substrates of AKR1D1 showed that the enzyme displayed a 20-fold range in its turnover number [19]. In the present study, we have investigated the detailed kinetic basis underlying the difference in steady-state kinetic parameters of five substrates (Figure 1C). Testosterone is a C19 steroid and active androgen, and has been used as substrate in standard assays of AKR1D1; aldosterone is a C21 steroid and active mineralocorticoid; cortisone is a C21 glucocorticoid and displayed the highest kcat; cholestenone is a C27 steroid and displayed the lowest kcat; and 7α-hydroxycholestenone is a C27 sterol and a precursor of bile acids. The discrete steps in the reaction of these substrates including the chemistry step and the product release steps were examined.
Transient multiple-turnover reactions
In stopped-flow multiple-turnover experiments, AKR1D1 was mixed with excess NADPH and was allowed to react with excess fast and slow steroid substrates. Since both pre-steady-state and steady-state turnover can be monitored under these conditions, these experiments provide clues on the relative contributions of product formation and release steps to kcat, the turnover number. Interestingly, two types of reaction time course for the depletion of NADPH catalysed by AKR1D1 were observed (Figure 3).
Figure 3. Representative kinetic traces for the multiple-turnover reactions catalysed by AKR1D1 in the transient state.

(A) Averaged progress curves of decreases in NADPH fluorescence for samples containing testosterone (red), cortisone (blue) and aldosterone (green). (B) Averaged progress curves of decreases in NADPH fluorescence for samples containing no steroid (black), cholestenone (magenta) or 7α-hydroxycholestenone (cyan). (C) Fitting of an averaged trace of cortisone multiple-turnover reaction to the burst equation. (D) Fitting of an averaged trace of 7α-hydroxycholestenone multiple-turnover reaction to the linear equation. The initial increase in fluorescence signal in the reaction of 7α-hydroxycholestenone was also observed in the sample containing no steroid and was not used in data fitting. Fitted lines are in grey and are mostly superimposed by the actual data points. Residual plots demonstrate the quality of the fit.
With substrates testosterone, aldosterone and cortisone, the fluorescence and absorption time courses of the NADPH depletion were curvilinear and were best fitted to the ‘burst’ equation (eqn 2), which is the sum of an exponential term and a linear term. Fitting data to a linear regression or single- and double-exponential functions were also attempted, but proved to be unsuitable based on the size of the residuals. Thus, in the transient state, multiple turnovers of these three steroid substrates catalysed by AKR1D1 occurred with an initial burst (exponential phase) corresponding to the first turnovers during which NADPH decayed more rapidly, followed by a slower rate of substrate consumption (linear phase) corresponding to steady-state turnovers. Data analysis revealed that at saturating steroid concentrations the initial burst (kburst) occurred at 0.71–1.98 s−1 with these substrates, whereas the steady-state turnovers (kss) occurred at 0.050–0.10 s−1 (Table 1). With aldosterone, the amplitude of the burst approached the concentration of the enzyme used in the experiment. Partial bursts were observed with testosterone and cortisone, possibly due to the fact that at the concentration used (25 μM) significant substrate inhibition occurs with both substrates in steady-state turnover.
Table 1.
Rate constants determined from transient multiple-turnover experiments with AKR1D1
| Parameters | Testosterone | Aldosterone | Cortisone | Cholestenone | 7α-Hydroxycholestenone |
|---|---|---|---|---|---|
| Aburst (%) | 45 ± 1 | 89 ± 1 | 53 ± 1 | No burst | No burst |
| kburst (s−1) | 0.71 ± 0.03 | 1.98 ± 0.05 | 1.39 ± 0.04 | ||
| kss (s−1) | 0.050 ± 0.002 | 0.094 ± 0.002 | 0.104 ± 0.002 | 0.0060 ± 0.0002 | 0.0160 ± 0.0002 |
| kcat (s−1) | 0.14 ± 0.03 | 0.15 ± 0.01 | 0.195 ± 0.002 | 0.0100 ± 0.0007 | 0.033 ± 0.002 |
kcat was determined by steady-state analysis [19]. Values are means± S.E.M.
The observed ‘burst’ indicated the presence of one or more slower product release steps following product formation (i.e. the chemistry step) with these steroids. The substrates that displayed burst kinetics were regarded as ‘fast’ substrates. Using cortisone as a representative fast substrate, the multiple-turnover reactions were further examined over a range of steroid concentrations in the fluorescence mode. Data yielded the limiting rate constants for the burst phase (klimB) and the linear phase (klimL), which were 1.73 s−1 and 0.16 s−1 respectively. The value of klimL was in agreement with the kcat of 0.19 s−1 determined by the steady-state turnover experiments.
With substrates cholestenone and 7α-hydroxycholestenone, the time courses of the NADPH depletion were linear (Figure 3). The slowness of the reaction only allowed detection in the fluorescence mode. Data analysis by fitting to the linear equation (eqn 3) yielded values of kss of 0.006 s−1 and 0.016 s−1 for cholestenone and 7α-hydroxycholestenone respectively (Table 1). Using 7α-hydroxycholestenone as a representative slow substrate, multiple-turnover reactions were further examined over a range of concentrations. This reaction required higher concentrations of enzyme, which in turn necessitated the use of higher concentrations of steroid to satisfy pseudo first-order conditions (i.e. [Sp] ≥ 8[E]). As a result, turnover of 7α-hydroxycholestenone was examined over a narrow range of concentrations. The maximum rate of kss (klimL) was determined to be 0.020 s−1. These values were in agreement with their kcat values determined in the steady-state experiments. The lack of burst kinetics under multiple-turnover conditions suggests that for these substrates the chemistry step was the major contributor to rate determination and that the chemistry step occurs at a much slower rate in comparison with the fast substrates, which display burst kinetics.
The switch from burst kinetics to linear kinetics appeared to be due to the significant difference in the rate of chemistry step (kchem), as kchem is represented by kburst for the fast substrate or kss for the slow substrate. Thus multiple-turnover experiments revealed a 330-fold difference in kchem from aldosterone to cholesteneone. Transient single-turnover experiments further confirmed the difference in kchem between representative substrates cortisone and 7α-hydroxycholestenone.
Transient single-turnover reactions
To monitor the reduction in steroids under single-turnover conditions, the enzyme was pre-mixed with a stoichiometric amount of NADPH so that the enzyme could not recycle. This pre-mixed enzyme–NADPH solution was rapidly mixed with the steroid substrate in the stopped-flow and changes in NADPH fluorescence signals were monitored upon mixing. In these experiments, the process of product formation is monitored directly and the rate of the chemistry step can be estimated. Representative stopped-flow traces of the single-turnover reactions of cortisone and 7α-hydroxycholestenone catalysed by AKR1D1 are shown in Figure 4. The progress curves of the cortisone reaction were best fitted to a double-exponential function. At saturating cortisone concentration, half of the single turnover occurred at 1.69 s−1 and half occurred at 0.21 s−1. Two populations of enzyme activity under single-turnover conditions were also observed with other fast substrates, such as aldosterone and testosterone (results not show). In each case, the faster ksto corresponded to the kburst observed in multiple-turnover reactions. The origin of the two kinetically distinct enzyme populations was not due to enzyme stability or cofactor status; AKR1D1 is a monomer. The molecular basis for the two populations of enzyme activity remains unknown. In contrast, the progress curves of the 7α-hydroxycholestenone reaction were best fitted to a single-exponential function. Comparison of the values of ksto for cortisone and 7α-hydroxycholestenone confirmed the strikingly large difference in kchem values of the two substrates, which is up to 84-fold.
Figure 4. Representative kinetic traces for the single-turnover reactions catalysed by AKR1D1.
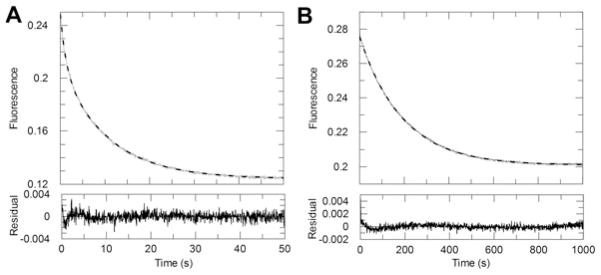
(A) The averaged progress curves for the reaction of the pre-mixed enzyme–NADPH solution with cortisone. Data were fitted to a double-exponential function. (B) The averaged progress curves for the reaction of the pre-mixed enzyme–NADPH solution with 7α-hydroxycholestenone. Data were fitted to a single-exponential function. Fitted lines are shown in grey. Residual plots demonstrate the quality of the fit. Note the different time window used for the two substrates.
Steroid-binding affinity to AKR1D1
Different steroid substrates may also have different rates of release for the steroid product (kr,Sp). Although this step could not be measured directly, comparison of the binding constants of different steroids can be informative. Binding of NADP+ or NADPH to AKR1D1 quenches the intrinsic fluorescence signal of the enzyme. In the case of the binding of NADPH to AKR1D1, a strong energy transfer signal which peaks at 460 nm can be observed. This energy transfer band can be further quenched by the binding of a steroid ligand. Taking advantage of this spectroscopic signal, the dissociation constants of three steroid ligands, including cortisone (a fast substrate), 7α-hydroxycholestenone (a slow substrate) and finasteride (an inhibitor) were determined (Figure 5 and Table 2). The experiments for cortisone and 7α-hydroxycholestenone were carried out at 4 °C, which ensured no substrate turnover occurred during the titration. The temperature did not appear to affect steroid-binding affinity, since similar Kd values obtained at 4 °C and room temperature for finasteride varied by only 2-fold. In general, the three different steroids displayed similar binding affinity to AKR1D1, suggesting a similar rate of steroid release for all steroid ligands. Specifically, the value of Kd for 7α-hydroxycholestenone is less than 2-fold higher than that for cortisone, which is a significantly smaller difference compared with their difference in kchem values.
Figure 5. Determination of cortisone dissociation constant (Kd) by fluorescence titration.
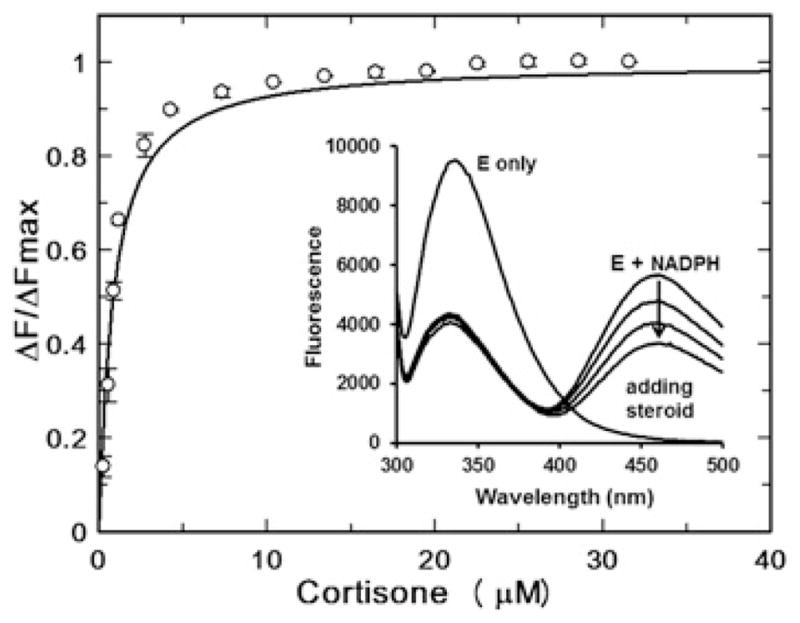
Formation of the AKR1D1–NADPH complex quenches intrinsic protein fluorescence and generates an energy transfer band at 460 nm. Addition of steroid quenches the energy transfer band, which was used to titrate the AKR1D1–NADPH–steroid ternary complex.
Table 2.
Dissociation constants for steroids
| Steroid | Kd (μM) |
|---|---|
| Cortisone (4 °C) | 0.80 ± 0.06 |
| 7α-Hydroxycholestenone (4 °C) | 1.3 ± 0.1 |
| Finasteride (4 °C) | 1.14 ± 0.09 |
| Finasteride (37 °C) | 2.26 ± 0.09 |
Values are means±S.E.M.
Molecular modelling of steroid binding in AKR1D1
Testosterone, cortisone and 7α-hydroxycholestenone were docked into the active site of AKR1D1 using an automated docking program. The lowest energy conformation found for cortisone overlaid well with that in the crystal structure of AKR1D1–NADP+ –cortisone complex, validating our docking procedure [37]. The lowest-energy conformations found for testosterone and 7α-hydroxycholestenone closely resembled that found for cortisone (Supplementary Figure S1 at http://www.biochemj.org/bj/462/bj4620163add.htm). These conformations represent a productive binding mode, as the steroid A-ring is buried deep at the bottom of the active site, whereas the D-ring and the C-17 side chain protrude towards the opening at the enzyme surface. No structural evidence was found from molecular modelling for different binding modes between the fast substrate cortisone and the slow substrate 7α-hydroxycholestenone to account for the vast difference in kchem values.
Rate of NADP + release
Tight binding of cofactors is a characteristic property of AKR enzymes such that the second product release step is often found limiting to overall catalysis [29–31]. Changes in the fluorescence signal of the energy transfer band were utilized to determine the off rate of NADP+ from the AKR1D1–NADP+ binary complex in stopped-flow binding-competition experiments. In the stopped-flow experiments, a pre-mixed solution of AKR1D1–NADP+ was mixed with a solution of NADPH (excess concentration). Upon mixing, NADPH competed off NADP+ to bind to AKR1D1, causing an increase in fluorescence signal due to the formation of the AKR1D1–NADPH complex. Under these experimental conditions, the binding of NADPH to AKR1D1 is a fast process, and is limited by the release of NADP+ from the AKR1D1–NADP+ complex to form free AKR1D1. Thus the kinetic transient observed in the competition experiment corresponds to the process of NADP+ release. The time course could be best fitted to a single-exponential increase (Figure 6), yielding a rate constant koff of 0.36 ± 0.01 s−1. The experiment was repeated using fixed concentrations of the AKR1D1–NADP+ complex and varied concentrations of NADPH while maintaining the concentration of NADPH in large excess. Kinetic traces yielded the same rate constant, whereas the amplitudes of the signal were affected by the concentration of the AKR1D1–NADP+ complex. This is consistent with the kinetic traces reflecting the release of NADP+, since the rate constant of this process is independent of the concentrations of AKR1D1, NADP+ or NADPH, but the absolute change in signal (or the amplitude) is dependent on the concentration of the AKR1D1–NADP+ complex. In the control experiment in which the NADP+ solution was mixed with NADPH, no kinetic transient was observed, indicating kinetic transients observed in experiments with AKR1D1 were enzyme dependent. In another set of control experiments, AKR1D1 was mixed with NADPH and the process of NADPH binding to AKR1D1 was directly monitored. The transient traces were dependent upon [NADPH] and best fitted by a double-exponential function. At saturating [NADPH], the rate constants were kobs1 = 258 ± 23 s−1 and kobs2 =5.8 ± 2.3 s−1, with the fast phase accounting for 81 % of fluorescence increase and the slower phase accounting for 19 % of the increase. Biphasic transient behaviours for cofactor binding were previously observed with other AKR enzymes and are consistent with kinetic binding mechanisms that comprise a fast binding step to form a loose complex followed by one or more isomerization steps to form the final tight complex [28,30].
Figure 6. Representative kinetic traces from competition of cofactor binding.
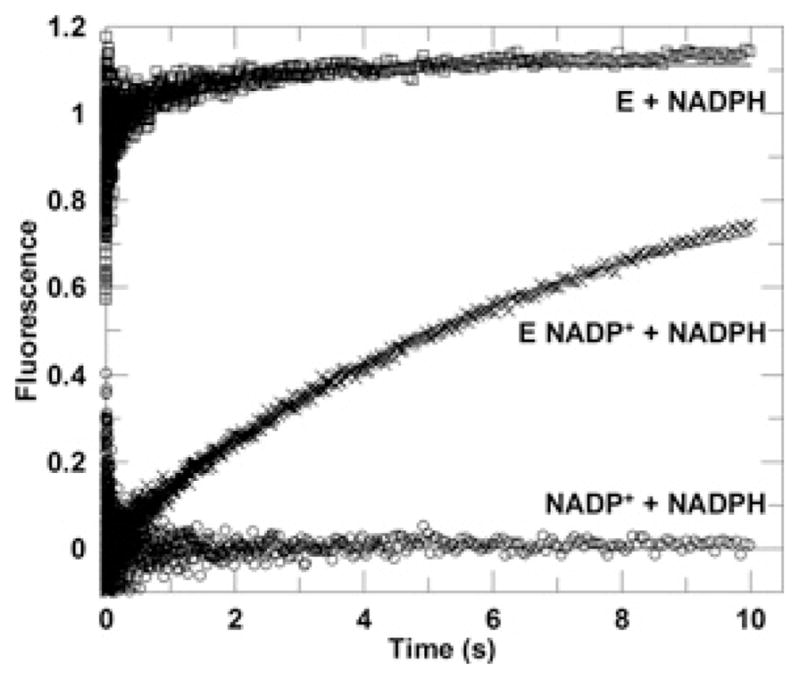
The averaged progress curve of the energy transfer fluorescence signal observed upon mixing an enzyme solution with excess NADPH solution. The final sample contained 0.5 μM AKR1D1 and 50 μM NADPH (□).The averaged progress curve observed upon mixing an enzyme–NADP + solution with excess NADPH solution. The final sample contained 0.5 μM AKR1D1, 2.5 μM NADP + and 50 μM NADPH (×). The averaged progress curve observed upon rapid mixing an NADP + solution with excess NADPH solution. The final sample contained 2.5 μM NADP + and 50 μM NADPH (○). Data for the E (enzyme) + NADPH line (□) were fitted to a double-exponential function, whereas data for E NADP + + NADPH line were fitted to a single-exponential function (×). The fitted lines are shown in grey.
Contributions of discrete steps to k cat
Using eqn (1), the rate of steroid product release (kr,Sp) and contribution of discrete steps to kcat can be calculated (Table 3). For the reaction of cortisone, kchem was determined to be 1.73 s−1 by transient-turnover experiments, which is 4-fold faster than the release of NADP+ (kr,NADP+, 0.36 s−1). The rate of the release of NADP+ is still significantly greater than kcat (0.19 s−1). Thus the overall turnover number is largely determined by the two product release steps, consistent with the observation of burst phase kinetics in multiple-turnover experiments. By contrast, for the reaction of 7α-hydroxycholestenone, kchem dominates kcat and the release of NADP+ is more than 10-fold faster than kchem. Although kr,Sp for this reaction cannot be calculated, it is most likely on the same scale as that for the cortisone reaction and contributes little to the overall kcat. Thus the different kinetic behaviour of AKR1D1 with the fast and slow substrates is due to the differences in their kchem values.
Table 3.
Contribution of discrete steps to rate determination for fast and slow substrates
| Parameters | Cortisone (fast substrate) | 7α-Hydroxycholestenone (slow substrate) |
|---|---|---|
| kcat (s−1) | 0.195 ± 0.002 | 0.033 ± 0.002 |
| kchem (s−1) | 1.7 ± 0.8 | 0.021 ± 0.002 |
| kr,Sp (s−1) | 0.55 | Not determined |
| kr,NADP+ (s −1) | 0.36 ± 0.01 | 0.36 ± 0.01 |
kcat was determined by steady-state analysis. kchem was the extrapolated limiting value of k burst measured in multiple-turnover experiments or ksto measured in single-turnover experiments. kr,NADP+ was determined in the cofactor binding competition experiment. kr,Sp was calculated using eqn (1), error was not extrapolated. Values are means±S.E.M.
DISCUSSION
In the present study, we have investigated the kinetic basis of the large variation in the turnover numbers of AKR1D1 with its various substrates. Transient kinetic analysis revealed an even larger difference in the kinetic rate constant of the chemistry step than observed in the steady-state kcat values. Striking differences of up to two orders of magnitude in the kchem values were observed, which resulted in different rate determining steps for the fast and slow substrates. For fast substrates, such as testosterone, aldosterone and cortisone, the chemical step occurs at a fast rate and the release of steroid and cofactor products are the major contributor to rate determination. For slow substrates, such as cholestenone and 7α-hydroxycholestenone, the chemistry step is significantly slower and determines the overall rate of turnover.
Our results show that the ability of AKR1D1 to carry out the chemical reaction is highly sensitive to steroid structure. Substantial differences in kchem do not correlate with the similarity in chemical structure. Significant differences in kchem may be caused by the difference in the positioning of the reactive groups at the active site with different steroid substrates, i.e. the ‘pre-organization’ of the enzyme, cofactor and the substrate, and/or the difference in dynamic properties of the enzyme, i.e. the ‘reorganization’ of the enzyme active site [38]. Both are possible with AKR1D1. The steroid-binding pocket of AKR1D1 resembles a cylindrical cavity lined largely by hydrophobic residues, with three flexible loops forming the entrance of the binding site [21–23,39,40]. The pocket is flexible such that alternative binding modes and ligand ‘induced fit’ are known to occur with AKR1D1 and the related AKR1C enzymes [33,39,41,42]. There is no apparent hindrance from the enzyme to limit the size of the ligand at the outer portion of the cavity, and this was also observed for the AKR1C enzymes [41]. The capability of the AKR1C steroid-binding site to accommodate large steroid substrates was also demonstrated by the reactivity of AKR1C enzymes with sulfate- and glucuronide-conjugated steroids. Moreover, using 5α-dihydrotestosterone-17β-glucuronide as a substrate, it was found that the stereochemistry of product formation catalysed by AKR1C2 was inverted compared with the free steroid, indicating that substitution at the distal 17-position can cause a significant change in the reaction of the steroid A-ring with an AKR enzyme [33]. It is thus possible that the long hydrophobic C-17 side chain of slow substrates of AKR1D1 (e.g. 7α-hydroxycholestenone) forms additional interactions with the enzyme, which affects the position of the Δ4-double bond relative to NADPH or the dynamic properties of the enzyme. Our attempt to model differential binding poses for fast and slow substrates with AKR1D1 yielded no significant spatial difference in the docked conformations of different steroid substrates. One limitation of our docking method is that the steroid-binding pocket from existing crystal structures of the AKR1D1 ternary complexes were used as template. The steroid-binding pocket was essentially fixed for the binding of the ligand even when the side chains of selected amino acid residues were allowed to move. This docking is unlikely to account for the possible induced-fit caused by the different C-17 side chains of steroids. Future crystal structures of AKR1D1 in complex with a slow steroid substrate or structural information on AKR1D1 dynamics may provide further clues on the structural basis of the difference in kchem.
The kinetic details of steroid hormone transformation catalysed by AKR1D1 obtained in the present study indicate that AKR1D1 is capable of very fast hydride transfer to steroid hormone substrates such as cortisone; however, the overall turnover is limited by cofactor release and steroid product release. In particular, cofactor release, which is the slowest step of catalysis, puts an upper limit on the overall turnover rate of AKR1D1 with steroid hormones. Such kinetic behaviour is consistent with AKR1D1’s role in the catabolism of steroid hormones in the liver. For this function, AKR1D1 works together with ketosteroid reductases to convert steroid hormones into THS metabolites (Figure 1A), which can then be conjugated and cleared. The ketosteroid reductases following AKR1D1 are the related AKR1C1–AKR1C4 enzymes. Interestingly, AKR1D1 displays a significant higher affinity for the cofactors than the related AKR1C enzymes, pointing to a slower cofactor release process than AKR1Cs. Thus the maximum rate of an AKR1C reaction would be higher than that of AKR1D1. Indeed, the liver-specific AKR1C4 displayed fast reactions with 5β-DHS substrates [43]. As such, despite the high kchem values of AKR1D1 with steroid hormone substrates, its slow product release ensures that AKR1D1 catalyses the slower step in the metabolic sequence and thus avoids the accumulation of 5β-DHS in the liver, some of which are active nuclear receptor ligands. With some steroid hormones (e.g. cortisol), comparable amounts of THS metabolites enter the circulation, suggesting a balance of the 5α- and 5β-pathways [4]. The kinetic control of AKR1D1 on steroid hormone transformation would be critical in maintaining this balance.
On the other hand, the ability of AKR1D1 to operate at capacity with steroid hormones allows the production of active 5β-DHS ligands in extra-hepatic tissues such as the uterus, placenta and brain. The AKR1C enzymes in these tissues (i.e. AKR1C1–AKR1C3) have significant lower ketosteroid reductase activities than the liver-specific AKR1C4, permitting the accumulation of 5β-DHS for the regulation on parturition and/or neurological activity.
In contrast with steroid hormones, we now show that the conversion of a bile acid precursor by AKR1D1 is inherently a slow reaction, as indicated by the low kchem value for the reaction of 7α-hydroxycholestenone. The action of AKR1D1 is required in each of the multiple pathways of bile acid synthesis known in humans [9] (Figure 1). The critical involvement of AKR1D1 in bile acid synthesis is highlighted by the clinical observation that a disorder in bile acid synthesis is associated with congenital deficiencies in the AKR1D1 gene [44,45]. We have shown five AKR1D1 mutations identified in patients to be causal for the condition as the mutations strongly affected AKR1D1 function [46]. With a low kcat value of 2 min−1, AKR1D1 is estimated to be abundantly expressed as 1 % of soluble protein in the liver to satisfy the demand of synthesizing several hundred milligrams of bile acids daily. This suggests that AKR1D1 not only catalyses the crucial step where the essential 5β-configuration is introduced into the steroid nucleus, but it may also serve as a point of regulation in bile acid synthesis. It is well accepted that the level of bile acids in the enterohepatic circulation regulates their own synthesis [8,9]. Multiple levels of regulation of bile acid synthesis in addition to the known feedback inhibition of cholesterol 7α-hydroxylase have been suggested [47,48]. AKR1D1 may be the enzyme downstream of cholesterol 7α-hydroxylase that provides additional kinetic control on the rate of bile acid synthesis by catalysing the slow 5β-reduction in bile acid precursors. In addition, the observation that chenodeoxycholate and ursodeoxycholate are potent non-competitive inhibitors of AKR1D1 also supports regulation at the level of AKR1D1 inhibition [19]. Taken together, AKR1D1 offers a potential point of regulation for all bile acid synthetic pathways.
Synthesis of bile acids from cholesterol constitutes a major mechanism for cholesterol degradation in the liver [13]. In addition, bile acids ensure proper absorption of cholesterol, promote bile flow for cholesterol excretion, and inhibit hepatic and intestinal cholesterol synthesis. Thus bile acids exert profound modulating effects at all phases of cholesterol metabolism. As a key enzyme for the synthesis of bile acids, AKR1D1 may play an unrecognized function in affecting cholesterol homoeostasis. Bile acids are increasingly implicated in pharmacological applications for pathological conditions such as dyslipidaemia and cardiovascular disease [49,50], the effect on AKR1D1 function in bile acid synthesis and, in turn, cholesterol metabolism and homoeostasis should also be explored.
Supplementary Material
Acknowledgments
FUNDING
This work was supported by the National Institutes for Health (NIH) [grant numbers R01-DK47015 and P30 ES013508 (to T.M.P) and F32-DK089827 (to M.C.)].
Abbreviations
- AKR1D1
steroid 5β-reductase/aldo-oxo reductase 1D1
- DHS
dihydrosteroid
- THS
tetrahydrosteroid
Footnotes
AUTHOR CONTRIBUTION
Yi Jin proposed the study, determined the rate constants in the multiple-turnover and cofactor release experiments, and drafted the paper. Mo Chen performed the single-turnover experiments, determined cofactor and steroid dissociation constants, and revised the paper. Trevor Penning oversaw the project, provided scientific guidance and input into result interpretation, and revised the paper.
References
- 1.Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol. 1997;54:639–647. doi: 10.1016/s0006-2952(97)84253-0. [DOI] [PubMed] [Google Scholar]
- 2.Kondo KH, Kai MH, Setoguchi Y, Eggertsen G, Sjoblom P, Setoguchi T, Okuda KI, Bjorkhem I. Cloning and expression of cDNA of human Δ4–3-oxosteroid 5β-reductase and substrate specificity of the expressed enzyme. Eur J Biochem. 1994;219:357–363. doi: 10.1111/j.1432-1033.1994.tb19947.x. [DOI] [PubMed] [Google Scholar]
- 3.Roberts S, Szego CM. Biochemistry of the steroid hormones. Annu Rev Biochem. 1955;24:543–596. doi: 10.1146/annurev.bi.24.070155.002551. [DOI] [PubMed] [Google Scholar]
- 4.Tomkins GM. Enzymatic mechanisms of hormone metabolism. I Oxidation-reduction of the steroid nucleus. Recent Prog Horm Res. 1956;12:125–133. [PubMed] [Google Scholar]
- 5.Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- 6.Perusquia M, Navarrete E, Gonzalez L, Villalon CM. The modulatory role of androgens and progestins in the induction of vasorelaxation in human umbilical artery. Life Sci. 2007;81:993–1002. doi: 10.1016/j.lfs.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Sheehan PM, Rice GE, Moses EK, Brennecke SP. 5β-Dihydroprogesterone and steroid 5β-reductase decrease in association with human parturition at term. Mol Hum Reprod. 2005;11:495–501. doi: 10.1093/molehr/gah201. [DOI] [PubMed] [Google Scholar]
- 8.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50:S120–S125. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992;31:4737–4749. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- 10.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khurana S, Raufman JP, Pallone TL. Bile acids regulate cardiovascular function. Clin Transl Sci. 2011;4:210–218. doi: 10.1111/j.1752-8062.2011.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28:220–224. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- 13.Wilson JD. The role of bile acids in the overall regulation of steroid metabolism. Arch Intern Med. 1972;130:493–505. [PubMed] [Google Scholar]
- 14.Clayton PT, Mills KA, Johnson AW, Barabino A, Marazzi MG. Δ4–3-Oxosteroid 5β-reductase deficiency: failure of ursodeoxycholic acid treatment and repsonse to chenodeoxycholic acid plus cholic acid. Gut. 1996;38:623–628. doi: 10.1136/gut.38.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzales E, Cresteil D, Baussan C, Dabadie A, Gerhardt MF, Jacquemin E. SRD5B1 (AKR1D1) gene analysis in Δ4–3-oxosteroid 5β-reductase deficiency: evidence for primary genetic defect. J Hepatol. 2004;40:716–718. doi: 10.1016/j.jhep.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 16.Lemonde HA, Custard EJ, Bouquet J, Duran M, Overmars H, Scambler PJ, Clayton PT. Mutations in SRD5B1 (AKR1D1), the gene encoding Δ4–3-oxosteroid 5β-reductase, in hepatitis and liver failure in infancy. Gut. 2003;52:1494–1499. doi: 10.1136/gut.52.10.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seki Y, Mizuochi T, Kimura A, Takahashi T, Ohtake A, Hayashi S, Morimura T, Ohno Y, Hoshina T, Ihara K, et al. Two neonatal cholestasis patients with mutations in the SRD5B1 (AKR1D1) gene: diagnosis and bile acid profiles during chenodeoxycholic acid treatment. J Inherit Metab Dis. 2012;36:565–573. doi: 10.1007/s10545-012-9526-6. [DOI] [PubMed] [Google Scholar]
- 18.Ueki I, Kimura A, Chen HL, Yorifuji T, Mori J, Itoh S, Maruyama K, Ishige T, Takei H, Nittono H, et al. SRD5B1 gene analysis needed for the accurate diagnosis of primary 3-oxo-Δ4-steroid 5β-reductase deficiency. J Gastroenterol Hepatol. 2009;24:776–785. doi: 10.1111/j.1440-1746.2008.05669.x. [DOI] [PubMed] [Google Scholar]
- 19.Chen M, Drury JE, Penning TM. Substrate specificity and inhibitor analyses of human steroid 5β-reductase (AKR1D1) Steroids. 2011;76:484–490. doi: 10.1016/j.steroids.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, Drury JE, Christianson DW, Penning TM. Conversion of human steroid 5β-reductase (AKR1D1) into a 3β-hydroxysteroid dehydrogenase by a single-point mutation E120H; an example of perfect enzyme-engineering. J Biol Chem. 2012;287:16609–16622. doi: 10.1074/jbc.M111.338780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Costanzo L, Drury JE, Penning TM, Christianson DW. Crystal structure of human liver Δ4–3-ketosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis. J Biol Chem. 2008;283:16830–16839. doi: 10.1074/jbc.M801778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drury JE, Di Costanzo L, Penning TM, Christianson DW. Inhibition of human steroid 5β-reductase (AKR1D1) by finasteride and structure of the enzyme-inhibitor complex. J Biol Chem. 2009;284:19786–19790. doi: 10.1074/jbc.C109.016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trauger JW, Jiang A, Stearns BA, LoGrasso PV. Kinetics of allopregnanolone formation catalyzed by human 3α-hydroxysteroid dehydrogenase type III (AKR1C2) Biochemistry. 2002;41:13451–13459. doi: 10.1021/bi026109w. [DOI] [PubMed] [Google Scholar]
- 25.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. Portland Press; London: 2002. Deriving steady-state rate equations; pp. 91–99. [Google Scholar]
- 26.Segal IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley and Sons; New York: 1975. Steady-state kinetics of multireactant enzymes; pp. 560–564. [Google Scholar]
- 27.Plapp B. On calculation of rate and dissociation constants from kinetic constants for the ordered Bi Bi mechanism of liver alcohol dehydrogenase. Arch Biochem Biophys. 1973;156:112–114. doi: 10.1016/0003-9861(73)90347-0. [DOI] [PubMed] [Google Scholar]
- 28.Cooper WC, Jin Y, Penning TM. Elucidation of a complete kinetic mechanism for a mammalian hydroxysteroid dehydrogenase (HSD) and identification of all enzyme forms on the reaction coordinate: the example of rat liver 3α-HSD (AKR1C9) J Biol Chem. 2007;282:33484–33493. doi: 10.1074/jbc.M703414200. [DOI] [PubMed] [Google Scholar]
- 29.Grimshaw CE, Lai CJ. Oxidized aldose reductase: in vivo factor not in vitro artifact. Arch Biochem Biophys. 1996;327:89–97. doi: 10.1006/abbi.1996.0096. [DOI] [PubMed] [Google Scholar]
- 30.Jin Y, Penning TM. Multiple steps determine the overall rate of the reduction of 5α-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: implications for the elimination of androgens. Biochemistry. 2006;45:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nidetzky B, Klimacek M, Mayr P. Transient-state and steady-state kinetic studies of the mechanism of NADH-dependent aldehyde reduction catalyzed by xylose reductase from the yeast Candida tenuis. Biochemistry. 2001;40:10371–10381. doi: 10.1021/bi010148a. [DOI] [PubMed] [Google Scholar]
- 32.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[α]pyrene to the activated o-quinone metabolite benzo[α]pyrene-7,8-dione. Biochemistry. 1999;38:10626. doi: 10.1021/bi995085z. [DOI] [PubMed] [Google Scholar]
- 33.Jin Y, Duan L, Lee SH, Kloosterboer HJ, Blair IA, Penning TM. Human cytosolic hydroxysteroid dehydrogenases of the aldo-ketoreductase superfamily catalyze reduction of conjugated steroids: implications for phase I and phase II steroid hormone metabolism. J Biol Chem. 2009;284:10013–10022. doi: 10.1074/jbc.M809465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson KA. Transient-state kinetic analysis of enzyme reaction pathways. In: Sigman DS, editor. The Enzymes. Vol. 20. Academic Press; San Diego: 1992. pp. 1–61. [Google Scholar]
- 35.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 37.Di Costanzo L, Drury JE, Christianson DW, Penning TM. Structure and catalytic mechanism of human steroid 5β-reductase (AKR1D1) Mol Cell Endocrinol. 2009;301:191–198. doi: 10.1016/j.mce.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry. 2011;50:10422–10430. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faucher F, Cantin L, Luu-The V, Labrie F, Breton R. Crystal structures of human Δ4–3-ketosteroid 5β-reductase (AKR1D1) reveal the presence of an alternative binding site responsible for substrate inhibition. Biochemistry. 2008;47:13537–13546. doi: 10.1021/bi801276h. [DOI] [PubMed] [Google Scholar]
- 40.Faucher F, Cantin L, Luu-The V, Labrie F, Breton R. Crystal structures of human Δ4–3-ketosteroid 5β-reductase defines the functional role of the residues of the catalytic tetrad in the steroid double bond reduction mechanism. Biochemistry. 2008;47:8261–8270. doi: 10.1021/bi800572s. [DOI] [PubMed] [Google Scholar]
- 41.Jin Y, Penning TM. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): insights into positional and stereochemical preferences. Steroids. 2006;71:380–391. doi: 10.1016/j.steroids.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin Y, Mesaros AC, Blair IA, Penning TM. Stereospecific reduction of 5β-reduced steroids by human ketosteroid reductases of the AKR (aldo-keto reductase) superfamily: role of AKR1C1-AKR1C4 in the metabolism of testosterone and progesterone via the 5β-reductase pathway. Biochem J. 2011;437:53–61. doi: 10.1042/BJ20101804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heubi JE, Setchell KD, Bove KE. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27:282–294. doi: 10.1055/s-2007-985073. [DOI] [PubMed] [Google Scholar]
- 45.Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456–468. doi: 10.1038/ncpgasthep1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drury JE, Mindnich R, Penning TM. Characterization of disease-related 5β-reductase (AKR1D1) mutations reveals their potential to cause bile acid deficiency. J Biol Chem. 2010;285:24529–24537. doi: 10.1074/jbc.M110.127779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertolotti M, Crosignani A, Del Puppo M. The use of stable and radioactive sterol tracers as a tool to investigate cholesterol degradation to bile acids in humas in vivo. Molecules. 2012;17:1939–1968. doi: 10.3390/molecules17021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crosignani A, Zuin M, Allocca M, Del Puppo M. Oxysterols in bile acid metabolism. Clin Chim Acta. 2011;412:2037–2045. doi: 10.1016/j.cca.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 49.Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53:1723–1737. doi: 10.1194/jlr.R024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma R, Long A, Gilmer JF. Advances in bile acid medicinal chemistry. Curr Med Chem. 2011;18:4029–4052. doi: 10.2174/092986711796957266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.