

Abstract
Polo-like kinase-1 (Plk1) belongs to a family of serine-threonine kinases and plays a critical role in mitotic progression. Plk1 involves in the initiation of mitosis, centrosome maturation, bipolar spindle formation, and cytokinesis, well-reported as traditional functions of Plk1. In this review, we discuss the role of Plk1 during DNA damage response beyond the functions in mitotsis. When DNA is damaged in cells under various stress conditions, the checkpoint mechanism is activated to allow cells to have enough time for repair. When damage is repaired, cells progress continuously their division, which is called checkpoint recovery. If damage is too severe to repair, cells undergo apoptotic pathway. If damage is not completely repaired, cells undergo a process called checkpoint adaptation, and resume cell division cycle with damaged DNA. Plk1 targets and regulates many key factors in the process of damage response, and we deal with these subjects in this review. [BMB Reports 2014; 47(5): 249-255]
Keywords: Cell cycle, DNA damage checkpoint, Polo-like kinase-1, p53
INTRODUCTION: BRIEF ON PLK1
The first polo-like kinase (Plk) was primarily discovered in polo gene of Drosophila melanogaster and functional mutation of this gene has various defects in mitosis (1,2). This polo gene has highly conserved from yeast to human and functions as a key regulator during mitosis, meiosis and cytokinesis (3). Plk1 is serine/threonine kinase and one of the polo-like kinase family members. Plk1 structurally has two functional domains; one is polo-box domain on C-terminal for targeting of substrate and involving in its subcellular localization (4), and the other is kinase domain regulated through phosphorylation by upstream kinases (5,6). The expression of Plk1 begins to increase from S/G2 phase, and its activity peaks at mitosis. Plk1 functions in various mitotic events such as centrosome maturation, assembly of the bipolar spindle, chromosome segregation, activation of the anaphase promoting complex (APC/C) and cytokinesis (3,7,8). Excessive or deregulated expression of Plk1 accelerates cell division abnormally and promotes tumorigenesis. Hence, Plk1 is usually over-expressed in various types of cancers (9), suggesting that Plk1 is considered as one of the strong candidate targets for anticancer therapy (10). As well as mitotic roles, it has been reported that Plk1 involves in checkpoint activation and recovery in response to DNA damage (11), even this mechanism has poorly been understood so far. In this review, we focus on the involvement of Plk1 in interphasic and mitotic DNA damage response. In addition, we refer to the relationship between Plk1 and p53 during DNA damage response.
DNA DAMAGE RESPONSE (DDR) DURING CELL CYCLE
Cells are continuously threatened with DNA damage by either endogenous reasons including by-product of metabolic pathway and replication stresses, or exogenous factor such as exposure to UV irradiation or genotoxic reagents. If DNA damage is allowed continuously, cells lose their own functions and could be developed to cancer (12). Fortunately, repair mechanisms have been evolutionally conserved and well-established for preservation of genetic stability under constantly being attacked conditions.
Once DNA double strands are broken, it can be repaired using two types of DNA repair mechanisms, non-homologous end-joining (NHEJ) and homologous recombination (HR) (13). During NHEJ, two broken ends of DNA templates are simply re-connected. However, NHEJ is prone to change total DNA integrity and to induce genomic error by ligation of incongruence DNA ends (14). When NHEJ is inevitably operated in response to DNA double strand breaks (DSBs), at first, DNA lesion sites are recognized by Ku70 and Ku80 sensor proteins for NHEJ repair mechanism (15). Then, DNA-protein kinase (DNA-PK) is recruited on impaired DNA site by interaction with the activated Ku proteins (16). DNA-PK is one of the PIKK family members. ATM (ataxia talangectasia mutated) and ATR (ATM and RAD3-related), the key protein kinases in response to DNA damage, belong to this family (17) and is mentioned below. Once DNA-PK is located on the DSB region, it phosphorylates the effector proteins, especially multimeric complex (DNA ligase IV-XRCC4-XLF) to connect two ends of broken DNA (18).
The other mechanism in response to DSB is HR which is restricted to S and G2 phase. Unlike NHEJ, HR repair pathway needs undamaged DNA template, and sister chromatid is used to repair by HR as a template (19). Broken DNA ends are recognized and bounded by Mre11-Rad51-Nbs1 (MRN) complexes (20). ATM kinase is then loaded on DNA lesion site and its activity is increased by interaction with the recruited MRN complexes. Subsequently, a large number of downstream target substrates such as Chk1/Chk2 are phosphorylated by the ATM. Activated ATM in the defected DNA regions can also phosphorylate γ-H2AX (21), and then Mdc1 is collected on the damaged site by γ-H2AX (22) and can amplify the γ-H2AX signals and form foci on the DSB regions (23). In the DSB foci, 5’ ends of two broken DNA are separated (24) by nucleases such as Mre11, Exo1, CtIP and BLM helicase (25,26). Exposed single strand DNA is then rapidly coated by RPA proteins (replication protein A) (27,28). In addition to ATM signals, ATR kinase signal pathway is also activated by ssDNA produced from 5’ ends (29). Following resection, RPA binding to ssDNA is replaced by Rad51 recombinase (30). Rad51 recruited on a damaged DNA combines the sister chromatid as an undamaged DNA template for recombination repair mechanism (31).
During the previously mentioned repair mechanisms, cells containing defective genome should be stopped in progressing to the next stage of the cell cycle. For this, when DNA damages were perceived, surveillance mechanisms called checkpoints are activated. G1/S boundary checkpoint inspects the DNA integrity for blockage before the replication of the damaged DNA (32). If there are defects on DNA at G1 phase, CDK2 is inactivated by ATM/Chk2 signaling, but not by ATR/Chk1. Also, interaction with p21, a downstream target of p53, results from the suppression of the S-phase entry. During intra-S checkpoint, Cdc25 phosphatases are predominantly regulated by ATR/Chk1 signaling (33). G2/M checkpoint supervises the genome integrity before the segregation of chromosomes in mitosis. For mitosis progression after damage recovery, cyclinB/CDK1 is up-regulated and activated (34). Under this DNA damage response, Plk1, consistently activated during mitosis, is known as a direct or an indirect regulator of the cell cycle-dependent proteins including cyclinB, CDK1, and Cdc25 phosphatases (3).
PLK1 AND DNA DAMAGE RESPONSE IN S/G2 INTERPHASE
Before mitotic entry, Aurora A cooperating with Bora phosphorylates threonine 210 residues of Plk1. This residue in inactive Plk1 is located inside of a closed structure by interacting with its own polo-box domain. The closed structure of Plk1, however, is opened by Bora, and is exposed for this residue to be phosphorylated by Aurora A for the activation of Plk1 during mitotic entry (35). When cells are attacked by DNA damage, especially in G2 phase, cells do not progress into mitosis to block the abnormal chromosomes thereafter. Usually, Plk1 loses its activity in response to DNA damage and cell division is arrested by G2 DNA damage checkpoint activity. ATM/ATR directly phosphorylates Bora at threonine 501 residue when the DSB is induced by UV irradiation. Phosphorylated Bora is recognized and degraded by E3 ubiquitin ligase SCF-β-TRCP (36). Threonine 501 of Bora is not phosphorylated in due to DNA damage by the UV irradiation when the ATR is down-regulated. Indeed, when the phosphorylation-defective mutant of Bora (T501A) is overexpressed in cells with UV irradiation, Bora cannot interact with SCF-β-TRCP and is not degraded (36). Once Bora is destructed, Plk1 cannot be activated and cell cycle is arrested in the G2 phase. Mitotic entry is also restricted by Cdk1/cyclinB inhibition resulting from Plk1 inactivation. In addition, Cdc25 phosphatase is inhibited by Chk1 and Chk2, resulting in the inactivation of Cdk1 by preventing elimination of inhibitory phosphorylation at tyrosine 14 and 15. Once damaged DNA is restored, Cdc25 is recovered by activated Plk1, following the approval of mitotic entry (37). Chk1 is one of the downstream target proteins of Claspin, an adaptor protein of ATR under damaged DNA (38). When DNA damage is recovered, activated Plk1 phosphorylates Claspin, and such phosphorylation induces ubiquitin-dependent degradation of Claspin and allows the dissociation from ATR. Finally, checkpoint pathway is terminated and cells enter into mitosis (39) (Fig. 1).
Fig. 1. Plk1 and DNA damage response in S/G2 phase.
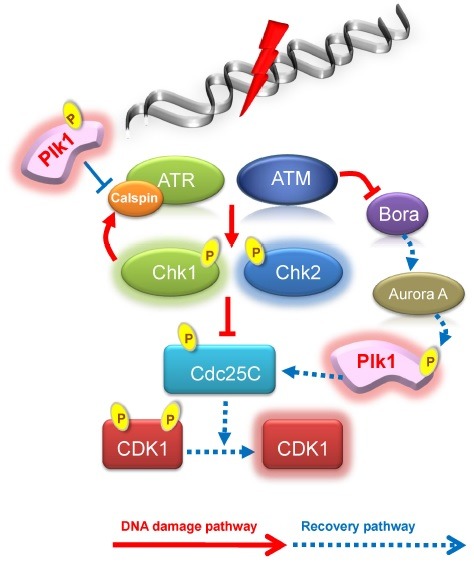
Interestingly, it has been reported that Plk1 directly phosphorylates the Rad51 recombinase and facilitates the homologous recombination (HR). In this mechanism, sensor protein Rad51 in response to DSB is phosphorylated at serine 14 residue, and sequentially threonine 13 residue of Rad51 is phosphorylated by Casein kinase 2 (CK2). These phosphorylations facilitate the HR repair mechanism by increased interaction with Nbs1 (40). However, it still remains questionable how Plk1 is activated through what signal in response to DSB, because Plk1 is originally inactivated in the S/G2-phase.
DNA DAMAGE RESPONSE ASSOCIATED WITH PLK1 IN MITOSIS
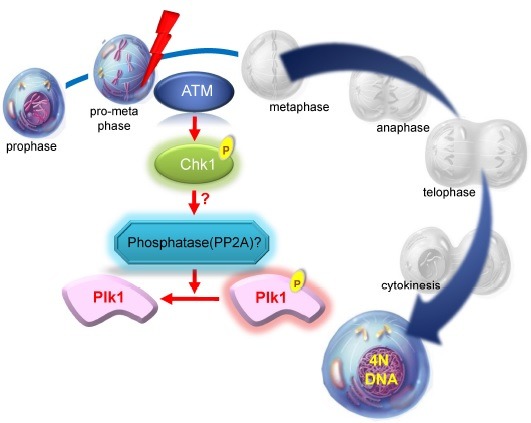
Although mitosis is a very short period than other stages of cell cycle, it is a crucial phase because chromosome replicated during S phase is separated and genetic information is inherited to the daughter cells. The occurrence of DNA damage during mitosis might induce mitotic arrest in the general concept of the checkpoint to avoid generating abnormal daughter cells containing aneuploidy or mitotic catastrophe. The studies about DNA damage response in mitosis suggest that cells containing DNA damage caused by exposure to irradiation in various stages of mitosis show the same phenotypic effects with non-irradiated cells (41). In addition, it has been reported that DNA damage response seems to be absent or is not effectively activated in irradiated mitotic cells (41,42). However, although these cells continually progress through cell division process, the positive γ-H2AX signal known as a DNA break marker still exists, and apparent delay is observed under this condition. Subsequent finding reveals that mitotic progression of these cells is delayed during metaphase by spindle assembly checkpoint, when cells incur severe DNA damage by irradiation or damaging agent (11,43). What has become of Plk1 under this situation? Whereas Plk1 activity should be increased for recovery from G2/M transition checkpoint, it might be principally suppressed by DNA damage occurring during mitotic phase where Plk1 is already activated. After the induction of DNA damage, the kinase activity of Plk1 is down- regulated through ATM/ATR-dependent fashion (44-47). Inactivation of Plk1 during the mitotic DNA damage response occurs by the dephosphorylation of phosphor-threonine 210, a requirement for protein phosphatase 2A activity. ATM-/- and Chk1-/- cells with mitotic DNA damage show a clear rescue of Plk1 dephosphorylation, and the addition of caffeine also completely rescues this effect, suggesting that protein phosphatase 2A activity on Plk1 dephophorylation is dependent on ATM-Chk1 pathway in mitotic DNA damage response (44,45) (Fig. 2). Importantly, overexpression of phospho-mimic mutant of Plk1, T210D, overcomes the arrest induced by the DNA damage (11). These reports suggest that Plk1 plays the roles in the communication between the DNA damage and proper checkpoint. It also suggests that down regulation of Plk1 is necessary for arresting cell cycle to allow ample time for DNA repair and maintenance of genome stability during mitotic DNA damage response. Very severe DNA damage is induced to bypass both late mitotic process and cytokinesis over a short delay of transition between metaphase and anaphase, in spite of the suppression of Plk1 activity from the results of time lapse microscopy and FACS analysis (48). After then, cells contain 4N DNA contents, even though cells have positive γ-H2AX signal. The condensed chromosome becomes loose and the nucleus size is increased comparably to that of G2 phase (45). In addition, the inhibitory phosphorylation of Cdk1 and the G2-specific phosphorylation of Cdc25C are increased without a remarkable effects on cyclin B levels under this condition (44), insisting that cells containing mitotic DNA damage enter into G2-like G1 phase with 4N DNA contents. Subsequently, within 24 hours, these cells containing 4N DNA contents become 8N DNA cells by re-replication process called as endoreplication. Finally, cells are removed by apoptotic cell death after one cycle of replication (48). Polyploidy and aneuploidy are common phenomena in cancer cells, and a thorough understanding of endoreplication in mitotic DNA damage response may provide important insights for cancer biology (49).
Fig. 2. Plk1 and DNA damage response in mitosis.
RELATIONSHIP BETWEEN PLK1 AND P53 IN DNA DAMAGE RESPONSE
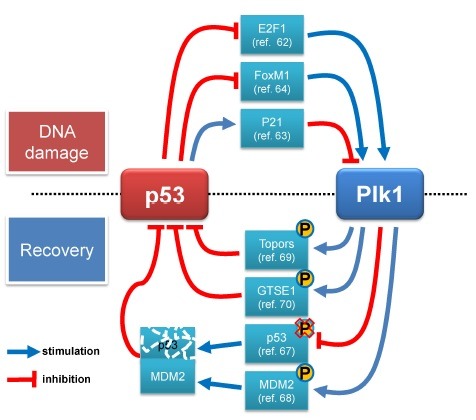
Tumor suppressor p53 is a key tumor suppressor and functions as genome guardian through the induction of cell cycle arrest, apoptosis and senescence in various stress signals (50,51). When stresses such as DNA damage are exerted to the cell, p53 is stabilized for its activation phosphorylation at Ser15 and Ser20 by ATM/ATR and Chk1/Chk2, respectively (52-55). The accumulation of phosphor-p53 protein occurs by suppression of the interaction between p53 and MDM2, which stimulates ubiquitin-mediated degradation of p53 (56). p53 up-regulates its downstream protein such as p21/waf1 inhibiting CDKs, Gadd14 and 14-3-3, and induces DNA damage checkpoint regulating downstream proteins (57). The tumor suppressor activity of p53 in most human cancer cells is impaired by either its mutation or inactivation of p53 signaling. As almost 95% mutation of p53 is detectable in central regions encoding for the DNA binding domain, p53 mutants lose the ability for sequence specific transactivation (51). Cells where p53 is mutated or deleted do not arrest even in a damaged condition and cell division cycle with damages is progressed (58,59). They can also promote tumorigenesis at high frequency in mice and human (60,61). The roles of p53 inducing cell cycle arrest rivals with Plk1 functions which promotes the progression of the cell cycle. Plk1 is inhibitied by ATM/ATR dependent fashion after DNA damage (11,44), and is transcriptionally regulated either directly or indirectly by p53 (62-64). It localizes to the Plk1 promoter and binds to E2F1 which induces an increment of transcriptional level of Plk1. Its binding forms p53-E2F1-Plk1 promoter DNA complex and suppresses the expression of Plk1 (62). Furthermore, the expression of Plk1 is indirectly repressed by p53 through either targeting a sequence of CDE/CHR within Plk1 promoter by p21/waf1 or a negative regulation of FoxM1 (forhead box M1), which stimulates Plk1 expression caused by p53 (63,64). Therefore, cells prevent abnormal overcoming of cell cycle arrest by suppressing Plk1 through p53 during DNA damage repair.
If DNA damage is not repaired completely, cell cycle is arrested consistently and finally cells undergo to death. In contrast, cells normally progress to next stage according to progression of cell cycle again when DNA damage is repaired completely (65). This recovery process by stoppage of checkpoint, especially G2/M transition arrest, requires the Plk1 activity which is essential for mitosis initiation (66). When cell cycle is recovered, re-activated Plk1 directly interacts with sequence specific DNA binding domain within p53, in return it is suppressed own transcriptional activity as well as pro-apoptotic function (67). Furthermore, Plk1 involves indirectly in decrement of phosphorylation on serine 15 of p53 interrupting interaction with MDM2, and reduction of p53 activity and MDM2 mediated degradation are increased (68). This interaction between MDM2 and p53 also is stimulated through phosphorylation at serine 260 of MDM2 by Plk1 (69). Furthermore, Plk1 phosphorylates serine 718 of Topors (topoisomerase 1 binding protein) which has ubiquitin and SUMO-1 E3 ligase activity and promotes degradation of p53 through inducing interaction of between Topors and p53 (69). In addition, Plk1 induces p53 export from nucleus during cell cycle arrest through activation of GSTE1 (G2 and S phase expressed 1 protein) (70). These results explain that the harmony of Plk1 and p53 protects cells from various stresses and aberrantly persistant cell cycle arrest (Fig. 3).
Fig. 3. Networks between p53 and Plk1 in DNA damage responses.
As mentioned above, Plk1 is a major regulator controlling cell cycle and deregulation of this kinase induces overriding checkpoint which leads genomic instability and promotes cell transformation and initiation of tumorigenesis (71). On the other hand, inhibition of Plk1 can induce cytotoxicity such as apoptosis and this effect is connected with cancer therapy. It has been reported that excessive expression of Plk1 is found in cancer cells (72), and that depletion of Plk1 does little or no affect to normal cells in comparison with cancer cells (73,74). Moreover, the cytotoxic effect such as suppression of proliferation and apoptosis resulting from depletion of Plk1 is increased in cancer cells which have a defective p53 (75,76). These indicate that Plk1 is a selective target for cancer therapy and p53 can be noticed as determiner of Plk1 inhibition effect in cancer cells. However, there are still arguments in relation between cytotoxic effect by Plk1 depletion and role of p53, and a number of studies have reported that inhibition of Plk1 induces similar cytotoxicity and sensitivity regardless of any type of cell or status of p53. Poloxin, a targeting inhibitor of polo-box domain, represses proliferation in primary/normal non-transformed cell as well as tumor (77). Another polo-box domain-targeting inhibitor, purpurogallin strongly induces apoptosis in normal rat kidney cells and NIH3T3 cells (78). BI2536, a Plk1 kinase inhibitor targeting kinase domain of Plk1 has been reported to suppress proliferation and to induce apoptosis in both normal cell and cancer cell lines (79). This efficacy is also shown regardless of p53 status in cells (80). A recent report by using p53+/+ and p53-/- cell lines also demonstrates that there is no obvious different cytotoxic response between cancer cells with and without functional p53 (81). Moreover, the arrest point in cell cycle by collaboration of Plk1 inhibition with status of p53 is not the same. While the p53 positive cells are arrested and undergo apoptosis at G1/S phase, the p53 negative cell is arrested at G2/M after the treatment of Plk1 inhibitor (81).
CONCLUSION
During the past 25 years after the discovery polo in Drosophila melanogaster, most of the studies on Plk1 have been focusing on its mitotic roles. Recently, accumulating evidences are opening a new era in Plk1 history beyond the traditional mitotic function. As well as DNA replication and chromosome/microtubule dynamics (82,83), Plk1 also plays a role in overall DNA damage response including DNA checkpoint activation, checkpoint maintenance, damage recovery and DNA repair. When DNA damage occurs in S/G2 phase before Plk1 activation, through ATM/ATR-Bora-SCF-β-TRCP ubiquitin ligase, Plk1 activation is blocked until the damage is repaired. When damaged DNA is repaired by HR mechanisms, Plk1 directly phosphorylates a sensor protein, Rad51 to increase the interaction with Nbs1, and finally the HR is activated. For recovery from DNA damage, Plk1 phosphorylates and induces the inhibition Claspin, which is necessary for Chk1 activation. In mitotic DNA damage after Plk1 activation, inactivation of Plk1 is induced by dephosphorylation through ATM-Chk1-PP2A pathway. The inactivation of Plk1 by dephosphoprylation beyond traditional degradation by anaphase-promoting complex is one more interesting subject of Plk1 study. Severe mitotic DNA damage causes continuous inactivation of Plk1 and other mitotic kinases, allowing cells to skip mitotic events and endoreplication. Additionally, Plk1 and p53 reciprocally regulate each other during DNA damage checkpoint activation and adaptation, and recovery process. Plk1 directly targets Topor and GSTE1 to down regulate p53. Most of all, new functions of Plk1 beyond mitotic roles are speculated by their potential substrates. Previously, Yaffe et al. (2007) reported that more than 600 proteins were identified as the interactome for polo box domain (84). Although much more detailed experiments are needed to validate these proteins, these are hints that Plk1 has various functions in DNA damage response and other cellular events beyond its mitotic functions.
Acknowledgments
We apologize to the scientists who made the contributions to this subject for any omission of their articles that we were unable to cite. This work was conducted by the research fund of Dankook University in 2014.
References
- 1.Sunkel C. E., Glover D. M. Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. (1988);89(Pt 1):25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 2.Llamazares S., Moreira A., Tavares A., Girdham C., Spruce B. A., Gonzalez C., Karess R. E., Glover D. M., Sunkel C. E. Polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes. Dev. (1991);5:2153–2165. doi: 10.1101/gad.5.12a.2153. [DOI] [PubMed] [Google Scholar]
- 3.Barr F. A., Sillje H. H., Nigg E. A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. (2004);5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 4.Lee K. S., Grenfell T. Z., Yarm F. R., Erikson R. L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl. Acad. Sci. U.S.A. (1998);95:9301–9306. doi: 10.1073/pnas.95.16.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jang Y. J., Ma S., Terada Y., Erikson R. L. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J. Biol. Chem. (2002);277:44115–44120. doi: 10.1074/jbc.M202172200. [DOI] [PubMed] [Google Scholar]
- 6.van de Weerdt B. C., Littler D. R., Klompmaker R., Huseinovic A., Fish A., Perrakis A., Medema R. H. Polo-box domains confer target specificity to the Polo-like kinase family. Biochim. Biophys. Acta. (2008);1783:1015–1022. doi: 10.1016/j.bbamcr.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 7.Glover D. M., Hagan I. M., Tavares A. A. Polo-like kinases: a team that plays throughout mitosis. Genes Dev. (1998);12:3777–3787. doi: 10.1101/gad.12.24.3777. [DOI] [PubMed] [Google Scholar]
- 8.van de Weerdt B. C., Medema R. H. Polo-like kinases: a team in control of the division. Cell Cycle. (2006);5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 9.Takai N., Hamanaka R., Yoshimatsu J., Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. (2005);24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 10.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug. Discov. (2010);9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 11.Smits V. A., Klompmaker R., Arnaud L., Rijksen G., Nigg E. A., Medema R. H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. (2000);2:672–676. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 12.Jackson S. P., Bartek J. The DNA-damage response in human biology and disease. Nature. (2009);461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao Z., Bozzella M., Seluanov A., Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) (2008);7:1765–1771. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieber M. R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. (2008);283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 15.Doherty A. J., Jackson S. P. DNA repair: how Ku makes ends meet. Curr. Biol. (2001);11:R920–924. doi: 10.1016/S0960-9822(01)00555-3. [DOI] [PubMed] [Google Scholar]
- 16.Featherstone C., Jackson S. P. Ku, a DNA repair protein with multiple cellular functions? Mutat. Res. (1999);434:3–15. doi: 10.1016/S0921-8777(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 17.Burma S., Chen D. J. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) (2004);3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 18.Calsou P., Delteil C., Frit P., Drouet J., Salles B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol. (2003);326:93–103. doi: 10.1016/S0022-2836(02)01328-1. [DOI] [PubMed] [Google Scholar]
- 19.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. (2003);532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 20.de Jager M., van Noort J., van Gent D. C., Dekker C., Kanaar R., Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell. (2001);8:1129–1135. doi: 10.1016/S1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) (2004);3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 22.Stucki M., Clapperton J. A., Mohammad D., Yaffe M. B., Smerdon S. J., Jackson S. P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. (2005);123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Capetillo O., Celeste A., Nussenzweig A. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle. (2003);2:426–427. doi: 10.4161/cc.2.5.509. [DOI] [PubMed] [Google Scholar]
- 24.Shibata A., Moiani D., Arvai A. S., Perry J., Harding S. M., Genois M. M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F., Ismail A., Ismalaj E., Petricci E., Neale M. J., Bristow R. G., Masson J. Y., Wyman C., Jeggo P. A., Tainer J. A. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell. (2014);53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nimonkar A. V., Ozsoy A. Z., Genschel J., Modrich P., Kowalczykowski S. C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U.S.A. (2008);105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You Z., Shi L. Z., Zhu Q., Wu P., Zhang Y. W., Basilio A., Tonnu N., Verma I. M., Berns M. W., Hunter T. CtIP links DNA double-strand break sensing to resection. Mol. Cell. (2009);36:954–969. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyman C., Kanaar R. Homologous recombination: down to the wire. Curr. Biol. (2004);14:R629–631. doi: 10.1016/j.cub.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 28.West S. C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. (2003);4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 29.Adams K. E., Medhurst A. L., Dart D. A., Lakin N. D. Recruitment of ATR to sites of ionising radiation- induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. (2006);25:3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumann P., Benson F. E., West S. C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. (1996);87:757–766. doi: 10.1016/S0092-8674(00)81394-X. [DOI] [PubMed] [Google Scholar]
- 31.McIlwraith M. J., Vaisman A., Liu Y., Fanning E., Woodgate R., West S. C. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell. (2005);20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Bartek J., Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. (2001);490:117–122. doi: 10.1016/S0014-5793(01)02114-7. [DOI] [PubMed] [Google Scholar]
- 33.Falck J., Mailand N., Syljuasen R. G., Bartek J., Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. (2001);410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 34.Heijink A. M., Krajewska M., van Vugt M. A. The DNA damage response during mitosis. Mutat. Res. (2013);750:45–55. doi: 10.1016/j.mrfmmm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Seki A., Coppinger J. A., Jang C. Y., Yates J. R., Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. (2008);320:1655–1658. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin B., Gao B., Yu J., Yuan J., Lou Z. Ataxia telangiectasia-mutated- and Rad3-related protein regulates the DNA damage-induced G2/M checkpoint through the Aurora A cofactor Bora protein. J. Biol. Chem. (2013);288:16139–16144. doi: 10.1074/jbc.M113.456780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donzelli M., Draetta G. F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. (2003);4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumagai A., Dunphy W. G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. (2000);6:839–849. doi: 10.1016/S1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 39.Mamely I., van Vugt M. A., Smits V. A., Semple J. I., Lemmens B., Perrakis A., Medema R. H., Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. (2006);16:1950–1955. doi: 10.1016/j.cub.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 40.Yata K., Lloyd J., Maslen S., Bleuyard J. Y., Skehel M., Smerdon S. J., Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell. (2012);45:371–383. doi: 10.1016/j.molcel.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison C., Rieder C. L. Chromosome damage and progression into and through mitosis in vertebrates. DNA Repair (Amst) (2004);3:1133–1139. doi: 10.1016/j.dnarep.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Giunta S., Jackson S. P. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle. (2011);10:1215–1221. doi: 10.4161/cc.10.8.15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikhailov A., Cole R. W., Rieder C. L. DNA damage during mitosis in human cells delays the metaphase/ anaphase transition via the spindle-assembly checkpoint. Curr. Biol. (2002);12:1797–1806. doi: 10.1016/S0960-9822(02)01226-5. [DOI] [PubMed] [Google Scholar]
- 44.Lee H. J., Hwang H. I., Jang Y. J. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle. (2010);9:2389–2398. doi: 10.4161/cc.9.12.11904. [DOI] [PubMed] [Google Scholar]
- 45.Jang Y. J., Ji J. H., Choi Y. C., Ryu C. J., Ko S. Y. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J. Biol. Chem. (2007);282:2473–2482. doi: 10.1074/jbc.M605480200. [DOI] [PubMed] [Google Scholar]
- 46.Tsvetkov L., Stern D. F. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle. (2005);4:166–171. doi: 10.4161/cc.4.1.1348. [DOI] [PubMed] [Google Scholar]
- 47.van Vugt M. A., Smits V. A., Klompmaker R., Medema R. H. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J. Biol. Chem. (2001);276:41656–41660. doi: 10.1074/jbc.M101831200. [DOI] [PubMed] [Google Scholar]
- 48.Hyun S. Y., Rosen E. M., Jang Y. J. Novel DNA damage checkpoint in mitosis: Mitotic DNA damage induces re-replication without cell division in various cancer cells. Biochem. Biophys. Res. Commun. (2012);423:593–599. doi: 10.1016/j.bbrc.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 49.Storchova Z., Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. (2004);5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 50.Brown C. J., Lain S., Verma C. S., Fersht A. R., Lane D. P. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer. (2009);9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 51.Vousden K. H., Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. (2002);2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 52.Cheng Q., Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. (2010);9:472–478. doi: 10.4161/cc.9.3.10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S. Y., Taya Y., Prives C., Abraham R. T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. (1999);13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chehab N. H., Malikzay A., Appel M., Halazonetis T. D. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. (2000);14:278–288. [PMC free article] [PubMed] [Google Scholar]
- 55.Shieh S. Y., Ahn J., Tamai K., Taya Y., Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. (2000);14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 56.Appella E. Modulation of p53 function in cellular regulation. Eur. J. Biochem. (2001);268:2763. doi: 10.1046/j.1432-1327.2001.02224.x. [DOI] [PubMed] [Google Scholar]
- 57.Meek D. W. Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer. (2009);9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 58.Lanni J. S., Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell Biol. (1998);18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minn A. J., Boise L. H., Thompson C. B. Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev. (1996);10:2621–2631. doi: 10.1101/gad.10.20.2621. [DOI] [PubMed] [Google Scholar]
- 60.Evans S. C., Lozano G. The Li-Fraumeni syndrome: an inherited susceptibility to cancer. Mol. Med. Today. (1997);3:390–395. doi: 10.1016/S1357-4310(97)01105-2. [DOI] [PubMed] [Google Scholar]
- 61.Attardi L. D., Jacks T. The role of p53 in tumour suppression: lessons from mouse models. Cell Mol. Life. Sci. (1999);55:48–63. doi: 10.1007/s000180050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Z., Cao J. X., Li S. Y., An G. S., Ni J. H., Jia H. T. p53 Suppresses E2F1-dependent PLK1 expression upon DNA damage by forming p53-E2F1-DNA complex. Exp. Cell Res. (2013);319:3104–3115. doi: 10.1016/j.yexcr.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 63.Lin Y. C., Sun S. H., Wang F. F. Suppression of Polo like kinase 1 (PLK1) by p21 (Waf1) mediates the p53-dependent prevention of caspase-independent mitotic death. Cell Signal. (2011);23:1816–1823. doi: 10.1016/j.cellsig.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 64.Pandit B., Halasi M., Gartel A. L. p53 negatively regulates expression of FoxM1. Cell Cycle. (2009);8:3425–3427. doi: 10.4161/cc.8.20.9628. [DOI] [PubMed] [Google Scholar]
- 65.Bartek J., Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. (2007);19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 66.van Vugt M. A., Bras A., Medema R. H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell. (2004);15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 67.Ando K., Ozaki T., Yamamoto H., Furuya K., Hosoda M., Hayashi S., Fukuzawa M., Nakagawara A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. (2004);279:25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 68.Chen J., Dai G., Wang Y. Q., Wang S., Pan F. Y., Xue B., Zhao D. H., Li C. J. Polo-like kinase 1 regulates mitotic arrest after UV irradiation through dephosphorylation of p53 and inducing p53 degradation. FEBS Lett. (2006);580:3624–3630. doi: 10.1016/j.febslet.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 69.Yang X., Li H., Zhou Z., Wang W. H., Deng A., Andrisani O., Liu X. Plk1-mediated phosphorylation of Topors regulates p53 stability. J. Biol. Chem. (2009);284:18588–18592. doi: 10.1074/jbc.C109.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X. S., Li H., Song B., Liu X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. (2011);11:626–632. doi: 10.1038/embor.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eckerdt F., Yuan J., Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. (2005);24:267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 72.Lu L. Y., Yu X. The balance of Polo-like kinase 1 in tumorigenesis. Cell Div. (2009);4:4. doi: 10.1186/1747-1028-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu X., Erikson R. L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. U.S.A. (2003);100:5789–5794. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spankuch-Schmitt B., Bereiter-Hahn J., Kaufmann M., Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J. Natl. Cancer Inst. (2002);94:1863–1877. doi: 10.1093/jnci/94.24.1863. [DOI] [PubMed] [Google Scholar]
- 75.Liu X., Lei M., Erikson R. L. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol. Cell Biol. (2006);26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guan R., Tapang P., Leverson J. D., Albert D., Giranda V. L., Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. (2005);65:2698–2704. doi: 10.1158/0008-5472.CAN-04-2131. [DOI] [PubMed] [Google Scholar]
- 77.Yuan J., Sanhaji M., Kramer A., Reindl W., Hofmann M., Kreis N. N., Zimmer B., Berg T., Strebhardt K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am. J. Pathol. (2011);179:2091–2099. doi: 10.1016/j.ajpath.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Watanabe N., Sekine T., Takagi M., Iwasaki J., Imamoto N., Kawasaki H., Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J. Biol. Chem. (2009);284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 79.Steegmaier M., Hoffmann M., Baum A., Lenart P., Petronczki M., Krssak M., Gurtler U., Garin-Chesa P., Lieb S., Quant J., Grauert M., Adolf G. R., Kraut N., Peters J. M., Rettig W. J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. (2007);17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 80.Lu B., Mahmud H., Maass A. H., Yu B., van Gilst W. H., de Boer R. A., Sillje H. H. The Plk1 inhibitor BI 2536 temporarily arrests primary cardiac fibroblasts in mitosis and generates aneuploidy in vitro. PLoS One. (2010);5:e12963. doi: 10.1371/journal.pone.0012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanhaji M., Kreis N. N., Zimmer B., Berg T., Louwen F., Yuan J. p53 is not directly relevant to the response of Polo-like kinase 1 inhibitors. Cell Cycle. (2012);11:543–553. doi: 10.4161/cc.11.3.19076. [DOI] [PubMed] [Google Scholar]
- 82.Liu X. S., Song B., Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein Cell. (2010);1:999–1010. doi: 10.1007/s13238-010-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mandal R., Strebhardt K. Plk1: unexpected roles in DNA replication. Cell Res. 23:1251–1253. doi: 10.1038/cr.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lowery D. M., Clauser K. R., Hjerrild M., Lim D., Alexander J., Kishi K., Ong S. E., Gammeltoft S., Carr S. A., Yaffe M. B. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. (2007);26:2262–2273. doi: 10.1038/sj.emboj.7601683. [DOI] [PMC free article] [PubMed] [Google Scholar]