

Abstract
Bortezomib has been known as the most promising anti-cancer drug for multiple myeloma (MM). However, recent studies reported that not all MM patients respond to bortezomib. To overcome such a stumbling-block, studies are needed to clarify the mechanisms of bortezomib resistance. In this study, we established a bortezomib-resistant cell line (U266/velR), and explored its biological characteristics. The U266/velR showed reduced sensitivity to bortezomib, and also showed crossresistance to the chemically unrelated drug thalidomide. U266/velR cells had a higher proportion of CD138 negative subpopulation, known as stem-like feature, compared to parental U266 cells. U266/velR showed relatively less inhibitory effect of prosurvival NF-κB signaling by bortezomib. Further analysis of RNA microarray identified genes related to ubiquitination that were differentially regulated in U266/velR. Moreover, the expression level of CD52 in U266 cells was associated with bortezomib response. Our findings provide the basis for developing therapeutic strategies in bortezomib-resistant relapsed and refractory MM patients. [BMB Reports 2014; 47(5): 274-279]
Keywords: Bortezomib resistance, Human multiple myeloma U266 cell line, NF-κB signaling, RNA microarray, Soft-agar forming assay
INTRODUCTION
The acquisition of anti-cancer drug resistance is a major issue with therapies in multiple myeloma (MM) (1). Studies focusing on the mechanisms of chemoresistance (2, 3) have helped us to understand the molecular pathogenesis of MM. Also, such efforts have led to bortezomib (PS-341, VelcadeTM) one of the most successful anti-cancer drugs, improving the clinical outcome of MM (4, 5). Although it has exhibited clinical success, some patients failed to respond to bortezomib, due to primary refractoriness, and acquisition of resistance (7).
The study of resistance to bortezomib has involved the elucidation of intrinsic mechanisms in cancer cells adapted to bortezomib in vitro. The mutation in the proteasome β5 subunit (PSMB5), and the increased expression of proteasome, have been shown in cancer cells with acquired resistance to bortezomib (8). Activation of NF-κB with inactivating abnormality of TNF receptor-associated factor 3 (TRAF3), in MM cells harboring genetic mutation of NF-κB pathways, correlated with bortezomib sensitivity (9). Extrinsic factors, bone marrow (BM) microenvironments can confer resistance to bortezomib, mediated by bone marrow stromal cells (BMSCs)-enhanced NF-κB activity (10, 11). However, to date, little is known about the mechanisms of bortezomib resistance. Therefore, it is necessary to identify the functional characteristics of resistant cells, to better understand the mechanisms.
In this study, we used soft agar assay, to isolate bortezomib-resistant U266 (U266/velR). The U266/velR had increased p-ERK and p-p65 following exposure to bortezomib, and less inhibitory effect of NF-κB, that resulted in the cells having less apoptotic effect by bortezomib. Moreover, the U266/velR cells showed an increased CD138 negative subpopulation, known as cancer-initiating cells with stem cell properties, characterized by quiescent cells and chemoresistance. We further analyzed the patterns of gene expressions, to identify molecular targets associated with bortezomib resistance. The expressions of proteasome subunit genes, including PSMB5, as known for the primary target of bortezomib, were not significantly changed in U266/velR; but genes involved in ubiquitination, such as transcription elongation factor B1 (TCEB1) and 2 (TCEB2), RING-box protein 1 (RBX1), anaphase promoting complex subunit 11 (ANAPC11), Von Hippel-Lindau tumor suppressor (VHL), and DNA damage-binding protein 1 (DDB1) were differently expressed in U266/velR. Interestingly, overexpression of CD52, one of the candidates related to bortezomib resistance in U266 cells, overcame bortezomib-induced apoptosis.
Our study provided insight into the mechanisms of how MM cells escape apoptosis by bortezomib; activation of NF-κB, increased CD138- population, and a changes of ubiquitination.
RESULTS
Establishment of bortezomib-resistant cell line (U266/velR)
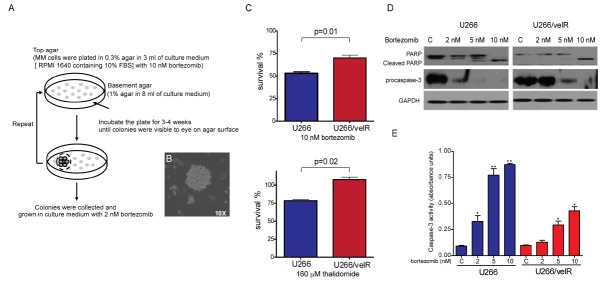
To establish bortezomib-resistant cell lines, three different MM cells (U266, RPMI-8226, and IM9) were grown in soft agar plates, in the presence of 10 nM bortezomib. Only U266 colonies were visible on the soft agar plate, after 3-4 weeks of incubation. Pooled U266 colonies were subsequently plated on new soft agar plate, with 10 nM bortezomib (Fig. 1A). After 2 weeks, the colonies promptly grew again in agar plate (Fig. 1B). The bortezomib-resistant cell line (U266/velR) was grown and maintained in culture medium (RPMI 1640 containing 10% FBS), with 2 nM bortezomib. First, to confirm resistance to bortezomib in U266/velR, we tested the parental U266 and U266/velR, for sensitivity to bortezomib. U266/velR had less sensitivity to bortezomib-induced cell cytotoxicity, compared to the parental cells. In addition, U266/velR showed cross-resistance to thalidomide. U266/velR was 1.5 fold more resistant to both bortezomib and thalidomide, than their parental cells were (Fig. 1C). We further examined the effect of bortezomib-induced apoptotic signal in U266/velR. Treatment of bortezomib led to PARP cleavage and reduction of procaspase-3 expression, as well as induction levels of caspase-3 activities in the parental cells. The effects were substantially reduced in U266/velR (Fig. 1D and E).
Fig. 1. U266/velR reduced sensitivity to bortezomib. (A) Flowchart for isolation of resistant clone to bortezomib. Detailed description about the flowchart can be found in Materials and Methods. (B) After U266 colonies were visible to the eye on the agar surface, colonies were observed, using an inverted microscope with a 10X objective lens. (C) U266 and U266/velR cells were treated for 72 h, with either 10 nM bortezomib, or 160 μM thalidomide. A cell proliferation assay was performed, as described in Materials and Methods. Data represented the mean ± SEM of 3 independent experiments. (D) U266 and U266/velR cells were treated with indicated concentrations of bortezomib for 24 h, and each 30 μg of whole-cell extracts was analyzed for PARP, procaspase-3 and GAPDH, by Western blot. (E) U266 and U266/velR cells were treated with indicated concentrations of bortezomib for 24 h, and the cells were then harvested, and lysed. These cells lysates were subjected to caspase-3 activity assay, using caspase-3 colorimeric activity assay kit. Data represented the mean ± SEM of 3 independent experiments (*P < 0.05; **P < 0.01).
NF-κB-mediated acquired bortezomib resistance in U266/velR
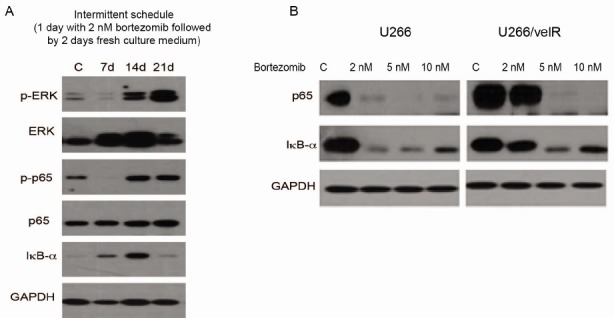
To assess changes in activation of cell signaling in U266, during acquisition of resistance to bortezomib, U266 cells that were cultured for 2 weeks with low dose of bortezomib (2 nM) were monitored for activation of ERK and NF-κB, by Western blotting. U266 cells that survived bortezomib pressure gradually increased pERK and p-p65 levels (Fig. 2A). Next, we further examined if the constitutive activation of NF-κB serves to blunt the efficacy of bortezomib. U266 cells dramatically reduced p65 and IκBα expression with exposure to bortezomib; whereas, their expression persisted in U266/velR that were treated with 2 nM bortezomib (Fig. 2B).
Fig. 2. NF-κB-mediated acquired bortezomib resistance in U266/velR. (A) U266 cells were treated with 2 nM bortezomib on an intermittent schedule (1 day with bortezomib, followed by 2 days without bortezomib). After 7, 14, and 21 days of post-treatment of bortezomib, Cells were lysed and subjected to immunoblotting with p-ERK, ERK, p-p65, IκBα, and GAPDH, respectively. (B) U266 and U266/velR cells were treated with the indicated concentrations of bortezomib for 24 h, and whole-cell extracts were subjected to immunoblotting with p65, IκBα, and GAPDH.
U266/velR cells contained higher proportion of CD138-
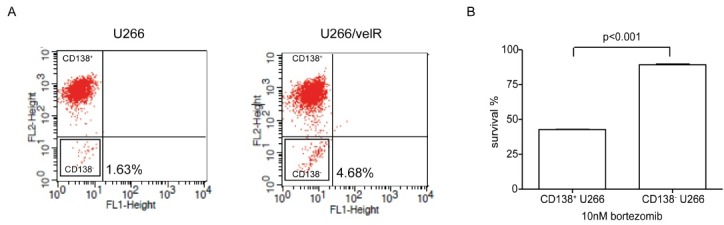
It has been postulated that a tumor-initiating population, known as cancer stem cells (CSCs), may be responsible for eventual relapses. CSCs are associated with chemoresistance, due to having high drug efflux capacity, and their properties, including the quiescent state. To determine whether bortezomib resistance enriched CD138- population, we performed FACS analysis, to confirm the CD138+ and CD138- fraction in U266 and U266/velR. The percentage of CD138- population was approximately 3-fold higher in U266/velR, than of that in U266 (Fig. 3A). We then evaluated the cytotoxic effect of sorted CD138+ and CD138- MM cells to bortezomib. Interestingly, the CD138- population of MM cells was shown to be resistant to bortezomib-induced apoptosis (Fig. 3B).
Fig. 3. U266/velR increased CD138- population. (A) After U266 and U266/velR cells were labeled with anti-human CD138, FACS analysis was performed, to determine CD138 expression of the cell lines. Percentage of CD138- U266 was represented by the mean ± SEM of 3 independent experiments. (B) CD138+ and CD138- U266 cells were isolated, using MACS. Cell proliferation was measured after CD138+ and CD138- cells were treated with 10 nM bortezomib for 72 h, using CCK-8 cell proliferation kit. Data represented the mean ± SEM of 3 independent experiments.
Genetic characterization in U266/velR
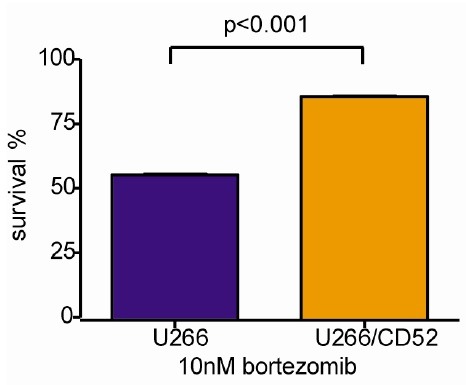
Gene expression analysis was performed on U266/velR, and the parental U266, to profile expression changes resulting from bortezomib resistance. We identified that 294 genes, among 16,298 genes, were differentially regulated (251 genes transcripts were increased, 42 genes transcripts were decreased). Expression of proteasome β5 subunit (PSMB5), known as a target related to bortezomib resistance, and other proteasome subunit genes were not differentially regulated in U266/velR (Table S1A). However, genes involved in ubiquitination, indicated in Table S1B, were significantly up-regulated and down-regulated. To find novel candidates for bortezomib resistance, we selected candidate genes not involved in mechanisms in the action of bortezomib (Table S1C). Among the candidate genes, we evaluated CD52 gene, for association with resistance. Overexpression of CD52 in U266 cells decreased bortezomib-induced apoptosis (Fig. 4).
Fig. 4. Genetic characterization in U266/velR. U266 and over-expression of CD52 in U266 (U266/CD52) were treated with 10 nM bortezomib for 72 h, using CCK-8 cell proliferation kit. Data represented the mean ± SEM of 3 independent experiments.
DISCUSSION
Despite bortezomib having been considered as a promising drug for multiple myeloma (MM), resistance still arises in MM patients, caused by refractoriness, and acquisition of resistance after treatment (7, 12, 13). Development of chemoresistance is a major problem in curing the disease. To overcome chemoresistance, the characteristics of cancer cells having resistant phenotype to anti-cancer drug need to be understood. We succeeded in establishing bortezomib-resistant U266 cells (U266/velR), from three different MM cell lines (U266, RPMI 8226, and IM9). Our preliminary data showed that IM9 was the most sensitive to bortezomib. In addition, the other two cell lines, U266 and RPMI 8226 responded to IL-6 and activated IL-6 downstream cell signaling, such as MAPK and STAT3 pathway; but IM9 was unresponsive to IL-6 (data not shown).
NF-κB is an important mediator in the activation of IL-6. Moreover, it has been reported that NF-κB activation has a crucial role in the regulation of bortezomib response to MM cells (10, 14, 15). Interestingly, U266 cells that were treated with a low dose of bortezomib (2 nM) on an intermittent schedule showed gradually increased expressions of p-ERK and p-p65, which are involved in NF-κB pathway. Our results indicated that NF-κB activity regulates bortezomib anti-cancer effect, as well as that its activity is required for acquisition of resistance to bortezomib. A previous study from our laboratory showed that 6-amino-4-quinazoline, an NF-κB activation inhibitor, combined with bortezomib, effectively inhibited IL-6 signaling in MM cells (14). Therefore, our data suggested that the targeting of NF-κB could be a possible strategy, to overcome bortezomib resistance.
The growth rate of U266/velR was slower, and their cell size was larger than that of the parental U266. Slower growth pattern and increase in cell size have been observed in cancer cells with acquired resistance in vitro (16). We hypothesized that their slow growth might be caused by an increased number of quiescent cancer cells, characterized by resistance to therapy, and stem-like properties. Studies have been exploring the role of cancer stem cells in chemoresistance (17, 18). We wondered whether cancer stem cells could escape from bortezomib-induced apoptosis, and enrich the population, during acquisition of resistance. Interestingly, U266/velR had a higher fraction of CD138-, compared to that of the parental U266. Moreover, CD138- population from the parental had decreased sensitivity to bortezomib. Its functional significance and mechanisms of resistance in the CD138- population to bortezomib require further investigation.
In further analysis for identifying targets related to bortezomib resistance, we found that several genes involved in ubiquitination were differentially expressed in U266/velR. However, the majority of proteoasome subunit genes directly associated with bortezomib treatment did not show a significant difference. Interestingly, the expression level of proteasome maturation protein (POMP), which is predicted to be a molecular partner of proteasome β5 subunit (PSMB5), in U266/velR was one-fold higher than that in U266 (data not shown). POMP is known to be a precursor intermediate during 20S proteasome biogenesis, and its expression is critical for de novo biogenesis of proteasomes (19, 20). Furthermore, Namalwaad cells with increased proteasomal proteolytic activities have recently been shown to exhibit increased POMP expression (21). CD52, a human leukocyte differentiation antigen, and one of the candidates in microarray data, was up-regulated in U266/velR. CD52 was detected on plasma cells, but its expression levels were lower than chronic lymphocytic leukemia (CLL) (22). It suggested that Alemtuzumab (Campath-1H), a humanized CD52 monoclonal antibody, currently used for CLL, may not be appropriate therapy for MM. However, our data suggested that elaborated CD52 levels on MM cells may be involved in acquired resistance to bortezomib. We are investigating the relationship between CD52 expression and bortezomib resistance in MM, and molecular mechanisms related to this.
Taken together, our findings indicated that U266 cells achieve survival, through combinational changes of NF-κB activation, ubiquitination, and CD138- population, following exposure to bortezomib. Also, these findings provide information for developing strategies for bortezomib resistant-MM patients.
MATERIALS AND METHODS
Cell culture
Multiple myeloma (MM) cell lines, U266 and RPMI 8226 were kindly provided by Dr. D Lee (Department of Laboratory of Medicine, Seoul National University College of Medicine). IM-9 was obtained from the Korean Cell Line Bank (KCLB). The cell lines were cultured in RPMI 1640 (Gibco-BRL, Gaithersburg, MD, USA), supplemented with 10% FBS, L-glutamine, and penicillin-streptomycin (GIBCO, Grand Island, NY, USA), at 37℃ and 5% CO2.
Antibodies and reagents
The specific proteasome inhibitor, bortezomib (VelcadeTM; formerly known as bortezomib), was generously provided by Janssen Korea, Ltd. (Seoul, Korea). PARP [rabbit polyclonal], p65 [mouse monoclonal], IκBα [mouse monoclonal], and GAPDH [rabbit polyclonal] (Santa Cruz, CA, USA), phospho-MAPK sampler kit, p-IκBα [mouse monoclonal] (Cell Signaling Technology, Beverly, MA, USA), and p-p65 [rabbit polyclonal] (Signalway Antibody Co., Ltd, Pearland, TX, USA) antibodies were used.
Establishment of bortezomib-resistant cell line
Soft agar was prepared, by autoclaving agarose in distilled water, before use. U266 cells (5×105 cells/100-mm plate) were spun down, resuspended in cooled 0.3% agar in culture media (RPMI 1640 medium containing 10% FBS) with 10 nM bortezomib, and then seeded onto a solidified base layer, containing 1% agar in the culture media. The plates were kept at room temperature for 30 min, and then transferred to a CO2 incubator. The plates were incubated for several weeks (3-4 weeks), until colonies were visible to the eye on the agar surface. We picked individual colonies growing on soft agar, and pooled colonies were grown in the culture media with 2 nM bortezomib, to avoid losing the resistant phenotype. To examine the response to bortezomib in the bortezomib resistant cell line (U266/velR), we performed experiments after the U266/velR were regrown in bortezomib-free culture media for 3 days.
Transfection
The human CD52 cDNA was purchased from OriGene (Rockville, MD, USA). The nucleofection protocols of the manufacturer program (for Nucleofactor II, Amaxa, Inc., Scientific Support (Walkersville, MD, USA) were followed, and the cell proliferation assay was performed.
Cell proliferation assay
Cell proliferation assay was performed, using Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer’s instructions (23).
Western blot analysis
Cells were washed once in ice-cold phosphate-buffered saline (PBS), and resuspended in lysis buffer [20 mM MOPS (pH 7.0), 2 mM EGTA, 5 mM EDTA, 30 mM sodium fluoride, 60 mM β-glycerophosphate (pH 7.2), 20 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1% Triton X-100, 1 mM PMSF, aprotinin, leupeptine, and 1 μg/ml pepstatin]. The proteins were resolved by 10% SDS-polyacrylamide gel electrophoresis, and electrotransferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 and 5% nonfat dry milk for 1 h at room temperature, and incubated with the appropriate primary antibodies for 2 h. Immunoreactive proteins were detected, using a horseradish peroxidase-conjugated secondary antibody (Jackson Immuno Research Laboratories, Inc., PA, USA), and enhanced with chemiluminescence reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA) (24).
FACS analysis
U266 and U266/velR cells (106 in 100 μl) were incubated with 20 μl of phycoerythrin-conjugated anti-CD138 (Becton Dickinson, San Jose, CA, USA) for 30 min at 4℃, washed, and then fixed with 2% paraformaldehyde. Then, the samples were analyzed by FACS Caliber flow cytometer (Becton Dickinson) (25).
Microarray analysis and data statistics
The total RNA was amplified and purified, using the Ambion RNA purification kit (Ambion, Austin, USA), to yield biotinylated cRNA. Labeled cRNA samples were hybridized to each human HT-12 expression bead array for 16-18 h at 58℃, according to the manufacturer’s instructions (Illumina, Inc., San Diego, USA). Detection of array signal was carried out using Amersham fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Little Chalfont, UK), according to the bead array manual. Arrays were scanned with an Illumina bead array reader confocal scanner, according to the manufacturer’s instructions. Array data export processing and analysis were performed, using Illumina Bead Studio v3.1.3 (Gene Expression Module v3.3.8). Raw data were extracted, using the software provided by the manufacturer (Illumina Bead Studio v3.1.3, Gene Expression Module v3.3.8). Gene signal value was transformed by logarithm, and normalized by the quantile method. The comparative analysis between U266 and U266/velR was carried out using t-test [adjusted Benjamini-Hochberg FDR (false discovery rate) was controlled at 5%], and fold-change calculation. Hierarchical cluster analysis was performed, using complete linkage, and Euclidean distance as a measure of similarity. K-means cluster analysis was performed, using differential method. All data analysis and visualization of differentially expressed genes were conducted using Array AssistⓇ (Stratagene, La Jolla, USA), and R statistical language v. 2.4.0. Biological ontology-based analysis was performed, by using the Panther database: http://www.pantherdb.org.
Statistic analysis
The statistical significance of differences was assessed, using Graphpad Prism 5 software.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No.2011-0029703), and by the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (PGM21-A111218).
References
- 1.Dalton W. S., Jove R. Drug resistance in multiple myeloma: approaches to circumvention. Semin Oncol. (1999);26:23–27. [PubMed] [Google Scholar]
- 2.Hideshima T., Chauhan D., Richardson P., Anderson K. C. Identification and validation of novel therapeutic targets for multiple myeloma. J. Clin. Oncol. (2005);23:6345–6350. doi: 10.1200/JCO.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 3.Podar K., Chauhan D., Anderson K. C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. (2009);23:10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams J. Proteasome inhibition in cancer: development of PS-341. Semin Oncol. (2001);28:613–619. doi: 10.1016/S0093-7754(01)90034-X. [DOI] [PubMed] [Google Scholar]
- 5.Jung L., Holle L., Dalton W. S. Discovery, Development, and clinical applications of bortezomib. Oncology (Williston Park) (2004);18:4–13. [PubMed] [Google Scholar]
- 6.Kisselev A. F., Goldberg A. L. Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. (2001);8:739–758. doi: 10.1016/S1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 7.Mateos M. V. Management of treatment-related adverse events in patients with multiple myeloma. Cancer Treat Rev. 36(Suppl 2):S24–32. doi: 10.1016/S0305-7372(10)70009-8. [DOI] [PubMed] [Google Scholar]
- 8.Oerlemans R., Franke N. E., Assaraf Y. G., Cloos J., van Zantwijk I., Berkers C. R., Scheffer G. L., Debipersad K., Vojtekova K., Lemos C., van der Heijden J. W., Ylstra B., Peters G. J., Kaspers G. L., Dijkmans B. A., Scheper R. J., Jansen G. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. (2008);112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 9.Keats J. J., Fonseca R., Chesi M., Schop R., Baker A., Chng W. J., Van Wier S., Tiedemann R., Shi C. X., Sebag M., Braggio E., Henry T., Zhu Y. X., Fogle H., Price-Troska T., Ahmann G., Mancini C., Brents L. A., Kumar S., Greipp P., Dispenzieri A., Bryant B., Mulligan G., Bruhn L., Barrett M., Valdez R., Trent J., Stewart A. K., Carpten J., Bergsagel P. L. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. (2007);12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markovina S., Callander N. S., O'Connor S. L., Kim J., Werndli J. E., Raschko M., Leith C. P., Kahl B. S., Kim K., Miyamoto S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res. (2008);6:1356–1364. doi: 10.1158/1541-7786.MCR-08-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hideshima T., Chauhan D., Hayashi T., Akiyama M., Mitsiades N., Mitsiades C., Podar K., Munshi N. C., Richardson P. G., Anderson K. C. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene. (2003);22:8386–8393. doi: 10.1038/sj.onc.1207170. [DOI] [PubMed] [Google Scholar]
- 12.Richardson P. G., Barlogie B., Berenson J., Singhal S., Jagannath S., Irwin D., Rajkumar S. V., Srkalovic G., Alsina M., Alexanian R., Siegel D., Orlowski R. Z., Kuter D., Limentani S. A., Lee S., Hideshima T., Esseltine D. L., Kauffman M., Adams J., Schenkein D. P., Anderson K. C. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. (2003);348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 13.Kastritis E., Palumbo A., Dimopoulos M. A. Treatment of relapsed/refractory multiple myeloma. Semin Hematol. (2009);46:143–157. doi: 10.1053/j.seminhematol.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Park J., Ahn K. S., Bae E. K., Kim B. S., Kim B. K., Lee Y. Y., Yoon S. S. Blockage of interleukin-6 signaling with 6-amino-4-quinazoline synergistically induces the inhibitory effect of bortezomib in human U266 cells. Anticancer Drugs. (2008);19:777–782. doi: 10.1097/CAD.0b013e32830c236a. [DOI] [PubMed] [Google Scholar]
- 15.Malara N., Foca D., Casadonte F., Sesto M. F., Macrina L., Santoro L., Scaramuzzino M., Terracciano R., Savino R. Simultaneous inhibition of the constitutively activated nuclear factor kappaB and of the interleukin-6 pathways is necessary and sufficient to completely overcome apoptosis resistance of human U266 myeloma cells. Cell Cycle. (2008);7:3235–3245. doi: 10.4161/cc.7.20.6832. [DOI] [PubMed] [Google Scholar]
- 16.Leuschner I., Heuer T., Harms D. Induction of drug resistance in human rhabdomyosarcoma cell lines is associated with increased maturation: possible explanation for differentiation in recurrences? Pediatr Dev Pathol. (2002);5:276–282. doi: 10.1007/s10024-001-0132-0. [DOI] [PubMed] [Google Scholar]
- 17.Matsui W., Wang Q., Barber J. P., Brennan S., Smith B. D., Borrello I., McNiece I., Lin L., Ambinder R. F., Peacock C., Watkins D. N., Huff C. A., Jones R. J. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. (2008);68:190–197. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jakubikova J., Adamia S., Kost-Alimova M., Klippel S., Cervi D., Daley J. F., Cholujova D., Kong S. Y., Leiba M., Blotta S., Ooi M., Delmore J., Laubach J., Richardson P. G., Sedlak J., Anderson K. C., Mitsiades C. S. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood. (2011);117:4409–4419. doi: 10.1182/blood-2010-02-267344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witt E., Zantopf D., Schmidt M., Kraft R., Kloetzel P. M., Kruger E. Characterisation of the newly identified human Ump1 homologue POMP and analysis of LMP7(beta 5i) incorporation into 20 S proteasomes. J. Mol. Biol. (2000);301:1–9. doi: 10.1006/jmbi.2000.3959. [DOI] [PubMed] [Google Scholar]
- 20.Kruger E., Kloetzel P. M., Enenkel C. 20S proteasome biogenesis. Biochimie. (2001);83:289–293. doi: 10.1016/S0300-9084(01)01241-X. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs D., Berges C., Opelz G., Daniel V., Naujokat C. Increased expression and altered subunit composition of proteasomes induced by continuous proteasome inhibition establish apoptosis resistance and hyperproliferation of Burkitt lymphoma cells. J. Cell Biochem. (2008);103:270–283. doi: 10.1002/jcb.21405. [DOI] [PubMed] [Google Scholar]
- 22.Rawstron A. C., Laycock-Brown G., Hale G., Davies F. E., Morgan G. J., Child J. A., Hillmen P., Owen R. G. CD52 expression patterns in myeloma and the applicability of alemtuzumab therapy. Haematologica. (2006);91:1577–1578. [PubMed] [Google Scholar]
- 23.Jee H., Lee S.-H., Park J.-W., Lee B.-R., Nam K.-T., Kim D.-Y. Connexin32 inhibits gastric carcinogenesis through cell cycle arrest and altered expression of p21Cip1 and p27Kip1. BMB Rep. (2013);46:25–30. doi: 10.5483/BMBRep.2013.46.1.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim E.-G., Shin E. Y. Nuclear Rac1 regulates the bFGF-induced neurite outgrowth in PC12 cells. BMB Rep. (2013);46:617–622. doi: 10.5483/BMBRep.2013.46.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng C., Zhou Y., Yang C, Chen J., Wang J., Zhang J., Zhao G. Detection of rare point mutation via allele-specific amplification in emulsion PCR. BMB Rep. (2013);46:270–275. doi: 10.5483/BMBRep.2013.46.5.155. [DOI] [PMC free article] [PubMed] [Google Scholar]