

Abstract
Endothelial cell protein C receptor (EPCR) plays important roles in blood coagulation and inflammation. EPCR activity is markedly changed by ectodomain cleavage and release as the soluble EPCR. EPCR can be shed from the cell surface, which is mediated by tumor necrosis factor-α converting enzyme (TACE). Oroxylin A (OroA), a major component of Scutellaria baicalensis Georgi, is known to exhibit anti-angiogenic, antiinflammation, and anti-invasive activities. However, little is known about the effects of OroA on EPCR shedding. Data showed that OroA induced potent inhibition of phorbol-12-myristate 13-acetate (PMA), tumor necrosis factor (TNF)-α, interleukin (IL)-1β and on cecal ligation and puncture (CLP)-induced EPCR shedding through suppression of TACE expression and activity. In addition, treatment with OroA resulted in reduced PMA-stimulated phosphorylation of p38, extracellular regulated kinases (ERK) 1/2, and c-Jun N-terminal kinase (JNK). These results demonstrate the potential of OroA as an anti-sEPCR shedding reagent against PMA and CLP-mediated EPCR shedding. [BMB Reports 2014; 47(6): 336-341]
Keywords: CLP, EPCR shedding, Oroxylin A, PMA
INTRODUCTION
Endothelial protein C receptor (EPCR) was initially described as an endothelial transmembrane glycoprotein capable of binding to protein C (PC) and activated protein C (APC) with high affinity (Kd = 30 nM) (1). EPCR belongs to the CD1/major histocompatibility complex superfamily, and is reasonably conserved between species, which suggests a prominent functional role; its expression was abundant in the endothelium of large vessels (2). The primary role of EPCR is anticoagulation because EPCR binds to protein C with high affinity and augments the activation of protein C by thrombin-thrombomodulin (TM) complex (3). Released activated protein C (APC) binds to cofactor protein S and degrades the coagulation factors Va and VIIIa (3).
Exposition of EPCR is strongly dependent on cleavage of EPCR and its release in the soluble form (sEPCR) (4, 5). Activity and function of EPCR, a cell surface component, are regulated through metalloproteinase-mediated shedding (1, 6). A number of in vitro-treatments can result in potent up-regulation of the shedding of EPCR and reduce the rate of protein C activation (5), and released sEPCR itself inhibits the function of protein C and its activated form (APC) through competition with APC/PC binding on membrane-associated EPCR (7). sEPCR can be detected in plasma, resulting from shedding of membrane EPCR, with a plasma concentration of approximately 100 ng/ml; high levels of sEPCR in systemic inflammatory diseases have been reported (8). In addition, the interaction of sEPCR with factor VII (FVII) and its activated form (FVIIa) was demonstrated, providing evidence that EPCR and its soluble form serve as binding sites for FVII/FVIIa and inhibit the procoagulant activity of FVIIa-tissue factor complex (9, 10). In vitro studies have reported a dramatic increase in EPCR shedding from the endothelium by a wide variety of inflammatory mediators (IL-1β, H2O2, and phorbol myristate acetate) and thrombin, and EPCR shedding is potentiated by the microtubule disrupting agent, nocodazole (6). In addition, phosphorylation of p38MAPK, ERK1/2, and JNK was increased by stimulation with PMA (11-13) and activation of TACE occurs upon activation of ERK or p38 (14, 15).
Oroxylin A (OroA) is one of the major flavonoids, the most common active ingredients in the root of Scutellaria baicalensis Georgi (16). OroA has been widely studied and proven to possess a wide spectrum of pharmacological properties, including anti-inflammatory, pro-apoptotic, and anti-invasive activities (17-19). A recent study reported that OroA had anti-inflammatory effects in rodents (20). Noting that sEPCR serves as a marker of vascular barrier integrity in vascular inflammatory disease and that sEPCR is involved in the pathophysiology of sepsis (8, 21), we hypothesized that OroA may have anti-sEPCR shedding activity. Therefore, in the current study, we investigated the effect of OroA against PMA-induced EPCR shedding in human endothelial cells and in a cecal ligation and puncture (CLP) model of septicemia in mice.
RESULTS AND DISCUSSION
Effect of OroA on PMA, TNF-α, or IL-1β-induced EPCR shedding
Previous studies have reported that PMA stimulates EPCR shedding from HUVECs (22, 23). In agreement with previous results, we found that PMA from 1 μM fully stimulated EPCR shedding from HUVECs (Fig. 1A) and cellular EPCR on HUVECs showed a dose-dependent decrease by PMA (Fig. 1B). EPCR shedding by TNF-α or interleukin (IL)-1β also showed an increase (Fig. 1C and D), in agreement with a previous study (11).
Fig. 1. Effect of OroA on PMA, TNF-α and IL-1β-induced EPCR shedding. The effects of various concentrations of OroA on PMA (1 μM, 1 h)-induced EPCR shedding were monitored by measurement of sEPCR (A) or cellular EPCR on HUVECs (B). (C and D) The same as A and B, except that HUVECs were incubated with TNF-α (25 ng/ml for 1 h, white bar) or IL-1β (25 ng/ml for 1 h, black bar). Results indicate the mean ± SEM of three separate experiments. **P < 0.01 vs. PMA alone (A, B) or TNF-α/IL-1β alone (C, D).
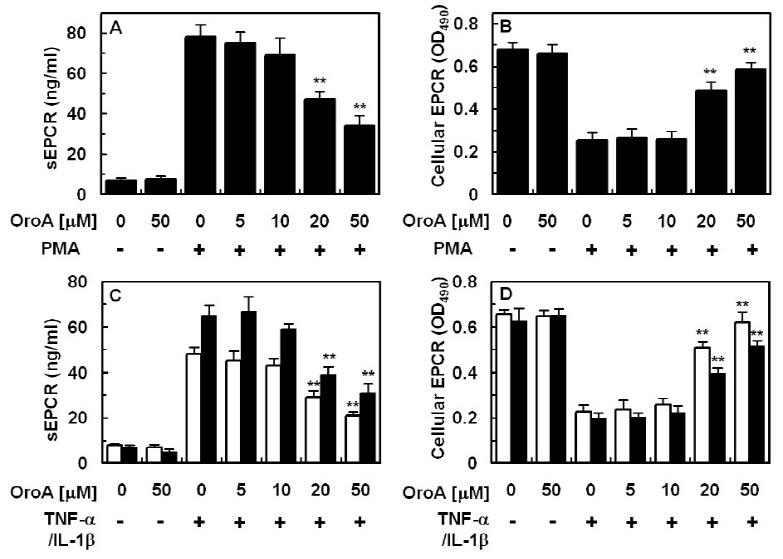
In order to investigate the effect of OroA on PMA-mediated EPCR shedding, endothelial cells were pretreated with increasing concentrations of OroA for 6 h, followed by stimulation with 1 μM PMA for 1 h. As shown in Fig. 1A and B, treatment with OroA resulted in inhibition of EPCR shedding induced by PMA in endothelial cells, with an optimal effect at 20-50 μM. As shown in Fig. 1A, OroA alone (50 uM) did not affect the shedding of EPCR. Therefore, OroA alone (50 uM) did not affect the expression of membrane bound EPCR (Fig. 1B). OroA also induced suppression of TNF-α or IL-1β-mediated EPCR shedding in HUVECs (Fig. 1C and D).
Effects of OroA on PMA-stimulated expression and activity of TACE
A previous study reported that PMA-stimulated EPCR shedding is mediated by tumor necrosis factor-α converting enzyme/ADAM17 (TACE) (23). In order to determine whether OroA could inhibit stimulation of TACE expression and activity, endothelial cells were pretreated with increasing concentrations of OroA for 6 h, followed by stimulation with 1 μM PMA for 1 h. Data showed that OroA inhibited TACE expression (Fig. 2A and Fig. 4D) and activity (Fig. 2B) induced by PMA in endothelial cells
Fig. 2. Effect of OroA on PMA-stimulated expression and activity of TACE. The effects of various concentrations of OroA on PMA (1 μM, 1 h)-induced expression (A) or activity (B) of TACE were monitored by measurement of TACE ELISA (A) or TACE activity assay Kit. All results indicate the mean ± SEM of three separate experiments. *P < 0.05 or **P < 0.01 vs. PMA alone.
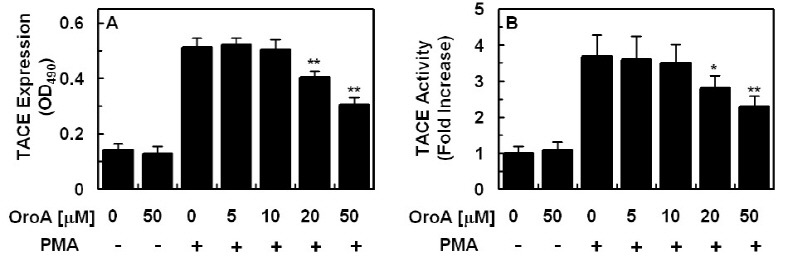
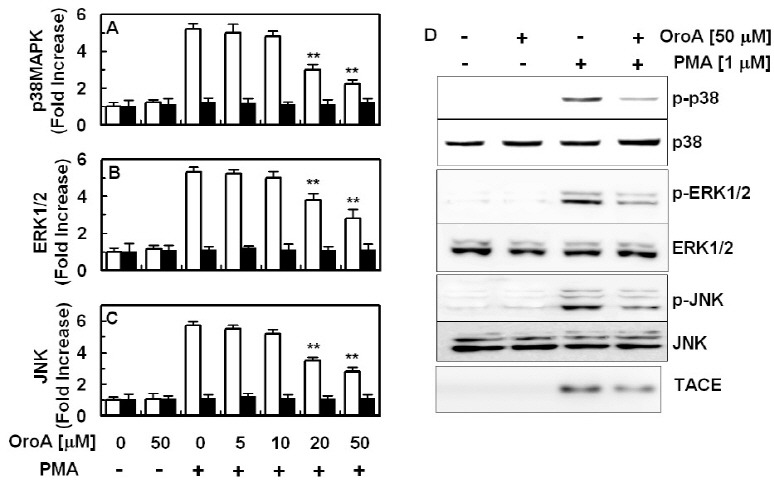
Fig. 4. Effect of OroA on PMA-induced phosphorylation of p38MAPK, ERK1/2, and JNK. PMA (1 μM, 1 h)-mediated phosphorylation of phosphop38MAPK (A, white bar) or total p38 MAPK (A, black bar), phospho-ERK1/2 (B, white bar) or total ERK1/2 (B, black bar) and phospho-JNK (C, white bar) or total JNK (C, black bar) were analyzed after treatment of cells with the indicated concentrations of OroA. Results are expressed as fold increase over control values. (D) Western blotting for phosphorylated or total p38, ERK1/2, JNK or TACE. All results indicate the mean ± SEM of three separate experiments. **P < 0.01 vs. PMA alone.
Effect of OroA on CLP-induced EPCR shedding
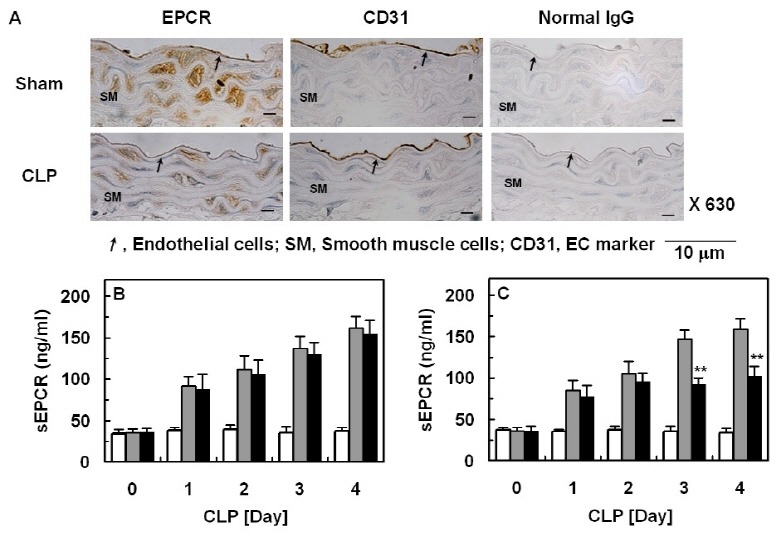
To confirm the inhibitory effects of OroA on EPCR shedding in vivo, we used a CLP mouse model, because this model more closely resembles human sepsis (24, 25). In CLP-induced septic mice, immunohistochemical analysis showed that expression of cellular EPCR was decreased compared to normal control (Fig. 3A). Administration of OroA at a single dose (28.4 μg, 12 h after CLP) did not result in prevention of CLP-induced EPCR shedding (Fig. 3B); therefore, it was administered twice (28.4 μg per mouse, once 12 h, then 50 h after CLP), resulting in a decrease in EPCR shedding (Fig. 3C). Assuming an average body weight of 20 g and an average blood volume of 2 ml, the amounts of OroA produced a concentration of approximately 50 μM in peripheral blood. This marked benefit achieved by administration of OroA suggested that inhibition of EPCR shedding provides a therapeutic strategy for management of severe vascular diseases.
Fig. 3. Effect of OroA on CLP-induced EPCR shedding. (A) Immunohistochemical stains of blood vessel for cellular EPCR from CLP-operated mice (four days after CLP), as indicated in the text. Staining results from sham operated mice are compared. Results are representative of three to six stainings from two independent experiments per condition. (B-C) Serum was obtained from sham-operated (white bar) or CLP-induced septic mice (gray bar) on the indicated day after CLP surgery (n=5) on different days for different mice. Or, OroA was administered once (B, black bar, 28.4 μg per mouse via i.v., once 12 h after CLP) or twice (C, black bar, 28.4 μg per mouse via i.v., once 12 h, then 50 h after CLP) on different days for different mice. EPCR shedding was then monitored by measurement of sEPCR. All results indicate the mean ± SEM of three separate experiments. **P < 0.01 vs. CLP alone.
Effects of OroA on PMA-stimulated phosphorylation of p38MAPK, ERK1/2, and JNK
Previous studies have reported involvement of p38MAPK, ERK1/2, and JNK in cytokine-induced EPCR shedding and phosphorylation of p38MAPK, ERK1/2, and JNK was known to be increased by stimulation with PMA (11-13). Therefore, in order to determine the molecular mechanisms of suppression of PMA-induced EPCR shedding by OroA, the effects of OroA on PMA-stimulated phosphorylation of p38MAPK, ERK1/2, and JNK were tested. As shown in Fig. 4, treatment with OroA resulted in reduction of PMA-stimulated phosphorylation of p38MAPK (Fig. 4A), ERK1/2 (Fig. 4B), and JNK (Fig. 4C). These results were also confirmed by western blotting (Fig. 4D). To confirm the involvement of MAPK on the EPCR shedding, a panel of pharmacological inhibitors of MAP kinases was used. As shown in supplementary figure, distinct attenuation of sEPCR release in HUVECs was observed after treatments with PD-98059 as pharmacological inhibitor of ERK 1/2 activity, SB-203580 as inhibitor of p38MAPK, and SP-600125 as inhibitor of JNK.
Tseng et al. recently reported that the active compound, Oroxylin A, an important component of Scutellaria baicalensis Georgi, had anti-inflammatory responses in lipopolysaccharide (LPS) treated rodents (20). They demonstrated that LPS-mediated inflammatory responses, such as altered white blood cell counts, elevated plasma tumor necrosis factor (TNF)-α and nitric oxide (NO), increased pulmonary edema, thickened alveolar septa, and decreased survival rate, were ameliorated by administration of OroA after LPS challenge (20). This post-treatment also resulted in significantly attenuated LPS-induced activation of nuclear factor-κB (NF-κB) and release of high mobility group box 1 (HMGB1) in lung tissues (20). In addition, post-treatment with OroA administered after LPS challenge in mice resulted in a significantly increased survival rate (20).
Phosphorylation of p38MAPK, ERK1/2, and JNK is known to be increased by stimulation with PMA (11-13) and activation of TACE occurs upon activation of ERK or p38 MAPK (14, 15); therefore, in order to define the processes responsible for inhibition of PMA-stimulated shedding of EPCR and expression of TACE by OroA, we investigated involvement of MAPK signaling pathways in PMA stimulated condition. MAPKs comprise a family of highly conserved serine/threonine protein kinases implicated as having key regulatory roles in mediation of inflammation (26). Three major classes of MAPKs are represented by ERK 1/2 and the two stress-activated protein kinase families, JNK and p38 MAPK. As shown in Figs. 2 and 4, PMA stimulated expression of TACE and phosphorylation of p38 MAPK, ERK1/2, JNK, and OroA inhibited these responses by PMA. Therefore, OroA inhibited shedding of EPCR by inhibition of PMA-stimulated expression of TACE and activation of MAPKs.
Noting that EPCR shedding is involved in the pathophysiological pathway in vascular inflammatory diseases, the hypothesis that OroA could be used as a candidate therapeutic for treatment of vascular inflammatory diseases has strengthened by the finding of a previous study (20) and the current finding. Collectively, the results of this study show that OroA induced potent inhibition of PMA and CLP-induced EPCR shedding by suppressing expression of TACE. In addition, OroA reduced PMA-stimulated phosphorylation of p38 MAPK, ERK 1/2, and JNK. A large number of cell membrane bound proteins were shed by TACE, and, in this study, EPCR shedding was mediated by TACE, which is inhibited by OroA. Even though the use of OroA for therapeutic purposes could have non-specific effects, the data presented in this study provide novel information on the role of OroA on EPCR shedding. Therefore, our findings indicate the potential of OroA as a candidate for treatment of severe vascular inflammatory diseases, such as sepsis and septic shock.
MATERIALS AND METHODS
Reagents
sEPCR and TNF-α were purchased from Abnova (Taiwan). Oroxylin A, phorbol-12-myristate 13-acetate (PMA) and IL-1β were purchased from Sigma (St. Louis, MO).
Cell culture
Primary human umbilical vein endothelial cells (HUVECs) were obtained from Cambrex Bio Science (Charles City, IA) and maintained as previously described (27). HUVECs of passage numbers 3 or 4 were used in the experiments.
Animals and husbandry
Male C57BL/6 mice (6-7-wks old, weighting 18-20 g) were purchased from Orient Bio Co. (Sungnam, KyungKiDo, Korea), and used after a 12-day acclimatization period. Animals were housed five per polycarbonate cage under controlled conditions (20-25℃/RH 40-45%) under a 12:12 hour light/dark cycle, and supplied a normal rodent pellet diet and water ad libitum. All animals were treated in accordance with the Guidelines for the Care and Use of Laboratory Animals issued by Kyungpook National University.
Enzyme-linked immunosorbent assay (ELISA) for cellular EPCR expression
Modified whole-cell ELISA was performed as previously described for determination of expression levels of EPCR on HUVECs (28). Briefly, confluent monolayers of HUVECs were treated with or without OroA for 6 h, followed by treatment with PMA, tumor necrosis factor (TNF)-α or interleukin (IL)-1β for 1 h. Media were then removed and cells were washed with PBS and fixed with 50 μl of 1% paraformaldehyde for 15 minutes at room temperature. After washing, 100 μl of EPCR antibodies (Abnova) was added, and, 1 h (37℃, 5% CO2) later, cells were washed three times, followed by treatment with 100 μl of 1:2,000 peroxidase-conjugated anti-rabbit IgG antibodies (Sigma) for 1 h. Cells were then washed three times and developed using o-phenylenediamine substrate (Sigma). Colorimetric analysis was performed by measurement of absorbance at 490 nm. All measurements were performed in triplicate wells.
Competitive Enzyme-linked immunosorbent assay (Competitive ELISA) for sEPCR and TACE
Ninety six-well plastic flat microtiter plates (Corning, NY) were coated with sEPCR or TACE protein in 20 mM carbonate-bicarbonate buffer (pH 9.6) containing 0.02% sodium azide overnight at 4℃. Lyophilized culture media were prepared for sEPCR and total cell lysates were prepared for TACE using lysis buffer containing (mM): Tris-HCl (20) pH 7.5, EGTA (0.5), EDTA (2), dithiothreitol (2), p-methylsulfonyl fluoride (0.5), and 10 μg/ml leupeptin. Plates were then rinsed three times in PBS-0.05% Tween 20 (PBS-T) and kept at 4℃. Prepared samples from cell culture media and mice plasma for sEPCR or from cell lysates for TACE were pre-incubated with anti-EPCR antibodies (1:500, Abnova) or anti-TACE antibodies (1:500, Santa Cruz) in 96-well plastic round microtiter plates for 90 min at 37℃, transferred to pre-coated plates, and incubated for 30 min at room temperature. Plates were then rinsed three times with PBS-T, incubated for 90 min at room temperature with peroxidase-conjugated anti-rabbit or anti-goat IgG antibodies (1:2,000, Amersham Pharmacia Biotech), rinsed three times in PBS-T, and incubated for 60 min at room temperature in the dark with 200 μl of substrate solution (100 μg/ml o-phenylenediamine containing 0.003% H2O2). The reaction was then stopped by addition of 50 μl of 8N H2SO4, and absorbances were read at 490 nm.
TACE activity assay
For TACE activity assay, commercially available TACE activity kit (Innozyme TACE activity assay kit, EMD Millipore, Billerica, MA) was used as described previously (29).
Cecal ligation and puncture (CLP)
For induction of sepsis, male mice were anesthetized with zoletil 50 and rompun. The CLP-induced sepsis model was prepared as previously described (30). In brief, a 2 cm midline incision was made in order to allow exposure of the cecum and adjoining intestine. The cecum was then tightly ligated using a 3.0-silk suture at 5.0 mm from the cecal tip, punctured once with a 22-gauge needle, gently squeezed in order to extrude a small amount of feces, and returned to the peritoneal cavity. The laparotomy site was then stitched with 4.0-silk. In sham controls, the cecum was exposed but not ligated or punctured and then returned to the abdominal cavity. This protocol was approved in advance by the Animal Care Committee at Kyungpook National University.
Immunohistochemistry
To analyze the expression pattern of EPCR, aortas from CLP-induced septic (Day 4) and sham operated mice were removed and fixed in 4% formaldehyde solution (Junsei, Japan) in PBS for 20 h at 4℃. After fixation, the aortas were dehydrated through an ethanol series, embedded in paraffin, and cut into 3 μm sections. Deparaffinized sections were quenched in 3% H2O2 in methanol, washed in PBS, placed in boiled 1 mM Tris solution (pH 9.0), supplemented with 0.5 mM EGTA solution in order to reveal the antigens, and blocked in PBS, supplemented with 1% bovine serum albumin, 0.2% gelatin, and 0.05% saponin for 1 h at RT. Sections were incubated with anti-EPCR antibody (Abcam) diluted 1:500 in PBS, and supplemented with 0.1% BSA and 0.3% Triton X 100 for 16 hours at 4℃ in a humidified chamber. After washing in PBS, supplemented with 0.1% BSA, 0.2% gelatin, and 0.05% saponin, the sections were incubated with peroxidase-conjugated anti-rabbit IgG antibody (DAKO, Glostrup, Denmark) for 1 h at RT and then developed using the Liquid DAB+ Substrate-Chromogen System (DAKO). Counterstaining was performed with 0.5% methyl green in ddH2O. Non-immune rabbit IgG (DAKO, at the same concentration instead of EPCR antibody) and anti-CD31 antibody (1:200, Abcam) was used as negative and positive control for immunohistochemistry, respectively.
ELISA for total and phospho p-38MAPK, ERK1/2, and JNK
HUVECs were cultured in 96-well microplates for quantitative determination of p38MAPK, ERK1/2, and JNK phosphorylation. On the day of experiments, culture medium was replaced by serum-free growth medium. Cells were then treated with or without OroA for 6 h, followed by treatment with PMA (1 μM) for 1 h. Activation of p38MAPK, ERK 1/2, and JNK was quantified in nuclear lysates using ELISA kits for total/phosphorylated p38MAPK (Invitrogen, for total p38MAPK or Cell Signaling Technology, for phosphorylated-p38MAPK), total/phospho ERK1/2 and JNK (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
Western blotting
Total cell extracts were prepared by lysing the cells and protein concentration was determined by using Bradford assay methods. Equal amounts of protein were separated by SDS-PAGE (10%) and electroblotted overnight onto Immobilon membrane (Millipore, Billerica, MA). The membranes were blocked for 1 h with 5% low-fat milk-powder TBS (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 0.05% Tween 20 and then incubated either for 1.5 h at room temperature with anti-phospho-p38 or total p38 (1:100, Abnova), anti-phospho-ERK1/2, total ERK1/2, anti-phospho-JNK or total JNK (1:1,000, Cell Signaling Tech.) or TACE anti-EPCR antibodies (1:500,) followed by incubation with horseradish-peroxidase-conjugated secondary antibody and ECL-detection according to the manufacturer's instructions.
Statistical Analysis
Results are expressed as mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical significance was determined using two-way ANOVA of variance (SPSS, version 14.0, SPSS Science, Chicago, Il, USA) and P values of < 0.05 (P < 0.05) were considered significant.
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) funded by the Korea government [MSIP] (Grant No. 2012R1A4A1028835 and 2012-000940).
References
- 1.Fukudome K., Esmon C. T. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J. Biol. Chem. (1994);269:26486–26491. [PubMed] [Google Scholar]
- 2.Fukudome K., Kurosawa S., Stearns-Kurosawa D. J., He X., Rezaie A. R., Esmon C. T. The endothelial cell protein C receptor. Cell surface expression and direct ligand binding by the soluble receptor. J. Biol. Chem. (1996);271:17491–17498. doi: 10.1074/jbc.271.29.17491. [DOI] [PubMed] [Google Scholar]
- 3.Mosnier L. O., Zlokovic B. V., Griffin J. H. The cytoprotective protein C pathway. Blood. (2007);109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 4.Esmon C. T. Structure and functions of the endothelial cell protein C receptor. Crit. Care. Med. (2004);32:S298–301. doi: 10.1097/01.CCM.0000126128.64614.81. [DOI] [PubMed] [Google Scholar]
- 5.Wang L., Bastarache J. A., Wickersham N., Fang X., Matthay M. A., Ware L. B. Novel role of the human alveolar epithelium in regulating intra-alveolar coagulation. Am. J. Respir. Cell Mol. Biol. (2007);36:497–503. doi: 10.1165/rcmb.2005-0425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J., Qu D., Esmon N. L., Esmon C. T. Metalloproteolytic release of endothelial cell protein C receptor. J. Biol. Chem. (2000);275:6038–6044. doi: 10.1074/jbc.275.8.6038. [DOI] [PubMed] [Google Scholar]
- 7.Kurosawa S., Stearns-Kurosawa D. J., Hidari N., Esmon C. T. Identification of functional endothelial protein C receptor in human plasma. J. Clin. Invest. (1997);100:411–418. doi: 10.1172/JCI119548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurosawa S., Stearns-Kurosawa D. J., Carson C. W., D A., Della Valle P., Esmon C. T. Plasma levels of endothelial cell protein C receptor are elevated in patients with sepsis and systemic lupus erythematosus: lack of correlation with thrombomodulin suggests involvement of different pathological processes. Blood. (1998);91:725–727. [PubMed] [Google Scholar]
- 9.Ghosh S., Pendurthi U. R., Steinoe A., Esmon C. T., Rao L. V. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J. Biol. Chem. (2007);282:11849–11857. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Sagaseta J., Montes R., Puy C., Diez N., Fukudome K., Hermida J. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J. Thromb. Haemost. (2007);5:1817–1824. doi: 10.1111/j.1538-7836.2007.02648.x. [DOI] [PubMed] [Google Scholar]
- 11.Menschikowski M., Hagelgans A., Eisenhofer G., Siegert G. Regulation of endothelial protein C receptor shedding by cytokines is mediated through differential activation of MAP kinase signaling pathways. Exp. Cell Res. (2009);315:2673–2682. doi: 10.1016/j.yexcr.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Han H., Du B., Pan X., Liu J., Zhao Q., Lian X., Qian M., Liu M. CADPE inhibits PMA-stimulated gastric carcinoma cell invasion and matrix metalloproteinase-9 expression by FAK/MEK/ERK-mediated AP-1 activation. Mol. Cancer Res. (2010);8:1477–1488. doi: 10.1158/1541-7786.MCR-10-0114. [DOI] [PubMed] [Google Scholar]
- 13.Leng Y., Steiler T. L., Zierath J. R. Effects of insulin, contraction, and phorbol esters on mitogen-activated protein kinase signaling in skeletal muscle from lean and ob/ob mice. Diabetes. (2004);53:1436–1444. doi: 10.2337/diabetes.53.6.1436. [DOI] [PubMed] [Google Scholar]
- 14.Huovila A. P., Turner A. J., Pelto-Huikko M., Karkkainen I., Ortiz R. M. Shedding light on ADAM metalloproteinases. Trends. Biochem. Sci. (2005);30:413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer. (2008);8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 16.Li C., Lin G., Zuo Z. Pharmacological effects and pharmacokinetics properties of Radix Scutellariae and its bioactive flavones. Biopharm. Drug. Dispos. (2011);32:427–455. doi: 10.1002/bdd.771. [DOI] [PubMed] [Google Scholar]
- 17.Li H. N., Nie F. F., Liu W., Dai Q. S., Lu N., Qi Q., Li Z. Y., You Q. D., Guo Q. L. Apoptosis induction of oroxylin A in human cervical cancer HeLa cell line in vitro and in vivo. Toxicology. (2009);257:80–85. doi: 10.1016/j.tox.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z., Lu N., Li C., Li F., Zhao K., Lin B., Guo Q. Oroxylin A inhibits matrix metalloproteinase-2/9 expression and activation by up-regulating tissue inhibitor of metalloproteinase-2 and suppressing the ERK1/2 signaling pathway. Toxicol. Lett. (2012);209:211–220. doi: 10.1016/j.toxlet.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y., Lu N., Ling Y., Gao Y., Chen Y., Wang L., Hu R., Qi Q., Liu W., Yang Y., You Q., Guo Q. Oroxylin A suppresses invasion through down-regulating the expression of matrix metalloproteinase-2/9 in MDA- MB-435 human breast cancer cells. Eur. J. Pharmacol. (2009);603:22–28. doi: 10.1016/j.ejphar.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Tseng T. L., Chen M. F., Tsai M. J., Hsu Y. H., Chen C. P., Lee T. J. Oroxylin-A rescues LPS-induced acute lung injury via regulation of NF-kappaB signaling pathway in rodents. PLoS One. (2012);7:e47403. doi: 10.1371/journal.pone.0047403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borgel D., Bornstain C., Reitsma P. H., Lerolle N., Gandrille S., Dali-Ali F., Esmon C. T., Fagon J. Y., Aiach M., Diehl J. L. A comparative study of the protein C pathway in septic and nonseptic patients with organ failure. Am. J. Respir. Crit. Care Med. (2007);176:878–885. doi: 10.1164/rccm.200611-1692OC. [DOI] [PubMed] [Google Scholar]
- 22.Qu D., Wang Y., Song Y., Esmon N. L., Esmon C. T. The Ser219- ->Gly dimorphism of the endothelial protein C receptor contributes to the higher soluble protein levels observed in individuals with the A3 haplotype. J. Thromb. Haemost. (2006);4:229–235. doi: 10.1111/j.1538-7836.2005.01676.x. [DOI] [PubMed] [Google Scholar]
- 23.Qu D., Wang Y., Esmon N. L., Esmon C. T. Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor-alpha converting enzyme/ADAM17. J. Thromb. Haemost. (2007);5:395–402. doi: 10.1111/j.1538-7836.2007.02347.x. [DOI] [PubMed] [Google Scholar]
- 24.Buras J. A., Holzmann B., Sitkovsky M. Animal models of sepsis: setting the stage. Nat. Rev. Drug. Discov. (2005);4:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 25.Yang H., Ochani M., Li J., Qiang X., Tanovic M., Harris H. E., Susarla S. M., Ulloa L., Wang H., DiRaimo R., Czura C. J., Roth J., Warren H. S., Fink M. P., Fenton M. J., Andersson U., Tracey K. J. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. U.S.A. (2004);101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thalhamer T., McGrath M. A., Harnett M. M. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford). (2008);47:409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 27.Bae J. S., Rezaie A. R. Activated protein C inhibits high mobility group box 1 signaling in endothelial cells. Blood. (2011);118:3952–3959. doi: 10.1182/blood-2011-06-360701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D. C., Lee W., Bae J. S. Vascular anti-inflammatory effects of curcumin on HMGB1-mediated responses in vitro. Inflamm. Res. (2011);60:1161–1168. doi: 10.1007/s00011-011-0381-y. [DOI] [PubMed] [Google Scholar]
- 29.Miller M. A., Meyer A. S., Beste M. T., Lasisi Z., Reddy S., Jeng K. W., Chen C. H., Han J., Isaacson K., Griffith L. G., Lauffenburger D. A. ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc. Natl. Acad. Sci. U.S.A. (2013);110:E2074–2083. doi: 10.1073/pnas.1222387110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., Liao H., Ochani M., Justiniani M., Lin X., Yang L., Al-Abed Y., Metz C., Miller E. J., Tracey K. J., Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. (2004);10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]