

Abstract
Cellular senescence is a physiological process of irreversible cell-cycle arrest that contributes to various physiological and pathological processes of aging. Whereas replicative senescence is associated with telomere attrition after repeated cell division, stress-induced premature senescence occurs in response to aberrant oncogenic signaling, oxidative stress, and DNA damage which is independent of telomere dysfunction. Recent evidence indicates that cellular senescence provides a barrier to tumorigenesis and is a determinant of the outcome of cancer treatment. However, the senescence-associated secretory phenotype, which contributes to multiple facets of senescent cancer cells, may influence both cancer-inhibitory and cancer-promoting mechanisms of neighboring cells. Conventional treatments, such as chemo- and radiotherapies, preferentially induce premature senescence instead of apoptosis in the appropriate cellular context. In addition, treatment-induced premature senescence could compensate for resistance to apoptosis via alternative signaling pathways. Therefore, we believe that an intensive effort to understand cancer cell senescence could facilitate the development of novel therapeutic strategies for improving the efficacy of anticancer therapies. This review summarizes the current understanding of molecular mechanisms, functions, and clinical applications of cellular senescence for anticancer therapy. [BMB Reports 2014; 47(2): 51-59]
Keywords: Anticancer therapy, Cellular senescence, Tumorigenesis, SASP
INTRODUCTION
Cellular senescence, first identified a half century ago, was originally characterized by the inability of human normal fibroblast to divide despite the presence of mitogens (1). Although senescent cells have irreversibly lost their capacity for cell division, they are viable and remain metabolically active, a phenotype referred to a replicative senescence (RS) (2). In general, RS is attributable to telomere erosion and has been suggested to represent a defense mechanism that limits the expansion of older cells containing potentially dangerous mutations (3). The other type of permanent growth arrest is stress-induced premature senescence (SIPS), which occurs rapidly in response to various stresses (4-6). Whereas the transformation of primary cells by oncogenes requires either the cooperation of other oncogenes or the inactivation of tumor suppressors, oncogenic Ras is known to promote premature senescence if accumulations of p53 or p16 are sufficient (5,7). Loss of tumor suppressors also leads to cellular senescence, both in vitro and in vivo (8-10). In agreement with these results, it has been suggested that SIPS functions as a barrier to tumor growth and recurrence. The signaling pathways that promote RS and SIPS appear to be more similar than different (6). However, whether upstream senescence sensors, mid-point transducers, and downstream effectors overlap in each of the senescence pathways is not clear.
Current research indicates that chemotherapeutic drugs and ionizing radiation induce SIPS in cancer cells (11,12). Since the amount of drug or dose of radiation required to induce senescence is much lower than that necessary to kill cells, senescence-inducing treatments offer the advantage of enhancing the efficacy and decreasing the side effects of anticancer therapy. Despite the tumor-suppressive potential of cellular senescence, senescent cancer cells secrete a characteristic profile of cytokines, growth factors and proteases, collectively termed the senescence-associated secretory phenotype (SASP). SASP influences tissue microenvironments and stimulates tumorigenesis, angiogenesis, epithelial-to-mesenchymal transition (EMT) and metastasis in vitro and in vivo (13-15). Conversely, the anti-cancer function of SASP contributes to tumor cell clearance by the immune system (16). Therefore, the implications of SASP for tumor progression can be both complex and subtle. Indeed, special attention must be paid to SASP in considering targeted approaches to inducing senescence in the clinical treatment of cancer. Here, we review the current understanding of the molecular mechanisms, functions, and clinical applications of cancer cell senescence for anticancer treatment.
CELLULAR SENESCENCE: A FUNDAMENTAL ASPECT OF CELL BIOLOGY
Traditionally, aging has been defined as a collection of degenerative pathologies that lead to losses of tissue or cells and the decline of physiological functions, thereby accounting for the majority of morbidities (17). It is well established that aging is also the biggest risk factor for cancer (18). Thus, understanding the cellular and molecular biology of the aging process and the development of age-associated human diseases is more important than ever. Nevertheless, a full characterization of aging mechanisms and a complete understanding of the underlying complexity of the aging process have remained out of reach. Recently, Lopez-Otin and colleagues revisited the concepts of aging, with a special emphasis on categorizing the hallmarks of aging illuminated by recent discoveries in the field of aging research (19). These modern hallmarks of aging include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunctions, stem cell exhaustion, altered intercellular communication, and cellular senescence.
Cellular senescence, which is caused by continual stresses and damage from various stimuli and gives rise to a state of permanent cell-cycle arrest, is a central event in the aging process. This observation was initially made by Hayflick and Moorhead, who reported that the proliferation potential of human embryonic fibroblasts in a cell culture system was limited (1). Although cultured cells reach a proliferative limit, termed the Hayflick limit, they remained viable and lost their original morphology, becoming enlarged and flattened with increased volume and granularity. This phenomenon, which follows an extended lifespan in cell culture, has been termed replicative senescence (RS) (3). The shortening of telomeres at each cell division suggested that the molecular mechanism of RS is telomere erosion, which is essential for recording the number of divisions. Following the discovery of the telomere-based mechanism of the Hayflick limit, subsequent studies have demonstrated that cells can undergo a rapid senescence process in response to numerous physiologic stresses. These studies revealed a second type of senescence, generally termed stress-induced premature senescence (SIPS). It has been suggested that SIPS is a telomere-independent process promoted by DNA-damaging agents, oxidative stress, chemotherapeutic drugs, irradiation, and overexpression of oncogenes in normal and cancer cells (4-6). Specifically, senescence-inducing stimuli are potentially oncogenic, and cancer cells acquire essential mutations that allow them to escape oncogene-induced senescence. For example, elevated proliferative signals by oncoproteins such as Ras, BRAF, and AKT facilitate oncogene-induced senescence (4,7,20,21). Loss of tumor suppressors, including PTEN (phosphatase and tensin homologue deleted on chromosome 10), von Hippel-Lindau (VHL), and retinoblastoma protein (pRb), are also phenotypic features of cellular senescence (8-10). Thus, both RS and SIPS almost certainly create barriers to tumorigenesis. Because cancer is an aging-associated disease, senescence can function in the opposite sense to promote tumor formation (13,15,16). However, it is currently impossible to unequivocally answer to the question of whether the SASP is pro-tumorigenic or anti-tumorigenic.
The fact that the expression of various genes is altered by cellular senescence and various cytokines, growth factors, and enzymes are concomitantly secreted creates an important opportunity for the development of biomarkers for cellular senescence (22). Recent improvements in genomics and proteomics have enabled the identification of senescence-specific proteins that may be of practical value in diagnosing senescence. However, although several candidate biomarkers have been discovered, a few specifically define the senescent state. The first senescence marker identified was senescence-associated β-galactosidase (SA-β-gal) (23). While there are some limitations on the use of activity assays for this enzyme, this marker can be sensitively detected by histochemical staining in most senescent cells and tissue. In addition to negative regulators of the cell cycle, such as p15IK4b (p15), p16INK4a (p16), ARF and p21Cip1/Waf1 (p21), differentiated embryonic chondrocyte expressed-1 (DEC1) and decoy death receptor-2 (DCR2) have been identified as senescence indicators (24,25). Recently, our group also identified cathepsin D and eukaryotic elongation factor 1 as promising markers for the detection of cellular senescence induced by a variety of treatments (26). The search for such markers continues, since a well-established set of definitive senescence biomarkers could clearly have a profound impact on the diagnosis, staging and prognosis-assessment of age-related diseases, providing a basis for therapeutic intervention in such diseases.
MOLECULAR PATHWAYS OF CELLULAR SENESCENCE
Considerable research effort has been devoted elucidating the molecular mechanisms of senescence, and many genes whose expression is altered in senescent cells have been identified (27-30). In most of these studies, it is not clear whether such altered gene expression patterns are unique to senescence, or instead are involved in limiting cell proliferation. Moreover, there is little concrete evidence to establish whether these changes in gene expression are a cause of senescence or merely a consequence of it. However, numerous recent studies have provided important insights into the molecular pathways that lead to cellular senescence (17,31,32).
Importantly, the combined activity of two central proteins in cellular senescence, p53 and pRb, could determine whether cells enter senescence or a cell death pathway (33,34). Both of these tumor-suppressor proteins are activated in response to telomere shortening and various stresses—the first step toward senescence. Recent evidence suggests that telomere shortening triggers a DNA-damage response, resulting in the activation of ATM/ATR and Chk1/Chk2; this, in turn, leads to activation of G1 checkpoint mechanisms, including phosphorylation of p53 (35). An important target of p53 is the cyclin-dependent kinase (CDK) inhibitor p21, which antagonizes the activity of several cyclin–CDK complexes and can arrest cells in both G1 and G2 phases of the cell cycle (36). The upregulation of p21 during RS or SIPS via both p53-dependent and -independent mechanism is associated with telomere shortening and DNA damage (37,38). Overexpression of p21 can induce a senescence-like cell-cycle arrest, whereas depletion of p21 can delay senescence-associated arrest. Collectively, these studies suggest that p53 functions in senescence, at least in part, by inducing p21. Interestingly, in contrast to human cells, mouse embryonic fibroblasts carrying a p21-null mutation undergo senescence normally, suggesting that p21 is not a critical downstream effector of p53 during senescence in mouse cells (39,40). Inactivation of pRb is often required to avert senescence in human cells, suggesting that pRb is crucially involved in senescence pathways (41). Cell-cycle progression is primarily regulated by CDK-mediated phosphorylation of pRb. Phosphorylated pRb releases the transcription factor E2F, allowing it to activate the transcription of genes necessary for DNA synthesis and cell-cycle progression. Therefore, the hyperphosphorylated, inactive form of pRb is required to allow entry into the cell cycle. In a senescent state, pRb protein is found in the active, hypophosphorylated form, which binds E2F protein and thereby blocks transcriptional activation of its targets, most of which are effectors of cell-cycle progression (41). pRb phosphorylation is prevented by inhibition of CDK activity by negative CDK regulators such as p21 and p16. Importantly, how the activation status of p53 and pRb determines whether a cell “decides” to senesce is a complex and incompletely understood process.
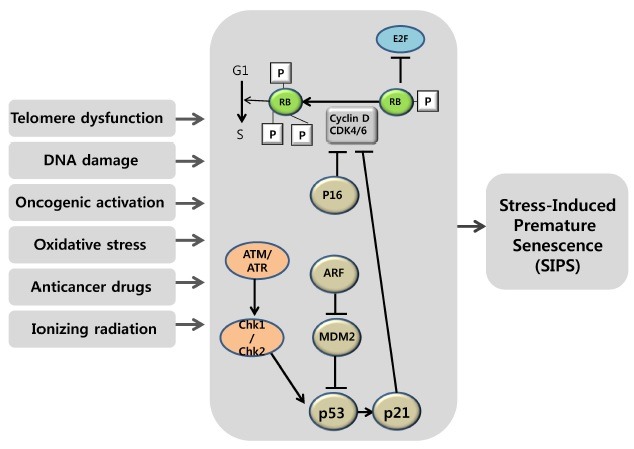
The INK4a/ARF/INK4b locus encodes three proteins, p16, ARF and p15, which are important inducers of cellular senescence (42). This locus is normally expressed at a very low level, but shows markedly increased expression with aging. Notably, there is an interplay between the proliferation-limiting functions of these proteins and pRb and p53 tumor suppressors that operates via diverse mechanisms. p16 was first found to be increased in most aged mammalian tissues and senescing fibroblasts, and later was found to be increased in oncogene-induced premature senescence (43). There is evidence that p16 might play a backup tumor-suppressor role for p53. p16 acts in the pRb pathway by inhibiting the activation of CDK4 and CDK6, which in most physiological situations is presumed to be the initial step in pRb phosphorylation (43). Thus, the function of p16 is to keep pRb in its active, growth-inhibitory state, which in turn blocks the expression of genes regulated by the E2F transcription factor and imposes a G1 cell-cycle arrest. Like p16, ARF is induced in cellular senescence by various stimuli. ARF sequesters the MDM2 protein, an E3 ubiquitin ligase for p53 that promotes p53 degradation; in so doing, ARF positively regulates p53 levels and thereby enhances its activity (44,45). The emerging notion is that the p53-p21 pathway is primarily responsible for senescence induced by telomere shortening or DNA damage, whereas the p16-pRb pathway is partly responsible for mediating SIPS. Cell populations in culture experience telomere shortening, as well as other stresses, to varying extents. Depending on cell type, culture conditions and the extent of stress, inactivating the p53-p21 or p16-pRb pathway, separately or together, is necessary to avert senescence (Fig. 1).
Fig. 1. Stress-induced premature senescence (SIPS) signaling pathway. A variety of stimuli, such as telomere erosion, DNA damage, oncogene activation, oxidative stress, anticancer drugs and ionizing radiation, induce cell-cycle arrest and transcription of senescence-associated genes. Senescence signaling and growth arrest-response signaling pathways have been shown to overlap. Upon sensing senescence signals, the cell cycle checkpoint machinery can force the cell to exit the cell cycle via the CDK4 inhibitor p16 and the MDM2 inhibitor ARF. Senescence-inducing stimuli lead to upregulation of p21, which blocks the cyclin D/CDK4-mediated hyperphosphorylation of pRb, and provokes cell cycle arrest and, ultimately, cellular senescence. Alternatively, these events induce ARF, which blocks the activity of the p53 inhibitor MDM2 and thereby activates p53. One of the characteristics of senescent cells is a change in morphology, such as enlargement, flattening, and increased granularity. In addition, senescent cells or tissue exhibit increased activity of senescence-associated β-galactosidase (SA-β-gal), a reliable biomarker of senescence.
THE SENESCENCE-ASSOCIATED SECRETORY PHENOTYPE: PRO-TUMORIGENIC OR ANTI-TUMORIGENIC?
Since senescence represents a permanent exit from cell proliferation, it might seem as if its contribution to age-related pathologies is passive. However, senescent cells express various secretory proteins, such as cytokines, chemokines, growth factors and matrix-remodeling associated proteases, that alter tissue microenvironments and/or are vigorously involved in tumorigenesis (13,15), giving rise to the innovative concept of the senescence-associated secretory phenotype (SASP), first proposed by Campisi and colleagues (13,14,16,17). Initially, it was reported that senescent fibroblasts increase the tumorigenesis of human mammary tumor cells, probably via molecules secreted by senescent cells (46). An examination of the cellular signaling pathways that activate the SASP suggests that the DNA-damage response and downstream activation of ATM are sufficient to activate several SASP-related factors. Furthermore, transcription factors such as NF-κB and C/EBPβ occupy the promoters of several cytokine genes in senescent cells (47). How these factors are activated in response to senescence-inducing stimuli and subsequently dictate transcriptional changes in senescence remains to be determined.
The SASP has been observed in a variety of both normal and cancerous senescent cells, and could affect the tumor microenvironment in an autocrine and paracrine manner. A number of reports have indicated that epithelial cells exposed to senescent human fibroblasts permanently lose their differentiated properties, become invasive, undergo neoplastic transformation, and stimulate tumor vascularization (46,48-51). On the basis these observations, it would be reasonable to infer that SASP factors are pro-tumorigenic. On the other hand, secreted proteins contribute an anti-tumorigenic action by promoting the elimination of senescent cells by the immune system. It was previously reported that senescent cells influence macrophage function and lymphocyte infiltration in the tumor environment by releasing SASP factors (52). These secretory molecules trigger an immune response that serves to eliminate senescent cells. In addition, the maintenance of senescence is dependent upon the cell-autonomous signaling of interleukin-6 (IL-6) and IL-8 (53). These contradictory effects of the SASP in cancer development pose a challenge for developing an understanding of its overall consequences based on separate, intensive examinations of its individual factors. However, these separate facets of the SASP do form a whole, and much of the complication arises from the dependence of the SASP on cell/tissue context. Within multicellular tissues, sophisticated lines of communication exist between senescent cells and their neighboring non-senescent cells, allowing for the specialized and harmonious activity of SASP factors. Elucidating the functional significance and molecular causes of the SASP is an important future research challenge.
Among typical SASP factors are interleukins, including IL-1, IL-6 and IL-8, which exert multiple phenotypic effects. It has been reported that members of the IL-1 family are upregulated and released by senescent endothelial cells and fibroblasts (54,55). Other pleiotropic inflammatory cytokines, namely IL-6 and IL-8, are secreted from cells that have senesced due to DNA damage, replicative exhaustion, and/or oncogenic insults (55-57). IL-1α regulates its own synthesis in an autocrine, receptor-mediated, positive-feedback loop via NF-κB, and is a key positive regulator of IL-6 and IL-8 expression in senescence (54). These reports indicate a biological hierarchy in the regulation of SASP components. Among the key players in the SASP are a variety of insulin-like growth factor-binding proteins (IGFBPs) and their regulators, IGFBP-rP1 and IGFBP-rP2 (58-60), which are expressed at high levels in senescent cells. Insulin-like growth factor (IGF) signaling via IGF receptors may contribute to the control of the mammalian life span (61). Additionally, and critically important, senescent fibroblasts secrete increased levels of matrix remodeling-associated enzymes, typically matrix metalloproteinases (MMPs) (16,62). An important concept that has emerged from accumulating evidence is that proteolysis of the extracellular matrix by senescent cells can stimulate epithelial cell growth. For example, it has been shown that MMPs secreted by senescent fibroblasts are responsible for increasing the tumorigenicity of mammary epithelial cells, most likely by facilitating mitogenic and chemotactic signals (16,46,62,63). Other protein families involved in the SASP are serine proteases and regulators of the plasminogen activation pathway, namely urokinase- or tissue-type plasminogen activators (uPA or tPA), the uPA receptor (uPAR), and inhibitors of these serine proteases (PAI-1 and -2) (16,64).
Remarkably, one of the most prominent effects of the SASP is to enhance the proliferation of epithelial cells. In the case of mammary epithelial cells, senescent human fibroblasts can stimulate the growth of pre-malignant and malignant mammary epithelial cells (46). Radiation-exposed stromal cells, which are presumed to be senescent, have been shown to disarrange the mammary epithelial microenvironment and promote abnormal epithelial cell proliferation in the mammary gland (63,65). Senescent fibroblasts from the human prostate gland have been shown to create a local tissue microenvironment that leads to hyperproliferation of prostate epithelial cells (50). Interestingly, malignant melanocytes express high levels of the C-X-C chemokine receptor-2 (CXCR2), and can be stimulated to grow by the CXCR2 ligands, GROα and IL-8 (66,67). Both GROα and IL-8 are part of the core SASP, suggesting that the senescent microenvironment may force or enable melanoma development. Importantly, vascular endothelial growth factor (VEGF), another SASP factor, can promote tumor progression by stimulating endothelial cell proliferation and increasing vessel formation (48,68). Thus, senescent cells support the differentiation of a new vasculature around and within a growing tumor.
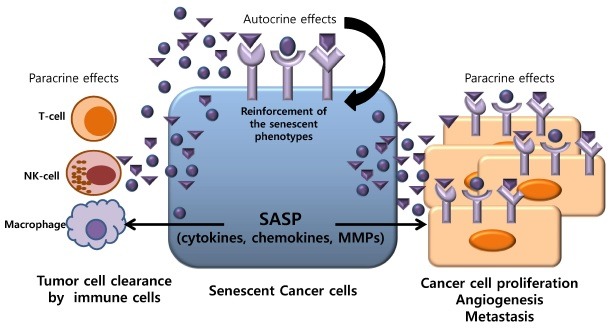
Excessive release of secretory proteins by senescent cells is important for promoting cell migration and invasion. This activity is largely attributable to the secretion of MMPs by senescent cells (16,69,70). In the case of breast cancer, high levels of IL-6 secreted by senescent fibroblasts are responsible for enhancing the invasiveness of cancer cells (71). Moreover, the secretion of MMPs by senescent fibroblasts can enhance the invasion of multiple epithelial cell types (69), and senescent stromal cells may promote EMT, a feature that is important in enabling cancer cells to metastasize (72,73). Recently, we investigated the effect of proteins secreted from senescent tumor cells induced by ionizing radiation on normal and tumor cells. A proteomics approach revealed a number of differentially secreted proteins, including Raf kinase inhibitor protein (RKIP), α-enolase, A-kinase anchoring protein 9 (AKAP9), and MAP/microtubule affinity-regulating kinase 4 (MARK4) (74). SASP factors secreted by senescent fibroblasts or hepatocytes have been implicated in the differentiation of circulating immune cells and their recruitment to tumor sites, which ultimately contribute to tumor immune surveillance (75-77). These secreted factors also lead to inadequate immune responses within close proximity of senescent cells. It has also been reported that senescent fibroblasts might affect lymphocytic infiltration of tumors (13,16,72). Either immune cells recruited by senescent cells or senescent cells per se secrete pro-angiogenic factors (72,74). Taken together, these observations indicate that, by stimulating neoplastic transformation and vascularization or enhancing tumor cell clearance through activation of the immune system, the SASP may negatively or positively affect cancer progression (Fig. 2).
Fig. 2. Pleiotropic nature of senescent cells. Senescent cancer cells positively and negatively affect the tumor microenvironment. In particular, the senescence-associated secretory phenotype (SASP) contributes to maintenance of the senescent state and the growth arrest (autocrine effect) of senescent cells. Senescent cells also exhibit pro-inflammatory responses through the production of cytokines and chemokines. In addition to inflammatory factors, matrix-remodeling factors alter the tissue microenvironment and enhance cancer cell proliferation, angiogenesis, and metastasis. Anti-inflammatory cytokines, which are released into the extracellular compartment by senescent cells, mediate the elimination of tumor cells by recruiting immune cells to the tumor tissues. On the other hand, pro-inflammatory cytokines exert a cancer-promoting activity on nearby cancer cells by enhancing tumorigenesis (paracrine effects).
SENESCENCE AS AN ANTICANCER THERAPY
The rationale behind current anticancer strategies is to kill rapidly dividing cancer cells by causing extensive DNA damage with high doses of drugs or irradiation. However, delayed side effects of anticancer treatment, such as recurrence, secondary cancers and normal tissue damage from chemotherapy and radiotherapy, pose clinical problems for the cancer survivor. Findings from the senescence research field suggest the possibility of harnessing the tumor-suppressive potential of senescence, giving rise to the hypothesis that pro-senescence therapy involving the induction of a fixed cytostatic condition could be an effective way to treat cancer while lessening side effects (78,79). Importantly, treatment of cancer cells with chemotherapy or radiotherapy has been clearly shown to produce a senescent state, termed therapy-induced senescence (TIS), which principally involves the p53/p21 and p16/pRb pathways (80). For example, a high concentration of doxorubicin induces apoptosis in human cancer cells, whereas a low concentration induces cellular senescence (26,81,82). Senescence-inducing agents used in the management of human cancer are of clinical interest (80,83). Ionizing radiation (IR) also induces cellular senescence in cancer cells. The impact of IR on self-renewal capacity appears to be mediated through the induction of senescence, as evidenced by the close correlation between the extent of radiation-induced senescence and radiation sensitivity (11,12,26,84). One of the main objectives of radiotherapy research has been to develop more efficient ways to increase the efficacy of radiotherapy without causing toxicity to normal tissue (11). Since low-dose IR exposure could efficiently induce SIPS, it could prove beneficial in radiotherapy by limiting damage to normal tissue. If key factors of TIS become inactivated in cancer, the result is acquired resistance to anticancer treatment and poor therapeutic outcome. Therefore, therapeutic approaches aimed at selectively inducing senescence could represent a promising strategy for cancer treatment.
One of the main concepts of prosenescence therapy that has emerged from recent reports is the importance of developing targeted therapeutics. A number of promising therapeutic approaches are currently under consideration, including inhibition of telomerase with the inhibitor, GRN 163L (85), and restoration of tumor suppressor function to mutant forms of p53 (75,86). These latter studies highlight the value of p53 as a target for prosenescence therapy, showing that restoring p53 function promotes tumor regression and tumor clearance via a senescence-inducing mechanism. These reports further support the concept that senescent tumor cells, but not non-senescent tumor cells, are efficiently eliminated by immune cells. Another interesting discovery is that p53-triggered senescence selectively affects tumor cells, leaving normal tissue totally unchanged (86). Novel p53-targeting compounds developed to date include Ellipticine and PRIMA-1, which restore wild-type activity to p53 mutants, and nutlins, which inhibit the binding of p53 to MDM2 (87-89).
Extrapolating from the current findings to clinical applications, it may be possible to trigger oncogene-induced senescence (OIS) and PTEN-loss-induced cellular senescence (PICS) in vivo as an anticancer therapy (90). It has been shown that oncogenic Ras expression is associated with enrichment of senescent cells in various cancer lesions (5,91). Similar to oncogenic Ras, the BRAFV600E mutant induces an increase in the number of senescent cells in melanoma (92). Since inactivation of c-Myc also triggers senescence in cancer cells, inhibition of c-Myc activity by small molecules could also prove to be an effective prosenescence therapy (93). Inactivation of tumor suppressors promotes senescence in premalignant lesions. Importantly, it has been demonstrated that the PTEN inhibitor, VO-OHpic, drives senescence and inhibits tumorigenesis (90). VO-OHpic thus represents an effective tool for evaluating the efficacy of drugs that modulate PICS-inducing pathways and developing protocols that improve responsiveness to current therapies. In particular, our data evidence that PTEN acts as a critical determinant of cell fate in the context of IR-induced senescence in human glioma cells (84). We also discovered that c-Jun N-terminal kinase (JNK) and Wig1 (wild-type p53-induced gene 1) are promising therapeutic targets for prosenescence therapy (94,95). Loss of another tumor suppressor, VHL, also results in marked senescence in premalignant lesions, with induction of senescence (9). Overall, a targeted prosenescence approach, keyed to the development of potential senescence inducing agents, could advance a therapeutic strategy that would ultimately improve cancer treatment outcome.
CONCLUDING REMARKS
Since the discovery of senescence as permanent growth arrest, with its potential implications for aging, the biological significance of senescence has grown in conjunction with the recognition that various stimuli, including oncogenes, oxidative stress and anticancer therapy, prematurely induce cellular senescence not only in normal cells, but also cancer cells. Our expanded understanding of senescence as both a barrier to tumorigenesis and a determinant of the outcome of cancer treatment takes practical form in the concept of prosenescence therapy, which could be an important alternative or adjunct to chemotherapy or radiotherapy. The suggestion is that prosenescence therapy could improve the efficacy of cancer therapies and decrease their side effects in cancer patients. Indeed, several compounds with potential prosenescence therapy applications are currently undergoing clinical trials. However, caution is warranted because the complicated phenotypes of senescent cancer cells, for example the SASP, as noted above, may create potential problems in prosenescence therapy. For example, it is conceivable that the induction of senescence might give rise to remnant dormant tumor cells that represent the potential for cancer recurrence, specifically in the case of cancer stem cells. Therefore, a deeper understanding of the mechanisms responsible for senescence is required before the promise of prosenescence therapy can be realized.
Acknowledgments
This work was supported by a National Research Foundation of Korea (NRF) grant, funded by the Korean government (MSIP) (2012-M2B2B-2012055637). None of the authors has a conflict of interest related to the work reported in this manuscript.
References
- 1.Hayflick L., Moorhead P. S. The serial cultivation of human diploid cell strains. Exp. Cell. Res. (1961);25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Campisi J. Replicative senescence: an old lives' tale? Cell. (1996);84:497–500. doi: 10.1016/S0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 3.Deng Y., Chang S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Invest. (2007);87:1071–1076. doi: 10.1038/labinvest.3700673. [DOI] [PubMed] [Google Scholar]
- 4.Toussaint O., Medrano E. E., von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. (2000);35:927–945. doi: 10.1016/S0531-5565(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 5.Gorgoulis V. G., Halazonetis T. D. Oncogene-induced senescence: the bright and dark side of the response. Curr. Opin. Cell. Biol. (2010);22:816–827. doi: 10.1016/j.ceb.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Porath I., Weinberg R. A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell. Biol. (2005);37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. (1997);88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z., Trotman L. C., Shaffer D., Lin H. K., Dotan Z. A., Niki M., Koutcher J. A., Scher H. I., Ludwig T., Gerald W., Cordon-Cardo C., Pandolfi P. P. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. (2005);436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young A. P., Schlisio S., Minamishima Y. A., Zhang Q., Li L., Grisanzio C., Signoretti S., Kaelin W. G. Jr. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. (2008);10:361–369. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- 10.Shamma A., Takegami Y., Miki T., Kitajima S., Noda M., Obara T., Okamoto T., Takahashi C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell. (2009);15:255–269. doi: 10.1016/j.ccr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki M., Boothman D. A. Stress-induced premature senescence (SIPS)--influence of SIPS on radiotherapy. J. Radiat. Res. (2008);49:105–112. doi: 10.1269/jrr.07081. [DOI] [PubMed] [Google Scholar]
- 12.Gewirtz D. A., Holt S. E., Elmore L. W. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. (2008);76:947–957. doi: 10.1016/j.bcp.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 13.Davalos A. R., Coppe J. P., Campisi J., Desprez P. Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis. Rev. (2010);29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campisi J., Andersen J. K., Kapahi P., Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin. Cancer Biol. (2011);21:354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young A. R., Narita M. SASP reflects senescence. EMBO Rep. (2009);10:228–230. doi: 10.1038/embor.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coppe J. P., Desprez P. Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. (2010);5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campisi J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. (2013);75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Magalhaes J. P. How ageing processes influence cancer. Nat. Rev. Cancer. (2013);13:357–365. doi: 10.1038/nrc3497. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Otin C., Blasco M. A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. (2013);153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oyama K., Okawa T., Nakagawa H., Takaoka M., Andl C. D., Kim S. H., Klein-Szanto A., Diehl J. A., Herlyn M., El-Deiry W., Rustgi A. K. AKT induces senescence in primary esophageal epithelial cells but is permissive for differentiation as revealed in organotypic culture. Oncogene. (2007);26:2353–2364. doi: 10.1038/sj.onc.1210025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhomen N., Reis-Filho J. S., da Rocha Dias S., Hayward R., Savage K., Delmas V., Larue L., Pritchard C., Marais R. Oncogenic Braf induces melanocyte senescene and melanoma in mice. Cancer Cell. (2009);15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 22.Kuilman T., Michaloglou C., Mooi W. J., Peeper D. S. The essence of senescence. Genes Dev. (2010);24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O., Peacocke M., Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. (1995);92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Canpo-Trapero J., Cano-Sánchez J., Palacios-Sánchez B., Llamas-Martínez S., Lo Muzio L., Bascones-Martínez A. Cellular senescence in oral cancer and precancer and treatment implications: A review. Acta. Oncologica. (2008);47:1464–1474. doi: 10.1080/02841860802183612. [DOI] [PubMed] [Google Scholar]
- 25.Collado M., Efeyan A., Guerra C., Schuhmacher A. J., Barradas M., Benguria A., Zaballos A., Flores J. M., Barbacid M., Beach D., Serrano M. Tumor biology: senescence in premalignant tumors. Nature. (2005);436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 26.Byun H. O., Han N. K., Lee H. J., Kim K. B., Ko Y. G., Yoon G., Lee Y. S., Hong S. I., Lee J. S. Cathepsin D and eukaryotic translation elongation factor 1 as promising markers of cellular senescence. Cancer Res. (2009);69:4638–4647. doi: 10.1158/0008-5472.CAN-08-4042. [DOI] [PubMed] [Google Scholar]
- 27.de Magalhaes J. P., Curado J., Church G. M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. (2009);25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mason D. X., Jackson T. J., Lin A. W. Molecular signature of oncogenic ras-induced senescence. Oncogene. (2004);23:9238–9246. doi: 10.1038/sj.onc.1208172. [DOI] [PubMed] [Google Scholar]
- 29.Wennmalm K., Wahlestedt C., Larsson O. The expression signature of in vitro senescence resembles mouse but not human aging. Genome. Biol. (2005);6:R109. doi: 10.1186/gb-2005-6-13-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calvanese V., Lara E., Kahn A., Fraga M. F. The role of epigenetics in aging and age-related diseases. Ageing. Res. Rev. (2009);8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Ohtani N., Hara E. Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci. (2013);104:525–530. doi: 10.1111/cas.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams P. D. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol. Cell. (2009);36:2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 33.Chandler H., Peters G. Stressing the cell cycle in senescence and aging. Curr. Opin. Cell Biol. (2013);25:765–771. doi: 10.1016/j.ceb.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Larsson L. G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol. (2011);21:367–376. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Reinhardt H. C., Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends. Genet. (2012);28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warfel N. A., El-Deiry W. S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. (2013);25:52–58. doi: 10.1097/CCO.0b013e32835b639e. [DOI] [PubMed] [Google Scholar]
- 37.Roninson I. B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. (2002);179:1–14. doi: 10.1016/S0304-3835(01)00847-3. [DOI] [PubMed] [Google Scholar]
- 38.Carnero A., Beach D. H. Absence of p21WAF1 cooperates with c-myc in bypassing Ras-induced senescence and enhances oncogenic cooperation. Oncogene. (2004);23:6006–6011. doi: 10.1038/sj.onc.1207839. [DOI] [PubMed] [Google Scholar]
- 39.Groth A., Weber J. D., Willumsen B. M., Sherr C. J., Roussel M. F. Oncogenic Ras induces p19ARF and growth arrest in mouse embryo fibroblasts lacking p21Cip1 and p27Kip1 without activating cyclin D-dependent kinases. J. Biol. Chem. (2000);275:27473–27480. doi: 10.1074/jbc.M003417200. [DOI] [PubMed] [Google Scholar]
- 40.Pantoja C., Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. (1999);18:4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 41.Indovina P., Marcelli E., Casini N., Rizzo V., Giordano A. Emerging roles of RB family: new defense mechanisms against tumor progression. J. Cell Physiol. (2013);228:525–535. doi: 10.1002/jcp.24170. [DOI] [PubMed] [Google Scholar]
- 42.Kim W. Y., Sharpless N. E. The regulation of INK4/ARF in cancer and aging. Cell. (2006);127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 43.Rayess H., Wang M. B., Srivatsan E. S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer. (2012);130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominguez-Brauer C., Brauer P. M., Chen Y. J., Pimkina J., Raychaudhuri P. Tumor suppression by ARF: gatekeeper and caretaker. Cell Cycle. (2010);9:86–89. doi: 10.4161/cc.9.1.10350. [DOI] [PubMed] [Google Scholar]
- 45.Wadhwa R., Sugihara T., Taira K., Kaul S. C. The ARF-p53 senescence pathway in mouse and human cells. Histol. Histopathol. (2004);19:311–316. doi: 10.14670/HH-19.311. [DOI] [PubMed] [Google Scholar]
- 46.Parrinello S., Coppe J. P., Krtolica A., Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. (2005);118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salminen A., Kauppinen A., Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. (2012);24:835–845. doi: 10.1016/j.cellsig.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 48.Coppe J. P., Kauser K., Campisi J., Beausejour C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. (2006);281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 49.Gosselin K., Martien S., Pourtier A., Vercamer C., Ostoich P., Morat L., Sabatier L., Duprez L., T'Kint de Roodenbeke C., Gilson E., Malaquin N., Wernert N., Slijepcevic P., Ashtari M., Chelli F., Deruy E., Vandenbunder B., De Launoit Y., Abbadie C. Senescence-associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res. (2009);69:7917–7925. doi: 10.1158/0008-5472.CAN-08-2510. [DOI] [PubMed] [Google Scholar]
- 50.Bavik C., Coleman I., Dean J. P., Knudsen B., Plymate S., Nelson P. S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. (2006);66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 51.Pazolli E., Luo X., Brehm S., Carbery K., Chung J. J., Prior J. L., Doherty J., Demehri S., Salavaggione L., Piwnica-Worms D., Stewart S. A. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. (2009);69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sagiv A., Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. (2013);14:617–628. doi: 10.1007/s10522-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 53.Acosta J. C., O'Loghlen A., Banito A., Raguz S., Gil J. Control of senescence by CXCR2 and its ligands. Cell Cycle. (2008);7:2956–2959. doi: 10.4161/cc.7.19.6780. [DOI] [PubMed] [Google Scholar]
- 54.Mariotti M., Castiglioni S., Bernardini D., Maier J. A. Interleukin 1 alpha is a marker of endothelial cellular senescent. Immun. Ageing. (2006);3:4. doi: 10.1186/1742-4933-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orjalo A. V., Bhaumik D., Gengler B. K., Scott G. K., Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. U. S. A. (2009);106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Novakova Z., Hubackova S., Kosar M., Janderova-Rossmeislova L., Dobrovolna J., Vasicova P., Vancurova M., Horejsi Z., Hozak P., Bartek J., Hodny Z. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene. (2010);29:273–284. doi: 10.1038/onc.2009.318. [DOI] [PubMed] [Google Scholar]
- 57.Coppe J. P., Patil C. K., Rodier F., Sun Y., Munoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008);6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Bermejo A., Buckway C. K., Devi G. R., Hwa V., Plymate S. R., Oh Y., Rosenfeld R. G. Characterization of insulin-like growth factor-binding protein-related proteins (IGFBP-rPs) 1, 2, and 3 in human prostate epithelial cells: potential roles for IGFBP-rP1 and 2 in senescence of the prostatic epithelium. Endocrinology. (2000);141:4072–4080. doi: 10.1210/endo.141.11.7783. [DOI] [PubMed] [Google Scholar]
- 59.Maki R. G. Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J. Clin. Oncol. (2010);28:4985–4995. doi: 10.1200/JCO.2009.27.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S., Moerman E. J., Jones R. A., Thweatt R., Goldstein S. Characterization of IGFBP-3, PAI-1 and SPARC mRNA expression in senescent fibroblasts. Mech. Ageing. Dev. (1996);92:121–132. doi: 10.1016/S0047-6374(96)01814-3. [DOI] [PubMed] [Google Scholar]
- 61.Puche J. E., Castilla-Cortazar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J. Transl. Med. (2012);10:224. doi: 10.1186/1479-5876-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu D., Hornsby P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. (2007);67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 63.Tsai K. K., Chuang E. Y., Little J. B., Yuan Z. M. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. (2005);65:6734–6744. doi: 10.1158/0008-5472.CAN-05-0703. [DOI] [PubMed] [Google Scholar]
- 64.Dass K., Ahmad A., Azmi A. S., Sarkar S. H., Sarkar F. H. Evolving role of uPA/uPAR system in human cancers. Cancer Treat. Rev. (2008);34:122–136. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 65.Barcellos-Hoff M. H., Ravani S. A. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. (2000);60:1254–1260. [PubMed] [Google Scholar]
- 66.Schadendorf D., Moller A., Algermissen B., Worm M., Sticherling M., Czarnetzki B. M. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J. Immunol. (1993);151:2667–2675. [PubMed] [Google Scholar]
- 67.Norgauer J., Metzner B., Schraufstatter I. Expression and growth-promoting function of the IL-8 receptor beta in human melanoma cells. J. Immunol. (1996);156:1132–1137. [PubMed] [Google Scholar]
- 68.Ksiazek K., Jorres A., Witowski J. Senescence induces a proangiogenic switch in human peritoneal mesothelial cells. Rejuvenation. Res. (2008);11:681–683. doi: 10.1089/rej.2008.0736. [DOI] [PubMed] [Google Scholar]
- 69.Hornebeck W., Maquart F. X. Proteolyzed matrix as a template for the regulation of tumor progression. Biomed. Pharmacother. (2003);57:223–230. doi: 10.1016/S0753-3322(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 70.Chaturvedi S., Hass R. Extracellular signals in young and aging breast epithelial cells and possible connections to age-associated breast cancer development. Mech. Ageing. Dev. (2011);132:213–219. doi: 10.1016/j.mad.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 71.Studebaker A. W., Storci G., Werbeck J. L., Sansone P., Sasser A. K., Tavolari S., Huang T., Chan M. W., Marini F. C., Rosol T. J., Bonafe M., Hall B. M. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. (2008);68:9087–9095. doi: 10.1158/0008-5472.CAN-08-0400. [DOI] [PubMed] [Google Scholar]
- 72.Laberge R. M., Awad P., Campisi J., Desprez P. Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. (2012);5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smit M. A., Peeper D. S. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging (Albany NY) (2010);2:735–741. doi: 10.18632/aging.100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han N. K., Kim B. C., Lee H. C., Lee Y. J., Park M. J., Chi S. G., Ko Y. G., Lee J. S. Secretome analysis of ionizing radiation-induced senescent cancer cells reveals that secreted RKIP plays a critical role in neighboring cell migration. Proteomics. (2012);12:2822–2832. doi: 10.1002/pmic.201100419. [DOI] [PubMed] [Google Scholar]
- 75.Xue W., Zender L., Miethng C., Dickins R. A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S. M. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. (2007);445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang T. W., Yevsa T., Woller N., Hoenicke L., Wuestefeld T., Dauch D., Hohmeyer A., Gereke M., Rudalska R., Potápova A., Iken M., Vucur M., Weiss S., Heikenwalder M., Khan S., Gil J., Bruder D., Manns M., Schirmacher P., Tacke F., Ott M., Luedde T., Longerich T., Kubicka S., Zender L. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. (2011);479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 77.Sica A., Schioppa T., Mantovani A., Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur. J. Cancer. (2006);42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 78.Nardella C., Clohessy J. G., Alimonti A., Pandolfi P. P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer. (2011);11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 79.Acosta J. C., Gil J. Senescence: a new weapon for cancer therapy. Trends. Cell Biol. (2012);22:211–219. doi: 10.1016/j.tcb.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 80.Ewald J. A., Desotelle J. A., Wilding G., Jarrard D. F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. (2010);102:1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwarze S. R., Fu V. X., Desotelle J. A., Kenowski M. L., Jarrard D. F. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia. (2005);7:816–823. doi: 10.1593/neo.05250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rebbaa A., Zheng X., Chu F., Mirkin B. L. The role of histone acetylation versus DNA damage in drug-induced senescence and apoptosis. Cell. Death. Differ. (2006);13:1960–1967. doi: 10.1038/sj.cdd.4401895. [DOI] [PubMed] [Google Scholar]
- 83.Nardella C., Clohessy J. G., Alimonti A., Pandolfi P. P. Pro-senescence therapy for cancer treatment. Nature. Rev. Cancer. (2011);11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 84.Lee J. J., Kim B. C., Park M. J., Lee Y. S., Kim Y. N., Lee B. L., Lee J. S. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death. Differ. (2011);18:666–677. doi: 10.1038/cdd.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Asai A., Oshima Y., Yamamoto Y., Uochi T. A., Kusaka H., Akinaga S., Yamashita Y., Pongracz K., Pruzan R., Wunder E., Piatyszek M., Li S., Chin A. C., Harley C. B., Gryaznov S. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. (2003);63:3931–3939. [PubMed] [Google Scholar]
- 86.Ventura A., Kirsch D. G., McLaughlin M. E., Tuveson D. A., Grimm J., Lintault L., Newman J., Reczek E. E., Weissleder R., Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. (2007);445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 87.Martinkova E., Maglott A., Leger D. Y., Bonnet D., Stiborova M., Takeda K., Martin S., Dontenwill M. alpha5beta1 integrin antagonists reduce chemotherapy-induced premature senescence and facilitate apoptosis in human glioblastoma cells. Int. J. Cancer. (2010);127:1240–1248. doi: 10.1002/ijc.25187. [DOI] [PubMed] [Google Scholar]
- 88.Bykov V. J., Issaeva N., Shilov A., Hultcrantz M., Pugacheva E., Chumakov P., Bergman J., Wiman K. G., Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. (2002);8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 89.Efeyan A., Ortega-Molina A., Velasco-Miguel S., Herranz D., Vassilev L. T., Serrano M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. (2007);67:7350–7357. doi: 10.1158/0008-5472.CAN-07-0200. [DOI] [PubMed] [Google Scholar]
- 90.Alimonti A., Nardella C., Chen Z., Clohessy J. G., Carracedo A., Trotman L. C., Cheng K., Varmeh S., Kozma S. C., Thomas G., Rosivatz E., Woscholski R., Cognetti F., Scher H. I., Pandolfi P. P. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. (2010);120:681–693. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benanti J. A., Galloway D. A. The normal response to RAS: senescence or transformation? Cell Cycle. (2004);3:715–717. doi: 10.4161/cc.3.6.948. [DOI] [PubMed] [Google Scholar]
- 92.Michaloglou C., Vredeveld L. C., Soengas M. S., Denoyelle C., Kuilman T., van der Horst C. M., Majoor D. M., Shay J. W., Mooi W. J., Peeper D. S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. (2005);436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 93.Delmore J. E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., Bergsagel P. L., Ghobrial I. M., Richardson P. G., Young R. A., Hahn W. C., Anderson K. C., Kung A. L., Bradner J. E., Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. (2011);146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee J. J., Lee J. H., Ko Y. G., Hong S. I., Lee J. S. Prevention of premature senescence requires JNK regulation of Bcl-2 and reactive oxygen species. Oncogene. (2010);29:561–575. doi: 10.1038/onc.2009.355. [DOI] [PubMed] [Google Scholar]
- 95.Kim B. C., Lee H. C., Lee J. J., Choi C. M., Kim D. K., Lee J. C., Ko Y. G., Lee J. S. Wig1 prevents cellular senescence by regulating p21 mRNA decay through control of RISC recruitment. EMBO J. (2012);31:4289–4303. doi: 10.1038/emboj.2012.286. [DOI] [PMC free article] [PubMed] [Google Scholar]