

Summary
ABCG2 is an ATP-binding cassette half-transporter initially identified in multidrug-resistant cancer cell lines and recently suggested to play an important role in pharmacokinetics. Here we report studies of a conserved arginine predicted to localize near the cytoplasmic side of TM1. First, we determined the effect of losing charge and bulk at this position via substitutions with glycine and alanine. The R383G mutant when transfected into HEK cells was not detectable on immunoblot or by functional assay, while the R383A mutant exhibited detectable but significantly decreased levels compared to wild-type, partial retention in the ER and altered glycosylation. Efflux of the ABCG2-substrates mitoxantrone and pheophorbide a was observed. Our experiments suggested rapid degradation of the R383A mutant by the proteasome via a kifunensine-insensitive pathway. Interestingly, overnight treatment of the R383A mutant with mitoxantrone assisted in protein maturation as evidenced by a shift to the N-glycosylated form. The R383A mutant when expressed in insect cells, though detected on the surface, had no measurable ATPase activity. In addition, substitution with the positively charged lysine resulted in significantly decreased protein expression levels in HEK cells, while retaining function. In conclusion, arginine 383 is a crucial residue for ABCG2 biogenesis, where even the most conservative mutations have a large impact.
Keywords: ABCG2, ABC transporter, membrane protein, mutagenesis, biogenesis
Introduction
ABCG2 is a member of the G subfamily of human ATP-binding cassette (ABC) transporters [1]. It was first identified almost a decade ago in multidrug-resistant carcinoma cell lines [2–4]. Consequently, the first substrates of ABCG2 described were chemotherapeutic agents, such as mitoxantrone, topotecan, daunorubicin, methotrexate, SN-38, flavopiridol, and more recently the tyrosine kinase-inhibitors imatinib and gefitinib [3–11]. Subsequent to the initial discovery of ABCG2, a wide range of substrates have been identified, among them fluorescent dyes, porphyrins, flavonoids, antibiotics, HMG-CoA-reductase inhibitors, and antiviral agents [12]. The list of inhibitors of ABCG2-mediated transport is also rapidly expanding; Fumitremorgin C (FTC) and its analog Ko143 are most frequently used in the laboratory [13, 14]. Despite the fact that several chemotherapeutic agents have been confirmed as substrates of ABCG2, its relevance in drug resistance is still unclear and no clinical trials have to date been initiated to evaluate its role in improving chemosensitivity. On the other hand, the expression pattern in normal tissues, namely its presence in the placenta, blood-brain barrier, and intestine suggests a physiologic role in protection against xenobiotics. The normal tissue distribution of ABCG2 and the fact that both dietary compounds and several drugs currently in clinical use are substrates, predict a major role for the protein in pharmacokinetics. ABCG2 has also been described on the surface of the so-called side population (SP) of stem cells, further suggesting an important physiologic role for the transporter [15].
ABCG2 is a 655-amino acid plasma membrane protein with six predicted transmembrane (TM) alpha helices. With its one nucleotide-binding domain (NBD) and one transmembrane domain (TMD) the protein is considered a half-transporter and is thought to homodimerize or homo-oligomerize for function [16–20]. Currently, only limited information is available regarding the structure of ABCG2. The protein is N-glycosylated at asparagine 596 [21], phosphorylated at threonine 362 [22], and it has been suggested that a disulfide bond forms between cysteines 603 in the homodimer [23]. The structure is most likely similar to the recently crystallized bacterial ABC transporters, yet the lack of significant sequence similarity and the reverse orientation with an N-terminal NBD and a C-terminal TMD, make it hard to predict the three dimensional structure of ABCG2.
Here we present studies aimed at understanding the role of a conserved arginine (R383) presumed to localize N-terminally to the first transmembrane segment (TM) of ABCG2 near the cytoplasmic side of the membrane. Most computer programs used to predict transmembrane helices identify glutamine 393 or alanine 394 as the first residue of TM1 of ABCG2 [24]. Our group recently published a homology model of ABCG2, in which, after considering various empirical rules we predicted a longer TM1 that starts at serine 384 [24]. The Msba structure we used to model the TMD region of ABCG2 has since been retracted together with four other structures by the same research group due the problems in the software used to analyze the data [25]. However, the NBD region (based on MalK from E. coli) and the predicted number and length of the TM segments in our model are not affected by these retractions. Mutational analysis of this arginine residue, as presented below, shows the importance of this residue in the proper function and biogenesis of ABCG2.
Experimental Procedures
Cell Culture
Human embryonic kidney (HEK) 293 cells (ATCC, Manassas, VA) were maintained in Minimal Essential Medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Invitrogen), 2 mM glutamine (BioFluids, Rockville, MD), and 100 units/L penicillin/streptomycin (BioFluids) at 37 °C in 5% CO2. Stably transfected cell lines were grown in 2 mg/mL G418 (Invitrogen).
Cells were incubated overnight at 37 °C with either 5 μM mitoxantrone (Sigma), or 10 nM bafilomycin (Sigma), or 3 μM MG132 (Calbiochem, San Diego, CA), or 20 μg/mL kifunensine (Cayman Chemical, Ann Arbor, Mi) followed by membrane preparation and immunoblotting as described below.
Mutagenesis and Transfection
The R383A, R383G, R383H, R383K, and R383G/S384R mutants were generated by site-directed mutagenesis in the pcDNA3.1/Myc-HisA(−) vector (Invitrogen) as previously described [26]. The mutations were confirmed by sequencing the vectors initially, followed by genomic DNA sequencing of one representative clone of each stably transfected mutant for the full-length ABCG2 insert.
Stable transfectants were generated in HEK 293 cells as previously described [26]. Transfections were performed using TransFast transfection reagent (Promega, Madison, WI). Colonies were selected in 2 mg/mL G418 with frequent removal of dead cells and were expanded prior to study. Cells previously transfected with wild-type ABCG2, R482G and pcDNA vector only were used as controls [27].
Membrane Preparation and Immunoblotting
Microsomal membrane preparation was performed as described previously [26]. Briefly, cells were disrupted by nitrogen cavitation (Parr Instrument, Moline, IL) in a hypotonic lysis buffer, and membranes were obtained by ultracentrifugation at 40 000 rpm. Protein concentrations were measured by the Bradford method with Bio-Rad’s Protein Assay Reagent (Bio-Rad, Hercules, CA) using BSA standards (Pierce, Rockford, IL).
Immunoblotting was performed as previously described [26]. Briefly, microsomal membrane proteins were loaded onto precast 7.5% (w/v) SDS-polyacrylamide gels (Bio-Rad), subjected to electrophoresis, and electrotransferred onto PVDF membranes (Millipore, Bedford, MA). Blots were probed with a 1:250 dilution of the monoclonal anti-ABCG2 antibody BXP-21 (Kamiya Biomedical, Seattle, WA) and visualized with the Odyssey System (LI-COR, Lincoln, NE) using a 1:2000 dilution of the IRDye 800CW goat anti-mouse secondary antibody (LI-COR). Membranes were stained with 0.1% Ponceau S (Sigma, St. Louis, MO) and checked for comparable loading.
For enzymatic deglycosylation, the Glyko® N-Glycanase® and Glyko® Endoglycosidase H kits were used (ProZyme, San Leandro, CA) following the manufacturer’s instructions. 50–100 μg of membranes were incubated with 2 μl PNGase F, or 6.7 μl Endo H overnight at 37 °C followed by immunoblotting as described above.
Northern Blotting
RNA was extracted from cells using RNA STAT-60 (Tel-Test Inc., Friendswood, TX). Northern blot analysis was performed by standard methods. Labeling of cDNAs was accomplished using Riboprobe in Vitro Transcription Systems (Promega). To compare the quality and quantities of RNA, 20 μg total RNA were electrophoretically separated in a 1% agarose, 6% formaldehyde gel and transferred onto a nitrocellulose membrane. Gels were stained with ethidium bromide and checked for comparable loading. Northern blot labeling was performed using a riboprobe generated from the first 662 bp of ABCG2 subcloned in a pCRII-TOPO vector (Invitrogen).
In vitro transcription and translation
In vitro transcription studies with T7 RNA polymerase and translation using the rabbit reticulocyte lysate system were carried out as described by Hegde et al [28]. PCR-amplified wild-type ABCG2 or the R383A mutant from the appropriate pcDNA3.1 vectors were used as templates. Transcription reactions were performed for 1 hr at 37 °C and translation reactions for 1 hr at 32 °C.
Flow Cytometry
Flow cytometry with the anti-ABCG2 antibody 5D3 (eBioscience, San Diego, CA), was performed as previously described [26]. Briefly, cells were trypsinized and resuspended in DPBS with 2% bovine serum albumin (BSA) to which phycoerythrin-conjugated 5D3 or phycoerythrin-conjugated mouse IgG was added for 30 min. For the transport studies, cells were trypsinized, resuspended in complete media (phenol red-free IMEM with 10% fetal calf serum containing 20 μM mitoxantrone (Sigma), or 1 μM pheophorbide a (Frontier Scientific, Logan, UT), or 250 nM BODIPY-prazosin (Molecular Probes, OR) with or without 10 μM of the ABCG2 blocker, Fumitremorgin C (FTC), and incubated for 30 min at 37 °C in 5% CO2. (FTC was synthesized by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, National Institutes of Health, Bethesda, MD). Cells were then incubated for 1 hr at 37 °C in substrate-free media, continuing with or without 10 μM FTC. Cells were analyzed on a FACSort flow cytometer, equipped with both a 488 nm argon laser and a 635 nm red diode laser.
Immunofluorescence
Immunofluorescence studies were performed as previously described [26, 29]. Briefly, cells were cultured for 3 days followed by fixation with 4% paraformaldehyde and permeabilization with prechilled (−20 °C) methanol. After blocking in a buffer containing 2 mg/mL BSA, 0.1% Triton X-100, and 5% goat serum, samples were incubated with a 1:100 dilution of the mouse monoclonal anti-ABCG2 antibody, BXP-21 (Kamiya Biomedical), and with a 1:1000 dilution of the rabbit polyclonal anti-calnexin antibody (Abcam, Cambridge, MA) for 1 h at room temperature followed by incubation with FITC-conjugated goat anti-mouse and rhodamine-conjugated goat anti-rabbit secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). After repeated washes, the staining was analyzed with an Olympus IX70 Laser Scanning Confocal Microscope.
Generation of Sf9 Cells Expressing the R383A and R383G Mutants
Generation of transfer vectors containing wild-type ABCG2 has been described previously [30, 31]. The transfer vectors carrying the R383A and R383G mutants were generated by cloning the SacI fragment of pcDNA 3.1/R383A or R383G into the corresponding site of the pAcUW21-L vector. Recombinant baculoviruses carrying the different human ABCG2 cDNAs were generated with the BaculoGold transfection kit (Pharmingen, San Diego, CA) according to the manufacturer’s instructions. Sf9 (Spodoptera frugiperda) cells were infected and cultured as described previously [32]. Individual virus clones, expressing high levels of the different human ABCG2 mutants, were obtained by end point dilution and subsequent amplification.
Membrane Preparation and Immunodetection of ABCG2 in Sf9 Cells
After being infected with virus for 3 days, Sf9 cells were harvested, and membranes were isolated. Membrane protein concentrations were determined by the modified Lowry method as previously described [32]. Immunoblotting was performed as described for the HEK 293 cells with a 1:2000 dilution of the monoclonal BXP-21 antibody.
Flow cytometry was performed by labeling the Sf9 cells at 37 °C using a final concentration of 1 μg/mL of the anti-ABCG2 monoclonal antibody 5D3. Binding was visualized by the addition of a secondary phycoerythrin-conjugated anti-mouse IgG (Immunotech, Marseille, France) at a final concentration of 1 μg/mL. Flow cytometric determination of the antibody reaction was carried out using a FACSCalibur cytometer with 488 nm excitation and 585/42 nm emission wavelengths.
ATP Hydrolysis
Sf9 membranes containing wild-type ABCG2, R383A, or R383G were harvested, and membranes were isolated and stored at −80 °C according to the method of Sarkadi et al. [33]. ATPase activity was measured as described previously by assessing the liberation of inorganic phosphate from ATP with a colorimetric reaction [16].
Results
Structure-function studies were undertaken to evaluate the role of arginine 383, which is predicted to immediately precede TM1 of ABCG2 [24]. This residue is highly conserved in the ABCG subfamily and in the Drosophila white protein, an ortholog of human ABCG2. Figure 1 shows an alignment of the six members of the ABCG subfamily and the Drosophila white protein created in the program ClustalX. Arginine 383 is also remarkably well conserved in all ABCG2 genes from other species that have so far been sequenced (Supplemental Table 1).
Figure 1.

Sequence alignment for members of the ABCG subfamily.
Arginine 383 of ABCG2 is well conserved in the ABCG subfamily and in the Drosophila white protein. Arginine 383 and other nearby positively charged residues in ABCG2 are highlighted. The program ClustalX was used to create the alignments.
To begin investigating the role of arginine 383 in ABCG2, HEK 293 cells were stably transfected with pcDNA3.1 vectors carrying the R383G and R383A mutants. We chose to substitute arginine with glycine or alanine to determine the impact of loss of charge and bulk at this position. Further, in the case of arginine 482 predicted to localize near the cytoplasmic interface of TM3, an arginine to glycine mutation results in marked alterations in substrate specificity with the addition of substrates including anthracyclines and rhodamine 123 [34]. HEK cells stably transfected with wild-type ABCG2 and pcDNA3.1 vector only served as controls. First, flow cytometry performed on nonpermeabilized cells using the 5D3 monoclonal antibody to recognize an extracellular epitope of ABCG2 demonstrated detectable levels of surface expression for some of the R383A clones, of which clones #11 and #24 exhibited the highest levels, though significantly lower compared to the wild-type (Figure 2/A). On the other hand, no surface expression was detected in any of the 24 glycine-substituted clones (Figure 2/A).
Figure 2.
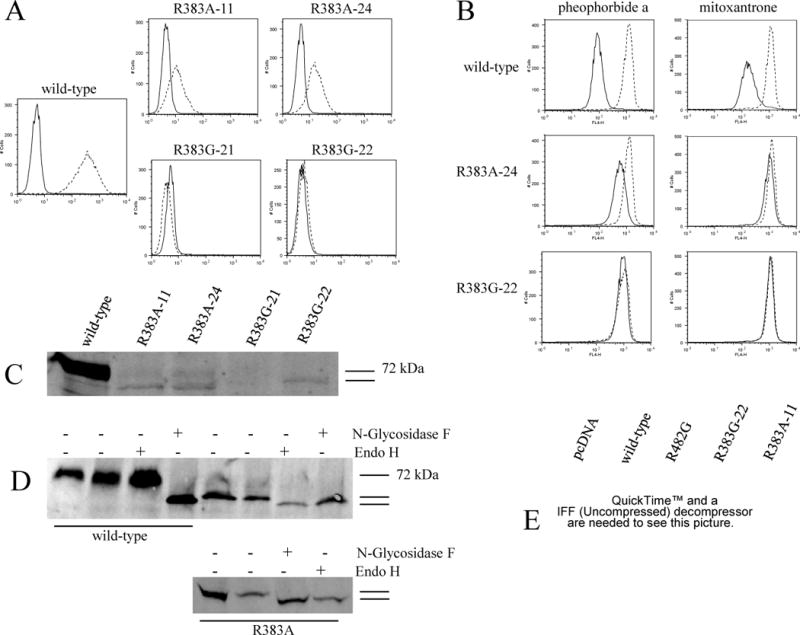
Surface expression, function, protein and RNA levels of the R383A and R383G mutants transfected into HEK 293 cells.
A: Flow cytometry with the 5D3 surface antibody for two clones of each mutant. Stably transfected HEK 293 cells were incubated for 30 min in phycoerythrin-labeled negative control antibody (solid line) or 5D3 antibody (dashed line) and analyzed in a FACSsort flow cytometer. B: Cells were incubated for 30 min in complete media containing 1 μM pheophorbide a or 20 μM mitoxantrone with or without 10 μM of the ABCG2-blocker FTC. (Accumulation without FTC – solid line, with FTC – dashed line.) C: Membrane proteins from the same mutant clones and wild-type were separated by SDS/PAGE (50 μg/lane), transferred onto a PVDF membrane, and probed with the monoclonal anti-ABCG2 antibody BXP-21. D: Immunoblot analysis of membrane proteins from wild-type (50 μg) and R383A (150 μg) transfectants with the BXP-21 following overnight treatment with Endo H, or with N-Glycosisdase F. The second lane for both the wild-type and the mutant represents overnight incubation in buffer with no enzyme. Two independent experiments are shown for the mutant. E: Northern blot showing RNA levels of one clone of each mutant compared to wild-type and empty vector transfected cells (pcDNA). Total RNA (20 μg/lane) from each transfectant was electrophoresed and transferred to a nitrocellulose membrane.
To test the functionality of the R383A and R383G mutants, flow cytometry was performed after incubating cells with the ABCG2-substrates mitoxantrone and pheophorbide a, with or without the ABCG2-inhibitor FTC (Figure 2/B). The magnitude of drug transport in this assay is indicated by the separation between histograms obtained from cells incubated with or without the inhibitor. The experiments were repeated several times, with limited efflux seen only in the R383A mutant as represented by a slight shift between the FTC-treated and non-treated histograms. Transport of pheophorbide a was more consistently observed in these experiments, most likely due to the fact that it is a “better” substrate than mitoxantrone, as evidenced by a larger shift in the wild-type transfectant, which was also observed in drug-selected cell lines [35]. The R383G mutant, as expected from its absence at the cell surface, did not display transport activity for either mitoxantrone or pheophorbide a.
Protein expression levels on immunoblot with the BXP-21 monoclonal anti-ABCG2 antibody were significantly decreased for the R383A mutant, and a band slightly lower than the expected 72 kDa was also visible (Figure 2/C). For subsequent experiments, clone #24 was used due to its slightly higher level of expression. The R383G mutant clones were virtually not detectable, only clone #22 displayed some level of expression and was also represented by a double band (Figure 2/C). In some of our subsequent experiments, the mutants were represented only by the lower molecular weight band on immunoblot under identical experimental conditions. To examine whether this lower molecular weight band corresponds to the non-glycosylated form of ABCG2, we performed overnight enzymatic deglycosylations of mutant and wild-type proteins with either N-glycosidase F, or endoglycosidase (endo) H. The molecular weight of the mutant proteins visualized on immunoblot following incubation with both enzymes were identical and slightly lower than without treatment, suggesting altered glycosylation of the mutant with endo H sensitivity, which would imply that the mutants did not reach the trans Golgi network (Figure 2/D, for the mutant two independent experiments are shown). To exclude the possibility that mutations at position 383 altered binding of the BXP-21 antibody, a commercially available rabbit polyclonal antibody and the 405 and 391 rabbit polyclonal antibodies with different epitopes previously generated in our laboratory [36] were also used on immunoblots of all the mutants described in this manuscript, none of which yielded better detection than BXP-21 (data not shown). Finally, Northern blotting was carried out to confirm that the reduced expression of these mutants was not due to poor transfection efficacy; significant amounts of ABCG2 RNA were noted in representative clones of the R383A and R383G mutants (Figure 2/E).
To determine whether the low protein levels were due to failure of protein translation or degradation mediated by ER quality control mechanisms, we performed in vitro transcription and translation in the presence, or absence of rabbit reticulocyte lysates. These experiments showed no difference between wild-type ABCG2 and the R383A mutant, suggesting that, similarly to the wild-type, the mutant protein is translated and inserted properly into the membrane in the in vitro system and implying that the ER quality control must promote rapid degradation in vivo (data not shown).
To investigate the mechanisms leading to the dramatic decrease in protein levels observed with the mutants, the R383A mutant was incubated overnight separately with either the lysosome inhibitor bafilomycin, or the proteasome inhibitor MG132 (Figure 3). Incubation with bafilomycin did not result in any changes in the amount of mutant protein detected. On the other hand, treatment with MG132 led to a 3 to 5-fold increase in the amount of the R383A mutant observed on immunoblot (Figure 4/A), indicating that the mutant is degraded by the ubiquitine-proteasome pathway. To further explore the ER quality control mechanism behind the degradation of the mutant, we incubated the R383A mutant overnight in kifunensine, a potent inhibitor of mannosidase I. Mannose trimming by mannosidase I is one of the known events leading to ubiquitination and proteasomal degradation via the so-called glycan-dependent pathway (Figure 3) [37]. Figure 4/B shows that kifunensine treatment did not increase the amount of the R383 mutant on immunoblot. In the case of the wild-type, a lower molecular weight band was visualized, suggesting that a fraction of the wild-type protein is degraded by a kifunensine-sensitive pathway. Overnight treatment with MG132 and kifunensine was also performed on HeLa cells transiently transfected with the wild-type and the R383A mutant providing results identical to the ones presented with the stable HEK transfectants (data not shown).
Figure 3.
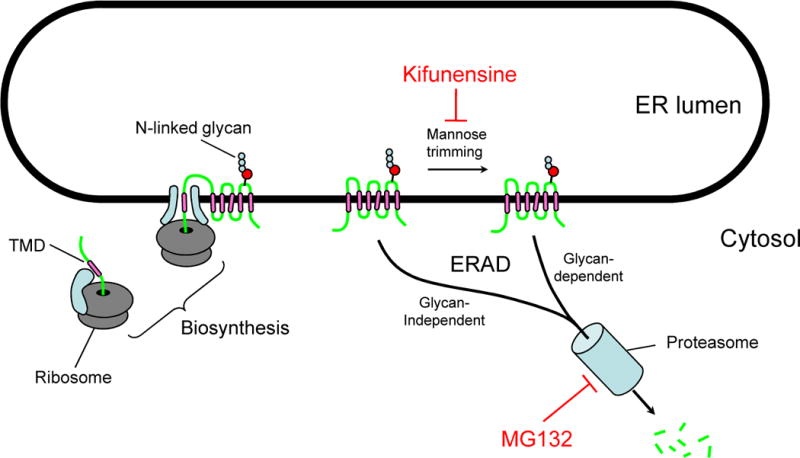
The mechanism of action of kifunensine and MG132
Schematic representation of the pathways blocked by kifunensine and MG132 as part of the Endoplasmic Reticulum Associated Protein Degradation (ERAD).
Figure 4.
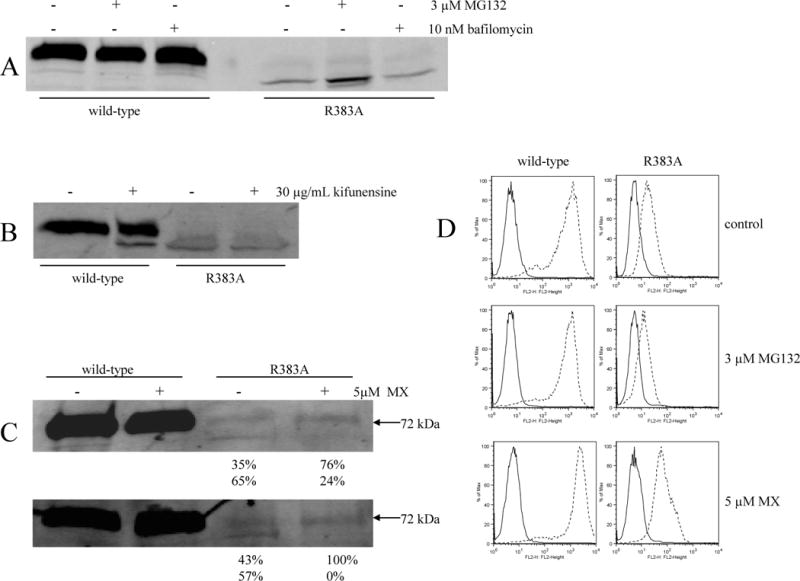
Proteasome/lysosome inhibition and “rescue” of the R383A mutant with mitoxantrone (MX).
Parts A, B, and C: membranes were harvested subsequent to overnight incubation with or without 3μM MG132, or 10 nM bafilomycin (part A), or 30 μg/mL kifunensine (part B), or 5 μM mitoxantrone (part C) followed by immunoblot analysis with the BXP-21 antibody for the wild-type (10 μg/lane) and R383A (10 μg/lane) transfectants. For part C, results of two experiments performed separaretly are shown. The numbers below represent the percentage of detected protein in the upper (N-glycosylated) and lower (non glycosylated) bands as determined by the Odyssey software. Part D: following overnight treatment with 3 μM MG132, or 5 μM mitoxantrone, cells were incubated for 30 min in phycoerythrin-labeled negative control antibody (solid line) or 5D3 antibody (dashed line) and analyzed in a FACSsort flow cytometer for surface expression of ABCG2. (The experiments were performed in triplicates.)
Next, we evaluated whether the mutant proteins could be “rescued” in the presence of a substrate or by decreased temperature. Treatment with substrate drugs has been reported to stabilize some P-glycoprotein mutants [38] and we have previously reported increased protein levels for the GXXXG motif-mutants of ABCG2 following overnight treatment with mitoxantrone [26]. The second approach, growing cells at lower temperature, was successful in increasing protein levels of certain temperature-sensitive CFTR mutants [39]. Overnight treatment of the stably transfected HEK cells with the ABCG2-substrate mitoxantrone failed to result in an increase in mutant protein levels on immunoblot and neither did culturing the transfected cells at 28 °C for two days (data not shown). Interestingly, in the case of the R383A mutant, though the overnight treatment with mitoxantrone did not result in increased protein expression level, a shift to the 72-kDa band was visible in the drug-treated lanes, suggesting that the drug did act as a chaperone and helped generate the mature, N-glycosylated form of the protein, although levels were not increased (Figure 4/C). Two separate experiments are shown in the figure, both demonstrating this shift in the lower band from constituting approximately 60% of total detectable ABCG2 in the untreated mutant to an average of 10% following treatment with mitoxantrone.
To determine whether the changes seen on immunoblot following treatment with MG132 and mitoxantrone are accompanied by parallel changes on the cell surface, flow cytometry experiments were performed with the 5D3 surface antibody subsequent to overnight incubation with either 3 μM MG132, or 5 μM mitoxantrone (Figure 4/D). While we previously observed an increase in the amount of the R383A mutant protein represented by the lower than 72-kDa band on immunoblot (Figure 4/A), we did not detect any increase on the cell surface with the 5D3 antibody, suggesting that by blocking the proteasomal degradation pathway the mutant protein accumulated intracellularly. On the other hand, in agreement with the shift observed in the case of the R383A mutant to the 72-kDa mature form on immunoblot (Figure 4/C), we detect increased cell surface expression of the mutant protein by flow cytometry following overnight treatment with mitoxantrone (Figure 4/D). Treatment with mitoxantrone resulted in a similar increase in the amount of the R383G mutant detected on the cell surface (data not shown).
To further analyze the localization of the R383A and R383G mutants in the mammalian cells, immunofluorescent staining followed by confocal microscopy was performed. In the case of the R383A mutant, colocalization with the ER marker calnexin was suggested, while some of this mutant also localized to the cell surface (Figure 5), which is in agreement with the results of flow cytometry shown in Figure 2. The expression level for the R383G mutant was too low to determine localization using confocal microscopy.
Figure 5.
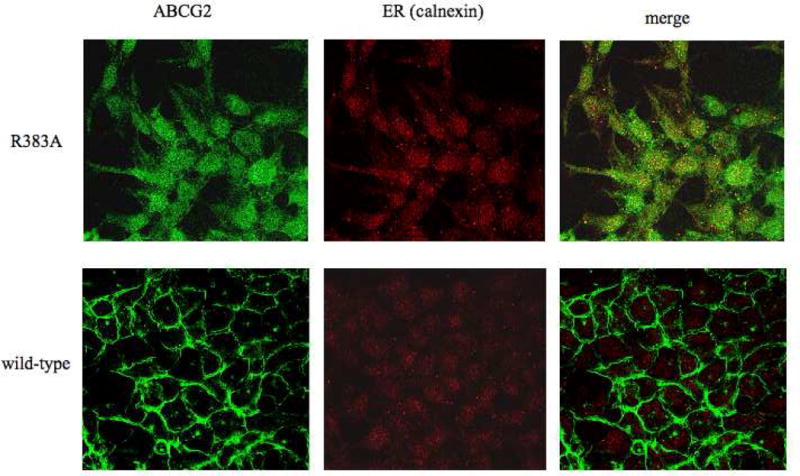
Localization of the R383A mutant in HEK 293 cells.
Confocal microscopy of stably transfected HEK 293 was performed following fixation with paraformaldehyde and permeabilization with methanol. Immunostaining was carried out for 1 hour at room temperature with the BXP-21 monoclonal anti-ABCG2 antibody (green) and the anti-calnexin polyclonal antibody (red).
Next, we evaluated the R383G and R383A mutations in a heterologous system, using Sf9 insect cells, a transfection system that generally yields high protein levels allowing the study of proteins with low expression levels in mammalian cells [40]. For both mutants, the amount of protein expressed on immunoblot in Sf9 cells was approximately one fifth of the wild-type levels (Figure 6/A), substantially more than seen in the HEK 293 cells (Figure 2/C). Flow cytometry with the 5D3 monoclonal anti-ABCG2 antibody revealed that, similar to the mammalian cells, the R383A mutant was detectable on the surface in the insect cells, while the R383G mutant was not (Figure 6/B). Neither mutant displayed any significant ATPase activity in the insect cells (Figure 6/C).
Figure 6.
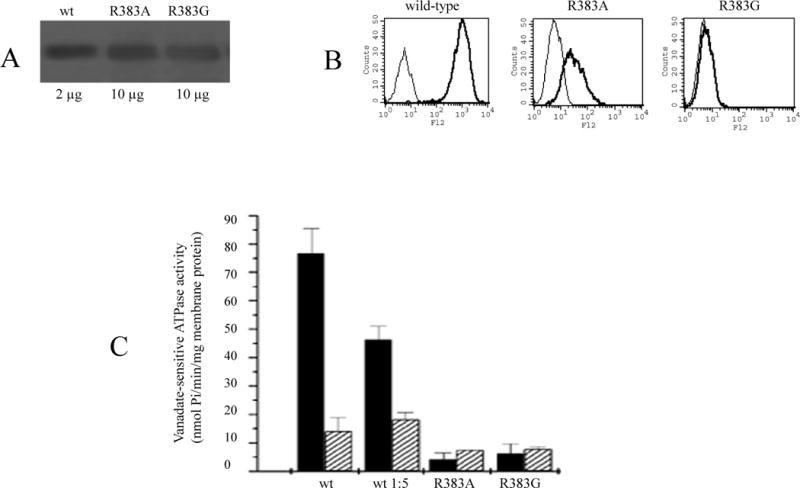
The R383A mutant in Sf9 insect cells.
A: Immunodetection of the indicated amounts of membrane proteins with the BXP-21 monoclonal anti-ABCG2 antibody. B: Flow cytometry with the 5D3 monoclonal anti-ABCG2 antibody using non-permeabilized Sf9 cells (as detailed in Figure 2). C: Basal ATPase activity of the wild-type, a 1:5 dilution of the wild-type, the R383A mutant, and the non-functional K86M mutant in Sf9 membranes. ATPase activity was measured determining the liberation of inorganic phosphate from ATP with a colorimetric reaction with (grey bars) or without (black bars) the ABCG2-specific inhibitor Ko143.
The results presented so far suggest a critical role for the conserved arginine 383 in the biogenesis of the ABCG2 protein. We postulated that this arginine might function as an anchor of the first transmembrane alpha helix, thus more conservative substitutions were performed: mutation to the positively charged lysine and introduction of arginine at position 384 instead of 383, creating the R383K and R383G/S384R mutants, followed by stable transfections in HEK 293 cells. Like R383A, the R383K mutant displayed some surface expression on flow cytometry (Figure 7/A) and the protein was detectable on immunoblot, though expression levels were still markedly reduced when compared to the wild-type transfectant (Figure 7/B). Just as observed for the alanine mutant, R383K was represented by a double band on immunoblot suggestive of altered glycosylation. The functionality of the R383K mutant was also assessed in flow cytometry-based transport assays. All three tested ABCG2 substrates, namely mitoxantrone, BODIPY-prazosin, and pheophorbide a, were effluxed from the cells by this mutant as evidenced by increased intracellular fluorescence after treatment with the ABCG2-inhibitor FTC (Figure 7/C). On the other hand, none of the R383G/S384R mutant clones were detectable either on the cell surface by flow cytometry with the 5D3 antibody or on immunoblot with the BXP-21 antibody (data not shown). Presence of the transfected vector was confirmed by PCR using pcDNA3.1 vector specific primers followed by sequencing.
Figure 7.
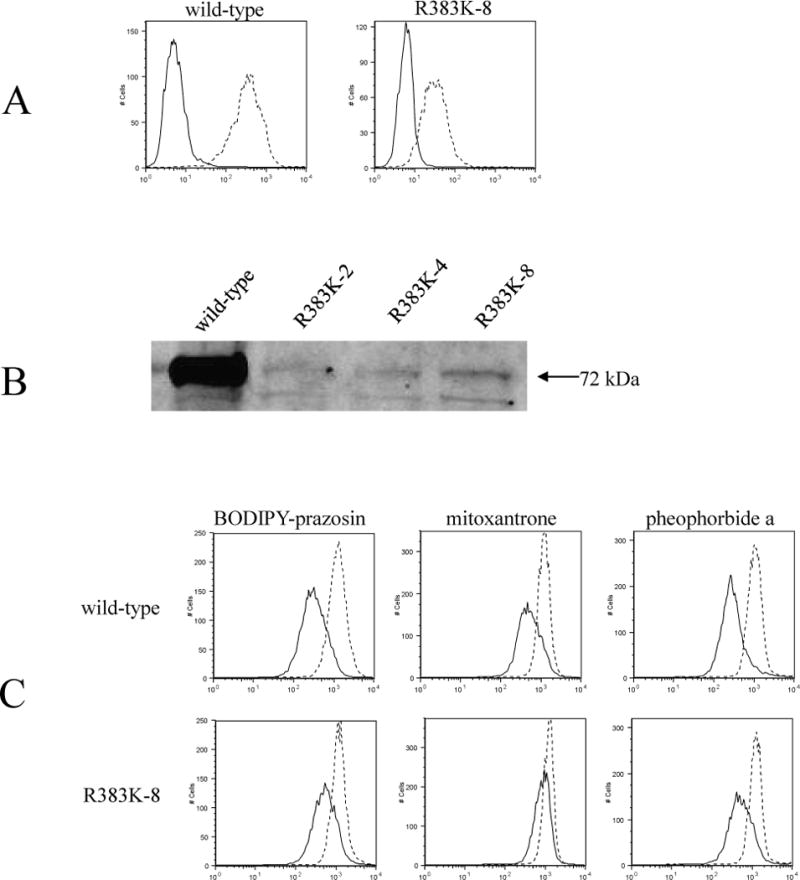
Surface expression, function and protein levels of the R383K mutant transfected into HEK 293 cells.
A: The R383K mutant (clone #8) detected on the cell surface with the 5D3 antibody by flow cytometry performed as described for Figure 2. (Negative control antibody – solid line, 5D3 antibody – dashed line) B: 30 μg of membrane proteins on immunoblot with the BXP-21 antibody. C: Flow cytometry following incubation in complete media containing 250 nM BODIPY-prazosin, or 20 μM mitoxantrone, or 1 μM pheophorbide a, with or without 10 μM of the ABCG2- blocker, FTC. (Accumulation without FTC – solid line, with FTC – dashed line)
Interestingly, we noted that an arginine to histidine mutation in the corresponding residue in ABCG8 (R405H) had been reported in a patient with sitosterolemia [41]. To study the effect of the same substitution in ABCG2, we created stable R383H clones in HEK 293 cells. This mutant was not detectable on the cell surface with the 5D3 antibody or on immunoblot with the BXP-21 antibody (data not shown). The presence of the mutant vector was confirmed by sequencing DNA from the transfected clones.
Discussion
ABCG2 is an ATP-binding cassette half-transporter with a potential role as a mediator of multidrug resistance in cancer. The protein is also of great interest as the determinant of the side population of stem cells; it most likely plays a major role in the pharmacokinetics of its substrate drugs and in protection against xenobiotics. In the present manuscript, we report mutational studies of a highly conserved arginine residue predicted to precede TM1 of ABCG2. We used an immortalized human cell system, HEK 293 that we and others have found to be a good model for mutational analysis based on the ease of transfection and the abundant protein generated in transfections of the wild-type vector. In this system mutating arginine 383 to glycine, alanine, histidine or lysine resulted in markedly reduced to no protein expression, impaired glycosylation and retention in the ER. The R383A mutant was shown to be degraded by the proteasome via a kifunensine-insensitive pathway. In insect cells, more tolerant of aberrant protein, more mutant protein was detected on immunoblot, but no ATPase activity could be detected. These results highlight the critical nature of this residue; the implications of the differences among the substitutions are discussed.
Resolving the three dimensional structure of transporters like ABCG2 will be critical in understanding how these proteins function and could aid in designing specific inhibitors as well as predicting whether particular molecules are transported by these pumps. Although it is estimated that as much as 30% of the human genome encodes membrane proteins [42], only a limited number of high-resolution structures are currently available. To date, the structure of little over 150 integral membrane proteins from different species is known to greater than 4-Å resolution. Only six of the known structures are those of ABC transporters, all from bacteria, while no human ABC transporter has been crystallized so far [43–48]. In the absence of available crystal structures, mutational analysis has proven to be a valuable tool in studying membrane proteins. An excellent example of this is P-glycoprotein, in the case of which extensive mutagenesis predicts a structure very similar to the recently crystallized bacterial homologue Sav1866 [45, 49].
The mutational analysis presented here emphasizes the importance of the conserved arginine 383 in ABCG2, where conservative and non-conservative mutations alike have a large impact on protein expression, trafficking and presumably folding. Elsewhere, non-conservative substitutions like arginine with alanine or glycine do not necessarily result in impaired protein folding and/or function. For example, mutating arginine 482 of ABCG2 to glycine, results in altered substrate specificity, often referred to as a gain-of-function, adding doxorubicin, daunorubicin, rhodamine 123, etc. to the list of substrates, although arginine 482 is predicted to localize near the cytoplasmic side in TM3, while arginine 383 is predicted to be on the outside of TM1 [24, 34]. Arginine 482 might to be involved in substrate binding and/or recognition, given that mutations not only to glycine but to almost any amino acid have the same gain-of-function effect [27, 50, 51]. On the other hand, recent studies suggest that that residue 482 does not play a direct role in drug binding; rather, it might be involved in energy coupling [52, 53]. Similarly positioned arginines and other positively charged residues have been implicated in substrate recognition of MRP1, another human ABC transporter associated with multidrug resistance [54–57]. For example, the R1249A mutant of MRP1 has been described as having altered substrate specificity. Arginine 1249 is on the cytoplasmic side of TM17 of MRP1 and might correspond to TM1 of ABCG2 since the order of the TMD and NBD in ABCG2 is reversed when compared to MRP1, a full ABC transporter with 17 TMs. [56]. Unlike arginine 482 of ABCG2 and the above-mentioned arginines of MRP1, the mutational analysis presented here does not suggest a role for arginine 383 in substrate binding, rather, the mutations described above seem to affect protein folding, and/or trafficking.
Mutants that fail to properly fold or insert into the membrane are most likely recognized by the cell’s quality control machinery and are thought to be rapidly degraded explaining the decreased expression levels observed with such mutants [58]. Degradation may occur through several pathways. Our results with the proteasome inhibitor MG132 indicate that the R383A mutant is degraded via the proteasome, while the wild-type protein is not. This result is in agreement with recent studies describing proteasomal degradation of certain ABCG2 mutants [59, 60]. Although, these reports suggest that the wild-type protein is degraded by the lysosome, our experiments show no significant increase in the amount of wild-type protein following inhibition of lysosomal degradation by bafilomycin. The quality control pathway by which the R383A mutant is targeted to the proteasome seems to be kifunensine insensitive. On the other hand, kifunensine treatment of the wild-type resulted in the appearance of a lower molecular weight band, most likely representing the small portion of the wild-type protein that is misfolded and degraded. During the complicated process of protein synthesis and folding multiple errors can occur resulting in proteins that are, similarly to mutants, recognized by the ER quality control machinery and targeted for degradation. A good example of this is the ABC transporter cystic fibrosis transmembrane conductance regulator (CFTR) protein, in the case of which, 40–60% of the wild-type protein is presumed to be misfolded, retained in the ER, and rapidly degraded [61, 62].
Our attempts, through overnight treatment with a substrate or through culture at 28 °C, to “rescue” the mutant proteins, did not succeed in increasing the levels of detected protein. Interestingly, in the case of the R383A mutant, mitoxantrone seemed to help generate the mature, glycosylated protein. Such an approach could be of clinical importance in “rescuing” disease-causing mutants, such as certain ABCG5 or ABCG8 mutants resulting in sitosterolemia. This strategy is of particular interest in the case of CFTR, mutations in which result in cystic fibrosis, a serious, sometimes fatal genetic disorder [63]. Other examples of misfolded proteins rescued by chemical chaperones are P-glycoprotein, MRP1, SUR1, and the dopamine D4 receptor as reviewed by Loo et al. [64].
The R383A mutant localized to the cell surface and displayed some function in the HEK cells. When transfected to Sf9 insect cells, R383A was also detected on the surface, yet was unable to hydrolyze ATP. Presumably, the insect cells are more tolerant of misfolded proteins; this and their ability to generate higher levels of a misfolded protein apparently leads to surface expression of a non-functional protein. We surmised that quality control in mammalian cells is very sensitive to proper folding and does not tolerate mutation at 383. If the process of folding during protein translation involves some trial and error, a small fraction may assume a normal conformation and fold together with its dimerization partner. These molecules reach the cell surface and are able to transport substrates normally. This difference in tolerance of misfolding between insect and human cells then leads to the observed discrepancy: more protein is present on the insect cell surface, yet it is not properly folded and the protein is non functional. Far less protein reaches the surface of a human cell, but it is properly folded and is functional.
The residue corresponding to arginine 383 of ABCG2 is very well conserved and predicted to localize near the cytoplasmic side of TM1 [24]. Charged residues are generally excluded from TM segments and positively charged ones tend to cluster on the intracellular side of the membrane serving as a type of membrane anchors, while negative ones are more likely to be found on the extracellular side [65]. This asymmetry in the distribution of charged residues, also called the “positive-inside rule” [66], is thought to promote correct orientation and insertion of transmembrane segments into the membrane bilayer. Indeed, residues 382 and 386 of ABCG2 are lysines and 378 is another arginine. With the exception of position 382, where the positive charge is present in other members of the ABCG subfamily, these residues are not conserved in the G subfamily. When arginine 383 was replaced by glycine, a small, neutral amino acid, the mutated protein was no longer expressed on the cell surface. Substitution with histidine, the residue associated with sitosterolemia, with weak positive charge at neutral pH, also resulted in loss of surface expression. On the other hand, the results with alanine at 383 showing a small amount of surface expression and function suggest that this amino acid of intermediate size between arginine and glycine may have allowed lysine 382 and arginine 378 to serve as the membrane anchor, whereas the glycine was too small. The R383K mutant, which preserved the strong positive charge at this position, was detectable on the cell surface and transported the tested substrates, though protein expression levels were significantly reduced. On the other hand, the differences observed with the various substitutions could, at least partially, be attributed to changes in the flexibility/motion of the surrounding sequence. Taken together, the results of the conservative lysine substitution and the in vitro translation experiments with the R383A mutant suggest that the role of arginine 383 is beyond that of simply anchoring the membrane.
In conclusion, we can say that arginine 383 plays a critical role in the proper expression, surface localization, and function of ABCG2. The fact that even the most conservative mutation at this position results in significant decrease in protein expression levels emphasizes the importance of this residue. It will be interesting to discover whether the corresponding R405H mutation in ABCG8 leads to similar effects on protein levels and trafficking as predicted by our data but not yet demonstrated in clinical samples, and if so, whether that mutant can be rescued in part or to any degree by pharmacological chaperones.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Tito Fojo for helpful advice and Elizabeth Finley and Julian Bahr for technical assistance with flow cytometry.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 2.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–5339. [PubMed] [Google Scholar]
- 3.Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, Brangi M, Greenberger L, Dean M, Fojo T, Bates SE. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- 4.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, Senderowicz AM, Ross DD, Bates SE. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (MXR/BCRP/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145–152. [PubMed] [Google Scholar]
- 6.Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, Ross DD, Bates SE, Kruh GD. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63:4048–4054. [PubMed] [Google Scholar]
- 7.Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, Kamihira S, Doyle LA, Ross DD, Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280:1216–1223. doi: 10.1006/bbrc.2001.4267. [DOI] [PubMed] [Google Scholar]
- 8.Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]
- 9.Ozvegy-Laczka C, Hegedus T, Varady G, Ujhelly O, Schuetz JD, Varadi A, Keri G, Orfi L, Nemet K, Sarkadi B. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65:1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- 10.Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, Szabo K, Homolya L, Varadi A, Buday L, Keri G, Nemet K, Sarkadi B. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 11.Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, Nooter K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamurthy P, Schuetz JD. Role of abcg2/bcrp in biology and medicine. Annu Rev Pharmacol Toxicol. 2006;46:381–410. doi: 10.1146/annurev.pharmtox.46.120604.141238. [DOI] [PubMed] [Google Scholar]
- 13.Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T, Greenberger LM. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58:5850–5858. [PubMed] [Google Scholar]
- 14.Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, Schellens JHM, Koomen GJ, Schinkel AH. Potent and Specific Inhibition of the Breast Cancer Resistance Protein Multidrug Transporter in Vitro and in Mouse Intestine by a Novel Analogue of Fumitremorgin C. Mol Cancer Ther. 2002;1:417–425. [PubMed] [Google Scholar]
- 15.Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99:12339–12344. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozvegy C, Litman T, Szakacs G, Nagy Z, Bates S, Varadi A, Sarkadi B. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem Biophys Res Commun. 2001;285:111–117. doi: 10.1006/bbrc.2001.5130. [DOI] [PubMed] [Google Scholar]
- 17.Henriksen U, Gether U, Litman T. Effect of Walker A mutation (K86M) on oligomerization and surface targeting of the multidrug resistance transporter ABCG2. J Cell Sci. 2005;118:1417–1426. doi: 10.1242/jcs.01729. [DOI] [PubMed] [Google Scholar]
- 18.Bhatia A, Schafer HJ, Hrycyna CA. Oligomerization of the human ABC transporter ABCG2: evaluation of the native protein and chimeric dimers. Biochemistry. 2005;44:10893–10904. doi: 10.1021/bi0503807. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. doi: 10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]
- 20.McDevitt CA, Collins RF, Conway M, Modok S, Storm J, Kerr ID, Ford RC, Callaghan R. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure. 2006;14:1623–1632. doi: 10.1016/j.str.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 21.Diop NK, Hrycyna CA. N-Linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry. 2005;44:5420–5429. doi: 10.1021/bi0479858. [DOI] [PubMed] [Google Scholar]
- 22.Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, Nakanishi T, Ross DD, Chen H, Fazli L, Gleave ME, Qiu Y. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349–3356. doi: 10.1074/jbc.M707773200. [DOI] [PubMed] [Google Scholar]
- 23.Henriksen U, Fog JU, Litman T, Gether U. Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J Biol Chem. 2005;280:36926–36934. doi: 10.1074/jbc.M502937200. [DOI] [PubMed] [Google Scholar]
- 24.Li YF, Polgar O, Okada M, Esser L, Bates SE, Xia D. Towards understanding the mechanism of action of the multidrug resistance-linked half-ABC transporter ABCG2: a molecular modeling study. Journal of molecular graphics & modelling. 2007;25:837–851. doi: 10.1016/j.jmgm.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Chang G. Retraction of “Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation” [J. Mol. Biol. (2003) 330 419–430] J Mol Biol. 2007;369:596. doi: 10.1016/j.jmb.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, Ambudkar SV, Tarasova N, Bates SE. Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry. 2004;43:9448–9456. doi: 10.1021/bi0497953. [DOI] [PubMed] [Google Scholar]
- 27.Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE. Mutations at amino acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–1978. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hegde RS, Voigt S, Lingappa VR. Regulation of protein topology by trans-acting factors at the endoplasmic reticulum. Mol Cell. 1998;2:85–91. doi: 10.1016/s1097-2765(00)80116-1. [DOI] [PubMed] [Google Scholar]
- 29.Polgar O, Ozvegy-Laczka C, Robey RW, Morisaki K, Okada M, Tamaki A, Koblos G, Elkind NB, Ward Y, Dean M, Sarkadi B, Bates SE. Mutational Studies of G553 in TM5 of ABCG2: a Residue Potentially Involved in Dimerization. Bichemistry. 2006 doi: 10.1021/bi0521590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozvegy C, Varadi A, Sarkadi B. Characterization of drug transport, ATP hydrolysis and nucleotide trapping by the human ABCG2 multidrug transporter: modulation of substrate specificity by a point mutation. J Biol Chem. 2002;277:47980–47990. doi: 10.1074/jbc.M207857200. [DOI] [PubMed] [Google Scholar]
- 31.Ozvegy-Laczka C, Varady G, Koblos G, Ujhelly O, Cervenak J, Schuetz JD, Sorrentino BP, Koomen GJ, Varadi A, Nemet K, Sarkadi B. Function-dependent conformational changes of the ABCG2 multidrug transporter modify its interaction with a monoclonal antibody on the cell surface. J Biol Chem. 2005;280:4219–4227. doi: 10.1074/jbc.M411338200. [DOI] [PubMed] [Google Scholar]
- 32.Muller M, Bakos E, Welker E, Varadi A, Germann UA, Gottesman MM, Morse BS, Roninson IB, Sarkadi B. Altered drug-stimulated ATPase activity in mutants of the human multidrug resistance protein. J Biol Chem. 1996;271:1877–1883. doi: 10.1074/jbc.271.4.1877. [DOI] [PubMed] [Google Scholar]
- 33.Sarkadi B, Price E, Boucher R, Germann U, Scarborough G. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated ATPase. J Biol Chem. 1992;267:4854–4858. [PubMed] [Google Scholar]
- 34.Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, Litman T, Dean M, Bates SE. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–6639. [PubMed] [Google Scholar]
- 35.Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- 36.Litman T, Jensen U, Hansen A, Covitz K, Zhan Z, Fetsch P, Abati A, Hansen P, Horn T, Skovsgaard T, Bates S. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim Biophys Acta. 2002;1565:6–16. doi: 10.1016/s0005-2736(02)00492-3. [DOI] [PubMed] [Google Scholar]
- 37.Spiro RG. Role of N-linked polymannose oligosaccharides in targeting glycoproteins for endoplasmic reticulum-associated degradation. Cell Mol Life Sci. 2004;61:1025–1041. doi: 10.1007/s00018-004-4037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loo TW, Clarke DM. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J Biol Chem. 1997;272:709–712. doi: 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- 39.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 40.Altmann F, Staudacher E, Wilson IB, Marz L. Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj J. 1999;16:109–123. doi: 10.1023/a:1026488408951. [DOI] [PubMed] [Google Scholar]
- 41.Lu K, Lee MH, Hazard S, Brooks-Wilson A, Hidaka H, Kojima H, Ose L, Stalenhoef AF, Mietinnen T, Bjorkhem I, Bruckert E, Pandya A, Brewer HB, Jr, Salen G, Dean M, Srivastava A, Patel SB. Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am J Hum Genet. 2001;69:278–290. doi: 10.1086/321294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 44.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 45.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 46.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 47.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 49.Zolnerciks JK, Wooding C, Linton KJ. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. Faseb J. 2007;21:3937–3948. doi: 10.1096/fj.07-8610com. [DOI] [PubMed] [Google Scholar]
- 50.Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int J Cancer. 2003;107:757–763. doi: 10.1002/ijc.11484. [DOI] [PubMed] [Google Scholar]
- 51.Ozvegy-Laczka C, Koblos G, Sarkadi B, Varadi A. Single amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition. Biochim Biophys Acta. 2005;1668:53–63. doi: 10.1016/j.bbamem.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Pozza A, Perez-Victoria JM, Sardo A, Ahmed-Belkacem A, Di Pietro A. Purification of breast cancer resistance protein ABCG2 and role of arginine-482. Cell Mol Life Sci. 2006;63:1912–1922. doi: 10.1007/s00018-006-6159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ejendal KF, Diop NK, Schweiger LC, Hrycyna CA. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006;15:1597–1607. doi: 10.1110/ps.051998406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conrad S, Kauffmann HM, Ito K, Leslie EM, Deeley RG, Schrenk D, Cole SP. A naturally occurring mutation in MRP1 results in a selective decrease in organic anion transport and in increased doxorubicin resistance. Pharmacogenetics. 2002;12:321–330. doi: 10.1097/00008571-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 55.Conseil G, Deeley RG, Cole SP. Functional importance of three basic residues clustered at the cytosolic interface of transmembrane helix 15 in the multidrug and organic anion transporter MRP1 (ABCC1) J Biol Chem. 2006;281:43–50. doi: 10.1074/jbc.M510143200. [DOI] [PubMed] [Google Scholar]
- 56.Ren XQ, Furukawa T, Aoki S, Sumizawa T, Haraguchi M, Nakajima Y, Ikeda R, Kobayashi M, Akiyama S. A positively charged amino acid proximal to the C-terminus of TM17 of MRP1 is indispensable for GSH-dependent binding of substrates and for transport of LTC4. Biochemistry. 2002;41:14132–14140. doi: 10.1021/bi026443s. [DOI] [PubMed] [Google Scholar]
- 57.Haimeur A, Conseil G, Deeley RG, Cole SP. Mutations of charged amino acids in or near the transmembrane helices of the second membrane spanning domain differentially affect the substrate specificity and transport activity of the multidrug resistance protein MRP1 (ABCC1) Mol Pharmacol. 2004;65:1375–1385. doi: 10.1124/mol.65.6.1375. [DOI] [PubMed] [Google Scholar]
- 58.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 59.Wakabayashi K, Nakagawa H, Tamura A, Koshiba S, Hoshijima K, Komada M, Ishikawa T. Intramolecular disulfide bond is a critical check point determining degradative fates of ATP-binding cassette (ABC) transporter ABCG2 protein. J Biol Chem. 2007;282:27841–27846. doi: 10.1074/jbc.C700133200. [DOI] [PubMed] [Google Scholar]
- 60.Nakagawa H, Tamura A, Wakabayashi K, Hoshijima K, Komada M, Yoshida T, Kometani S, Matsubara T, Mikuriya K, Ishikawa T. Ubiquitin-mediated proteasomal degradation of non-synonymous SNP variants of human ABC transporter ABCG2. Biochem J. 2008;411:623–631. doi: 10.1042/BJ20071229. [DOI] [PubMed] [Google Scholar]
- 61.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 62.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 63.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 64.Loo TW, Bartlett MC, Clarke DM. Rescue of folding defects in ABC transporters using pharmacological chaperones. J Bioenerg Biomembr. 2005;37:501–507. doi: 10.1007/s10863-005-9499-3. [DOI] [PubMed] [Google Scholar]
- 65.Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.