

Abstract
The management of congenital adrenal hyperplasia involves suppression of adrenal androgen production, in addition to treatment of adrenal insufficiency. Management of adolescents with congenital adrenal hyperplasia is especially challenging because changes in the hormonal milieu during puberty can lead to inadequate suppression of adrenal androgens, psychosocial issues often affect adherence to medical therapy, and sexual function plays a major part in adolescence and young adulthood. For these reasons, treatment regimen reassessment is indicated during adolescence. Patients with non-classic congenital adrenal hyperplasia require reassessment regarding the need for glucocorticoid drug treatment. No clinical trials have compared various regimens for classic congenital adrenal hyperplasia in adults, thus therapy is individualised and based on the prevention of adverse outcomes. Extensive patient education is key during transition from paediatric care to adult care and should include education of females with classic congenital adrenal hyperplasia regarding their genital anatomy and surgical history. Common issues for these patients include urinary incontinence, vaginal stenosis, clitoral pain, and cosmetic concerns; for males with classic congenital adrenal hyperplasia, common issues include testicular adrenal rest tumours. Transition from paediatric to adult care is most successful when phased over many years. Education of health-care providers on how to successfully transition patients is greatly needed.
Introduction
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is an autosomal recessive disease of the adrenal cortex caused by mutations in the CYP21A2 gene. Impairment of cortisol biosynthesis results in overstimulation of the adrenal glands and excessive androgen production. There is a wide range of disease severity with good genotype-phenotype correlation. Genetic mutations that significantly impair synthesis of the 21-hydroxylase enzyme result in the classic or severe form of congenital adrenal hyperplasia, whereas genetic mutations that mildly impair the 21-hydroxylase enzyme result in a mild non-classic form of the disease.1 Classic congenital adrenal hyperplasia has an estimated worldwide prevalence of 1 in 16 000 births and presents in newborn girls as genital ambiguity.1,2 About 75% of patients with classic congenital adrenal hyperplasia also have severe aldosterone deficiency, which can result in a life-threatening salt-wasting adrenal crisis in the neonate. Patients with the classic form of the disease who produce small amounts of aldosterone and therefore escape the neonatal adrenal crisis are termed classic simple virilising. The prevalence of non-classic congenital adrenal hyperplasia is estimated as 0·1%, but can be higher (1–2%) in some ethnic groups (eg, Ashkenazi Jewish, Hispanic, and Yugoslav ethnic groups).3,4
The clinical management of congenital adrenal hyperplasia (CAH) involves suppression of adrenal androgen production, in addition to treatment of adrenal insufficiency. Supraphysiological doses of glucocorticoid drugs are often needed to adequately suppress adrenal androgens.5 However, glucocorticoid excess can result in obesity, growth suppression in children, decreased bone mineral density, cardiovascular risk, and adverse psychological effects.6,7 Absence of adequate androgen suppression in congenital adrenal hyperplasia can result in early puberty, virilisation in females, infertility, adrenal and adrenal rest tumour formation (classic disease only), and potential adverse psychological effects. Patient management focuses on the control of excess androgen production, while avoiding the side-effects of hypercortisolism. Generally, clinicians need to provide individualised therapy because a standardised approach fails to account for individual variability in disease severity and response to treatment.
Management of adolescents with congenital adrenal hyperplasia presents unique challenges. Changes in the hormonal milieu during puberty can lead to inadequate suppression of adrenal androgens at a time when psychosocial issues often affect adherence to medical therapy. Once epiphyseal closure has occurred and growth regulation is no longer a concern, a shift in treatment goals from optimisation of growth and development to prevention of long-term adverse outcomes and optimisation of fertility and sexual function is needed. Sexual maturation occurs during adolescence, and clinicians caring for children with congenital adrenal hyperplasia play a major part in promoting a healthy self-image. For women with classic congenital adrenal hyperplasia, comprehension of their genital anatomy and surgical history is essential. Health professionals need to be educated about the unique needs of adolescents with congenital adrenal hyperplasia as they become increasingly independent from parental supervision and transfer from paediatric care to adult care (termed here transition). In this Review, we discuss several challenges faced by adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency, and provide guidance to health-care professionals in helping patients to navigate a path towards self-reliance and successful transfer of care to an adult practitioner.
Genetics
The 21-hydroxylase gene CYP21A2 is located within the HLA class 3 region of chromosome 6p21.3, a region with complex genetic variation and gene duplications. Most mutations causing 21-hydroxylase deficiency arise from unequal recombination events between the active CYP21A2 gene and a highly homologous CYP21A1P pseudogene. The expected phenotype resulting from a known CYP21A2 mutation is based on published in-vitro studies of 21-hydroxylase activity.8 Most patients are compound heterozygotes, harbouring various mutations on different alleles, and the phenotype is determined by the milder mutation (ie, if a patient has both a mild and a severe mutation, the phenotype will be mild). About two-thirds of patients with non-classic congenital adrenal hyperplasia are carriers of a classic mutation (ie, a mutation that results in a classic phenotype).9,10
Generally, large cohort studies have reported good phenotype-genotype correlation.9,11,12 However, divergence between the reported and predicted phenotype exists because of variations in the CYP21A2 gene and the effects of other genes.13,14 Absence of genotype-phenotype correlation has been associated with specific CYP21A2 mutations, such as the IVS2-13A/C>G splice site mutation and the p.172N simple virilising mutation.9,11,12,15 Variations in large deletions can result in a partly functional gene and thus a milder phenotype, or a contiguous gene-deletion syndrome associated with hypermobility type Ehlers-Danlos syndrome.16,17 Genetic sequencing is necessary to identify rare mutations that are estimated to occur in 10% of affected alleles.9 Although the CYP21A2 gene was identified more than 30 years ago, novel mutations and new aspects of the genetics of CYP21A2 are continually being defined.9,16–18 Thus, molecular diagnosis should only be undertaken by a laboratory familiar with the genetic complexities of CYP21A2 and able to offer a combination of diagnostic strategies.
There is a continuum of disease severity. Although the distinction between classic and non-classic congenital adrenal hyperplasia is important for patient management, the distinction between the subtypes of classic congenital adrenal hyperplasia—salt-wasting versus simple virilising—becomes less important as the child ages.6 Genotype is not related to the development of osteoporosis or other long-term adverse health outcomes, and in adults with classic congenital adrenal hyperplasia, clinical status is mostly acquired as a result of long-term quality of hormonal control treatment effects.19,20 If genotyping has been done during childhood, this information should be passed on when transfer to adult care occurs. Genotyping is most useful if the diagnosis of congenital adrenal hyperplasia is equivocal after hormonal testing or if family planning advice is being sought,1,21 both of which are not typical issues during adolescence.
General genetic information should be given to the patient in preparation for the transition to adult care. Counselling should be aimed at achieving an understanding that congenital adrenal hyperplasia is a genetic disease and the future risks of having an affected child. Generally, the chance of a patient with classic congenital adrenal hyperplasia having a child with classic congenital adrenal hyperplasia is 1 in 120 (chance of partner being a carrier [1/60] × 1/2).1 The chance of a patient with non-classic congenital adrenal hyperplasia having a child with classic congenital adrenal hyperplasia is 1 in 360 (chance of partner being a carrier × chance of non-classic patient being a carrier of classic mutation × 1/4 = 1/60 × 2/3 × 1/4). Determination of patient-specific risks are dependent on the carrier status of the patient’s partner. A patient with congenital adrenal hyperplasia could have a child with the disease only if their partner carries one or more CYP21A2 mutations, and then all of the children will at least be a carrier (figure 1). De-novo germline mutations occur in 1–2% of congenital adrenal hyperplasia alleles.22
Figure 1. Pedigree displaying the genetic risk to the offspring of a patient with congenital adrenal hyperplasia.
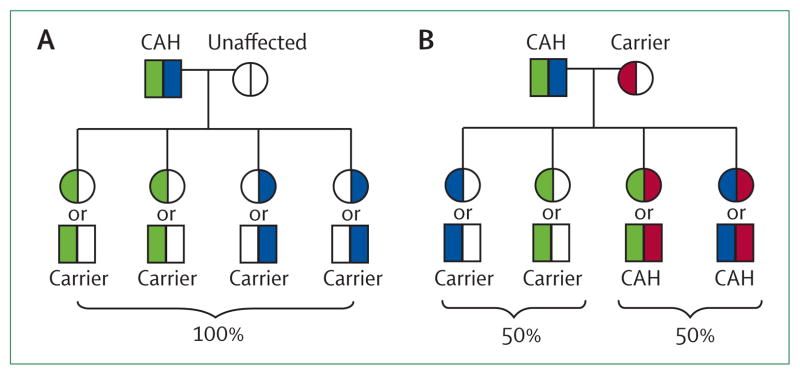
CAH=congenital adrenal hyperplasia. Risks are the same for men and women, a representative affected male is shown as the patient. (A) All children are carriers when one parent is affected and the other parent is unaffected. (B) Half of children are affected when one parent is affected and the other parent is a carrier. Most patients are compound heterozygotes. Square=male. Circle=female. White=unaffected (wild type). Blue=mutation #1. Green=mutation #2. Pink=mutation #3.
Changes in the hormonal milieu during puberty
Patients with classic congenital adrenal hyperplasia have cortisol deficiency, epinephrine deficiency, varying degrees of aldosterone deficiency, and androgen excess, whereas patients with non-classic congenital adrenal hyperplasia patients mostly have androgen excess, with normal or mildly impaired cortisol production.1 All individuals with congenital adrenal hyperplasia (male or female with classic or non-classic disease) have greater insulin resistance compared with BMI-matched controls.23–25 The role of androgens in promoting insulin resistance, and conversely the role of insulin resistance in promoting androgen production, in congenital adrenal hyperplasia is unknown. However, in women with hyperandrogenism due to other causes, an acute increase in insulin increases adrenocorticotropic hormone-mediated adrenal androgens.26
The block in the biosynthetic pathway of cortisol production due to 21-hydroxylase enzyme deficiency results in a build-up of precursors, most notably 17-hydroxyprogesterone and androstenedione. These precursors can be converted to androgens through the classic adrenocortical steroid pathway or through an alternative pathway (figure 2).27 Increased expression of cytochrome b5 at puberty increases 17,20 lyase activity leading to increased adrenal androgen production, mostly via the classic pathway.28 A decrease in insulin sensitivity usually occurs during puberty, also promoting adrenal androgen production.
Figure 2. Adrenocortical biosynthetic pathway.

(A) In normal physiology, the classic pathway of androgen biosynthesis occurs from conversion of 17-hydroxypregnenolone to DHEA and 17-hydroxyprogesterone to androstenedione. (B) In the absence of 21-hydroxylase activity, 17-hydroxyprogesterone accumulates and can be converted to testosterone and dihydrotestosterone through two pathways: the classic pathway and an alternative pathway. Increased expression of cytochrome b5 occurs during adolescence, promoting adrenal androgen production via the classic pathway. 17-OH activity in the alternative pathway is less affected by cytochrome b5 activity. 17-OHPreg=17-hydroxypregnenolone. 17-OHProg=17-hydroxyprogesterone. DHEA=dehydroepiandrosterone. DHT=dihydrotestosterone. P450scc+StAR=P450 side chain cleavage plus steroidogenic acute regulatory protein. 3βHSD=3-beta-hydroxysteroid dehydrogenase. 17-OH=17-hydroxylase/17,20 lyase. b5=cytochrome b5. 21-OH=21-hydroxylase. 11-OH=11-hydroxylase. AS=aldosterone synthase. 17βHSD=17-beta-hydroxysteroid dehydrogenase. 5αR2=5-alpha-reductase type 2. Adapted from J Clin Endocrinol Metab: Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline 2010, figure 1.1
Pubertal activation of the hypothalamic–pituitary–gonadal axis and increases in growth hormone and IGF-1 secretion indirectly affect management of congenital adrenal hyperplasia. Pubertal increases in growth hormone and IGF-1 have been shown to decrease 11β-hydroxysteroid dehydrogenase type 1 activity and increase glomerular filtration rate, resulting in increased cortisol clearance.29 These changes in the pharmacokinetics of cortisol can decrease glucocorticoid drug effectiveness. Additionally, inconsistent compliance, a common occurrence during adolescence, can lead to under-replacement of cortisol.
Puberty is a time of exacerbation of hormonal imbalances, with possible adverse outcomes for patients with congenital adrenal hyperplasia (figure 3). Management is targeted towards prevention of long-term adverse outcomes such as adrenal rest tumours (figure 4A), infertility, metabolic syndrome, obesity, and osteoporosis. Adrenal hyperplasia and nodularity occur in patients with longstanding and poorly controlled disease (figure 4B), and should be prevented. Adrenal enlargement has been associated with adrenal tumour formation, most often myelolipomas.30,31 Adrenal rest tumours and myelolipomas are not precancerous diseases, but are hyperplastic cells responsive to adreno-corticotropic hormone stimulation. Enlargement of testicular adrenal rest tissue results in infertility.32,33 Massive growth of myelolipoma can cause symptoms of mass effect.34 Clinical status during adolescence and young adulthood largely determines long-term outcome.
Figure 3. Common hormonal imbalances during adolescence in patients with congenital adrenal hyperplasia and clinical consequences.

Normal pubertal increases in growth hormone increases cortisol clearance, which decreases glucocorticoid effectiveness. The age-related physiological increase in adrenal androgens and insulin resistance are magnified in patients with congenital adrenal hyperplasia and contribute to the management challenges. Treatment is aimed at controlling the adverse effects of androgen excess and insulin resistance while avoiding the complications of iatrogenic Cushing’s syndrome.
Figure 4. Tumour formation in congenital adrenal hyperplasia.

(A) Ultrasound (transverse view) shows testicular adrenal rest tumour (arrows) surrounding the mediastinum testes (arrowhead). (B) Axial enhanced CT section through the left adrenal gland shows hypertrophy (arrows) with nodules (arrowhead) and fatty adrenal masses representing myelolipomas (curved arrow).
Management of adolescents with non-classic congenital adrenal hyperplasia
A subset of patients with non-classic congenital adrenal hyperplasia is diagnosed during childhood when signs of precocious puberty are detected. These children are often treated with hydrocortisone to suppress adrenal hormones and prevent rapid advancement of bone age that could adversely affect adult height. Glucocorticoid drug therapy might be indicated at specific times during adulthood, but lifetime glucocorticoid therapy is not warranted. In 2010, the Endocrine Society1 developed evidence-based clinical practice guidelines that recommended that treated patients with non-classic disease be given the option of discontinuing therapy when symptoms resolve, and that adult men do not require daily glucocorticoid treatment. Similarly, treatment options vary for adult women (table), many of whom do not need glucocorticoid therapy. However, guidelines do not exist for discontinuation of glucocorticoid therapy in children who began glucocorticoid treatment during childhood.
Table.
| Physiological effect | Clinical effect | Adverse effects | |
|---|---|---|---|
| Glucocorticoids1 | Cortisol replacement; suppression of hypothalamic–pituitary–adrenal axis, reduces adrenal androgen secretion | Treatment of adrenal insufficiency, prevention of female virilisation, prevention and treatment of infertility | Iatrogenic Cushing’s syndrome (dose related) |
| Fludrocortisone* (mineralocorticoid)1 | Mineralocorticoid replacement; suppression of renin– angiotensin–aldosterone axis, reduces vasopressin and adrenocorticotropic hormone concentrations, reduces adrenal androgen secretion | Maintenance of sodium and potassium balance, prevention of intravascular volume depletion, allows for reduced glucocorticoid dosing | Hypertension (dose related) |
| Oral contraceptive pills1,6,35 | Stimulation of hepatic sex hormone binding globulin, reduces serum free androgen; suppression of hypothalamic–pituitary–ovarian axis, reduces ovarian androgen secretion | Regulation of menstrual cycle, reduced hirsutism | NA |
| Spironolactone1,6,35† | Mineralocorticoid antagonist; competitive inhibitor androgen receptor; inhibition of 5α-reductase activity, reduces androgen production | Reduced hirsutism | Diuresis (dose related) |
NA=not applicable.
Used for treatment of classic congenital adrenal hyperplasia patients.
Commonly used for treatment of non-classic women; rarely used for classic women because of anti-mineralocorticoid effect.
No specific data exists regarding the timing of regimen changes or cessation of glucocorticoid therapy, but healthy puberty (ie, the occurrence of normal puberty in a normal timeframe) provides guidance. In healthy boys, a testicular volume of 4–6 mL marks the beginning of testicular testosterone production.36,37 Thus, the need to suppress adrenal androgens beyond midpuberty is questionable. Based on our experience, we recommend weaning male adolescents with non-classic congenital adrenal hyperplasia off glucocorticoid drugs by gonadal Tanner stage 3 (testicular volume 8–10 mL), before the pubertal growth spurt (figure 5). In healthy women, early menstrual life is characterised by anovulatory cycles, and by the third year after menarche, most women have regular menstrual cycles typical of an adult.38 Therefore, for women with non-classic disease treated during childhood, continuation of hydrocortisone drug therapy until at least 2–3 years postmenarche is prudent to establish menstrual cyclicity.
Figure 5. Transition of care guidelines for patients with non-classic and classic congenital adrenal hyperplasia.

OCP=oral contraceptive pill. TART=testicular adrenal rest tissue. DXA=dual-energy X-ray absorptiometry. *A subset of patients with non-classic congenital adrenal hyperplasia have subnormal cortisol response with unknown clinical significance; stress dose glucocorticoids should be considered.
Adrenal function is iatrogenically suppressed in children with non-classic disease treated with glucocorticoids, thus increased doses during periods of stress are indicated until the patient is successfully weaned off therapy and adrenal function is assessed and normal. About a third of patients with non-classic disease might have subnormal cortisol responses to stimulation with tetracosactide (Synacthen, Novartis) in the untreated state.10,39 Although there is no evidence of adrenal crisis occurring in these patients, additional glucocorticoid therapy during periods of stress might be warranted once regular therapy has been discontinued.
Adult men with non-classic congenital adrenal hyperplasia do not warrant daily therapy and most are asymptomatic.1 However, rare isolated case reports have shown reversal of oligospermia with glucocorticoid treatment.4,40 Most adult males with non-classic disease do not require endocrine care, but evidence-based longitudinal outcome data from a large cohort of patients with non-classic congenital adrenal hyperplasia are not available. Endocrine assessment every 5 years should be considered (figure 5).
Women with non-classic congenital adrenal hyperplasia sometimes do not require treatment, or can be treated successfully with oral contraceptive pills with or without an anti-androgen treatment (eg, spironolactone) to combat virilising features.1,35,41 Some female patients might choose to continue glucocorticoid therapy if oral contraceptive pills, anti-androgens, or both are not tolerated or desired, or pronounced hyperandrogenism is present.4,35 Additionally, glucocorticoid therapy might be indicated at specific times during adulthood for women with non-classic disease, for example as an effective treatment for infertility and recurrent miscarriages.42 Women with non-classic disease should be transferred to an adult endocrine caregiver because they can have hyperandrogenism as an adult (figure 5).
Management of classic congenital adrenal hyperplasia
In children, hydrocortisone is the glucocorticoid of choice because longer acting glucocorticoids are more likely to impair growth.1,43 Minimisation of glucocorticoid dose during puberty is important because pubertal height gain is a predictor of final height.44 In a retrospective analysis of 92 patients with classic congenital adrenal hyperplasia given hydrocortisone, a decrease in pubertal growth was associated with a glucocorticoid dose greater than 17 mg hydrocortisone per m2 body surface area per day.45 Conversely, too low a glucocorticoid dose with subsequent hyperandrogenism can induce premature epiphyseal closure and can also contribute to height loss. Thus, close monitoring during puberty is especially important (panel 1). Hoepffner and colleagues46 showed that strict adherence to hydro-cortisone three times daily with fludrocortisone therapy and monitoring every 3 months resulted in 26 patients with congenital adrenal hyperplasia achieving target height.
Once growth is less than 1 cm per year, the glucocorticoid regimen should be re-assessed. Cross-sectional studies from both the UK and the USA show the use of varied glucocorticoid regimens in adults, with about a third of adult patients with congenital adrenal hyperplasia given hydrocortisone and two-thirds given long-acting glucocorticoid preparations such as dexamethasone, prednisone, or prednisolone.7,49 No randomised controlled trials have assessed these varied regimens; thus, the glucocorticoid choice remains individualised. However, the goal is always to use the lowest dose that adequately suppresses adrenal androgens, optimises compliance based on lifestyle, and maintains good general health. Once growth is complete, the management goals change and the focus becomes prevention of long-term adverse outcomes. For adolescent males testicular ultrasound is recommended to assess or screen for testicular adrenal rest tissue and to help to guide management (figures 4A, 5). For adolescent females addition of oral contraceptive pills to the regimen might help to reduce androgen concentrations (table). Although most children with congenital adrenal hyperplasia have normal bone mineral density, they are at risk of reduced bone density with increasing age and with high cumulative glucocorticoid treatment.7,50–53 Baseline dual-energy X-ray absorptiometry to measure bone density should be considered before adult practitioner referral. Osteoporosis prophylaxis, including vitamin D and calcium supplementation, should begin in childhood.7
Panel 1: Laboratory assessment of patients with congenital adrenal hyperplasia1,6.
1 Standard of care for monitoring disease
17-Hydroxyprogesterone and androstenedione as biomarkers of adrenocortical activation and adequacy of the glucocorticoid regimen
Plasma renin activity to assess the adequacy of fl udrocortisone replacement
Testosterone, free testosterone, and sex-hormone binding globulin for women with possible secondary polycystic ovary syndrome
Testosterone and gonadotropins in young adult males with poorly controlled congenital adrenal hyperplasia; very raised adrenal androgens can result in hypogonadotropic hypogonadism, which is reversible with glucocorticoid therapy46
2 Alternative laboratory assessments
3 Timing of laboratory assessment
Every 3–4 months during pubertal growth
4–6 weeks after major regimen changes
Every 6 months or yearly after completion of growth once receiving an optimum glucocorticoid regimen
Best undertaken in a consistent manner with the timing of drugs taken into account
Adrenal androgen concentrations will be lower after drugs and later in the day
4 Laboratory goals
Laboratory assessment should guide, but not define treatment
Normalisation or near normalisation of androstenedione, with recognition that females with concurrent polycystic ovary syndrome might have higher values
Raised 17-hydroxyprogesterone values to at least twice the normal range, suppression of 17-hydroxyprogesterone to the low normal or undetectable range indicates excess glucocorticoid treatment
Plasma renin activity in the midnormal range
Target hormone concentrations should be individualised; the young adult male with no evidence of testicular adrenal rest tissue could be maintained at higher 17-hydroxyprogesterone and androstenedione concentrations than the young adult female
Transition of care is a time when many patients with congenital adrenal hyperplasia stop adherence to their medication; male patients especially might not comprehend the long-term implications of non-adherence, because male adults are already fully virilised.54 Thus, provision of detailed congenital adrenal hyperplasia-related education by the paediatric practitioner to the patient before the transfer to an adult practitioner in a new setting is of utmost importance. For practical reasons and to limit non-compliance, most patients with classic congenital adrenal hyperplasia are changed to a longer-acting glucocorticoid when growing is complete; hydrocortisone is best given three times daily but this frequency of daily medication is not appealing to most active adolescents and young adults. Therefore, continuation of three times daily hydrocortisone should be reserved for the highly compliant patient whose disease is well controlled. Switching back to hydrocortisone later in life for the mature adult patient is always an option.
One method to estimate the long-acting glucocorticoid dose is to calculate an approximate equivalency dose based on the present hydrocortisone dose. This estimation should be done with caution because no accurate formula exists and anti-inflammatory equivalencies are not appropriate in this setting.55 The glucocorticoid dose required to control congenital adrenal hyperplasia-mediated androgen excess is estimated to have a hydrocortisone equivalency ratio of 5:1 for prednisone and prednisolone and of 70–80:1 for dexamethasone.56,57 These calculations provide estimates; however, starting with a low dose is always best, and once growth is complete many patients can be treated with reduced glucocorticoid doses. Long-acting glucocorticoid medications have reduced mineralocorticoid effects, but monitoring of plasma renin activity and blood pressure is always indicated. Fludrocortisone dose should be adjusted to maintain plasma renin activity within the normal range.
Dexamethasone is the preferred treatment for testicular adrenal rest tissue, but high-dose treatment should be avoided.58 Dexamethasone 250–375 μg given once daily is a good starting dose. Some patients are fast metabolisers of dexamethasone and might require twice daily dosing. Dexamethasone should be avoided in young adult women who are heterosexually active because it is not inactivated by placental 11β-hydroxysteroid dehydrogenase type 2 and would result in fetal exposure should pregnancy occur.1 Prednisone or prednisolone 4–6 mg per day divided into two doses are appropriate starting doses for both men and women.1 Substantial inter-individual variability in the metabolism of glucocorticoid medication exists, thus starting of treatment at a low dose is best, with laboratory assessment of adrenal metabolites 4–6 weeks after a change in medication (panel 1). Starting with too high a dose will inevitably cause weight gain and might encourage non-adherence, especially in adolescents.
Giving the highest dose of glucocorticoid at night is sometimes tried in an attempt to suppress the early morning adrenocorticotropic hormone surge, which occurs as part of the diurnal variation of the hypothalamic–pituitary–adrenal axis.5,7,49 A randomised trial compared high-morning dose of hydrocortisone with high-evening dose in 15 children and showed no differences in disease control or sleep quality.59 Giving a higher night-time dose can cause sleep disturbances, especially in adult patients. If sleep disturbances are reported, the highest dose of glucocorticoid should be given in the morning.
Female specific issues in classic congenital adrenal hyperplasia
Joint guidelines by Pediatric Endocrine Society, formerly the Lawson Wilkins Pediatric Endocrine Society, the European Society for Pediatric Endocrinology (ESPE) CAH Working Group,60 and the Congenital Adrenal Hyperplasia Research Education and Support (CARES) Foundation Initiative61 included a survey of specialists who treated patients with congenital adrenal hyperplasia. This survey showed that 78% supported early surgery (patient aged younger than 2 years), which included clitoroplasty, vaginoplasty, and labioplasty62 early surgical treatment for females with classic congenital adrenal hyperplasia and specific degrees of genital masculinisation. These guidelines do not recommend delayed or staged approaches. Once these guidelines have been implemented, one of the greatest challenges facing paediatric endocrinologists is how to follow up the surgical-specific issues these patients can have as they transition from childhood to adolescence.54 Menarche and sexual function play a major part in adolescence and young adulthood. During this transition from childhood to adolescence, many issues relating to early genital surgery might become evident. Thus, it is important that the treating endocrinologist knows what questions to ask, what to look for on examination, and when to refer the patient to an experienced surgeon for further assessment and possible treatment. Concerns after early surgical intervention are varied and relate to specific procedures, the techniques used, and the experience of the surgeon. Adolescents who have had early genital surgery can present with several issues. In our experience, the most common issues arising in adolescence are related to urinary incontinence, clitoral pain, vaginal stenosis, and cosmetic concerns.
Unique causes of urinary incontinence in adolescents with congenital adrenal hyperplasia who had early genital reconstruction are usually related to anatomical rather than neurological issues.63 Neurological causes of urinary incontinence after genital surgery are rare in congenital adrenal hyperplasia, with the exception of patients who had total urogenital sinus mobilisation.64,65 There is some concern that aggressive mobilisation anterior to the bladder neck and release of the pubovescical ligament could result in incontinence.66 Patients suspected of having this issue are likely to have global urinary incontinence (constant leaking throughout the day without urgency or frequency symptoms) and should be referred to a urologist for comprehensive assessment.
Vaginal voiding is one of the most common causes of incontinence in patients with congenital adrenal hyperplasia and presents with day time wetting and dampness. Soaking incontinence (wetting through underwear) and nocturia are rare. Vaginal voiding occurs when urine exiting the urethral meatus enters the vagina, which can happen when patients sit with their legs together during urination; this compresses the labia and prevents a free flow of urine from the urethral meatus and redirects it into the vagina. Once the patient completes voiding and stands up, the urine in the vagina drains out, resulting in dampness or wetting limited to underwear. Wetting usually occurs within the first hour after voiding. This type of incontinence is often overlooked, leading to unnecessary testing and patient frustration. The best way to confirm a diagnosis of vaginal voiding is to ask the patient to sit facing backward on the toilet each time they void for 2 days. If they are dry during this test period, then a diagnosis of vaginal voiding can usually be confirmed.
Vaginal voiding can also be caused by a hypospadiac urethra. Urine is released directly into the vagina when the urethral meatus opens proximally along the anterior vaginal wall. In some minor forms of female hypospadias, instructing the patient to void with her legs wide apart will solve the problem. However, surgical repositioning of the urethral meatus might be required if the meatus is located proximally. In these patients, a genital examination will reveal a single opening below the clitoris as opposed to a separate opening for the urethra and vagina.
Patients with urinary incontinence are best managed by a surgeon with experience in voiding dysfunction and feminising genital surgery. Clitoral pain during sexual arousal in patients with congenital adrenal hyperplasia who have had clitoral surgery is a challenging issue. Knowledge of the type of clitoral surgery that the patient has had is important. In the past three decades, clitoral recession was often undertaken. This technique involves mobilising and repositioning the erectile bodies of the clitoris under the pubis. In patients with poor endocrine control, an increase in androgen concentrations can result in growth of the erectile bodies. As a result, large, trapped erectile bodies that engorge with arousal can produce pain. In such women, the erectile bodies can usually be palpated. The only treatment option is to have the clitoral recession converted to a clitoroplasty with partial removal of the erectile bodies. The nerve-sparing approach to clitoroplasty based on Baskin and colleagues’67 description of clitoral anatomy used since 1996 should be considered for all patients with congenital adrenal hyperplasia undergoing clitoral surgery, regardless of age.68
Some young girls and adolescents who did not have clitoral surgery as infants will develop an enlarged clitoris later in life as a result of poor endocrine control or non-compliance with medication. These patients might present with embarrassing clitoral erections that become obvious when wearing a bathing suit or gymnastics clothing. For these patients, counselling, clitoroplasty, or both, are options.
Decreased or absent clitoral sensation in adolescents and young adult is a challenging issue for which there is no surgical correction. Loss of sensation might be caused by damage to the neurovascular bundles during surgery or from having had a clitorectomy.69–71 Nowadays, clitorectomy should never be done, and techniques used for clitoroplasty should provide improved outcome compared with older surgical methods. Unfortunately, long-term outcome data from patients who have had the new techniques are not available because these patients are now becoming adolescents and young women. Future studies assessing these women will be critical to our understanding of surgical outcome.
Patients who had vaginal surgery in infancy might present in adolescence with several concerns including vaginal stenosis, hairy vaginas, short vaginal length, or incomplete separation of the urogenital sinus. By far the most common is vaginal stenosis.72,73 In our experience, these girls typically present for the first time after they have begun menarche and attempt to use a tampon for the first time. They will complain of pain on insertion or an inability to insert the tampon. For these patients, a thorough genital examination should be done. If the vaginal opening is confirmed to be narrow or a single opening is identified, they should be referred to an experienced paediatric urologist or surgeon for assessment and management.
Some women who have had early vaginal surgery are able to use a tampon without difficulty but might have difficulty during penetrative sexual intercourse. These patients should be referred to an experienced surgeon or gynaecologist for assessment and treatment. In most cases, these women will require oestrogen cream and vaginal dilation therapy. The use of dilators should be considered only when the patient is psychologically ready and personally motivated, which is usually during early adulthood. In more severe cases, vaginal surgery might be required to release the constricting scar tissue or rarely, more extensive surgery might need to be considered.
Most vaginal surgery undertaken in infants with congenital adrenal hyperplasia will involve the use of a perineal vascularised flap.74 In infancy, these flaps do not have hair; however, during adolescence the flap can develop hair, which can be a concern for patients. On examination, hair can be seen along the posterior vaginal wall where the flap was placed. In some cases, this can be corrected with professional depilation. In more extensive cases or when associated with vaginal stenosis, flap removal and replacement with a buccal mucosal graft has been successful.75,76 Review of surgical history, patient education, and determination of the possible need for future surgery are important in adolescence.
Male specific issues in classic congenital adrenal hyperplasia
Men with classic congenital adrenal hyperplasia are at risk of infertility, mostly because of risk of testicular adrenal rest tissue. Ultrasound is the method of choice for detection of testicular adrenal rest tissue, which typically presents as bilateral hypoechoic masses located in the mediastinum testes (figure 4A).77 This tissue can cause compression of the seminiferous tubules leading to obstructive azoospermia. Testicular venous sampling has shown that testicular adrenal rest tissue also produces adrenal hormones, which can act locally to contribute to subfertility.78 The prevalence of testicular adrenal rest tissue is approximately one in three paediatric male patients with congenital adrenal hyperplasia in Europe and the USA, and the prevalence increases with age.7,79 Luteinising hormone receptors have been identified in testicular adrenal rest tissue and therefore the rise in luteinising hormone during puberty might contribute to tumour growth.80 Progressive testicular adrenal rest tissue growth can eventually result in fibrosis and irreversible testicular damage,81 and reduced fecundity has been reported. In a cross-sectional study of 22 men with congenital adrenal hyperplasia in Germany, more than half had impaired spermatogenesis.33 Reduced fertility can also be due to hypogonadotropic hypogonadism, which has been noted in patients with adrenal androgen excess and in patients with glucocorticoid excess.33,82 Adolescence is an ideal time to screen patients for testicular adrenal rest tissue. Tumour shrinkage is possible with tight metabolic control at early stages,83 but prevention of tumour enlargement is key to optimising long-term fertility potential.
Transition goals
Disclosure of a patient’s medical and surgical history should occur in an age-appropriate manner with facts and explanations geared towards the developmental level of the child. Ideally, as the child grows up, both the paediatric endocrine team and the family should provide medical information to the child, with full disclosure achieved by age 16 years.84 If or when a patient reads his or her medical records as an adult, there should be no surprising information; learning about their diagnosis in this way can be distressing to patients.85,86 Openness promotes a trusting relationship between patients, doctors, and families, and encourages the development of a positive self-image. The paediatric endocrinologist sometimes replaces the general paediatrician as the primary caregiver and should be prepared to openly discuss sexual issues. Female patients often wish to know whether they can have normal sexual intercourse. Parents who wish to withhold information from girls with congenital adrenal hyperplasia regarding their surgical history because of cultural concerns or their own discomfort should be offered counselling.
In 2006, the European Society for Paediatric Endocrinology and the Pediatric Endocrine Society jointly published a consensus statement on the management of disorders of sex development.86,87 Classic congenital adrenal hyperplasia in women was one of the many disorders considered. An important aspect of the psychosocial management of disorders of sex development is disclosure of information to the child. The consensus recommends an early planning strategy with the parents and the development of a gradual process of reinforced education, coupled with professional counselling. Children with congenital adrenal hyperplasia are at risk of anxiety disorders,88 and adolescent girls are particularly vulnerable to developing a negative body image, especially if puberty occurs at a rate different from the norm.88,89 Reinforced education over years in conjunction with the family optimises self-confidence and coping mechanisms. Before transitioning to an adult caregiver, adolescents with classic congenital adrenal hyperplasia should have achieved specific knowledge goals (panel 2); for females with classic congenital adrenal hyperplasia, one goal is for the patient to have knowledge about their surgical history and the physical implications. In a study of adolescent patients with either congenital adrenal hyperplasia or phenylketonuria who had not previously received genetic counselling, most patients wanted to understand the science behind their disorder, and (65%) preferred to receive information with their parents before 12 years of age, thus emphasising the importance of openness at a young age.90
Panel 2: Knowledge goals for adolescents with classic congenital adrenal hyperplasia.
Understand that congenital adrenal hyperplasiais a lifelong disease that is genetically inherited
Understand the medication regimen, including the results of too much or too little medication
Be able to self-manage stress doses of glucocorticoid during illness, including self-administration of intramuscular hydrocortisone
Knowledge of surgical history and physical implications
Knowledge of infertility risks but also that fertility and satisfying sexual experiences can be achieved
Many international groups have suggested that patients with congenital adrenal hyperplasia are best managed by multidisciplinary teams in tertiary care centres. Joint clinics with paediatric and adult endocrinologists have been suggested to optimise communication during the transition from paediatric to adult care, but for most patients worldwide this is not available.1,61,91–93 Moreover, a study in the UK showed that half of all paediatric patients with congenital adrenal hyperplasia were lost to follow up, despite introduction of a joint clinic.54 Patients with fewer appointments were at risk of being lost to follow up, which emphasises the importance of engaging with the patient early on. A transition clinic in Germany, run jointly by paediatric and adult endocrinologists, has reported better results.48 Transition is most successful when not viewed as a one-time event, but as a process that occurs over many years. Young adults need to understand the pathophysiology and genetics of congenital adrenal hyperplasia. They also need to have access to their past medical records. Knowledge regarding sexuality, fertility, navigating the health-care system, long-term concerns, and potential issues in adulthood need to be thoroughly addressed.
Search strategy and selection criteria.
We searched the Cochrane Library, Medline, and Embase from January, 2005, to December, 2013, and PubMed and Google Scholar, with the search terms “congenital adrenal hyperplasia” or “CAH” in combination with the terms “adolescent”, “CYP21A2”, “transition adolescent”, “genital surgery”, “clitoroplasty”, and “surgical outcomes”. We did not exclude commonly referenced or highly regarded older publications. We also searched the reference lists of articles identified by this search strategy and selected those we judged relevant.
Lessons learned from other childhood chronic diseases
Children with other chronic diseases face similar challenges. Paediatric patients with diabetes, cystic fibrosis, and sickle-cell disease are often lost to follow-up during the transition period.94–98 In a systematic review of studies assessing adolescent transition programmes, successful interventions were reported in studies of diabetes and included extensive patient education regarding self-management skills and having a dedicated young adult clinic or a specific transition clinic that was jointly staffed by paediatric and adult practitioners. Comparison of studies is complicated by the many different interventions; however, an extended period of support in preparation for, during, and after transition, and training and education of health-care professionals are all recommended, recognising that additional personnel are needed to meet these needs, which might be costly.99,100
Conclusions
Care of adolescents with congenital adrenal hyperplasia has unique challenges. Health-care providers play a crucial part in helping the child and their family to transition to adult life. There is no standard approach, but a defined referral pathway should exist so that each patient has the opportunity to access quality adult care. Transition of adolescents to adult care involves changes in treatment and a shift in management goals that become targeted towards prevention of long-term complications and optimisation of adult quality of life. Further improvements depend on practitioners, patients, and their families working together to educate children to become self-reliant adults knowledgeable about their disease. Children with rare congenital diseases are now living full, productive lives and the issue of effectively transitioning these children to adulthood is a major public health problem. Building an infrastructure that includes improved education of paediatric and adult practitioners regarding how best to transition patients is greatly needed.
Acknowledgments
This work was supported (in part) by the Intramural Research Programs of the National Institutes of Health Clinical Center and of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
Footnotes
Contributors
DPM and DPP did the literature searches and participated in the writing of the Review. DPM designed the figures.
Conflicts of interest
DPM receives research funding from Diurnal Limited. DPP is a shareholder and an officer of Promethean Surgical Devices.
References
- 1.Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133–60. doi: 10.1210/jc.2009-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol. 2004;151 (suppl 3):U71–75. doi: 10.1530/eje.0.151u071. [DOI] [PubMed] [Google Scholar]
- 3.Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37:650–67. [PMC free article] [PubMed] [Google Scholar]
- 4.New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:4205–14. doi: 10.1210/jc.2006-1645. [DOI] [PubMed] [Google Scholar]
- 5.Merke DP. Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2008;93:653–60. doi: 10.1210/jc.2007-2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98:2645–55. doi: 10.1210/jc.2013-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finkielstain GP, Kim MS, Sinaii N, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:4429–38. doi: 10.1210/jc.2012-2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speiser PW, Dupont J, Zhu D, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90:584–95. doi: 10.1172/JCI115897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finkielstain GP, Chen W, Mehta SP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96:E161–72. doi: 10.1210/jc.2010-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab. 2009;94:1570–78. doi: 10.1210/jc.2008-1582. [DOI] [PubMed] [Google Scholar]
- 11.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85:1059–65. doi: 10.1210/jcem.85.3.6441. [DOI] [PubMed] [Google Scholar]
- 12.Stikkelbroeck NM, Hoefsloot LH, de Wijs IJ, Otten BJ, Hermus AR, Sistermans EA. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J Clin Endocrinol Metab. 2003;88:3852–59. doi: 10.1210/jc.2002-021681. [DOI] [PubMed] [Google Scholar]
- 13.Gomes LG, Huang N, Agrawal V, Mendonca BB, Bachega TA, Miller WL. Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4: effect on 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2009;94:89–95. doi: 10.1210/jc.2008-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaupert L, Lemos-Marini S, De Mello M, et al. The effect of fetal androgen metabolism-related gene variants on external genitalia virilization in congenital adrenal hyperplasia. Clin Genet. 2012 doi: 10.1111/cge.12016. published online Sept 14. [DOI] [PubMed] [Google Scholar]
- 15.Dolzan V, Solyom J, Fekete G, et al. Mutational spectrum of steroid 21-hydroxylase and the genotype-phenotype association in Middle European patients with congenital adrenal hyperplasia. Eur J Endocrinol. 2005;153:99–106. doi: 10.1530/eje.1.01944. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Xu Z, Sullivan A, et al. Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin Chem. 2012;58:421–30. doi: 10.1373/clinchem.2011.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merke DP, Chen W, Morissette R, et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98:E379–87. doi: 10.1210/jc.2012-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Concolino P, Mello E, Minucci A, Giardina B, Capoluongo E. Genes, pseudogenes and like genes: the case of 21-hydroxylase in Italian population. Clin Chim Acta. 2013;424:85–89. doi: 10.1016/j.cca.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 19.Falhammar H, Filipsson H, Holmdahl G, et al. Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:4643–49. doi: 10.1210/jc.2007-0744. [DOI] [PubMed] [Google Scholar]
- 20.Krone N, Rose IT, Willis DS, et al. Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab. 2013;98:E346–54. doi: 10.1210/jc.2012-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.New MI, Abraham M, Gonzalez B, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 2013;110:2611–16. doi: 10.1073/pnas.1300057110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Speiser PW, White PC. Congenital adrenal hyerplasia. N Engl J Med. 2003;349:776–88. doi: 10.1056/NEJMra021561. [DOI] [PubMed] [Google Scholar]
- 23.Falhammar H, Filipsson H, Holmdahl G, et al. Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:110–16. doi: 10.1210/jc.2006-1350. [DOI] [PubMed] [Google Scholar]
- 24.Charmandari E, Weise M, Bornstein SR, et al. Children with classic congenital adrenal hyperplasia have elevated serum leptin concentrations and insulin resistance: potential clinical implications. J Clin Endocrinol Metab. 2002;87:2114–20. doi: 10.1210/jcem.87.5.8456. [DOI] [PubMed] [Google Scholar]
- 25.Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1992;75:1421–24. doi: 10.1210/jcem.75.6.1464643. [DOI] [PubMed] [Google Scholar]
- 26.Tosi F, Negri C, Brun E, et al. Insulin enhances ACTH-stimulated androgen and glucocorticoid metabolism in hyperandrogenic women. Eur J Endocrinol. 2011;164:197–203. doi: 10.1530/EJE-10-0782. [DOI] [PubMed] [Google Scholar]
- 27.Kamrath C, Hochberg Z, Hartmann MF, Remer T, Wudy SA. Increased activation of the alternative “backdoor” pathway in patients with 21-hydroxylase deficiency: evidence from urinary steroid hormone analysis. J Clin Endocrinol Metab. 2012;97:E367–75. doi: 10.1210/jc.2011-1997. [DOI] [PubMed] [Google Scholar]
- 28.Kamrath C, Hartmann MF, Remer T, Wudy SA. The activities of 5α-reductase and 17,20-lyase determine the direction through androgen synthesis pathways in patients with 21-hydroxylase deficiency. Steroids. 2012;77:1391–97. doi: 10.1016/j.steroids.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Charmandari E, Brook CG, Hindmarsh PC. Classic congenital adrenal hyperplasia and puberty. Eur J Endocrinol. 2004;151 (suppl 3):U77–82. doi: 10.1530/eje.0.151u077. [DOI] [PubMed] [Google Scholar]
- 30.Nermoen I, Rorvik J, Holmedal SH, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 2011;75:753–59. doi: 10.1111/j.1365-2265.2011.04151.x. [DOI] [PubMed] [Google Scholar]
- 31.Reisch N, Scherr M, Flade L, et al. Total adrenal volume but not testicular adrenal rest tumor volume is associated with hormonal control in patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:2065–72. doi: 10.1210/jc.2009-1929. [DOI] [PubMed] [Google Scholar]
- 32.Claahsen-van der Grinten HL, Otten BJ, Hermus AR, Sweep FC, Hulsbergen-van de Kaa CA. Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertil Steril. 2008;89:597–601. doi: 10.1016/j.fertnstert.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 33.Reisch N, Flade L, Scherr M, et al. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2009;94:1665–70. doi: 10.1210/jc.2008-1414. [DOI] [PubMed] [Google Scholar]
- 34.McGeoch SC, Olson S, Krukowski ZH, Bevan JS. Giant bilateral myelolipomas in a man with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:343–44. doi: 10.1210/jc.2011-2373. [DOI] [PubMed] [Google Scholar]
- 35.Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1105–20. doi: 10.1210/jc.2007-2437. [DOI] [PubMed] [Google Scholar]
- 36.Albertsson-Wikland K, Rosberg S, Lannering B, Dunkel L, Selstam G, Norjavaara E. Twenty-four-hour profiles of luteinizing hormone, follicle-stimulating hormone, testosterone, and estradiol levels: a semilongitudinal study throughout puberty in healthy boys. J Clin Endocrinol Metab. 1997;82:541–49. doi: 10.1210/jcem.82.2.3778. [DOI] [PubMed] [Google Scholar]
- 37.Ankarberg-Lindgren C, Norjavaara E. Changes of diurnal rhythm and levels of total and free testosterone secretion from pre to late puberty in boys: testis size of 3 ml is a transition stage to puberty. Eur J Endocrinol. 2004;151:747–57. doi: 10.1530/eje.0.1510747. [DOI] [PubMed] [Google Scholar]
- 38.Diaz A, Laufer MR, Breech LL. Menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Pediatrics. 2006;118:2245–50. doi: 10.1542/peds.2006-2481. [DOI] [PubMed] [Google Scholar]
- 39.Nandagopal R, Sinaii N, Avila NA, et al. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: findings in 145 unrelated families. Eur J Endocrinol. 2011;164:977–84. doi: 10.1530/EJE-11-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonaccorsi AC, Adler I, Figueiredo JG. Male infertility due to congenital adrenal hyperplasia: testicular biopsy findings, hormonal evaluation, and therapeutic results in three patients. Fertil Steril. 1987;47:664–70. doi: 10.1016/s0015-0282(16)59119-5. [DOI] [PubMed] [Google Scholar]
- 41.Spritzer P, Billaud L, Thalabard JC, et al. Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. J Clin Endocrinol Metab. 1990;70:642–46. doi: 10.1210/jcem-70-3-642. [DOI] [PubMed] [Google Scholar]
- 42.Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:1182–90. doi: 10.1210/jc.2009-1383. [DOI] [PubMed] [Google Scholar]
- 43.Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. 2007;92:1635–39. doi: 10.1210/jc.2006-2109. [DOI] [PubMed] [Google Scholar]
- 44.Manoli I, Kanaka-Gantenbein C, Voutetakis A, Maniati-Christidi M, Dacou-Voutetakis C. Early growth, pubertal development, body mass index and final height of patients with congenital adrenal hyperplasia: factors influencing the outcome. Clin Endocrinol (Oxf) 2002;57:669–76. doi: 10.1046/j.1365-2265.2002.01645.x. [DOI] [PubMed] [Google Scholar]
- 45.Bonfig W, Pozza SB, Schmidt H, Pagel P, Knorr D, Schwarz HP. Hydrocortisone dosing during puberty in patients with classical congenital adrenal hyperplasia: an evidence-based recommendation. J Clin Endocrinol Metab. 2009;94:3882–88. doi: 10.1210/jc.2009-0942. [DOI] [PubMed] [Google Scholar]
- 46.Hoepffner W, Kaufhold A, Willgerodt H, Keller E. Patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency can achieve their target height: the Leipzig experience. Horm Res. 2008;70:42–50. doi: 10.1159/000129677. [DOI] [PubMed] [Google Scholar]
- 47.Hoeffner W, Kratzsch J, Willgerodt H, Keller E, Pfaffle R. 17-Hydroxyprogesteron im speichel. Monatsschr Kinderheilkd. 2012;160:565–71. [Google Scholar]
- 48.Kruse B, Riepe FG, Krone N, et al. Congenital adrenal hyperplasia—how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. 2004;112:343–55. doi: 10.1055/s-2004-821013. [DOI] [PubMed] [Google Scholar]
- 49.Arlt W, Willis DS, Wild SH, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95:5110–21. doi: 10.1210/jc.2010-0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachelot A, Chakhtoura Z, Samara-Boustani D, Dulon J, Touraine P, Polak M. Bone health should be an important concern in the care of patients affected by 21 hydroxylase deficiency. Int J Pediatr Endocrinol. 2010 doi: 10.1155/2010/326275. published online Sept 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bachelot A, Plu-Bureau G, Thibaud E, et al. Long-term outcome of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res. 2007;67:268–76. doi: 10.1159/000098017. [DOI] [PubMed] [Google Scholar]
- 52.Jaaskelainen J, Voutilainen R. Bone mineral density in relation to glucocorticoid substitution therapy in adult patients with 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 1996;45:707–13. doi: 10.1046/j.1365-2265.1996.8620871.x. [DOI] [PubMed] [Google Scholar]
- 53.King JA, Wisniewski AB, Bankowski BJ, Carson KA, Zacur HA, Migeon CJ. Long-term corticosteroid replacement and bone mineral density in adult women with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91:865–69. doi: 10.1210/jc.2005-0745. [DOI] [PubMed] [Google Scholar]
- 54.Gleeson H, Davis J, Jones J, O’Shea E, Clayton PE. The challenge of delivering endocrine care and successful transition to adult services in adolescents with congenital adrenal hyperplasia: experience in a single centre over 18 years. Clin Endocrinol (Oxf) 2013;78:23–28. doi: 10.1111/cen.12053. [DOI] [PubMed] [Google Scholar]
- 55.Rivkees SA. Lost lessons of glucocorticoid potency and the treatment of children with congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. 2008;21:297–99. doi: 10.1515/jpem.2008.21.4.297. [DOI] [PubMed] [Google Scholar]
- 56.Hindmarsh PC. Management of the child with congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:193–208. doi: 10.1016/j.beem.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Rivkees SA, Crawford JD. Dexamethasone treatment of virilizing congenital adrenal hyperplasia: the ability to achieve normal growth. Pediatrics. 2000;106:767–73. doi: 10.1542/peds.106.4.767. [DOI] [PubMed] [Google Scholar]
- 58.Claahsen-van der Grinten HL, Otten BJ, Sweep FC, Hermus AR. Repeated successful induction of fertility after replacing hydrocortisone with dexamethasone in a patient with congenital adrenal hyperplasia and testicular adrenal rest tumors. Fertil Steril. 2007;88:705e5–8. doi: 10.1016/j.fertnstert.2006.11.148. [DOI] [PubMed] [Google Scholar]
- 59.German A, Suraiya S, Tenenbaum-Rakover Y, Koren I, Pillar G, Hochberg Z. Control of childhood congenital adrenal hyperplasia and sleep activity and quality with morning or evening glucocorticoid therapy. J Clin Endocrinol Metab. 2008;93:4707–10. doi: 10.1210/jc.2008-0519. [DOI] [PubMed] [Google Scholar]
- 60.Joint LWPES/ESPE CAH Working Group. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab. 2002;87:4048–53. doi: 10.1210/jc.2002-020611. [DOI] [PubMed] [Google Scholar]
- 61.Auchus RJ, Witchel SF, Leight KR, et al. Guidelines for the development of comprehensive Care Centers for Congenital Adrenal Hyperplasia: Guidance from the CARES Foundation Initiative. Int J Pediatr Endocrinol. 2010;2010:275213. doi: 10.1155/2010/275213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yankovic F, Cherian A, Steven L, Mathur A, Cuckow P. Current practice in feminizing surgery for congenital adrenal hyperplasia; a specialist survey. J Pediatr Urol. 2013 doi: 10.1016/j.jpurol.2013.03.013. published online May 18. [DOI] [PubMed] [Google Scholar]
- 63.Davies MC, Crouch NS, Woodhouse CR, Creighton SM. Congenital adrenal hyperplasia and lower urinary tract symptoms. BJU Int. 2005;95:1263–66. doi: 10.1111/j.1464-410X.2005.05516.x. [DOI] [PubMed] [Google Scholar]
- 64.Pena A. Total urogenital mobilization—an easier way to repair cloacas. J Pediatr Surg. 1997;32:263–67. doi: 10.1016/s0022-3468(97)90191-3. [DOI] [PubMed] [Google Scholar]
- 65.Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int. 2008;102:1182–97. doi: 10.1111/j.1464-410X.2008.08091.x. [DOI] [PubMed] [Google Scholar]
- 66.Palmer BW, Trojan B, Griffin K, et al. Total and partial urogenital mobilization: focus on urinary continence. J Urol. 2012;187:1422–26. doi: 10.1016/j.juro.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 67.Baskin LS, Erol A, Li YW, Liu WH, Kurzrock E, Cunha GR. Anatomical studies of the human clitoris. J Urol. 1999;162:1015–20. doi: 10.1016/S0022-5347(01)68052-2. [DOI] [PubMed] [Google Scholar]
- 68.Poppas DP, Hochsztein AA, Baergen RN, Loyd E, Chen J, Felsen D. Nerve sparing ventral clitoroplasty preserves dorsal nerves in congenital adrenal hyperplasia. J Urol. 2007;178:1802–06. doi: 10.1016/j.juro.2007.03.186. [DOI] [PubMed] [Google Scholar]
- 69.Crouch NS, Minto CL, Laio LM, Woodhouse CR, Creighton SM. Genital sensation after feminizing genitoplasty for congenital adrenal hyperplasia: a pilot study. BJU Int. 2004;93:135–38. doi: 10.1111/j.1464-410x.2004.04572.x. [DOI] [PubMed] [Google Scholar]
- 70.Minto CL, Liao LM, Woodhouse CR, Ransley PG, Creighton SM. The effect of clitoral surgery on sexual outcome in individuals who have intersex conditions with ambiguous genitalia: a cross-sectional study. Lancet. 2003;361:1252–57. doi: 10.1016/S0140-6736(03)12980-7. [DOI] [PubMed] [Google Scholar]
- 71.Schober JM, Pfaff D. The neurophysiology of sexual arousal. Best Pract Res Clin Endocrinol Metab. 2007;21:445–61. doi: 10.1016/j.beem.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Krege S, Walz KH, Hauffa BP, Korner I, Rubben H. Long-term follow-up of female patients with congenital adrenal hyperplasia from 21-hydroxylase deficiency, with special emphasis on the results of vaginoplasty. BJU Int. 2000;86:253–58. doi: 10.1046/j.1464-410x.2000.00789.x. [DOI] [PubMed] [Google Scholar]
- 73.Gastaud F, Bouvattier C, Duranteau L, et al. Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007;92:1391–96. doi: 10.1210/jc.2006-1757. [DOI] [PubMed] [Google Scholar]
- 74.Fortunoff S, Lattimer JK, Edson M. Vaginoplasty technique for female pseudohermaphrodites. Surg Gynecol Obstet. 1964;118:545–58. [PubMed] [Google Scholar]
- 75.Lin WC, Chang CY, Shen YY, Tsai HD. Use of autologous buccal mucosa for vaginoplasty: a study of eight cases. Hum Reprod. 2003;18:604–07. doi: 10.1093/humrep/deg095. [DOI] [PubMed] [Google Scholar]
- 76.Samuelson ML, Baker LA. Autologous buccal mucosa vulvovaginoplasty for high urogenital sinus. J Pediatr Urol. 2006;2:486–88. doi: 10.1016/j.jpurol.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 77.Avila NA, Premkumar A, Merke DP. Testicular adrenal rest tissue in congenital adrenal hyperplasia: comparison of MR imaging and sonographic findings. AJR Am J Roentgenol. 1999;172:1003–06. doi: 10.2214/ajr.172.4.10587136. [DOI] [PubMed] [Google Scholar]
- 78.Claahsen-van der Grinten HL, Otten BJ, Sweep FC, et al. Testicular tumors in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency show functional features of adrenocortical tissue. J Clin Endocrinol Metab. 2007;92:3674–80. doi: 10.1210/jc.2007-0337. [DOI] [PubMed] [Google Scholar]
- 79.Claahsen-van der Grinten HL, Sweep FC, Blickman JG, Hermus AR, Otten BJ. Prevalence of testicular adrenal rest tumours in male children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. 2007;157:339–44. doi: 10.1530/EJE-07-0201. [DOI] [PubMed] [Google Scholar]
- 80.Benvenga S, Smedile G, Lo Giudice F, Trimarchi F. Testicular adrenal rests: evidence for luteinizing hormone receptors and for distinct types of testicular nodules differing for their autonomization. Eur J Endocrinol. 1999;141:231–37. doi: 10.1530/eje.0.1410231. [DOI] [PubMed] [Google Scholar]
- 81.Claahsen-van der Grinten HL, Hermus AR, Otten BJ. Testicular adrenal rest tumours in congenital adrenal hyperplasia. Int J Pediatr Endocrinol. 2009;2009:624823. doi: 10.1155/2009/624823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stikkelbroeck NM, Otten BJ, Pasic A, et al. High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86:5721–28. doi: 10.1210/jcem.86.12.8090. [DOI] [PubMed] [Google Scholar]
- 83.Aycan Z, Bas VN, Cetinkaya S, Yilmaz Agladioglu S, Tiryaki T. Prevalence and long-term follow-up outcomes of testicular adrenal rest tumours in children and adolescent males with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2013;78:667–72. doi: 10.1111/cen.12033. [DOI] [PubMed] [Google Scholar]
- 84.Consortium on the Management of Disorders of Sex Differentiation. Accord Alliance. Rohnert Park: Intersex Society of North America; 2006. Clinical Guidelines for the Management of Disorders of Sex Development in Childhood. [Google Scholar]
- 85.Hughes IA, Nihoul-Fekete C, Thomas B, Cohen-Kettenis PT. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2007;21:351–65. doi: 10.1016/j.beem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 86.Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554–63. doi: 10.1136/adc.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Houk CP, Hughes IA, Ahmed SF, Lee PA. Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference. Pediatrics. 2006;118:753–57. doi: 10.1542/peds.2006-0737. [DOI] [PubMed] [Google Scholar]
- 88.Mueller SC, Ng P, Sinaii N, et al. Psychiatric characterization of children with genetic causes of hyperandrogenism. Eur J Endocrinol. 2010;163:801–10. doi: 10.1530/EJE-10-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dorn LD, Hitt SF, Rotenstein D. Biopsychological and cognitive differences in children with premature vs. on-time adrenarche. Arch Pediatr Adolesc Med. 1999;153:137–46. doi: 10.1001/archpedi.153.2.137. [DOI] [PubMed] [Google Scholar]
- 90.Szybowska M, Hewson S, Antle BJ, Babul-Hirji R. Assessing the informational needs of adolescents with a genetic condition: what do they want to know? J Genet Couns. 2007;16:201–10. doi: 10.1007/s10897-006-9060-5. [DOI] [PubMed] [Google Scholar]
- 91.Clayton PE, Miller WL, Oberfield SE, Ritzen EM, Sippell WG, Speiser PW. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm Res. 2002;58:188–95. doi: 10.1159/000065490. [DOI] [PubMed] [Google Scholar]
- 92.Lee PA, Fuqua JS, Nebesio TD. Treatment and outcome of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Int J Pediatr Endocrinol. 2010;2010:276843. doi: 10.1155/2010/276843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brain CE, Creighton SM, Mushtaq I, et al. Holistic management of DSD. Best Pract Res Clin Endocrinol Metab. 2010;24:335–54. doi: 10.1016/j.beem.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakhla M, Daneman D, Frank M, Guttmann A. Translating transition: a critical review of the diabetes literature. J Pediatr Endocrinol Metab. 2008;21:507–16. [PubMed] [Google Scholar]
- 95.Peters A, Laffel L for the American Diabetes Association Transitions Working Group. Diabetes care for emerging adults: recommendations for transition from pediatric to adult diabetes care systems: a position statement of the American Diabetes Association, with representation by the American College of Osteopathic Family Physicians, the American Academy of Pediatrics, the American Association of Clinical Endocrinologists, the American Osteopathic Association, the Centers for Disease Control and Prevention, Children with Diabetes, The Endocrine Society, the International Society for Pediatric and Adolescent Diabetes, Juvenile Diabetes Research Foundation International, the National Diabetes Education Program, and the Pediatric Endocrine Society (formerly Lawson Wilkins Pediatric Endocrine Society) Diabetes Care. 2011;34:2477–85. doi: 10.2337/dc11-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hanna KM, Woodward J. The transition from pediatric to adult diabetes care services. Clin Nurse Spec. 2013;27:132–45. doi: 10.1097/NUR.0b013e31828c8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tuchman LK, Schwartz LA, Sawicki GS, Britto MT. Cystic fibrosis and transition to adult medical care. Pediatrics. 2010;125:566–73. doi: 10.1542/peds.2009-2791. [DOI] [PubMed] [Google Scholar]
- 98.Jordan L, Swerdlow P, Coates TD. Systematic review of transition from adolescent to adult care in patients with sickle cell disease. J Pediatr Hematol Oncol. 2013;35:165–69. doi: 10.1097/MPH.0b013e3182847483. [DOI] [PubMed] [Google Scholar]
- 99.Crowley R, Wolfe I, Lock K, McKee M. Improving the transition between paediatric and adult healthcare: a systematic review. Arch Dis Child. 2011;96:548–53. doi: 10.1136/adc.2010.202473. [DOI] [PubMed] [Google Scholar]
- 100.Van Lierde A, Menni F, Bedeschi MF, et al. Healthcare transition in patients with rare genetic disorders with and without developmental disability: neurofibromatosis 1 and Williams-Beuren syndrome. Am J Med Genet A. 2013;161:1666–74. doi: 10.1002/ajmg.a.35982. [DOI] [PubMed] [Google Scholar]