

Abstract
Background
A clinical study to investigate the leukotriene B4 (LTB4)-receptor antagonist BIIL 284 in cystic fibrosis (CF) patients was prematurely terminated due to a significantly increased risk of adverse pulmonary events. We aimed to establish the effect of BIIL284 in models of Pseudomonas aeruginosa lung infection, thereby contributing to a better understanding of what could have led to adverse pulmonary events in CF patients.
Methods
P. aeruginosa DNA in the blood of CF patients during and after acute pulmonary exacerbations and in stable patients with non-CF bronchiectasis (NCFB) and healthy individuals was assessed by PCR. The effect of BIIL 284 treatment was tested in an agar beads murine model of Pseudomonas aeruginosa lung infection. Bacterial count and inflammation were evaluated in lung and other organs.
Result
Most CF patients (98%) and all patients with NCFB and healthy individuals had negative P. aeruginosa DNA in their blood. Similarly, the P. aeruginosa-infected mice showed bacterial counts in the lung but not blood or spleen. BIIL 284 treatment decreased pulmonary neutrophils and increased P. aeruginosa numbers in mouse lungs leading to significantly higher bacteremia rates and lung inflammation compared to placebo treated animals.
Conclusions
Decreased airway neutrophils induced lung proliferation and severe bacteraemia in a murine model of P. aeruginosa lung infection. These data suggest that caution should be taken when administering anti-inflammatory compounds to patients with bacterial infections.
Keywords: cystic fibrosis, pulmonary infection, anti-inflammatory treatment
1. Introduction
In cystic fibrosis (CF), chronic lung infection with opportunistic pathogens, particularly Pseudomonas aeruginosa, causes a chronic inflammatory response dominated by neutrophils (1) which contributes to the decline in lung function, extensive tissue destruction (2), and the shortened life expectancy (3). Therefore, in addition to strategies that minimize infection (4), strategies which reduce chronic inflammation have been put forward (5). Furthermore, episodes of acute pulmonary exacerbations (6, 7) have a detrimental effect on life expectancy in CF (8, 9). Increased bacterial proliferation in sputum samples from CF patients during acute pulmonary exacerbations (10) raise the possibility that P. aeruginosa may translocate to the blood stream, contributing to symptoms of acute pulmonary exacerbations. However, to date this theory has not been adequately addressed.
To limit inflammation in CF airways, inhibition of the leukotriene B4 (LTB4)-receptor has been tested in CF patients using the LTB4-receptor antagonist BIIL 284 (11). LTB4, a dihydroxy fatty acid formed from arachidonic acid by the 5-lipoxygenase pathway, is produced mainly by activated neutrophils and macrophages (12). When binding to its receptor (13), LTB4 stimulates its own production in an autocrine manner, thereby augmenting inflammation via NFκB (14, 15). The main biological functions of LTB4 are recruitment and activation of inflammatory cells, particularly neutrophils, but also macrophages, monocytes, eosinophils and lymphocytes (12, 16). Thus, LTB4 has been demonstrated to be an important participant in the pathophysiology of inflammatory diseases including CF (17). However, a 24 week, placebo-controlled phase II trial, assessing the efficacy of BIIL 284 in children and adults with CF was terminated prematurely in 2004, due to a significantly increased risk of adverse pulmonary events in the the adult patients receiving active treatment compared to those receiving placebo (see Konstan et al, this Journal). These adverse pulmonary events were characterized by increased presentation of respiratory signs and/or symptoms associated with pulmonary exacerbation and resulted in hospital admission and/or administration of IV antibiotics.
A potential cause for these adverse pulmonary events has not been previously explored. We hypothesized that neutrophils migrating into CF airways in response to chronic P. aeruginosa infection would usually be sufficient to prevent proliferation of bacteria in the lung and dissemination to the bloodstream. Therefore, the inhibitory effect of BIIL 284 on neutrophil migration and activation in CF airways could conceivably deplete neutrophil cell numbers to such an extent that the infecting micro-organisms could replicate in the lung and enter the blood stream and contribute to the symptoms of acute pulmonary exacerbations.
Therefore, our aim was to understand the mechanism(s) by which BIIL 284 could have induced adverse pulmonary events in CF patients; we employed the agar bead mouse model of lung infection model which resembles in vivo conditions in the CF airways (18). In the agar bead model, bacteria are protected from an immediate neutrophil attack in the airways by the agar beads and the microaerobic/anaerobic growth conditions present in these beads rapidly induces a switch from a completely alginate-negative to an alginate-positive phenotype of P. aeruginosa. The agar bead model has been previously used as a model for chronic model of P. aeruginosa lung infection mimicking the CF pathophysiology.
Here we show that translocation of P. aeruginosa from the airways into the bloodstream is a rare event even during acute pulmonary exacerbations, and may be controlled by the high neutrophil numbers typically found in the airways of infected CF patients. Similarly to human, bacteremia in P. aeruginosa-infected mice does not occur in the mouse model established in this study. However, we report that treatment of P. aeruginosa-infected mice with high doses of BIIL 284 significantly decreased pulmonary neutrophil numbers but also increased pulmonary P. aeruginosa numbers, leading to higher bacteremia rates and higher lung inflammation compared to placebo treated animals.
Methods
2.1 Patients
To assess whether acute pulmonary exacerbations in CF patients would lead to translocation of P. aeruginosa from the airways to the bloodstream, we determined the relative frequency of DNA in plasma samples from CF patients with and without acute pulmonary exacerbations. From November 9, 2010 to November 30, 2012, 44 adolescent and adult CF patients (21 females, 23 males, mean age: 35.7 years) were recruited for a prospective screening study for P. aeruginosa DNA in blood samples. Patients were eligible for enrolment if they had CF that had been diagnosed according to conventional criteria, an age ≥18 years, chronic P. aeruginosa lung infection and an acute pulmonary exacerbation, defined by at least 4/12 positive Fuchs criteria (6). Patients were excluded if they had an acute asthmatic complication. The primary aim of the study was to determine the relative frequency of P. aeruginosa DNA in plasma samples from CF patients with and without acute pulmonary exacerbation. Blood samples were stored at -20°C prior to examination. The patients attended the Comprehensive CF Centre Tübingen-Stuttgart (CCFC) at the clinic Schillerhöhe, Robert-Bosch Krankenhaus, Gerlingen, Germany (n=1) and the Universitätsklinikum Frankfurt, Frankfurt, Germany (n=43). Additionally, blood samples from 17 CF patients (10 females, 7 males, mean age: 33.3 years) attending the Queens University Belfast, Northern Ireland (n=11) and the Case Western Reserve University, UH Rainbow Babies & Children’s Hospital, Cleveland, USA (n=6) and blood samples from 10 patients with non-CF bronchiectasis, suffering from chronic P. aeruginosa lung infection (8 females, 2 males, mean age: 59.1 years) attending the Royal Brompton Hospital, London, UK, and from 17 healthy individuals (9 females, 8 males, mean age: 34.3 years) from the Queens University Belfast, Northern Ireland (7) and the Institute of Medical Microbiology and Hygiene, Universitätsklinikum Tübingen, Tübingen, Germany (n=10), were examined for P. aeruginosa DNA. Informed consent was obtained from all patients and the study protocols were approved by the institutional review boards in the respective jurisdictions.
2.2 Quantitative real-time PCR (qPCR)
DNA was extracted from whole blood samples using the Qiagen DNeasy Maxi kit (Qiagen) according to the manufacturer’s instructions and stored at -20°C until required. P. aeruginosa DNA was quantitated in blood samples from CF patients, patients with non-CF bronchiectasis and healthy individuals using a qPCR TaqMan assays based on the PA4331 gene, which is ubiquitously present in a collection of 117 isolates from 14 CF patients and therefore a stable marker of P. aeruginosa. A standard curve was used to determine the absolute copy number of the target. DNA was amplified in duplicate using an iCycler (BioRad) with cycle threshold values obtained using iCycler CFX Manager 1.6. Baseline cycles and threshold positions were manually adjusted. The RT-PCR mixture (15 μl final volume) contained: 1x KAPA PROBE FAST Universal qPCR Master Mix (Mix contains TaqHotStart DNA polymerase, qPCR Buffer, dNTPs, MgCl2 and stabilizers) (peQlab, Erlangen, Germany), 0.3μM Primer PA4331 forward (5′-GTGTTGCAGCCTTTCGATCC3′-), 0.3 μM Primer PA4331 reverse (5′- AACTCCAGCCATGGGTCCTC 3′-), 0.3 μM qPCR PA4331 Probe (5′-FAM GCAGCACCTGCTGCTGTGGA 3′-TAM) (Eurofins MWG Operon, Ebersberg, Germany). PCR conditions were 3 min at 95°C, followed by 50 cycles of 10 sec at 95°C and 30 sec at 64°C. Fluorescent data were collected at the 64°C step.
2.3 Mouse model, bacterial numbers and markers of lung inflammation
Groups of 6-14 C57Bl/6 male mice (Charles River, Sulzfeld, Germany; Charles River, Calco, Italy), 8-10 weeks old (24-26 g), received orally by gavage 100 μl of suspensions containing 0.3, 50 or 100 mg/kg body weight of BIIL 284 (Novartis, Horsham, UK) in phosphate buffered saline, pH 7.2 (PBS), supplemented with polyethylenglycol (PEG) 200 in a 1:1 (vol/vol) ratio. Control mice were treated with placebo. One h later the mice were injected intratracheally with either 50 μl of PBS, or 50 μl of a suspension of P. aeruginosa, strain PAO1, containing 5×105 - 5×107 colony forming units (cfu), embedded in agar beads (18). After 24 h, mice were killed, with an overdose of CO2. Lungs and spleens were removed and homogenized in 1ml PBS with Ca2+/Mg2+ containing the protease inhibitor PMSF (Sigma). Bacterial counts (cfu) were determined in total lung and spleen homogenates as well as in blood by routine dilution methods on agar plates. Additionally, at this time point, neutrophil and macrophage numbers were assessed in bronchoalveolar (BAL) fluids. BAL was performed three times using 1 ml of RPMI 1640 (Complete tablets, Roche Diagnostic) with a 22-gauge venous catheter. Total cells present in the BAL fluid were counted and a differential cell count was performed on cytospins stained with Diff Quick (Dade, Biomap, Italy). BALF was serially diluted 1:10 in PBS and plated for bacterial counts. BALF were then centrifuged and supernatants were collected for quantification of total protein content using the Bradford assay (Bio-RAD). After erythrocyte lysis with ACK lysis solution pH 7.2 (NH4Cl 0.15 M, KHCO3 10 mM, EDTA 0.1 mM), cells were re-suspended in cetyltrimethylammonium chloride 0.5% (CTAC, Sigma Aldrich) (250 μI/mouse) and centrifuged.
Murine cytokines (IL-1ß, KC, MIP-2 and JE) were measured by ELISA (R&D DuoSet ELISA Development System, USA) according to manufacturer’s instructions in supernatant fluids of total lung homogenates after centrifugation (14,000 rpm/ 30 min/ 4°C). MIP-2 and KC affect neutrophil recruitment, while JE promotes monocytes/macrophages recruitment and IL-1 ß is involved in inflammation and cell death.
To localize P. aeruginosa in lungs of untreated and BIIL 284 treated C57Bl/6 mice, infected with P. aeruginosa as described above, mice from each group were sacrificed after 24 h, the lungs were removed en bloc, rinsed in PBS and prepared for detection of P. aeruginosa by indirect immunofluorescence as described previously (2), using a specific rabbit antibody against whole cell P. aeruginosa. The animal study protocols were reviewed and approved by the Animal ethics committee of the H. S. Raffaele Animal Care and Use Committee, Milano, Italy.
2.4 Statistical analysis
CFU data, cell count and cytokines are expressed as mean ± SEM. Two-tailed Student’s t and Chi-square test was used. p ≤ 0.05 was considered significant. Kendall’s rank-correlation, tau was used to assess the relationship between the ordinal variable BIIL 284 and the continuous variable ratio (cfu/neutrophils) (19).
3. Results
3.1. Assessment of P. aeruginosa bacteraemia in patients with acute pulmonary exacerbations and in the mouse model
First, we assessed the incidence of bacteraemia in P. aeruginosa chronically infected CF patients during episodes of acute pulmonary exacerbation (n=17) or when clinically stable (n=44) by employing qPCR for the gene PA4331 in 61 CF patients. Regardless of whether clinically stable or presenting with an acute pulmonary exacerbation, all but one blood sample from CF patients (60/61; 98%) yielded negative qPCR for P. aeruginosa (Fig. 1). The single qPCR positive CF patient who had a forced expiratory volume in 1 sec, (FEV1%), predicted, of 44%, and did not recover clinically following treatment of an acute pulmonary exacerbation, had 4×103 cells of P. aeruginosa per ml in his blood. Mean lung function, measured as FEV1% was 56% (range: 21%-76.1%) in the 17 CF patients suffering from acute pulmonary exacerbation and 59% (range: 26%-116%) in the 44 clinically stable CF patients. These data reveal that even in CF patients with severe lung disease P. aeruginosa is not translocating from the airways to the circulation. Similarly, we were unable to detect P. aeruginosa DNA by qPCR in blood samples from a cohort of 10 patients with non-CF bronchiectasis and P. aeruginosa lung infection and in blood samples from 17 healthy individuals.
Fig. 1. Bacterial quantification of P. aeruginosa DNA in the blood of a CF patient.
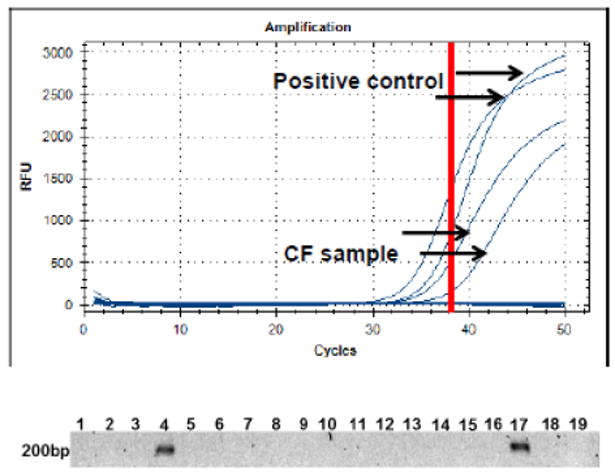
TaqMan quantitative PCR was performed on DNA extracts of blood samples from CF patients, patients with non-CF bronchiectasis and healthy individuals. A. Agarose gel with qPCR products results of 16 CF patients revealing one positive sample with the amplication of the 200 bp PA4331. 17: positive control (plasmid DNA including PA 4331, spiked with human DNA), 18: negative control (human DNA) 19: water control; B. qPCR amplification curves of the PA4331 and controls in the blood sample from CF patient #4. Red bar: cut-off of the assay.
Next, the P. aeruginosa agar bead mouse model was used to simulate lung infection of CF patients and determine the effect of BILL 284 on bacterial counts and inflammation. We assessed respiratory infection and bacteraemia by challenging via intratracheal administration C57Bl/6 mice with different doses of P. aeruginosa PAO1 strain (Table 1S). A dose of 5*107 cfu of P. aeruginosa was found to cause high mortality during the first 3 days (100%) due to high bacterial load in the airways and bloodstream. Doses of 5*106 and 5*105 decreased mortality to 25 and 0%, respectively, while maintained chronic infection at 7 days (25%). Since bacteraemia did not occur in mice treated with P. aeruginosa 5*105 cfu, this dose was selected for testing the effect of BILL 284 treatment as described below.
3.2. BIIL 284 decreases numbers of phagocytic cells in lungs of mice infected with P. aeruginosa
To assess whether BIIL 284, administered orally to C57Bl/6 mice in doses of 0.3, 50 or 100 mg/kg body weight, would decrease neutrophil and macrophage numbers in P. aeruginosa infected airways, we challenged groups of six to eight mice by intratracheal administration with 5×105 cfu of P. aeruginosa one h after BIIL 284 oral administration. Twenty-four h after treatment, neutrophil numbers had decreased ≥50% in BAL fluids compared to untreated control animals. Untreated mice had a mean influx of 3.42×106 neutrophils in their P. aeruginosa infected lungs; however treatment with 0.3 mg/kg, 50 mg/kg or 100 mg/kg BIIL 284 resulted in a significant decrease in neutrophils to 1.37×106, 1.26×106 and 1.55×106 neutrophils per ml BAL, respectively (Fig. 2A). We also observed a trend towards, decreases in macrophage numbers, though this was not significant compared to control animals (p<0.05) (Fig. 2B). The data demonstrated the expected biological efficacy of BIIL 284 to inhibit neutrophil migration to pulmonary sites of infection.
Fig. 2. BIIL 284 treatment changes the ratio between phagocytic cells and bacteria in lungs of mice infected with P. aeruginosa.
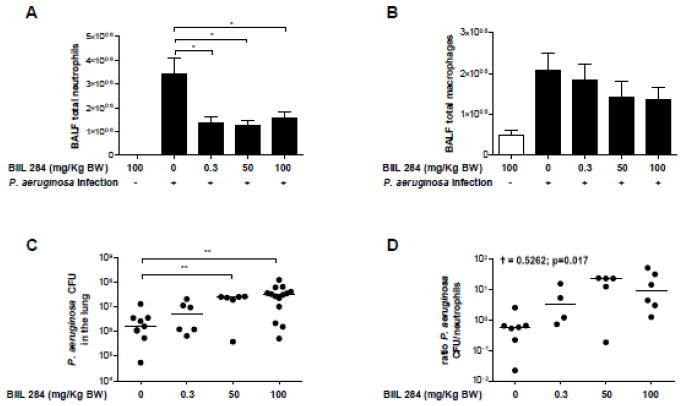
A, B, BIIL 284 treatment decreases neutrophil but not macrophage numbers in bronchoalveolar lavage fluids (BALF) of infected mice. Groups of six to eight mice were challenged intratracheally with 5×105 cfu of P. aeruginosa and one h therafter with different doses of BIIL 284. Neutrophil (A) and macrophage (B) numbers were determined in BALF of infected animals and uninfected controls by routine methods. C, BIIL 284 promotes growth of P. aeruginosa in lungs of infected mice. Groups of seven to 14 mice were challenged with different doses of BIIL 284 one h before intratracheal challenge with 5×105 cfu of P. aeruginosa and bacterial colony forming units (cfu) were determined in total lung tissues of infected and uninfected animals after 24 h using routine methods. D, BIIL 284 treatment changes the ratio of P. aeruginosa cfus/neutrophils. Kendall’s rank-correlation, † was used to assess the relationship between the ordinal variable BIIL 284 and the continuous variable ratio (cfu/neutrophils). * p≤0.05; ** p<0.01;
3.3. BIIL 284 allows growth of P. aeruginosa in lungs of infected mice and causes bacteraemia at high doses
Next we determined whether the effect of BIIL 284 on neutrophil levels in the lungs would affect the growth of P. aeruginosa in the infected mouse lung. P. aeruginosa cfus were determined in groups of seven or 14 mice challenged with 5×105 cfu of P. aeruginosa and treated with 0.3, 50 or 100 mg/kg body weight of BIIL 284, respectively. While bacterial numbers remained stable in total lung homogenates of animals treated with 0.3 mg/kg body weight of BIIL 284 after 24 h, P. aeruginosa cfu’s increased significantly (p<0.01) when 50 or 100 mg/kg body weight of BIIL 284 was administered compared to untreated control animals (Fig. 2C). A significant correlation between the ratio of P. aeruginosa cfus/neutrophils and BIIL 284 concentration was found (ϯ = 0.5262; p=0.017) (Fig. 2D).
To assess whether excessive bacterial growth would lead to sepsis, we also determined P. aeruginosa levels in other mouse organs. Two of 12 BIIL 284-untreated animals (17%) had between 101 and 102 cfu of P. aeruginosa in the blood and spleens, while in 4/6 (67%) animals treated with 0.3 mg/kg body weight of BIIL 284, cfus between 102 and 103 cfus were detected in the blood and spleens (Table 2, Table 3). Furthermore, in 5/6 (83%) animals treated with 50 mg/kg of BIIL 284, between 102 and 103 cfus were detected and in 11/14 (78%) animals treated with 100 mg/kg of BIIL 284 between 102 and 103 were detected (Table 2, Table 3). Thus, BIIL 284 treatment significantly increased the risk of sepsis in a dose dependent manner when compared to untreated mice (0.3 mg/kg vs untreated: p=0.1; 50 mg/kg: p=0.01; 100 mg/kg: p=0.005).
3.4. BIIL 284 promotes increased cytokine expression in lungs of infected mice and allows P. aeruginosa to reach the airway epithelium
We determined whether the increased bacterial growth in BIIL 284-treated mice would stimulate respiratory epithelial cells to increase production of cytokines and chemokines. At doses of 50 and 100 mg/kg body weight of BIIL 284, 24 h after challenge, IL-1ß, KC (equivalent to IL-8 in humans), MIP-2 and JE were found all to be significantly increased in lung homogenates from treated/infected mice versus untreated/infected mice. At doses of 0.3 mg/kg body weight of BIIL 284, only IL-1ß and JE were significantly increased (Fig. 3). Next, we localized the pathogen in infected airways by indirect immunofluorescence. While in untreated mice P. aeruginosa was detected exclusively in the bronchial lumen (Fig. 4), treatment with a single dose of 50 or 100 mg/kg of BIIL 284 led to additional detection of P. aeruginosa at the bronchial epithelium as well as in the alveoli (Fig. 4). These data suggest that immunosuppression allows P. aeruginosa to elicit increased cytokine expression from airway epithelial cells and to enter the blood stream via the bronchial epithelium or the alveoli. Taken together, our data demonstrate that immune suppression by BIIL 284 at high doses can provoke inflammation and bacteraemia in mice whose airways are infected with P. aeruginosa.
Fig. 3. BIIL 284 treatment increases lung inflammation infected mice.
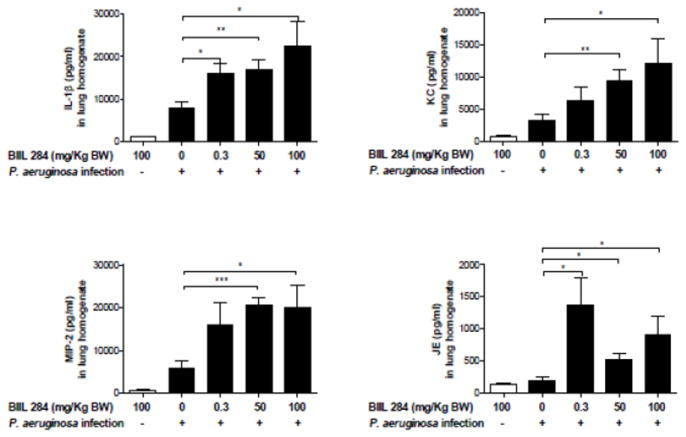
A, BIIL 284 promotes increased cytokine expression in lungs of infected mice. The chemokines IL-1ß, KC, MIP-2 and JE were quantitatively determined by ELISA in lung homogenates of uninfected mice, treated with a single dose of 100 mg/kg BIIL 284, and in untreated infected mice (CTR) and in infected mice, treated with the doses of 0.3, 50 and 100 mg/kg BIIL 284. *p≤0.05; **p<0.01; p<0.001
Fig. 4. BIIL 284 treatment increases P. aeruginosa translocation via the alveoli in infected mice.
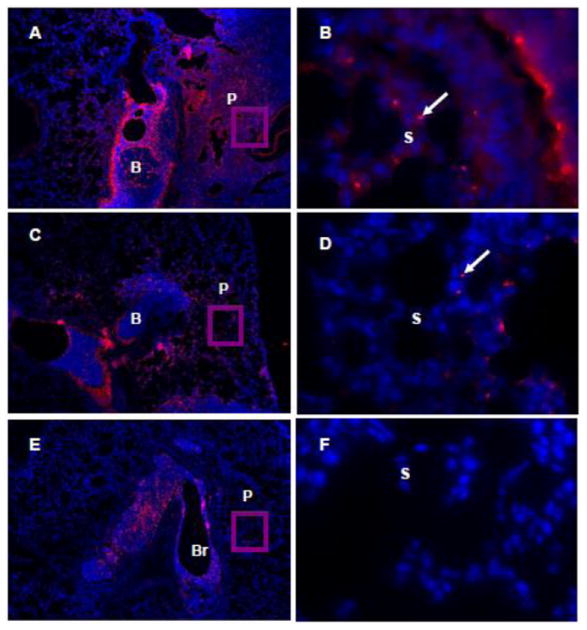
BIIL 284 treatment allows P. aeruginosa to reach the airway epithelium. P. aeruginosa was stained with a specific antibody in 5 μm lung tissue sections of infected mice treated with a single dose of 100 mg/kg BIIL 284 (A, B), or 50 mg/kg of BIIL 284 (C, D) or untreated animals (E, F). Br: Bronchus, P: Periphery, S: septa, red color and arrow: P. aeruginosa. Original magnifications: A, C, E × 100; inserts B, D, F × 1,000.
4. Discussion
The oral long-acting LTB4 receptor antagonist BIIL 284 has been shown previously to inhibit LTB4-induced neutrophil cell activation as monitored by intracellular Ca release, and to inhibit LTB4-induced neutrophil chemotaxis, possibly by inhibition of expression of the integrin Mac-1 (αMβ2, CR3) (20). In a previous study, a single oral dose of 0.3 mg/kg p.o. completely inhibited LTB4-induced Mac-1 expression over 24 h in monkeys (11). Because Mac-1 also mediates phagocytosis of bacteria and other particles (21), BIIL 284 treatment most probably also interferes with this neutrophil function.
Here we demonstrate that administration of BIIL 284 to mice at a dose of 0.3 to 100 mg/kg body weight significantly reduces the numbers of phagocytic cells in airways and lung tissues of P. aeruginosa-infected animals thereby confirming previous results with this anti-inflammatory drug (11, 21). Moreover, reducing the neutrophil influx into P. aeruginosa infected airways of mice by ≥50% highlighted the extreme potency of this compound.
In the presence of an established P. aeruginosa infection, the reduction in neutrophil numbers in conjunction with the established down-regulation of Mac-1 (CR3) by BIIL 284 was associated with a profound proliferation of P. aeruginosa in the lungs of the infected mice, as well as entry of this pathogen into the blood stream. Our data also demonstrated that BIIL treatment resulted in increased pro-inflammatory cytokine production in this mouse model, suggesting that the bacterial proliferation led to stimulation of lung epithelial cells to enhance cytokine production, probably via Toll-like receptor activation. This notion is also supported by our finding that P. aeruginosa cells were detectable in the lung periphery of BIIL 284 treated animals but not in untreated, infected animals and that BIIL 284 treatment resulted in increased sepsis rates in animals treated with 50 and 100 mg/kg BIIL 284. Both KC and MIP-2 are selectively chemotactic for neutrophils and expressed in various cell types including lung epithelial cells (22). Their up-regulation during BIIL 284 treatment creates the paradoxical situation that an anti-inflammatory drug induces inflammation in the infected murine airways. In contrast to BIIL 284, the macrolide azithromycin given at a concentration of 20 mg/kg has been shown to reduce airway neutrophil numbers, inflammatory cytokine levels and P. aeruginosa cell numbers significantly in a similar model of murine lung infection (23).
Our results may help explain the termination of a phase II study in which the efficacy and safety of BIIL 284 was evaluated in patients with CF. In this study, pediatric subjects received 1.75 mg/kg BIIL 284 for a median of 102 days, while adults received 1.23 mg/kg or 2.35 mg/kg BIIL 284 for a median of 149 days. The study was terminated due to a significantly higher incidence of adverse pulmonary events in the treatment group. Although it is not clear at present how and whether these adverse pulmonary events are related to sepsis, proliferation of bacterial cell numbers is a consistent finding in such episodes (8). Because BIIL 284 inhibited Mac-1 expression in monkey studies for more than 24 h (11), the repeated daily BIIL 284 dosing schedule in the phase II clinical study may have led to an accumulation of functional impairments in neutrophils during the study period.
Here we have shown that translocation of P. aeruginosa from the airways into the bloodstream is a rare event in CF and NCFB, and may be controlled by the neutrophil response to chronic bacterial lung infection. Similarly to humans, we also demonstrated in a mouse model of P. aeruginosa lung infection, optimised to test the effect of BIIL 284, that bacteremia did not occur. Based on our animal data, we propose that adverse pulmonary events in the BIIL 284 CF patient study may have been caused by unrestrained bacterial multiplication, followed by septic episodes due to reduced neutrophil activity and cell numbers. Unfortunately, the causal relationship between neutrophil response and sepsis in the original BIIL 284 study can not been demonstrated directly, due to the lack of blood samples from CF patients.
A number of effector defence mechanisms have been implicated in the host fight against P. aeruginosa, including immune cells, as macrophages and natural killer cells, or molecules, as antimicrobial peptides. However, neutrophils have been shown to be the dominant effector cells which control P. aeruginosa infection (24). Our data support the conclusions of others (25), that a critical neutrophil concentration (CNC) is necessary to control bacterial proliferation: for example for killing ~5×107 cfus of E. coli, a CNC of ~4×106 neutrophils are needed (25). Clearly this CNC was not reached during treatment of infected mice with 100 mg/kg BIIL 284, where only 1.55×106 neutrophils are confronted with ~5×107 cfus of P. aeruginosa. In contrast, in untreated animals 3.42×106 neutrophils are confronted with ~2×106 cfus of P. aeruginosa. In the latter case, septic episodes were significantly lower than in BIIL 284 treated animals. Our results revealing that P. aeruginosa does not enter the blood stream in chronically infected CF patients under clinically stable conditions or under conditions of acute pulmonary exacerbations therefore demonstrates the importance of the considerable neutrophil influx into the infected CF airways. Only one out of 61 CF blood samples yielded a positive qPCR for P. aeruginosa. Thus, P. aeruginosa bacteraemia is a rare event even in CF patients with severe lung disease. This notion is also true for patients with NCFB infected with P. aeruginosa.
Taken together, these results show that caution should be taken when administering potent anti-inflammatory compounds as there is a potential to suppress the immune system thus increasing the risk of exacerbation in patients with acute or chronic infections.
Supplementary Material
Acknowledgments
The authors would like to thank Marie Haug for excellent technical support. BIIL 284 BS was a kind gift of Novartis, Horsham, UK. This study is dedicated to the memory of Gerd Döring and his commitment to CF research.
Footnotes
Competing Interest
Competing Interest: None to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Döring G, Ratjen F. Immunology of cystic fibrosis. In: Hodson ME, Geddes D, Bush A, editors. Cystic Fibrosis. London, England: Arnold Hammer; 2000. pp. 69–80. [Google Scholar]
- 2.Ulrich M, Worlitzsch D, Viglio S, et al. Alveolar inflammation in cystic fibrosis. J Cyst Fibros. 2010;9:217–27. doi: 10.1016/j.jcf.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cystic Fibrosis Foundation Cystic Fibrosis Foundation Patient Registry 2005 Annual Data Report. Bethesda: Cystic Fibrosis Foundation; 2006. [Google Scholar]
- 4.Döring G, Flume P, Heijerman H, Elborn JS for the Consensus Study Group. Treatment of Lung Infection in Patients with Cystic Fibrosis: Current and Future Strategies. J Cyst Fibros. 2012;11:461–79. doi: 10.1016/j.jcf.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Döring G, Hoiby N for the Consensus Study Group. Early intervention and prevention of lung disease in cystic fibrosis: a European consensus. J Cyst Fibros. 2004;3:67–91. doi: 10.1016/j.jcf.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637–42. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- 7.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax. 2007;62:360–7. doi: 10.1136/thx.2006.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstan MW, Morgan WJ, Butler SM, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151:134–9. doi: 10.1016/j.jpeds.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 9.McCormick J, Mehta G, Olesen HV, Viviani L, Macek M, Jr, Mehta A. Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet. 2010;375:1007–13. doi: 10.1016/S0140-6736(09)62161-9. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin FJ, Matthews WJ, Jr, Strieder DJ, et al. Clinical and bacteriological responses to three antibiotic regimens for acute exacerbations of cystic fibrosis: ticarcillin-tobramycin, azlocillin-tobramycin, and azlocillin-placebo. J Infect Dis. 1983;147:559–67. doi: 10.1093/infdis/147.3.559. [DOI] [PubMed] [Google Scholar]
- 11.Birke FW, Meade CJ, Anderskewitz R, Speck GA, Jennewein HM. In vitro and in vivo pharmacological characterization of BIIL 284, a novel and potent leukotriene B(4) receptor antagonist. J Pharmacol Exp Ther. 2001;297:458–66. [PubMed] [Google Scholar]
- 12.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–5. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 13.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–4. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 14.Back M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, Hansson GK. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci USA. 2005;102:17501–6. doi: 10.1073/pnas.0505845102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serio KJ, Baker JR, Ring WL, Riddick CA, Bigby TD. Leukotriene B4 costimulates 5-lipoxygenase activity in neutrophils via increased 5-lipoxygenase translocation. Am J Physiol. 1997;272:C1329–C1334. doi: 10.1152/ajpcell.1997.272.4.C1329. [DOI] [PubMed] [Google Scholar]
- 16.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science. 1987;237:1171–6. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 17.Konstan MW, Walenga RW, Hilliard KA, Hilliard JB. Leukotriene B4 is markedly elevated in epithelial lining fluid of patients with cystic fibrosis. Am Rev Respir Dis. 1993;148:896–901. doi: 10.1164/ajrccm/148.4_Pt_1.896. [DOI] [PubMed] [Google Scholar]
- 18.Bragonzi A, Worlitzsch D, Timpert P, et al. Non-mucoid Pseudomonas aeruginosa expresses alginate in lungs of patients with cystic fibrosis and in a murine model. J Infect Dis. 2005;192:410–9. doi: 10.1086/431516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kendall M. A New Measure of Rank Correlation. Biometrika. 1938;30:81–89. [Google Scholar]
- 20.Ehlers MRW. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microb Infect. 2000;2:289–94. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 21.Alten R, Gromnica-Ihle E, Pohl C, et al. Inhibition of leukotriene B4-induced CD11B/CD18 (Mac-1) expression by BIIL 284, a new long acting LTB4 receptor antagonist, in patients with rheumatoid arthritis. Ann Rheum Dis. 2004;63:170–6. doi: 10.1136/ard.2002.004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Driscoll KE, Hassenbein DG, Carter J, et al. Macrophage inflammatory proteins 1 and 2: expression by rat alveolar macrophages, fibroblasts, and epithelial cells and in rat lung after mineral dust exposure. Am Rev Respir Cell Mol Biol. 1993;8:311–8. doi: 10.1165/ajrcmb/8.3.311. [DOI] [PubMed] [Google Scholar]
- 23.Tsai WC, Hershenson MB, Zhou Y, Sajjan U. Azithromycin increases survival and reduces lung inflammation in cystic fibrosis mice. Inflamm Res. 2009;58:491–501. doi: 10.1007/s00011-009-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun. 2009;77:5300–10. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Karlin A, Loike JD, Silverstein SC. Determination of the critical concentration of neutrophils required to block bacterial growth in tissues. J Exp Med. 2004;200:613–22. doi: 10.1084/jem.20040725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.