

Abstract
Background
This study had two goals (1) to evaluate changes in neuropsychological performance among cognitively normal individuals that might precede the onset of clinical symptoms, and (2) to examine the impact of Apolipoprotein E (ApoE) genotype on these changes.
Methods
Longitudinal neuropsychological, clinical assessments and consensus diagnoses were completed prospectively in 268 cognitively normal individuals. The mean duration of follow-up was 9.2 years (+/− 3.3). 208 participants remained normal and 60 developed cognitive decline, consistent with a diagnosis of MCI or dementia. Cox regression analyses were completed, for both baseline scores and rate of change in scores, in relation to time to onset of clinical symptoms. Analyses were completed both with and without ApoE-4 status included. Interactions with ApoE-4 status were also examined.
Results
Lower baseline test scores, as well as greater rate of change in test scores, were associated with time to onset of clinical symptoms (p<0.001). The mean time from baseline to onset of clinical symptoms was 6.15 (+/− 3.4) years. The presence of an ApoE-4 allele doubled the risk of progression. The rate of change in two of the test scores was significantly different in ApoE-4 carriers vs. non-carriers.
Conclusions
Cognitive performance declines prior to the onset of clinical symptoms that are a harbinger of a diagnosis of MCI. Cognitive changes in normal individuals who will subsequently decline may be observed at least 6.5 years prior to symptom onset. In addition, the risk of decline is doubled among individuals with an ApoE-4 allele.
Keywords: preclinical Alzheimer's disease, cognitive decline, Apolipoprotein E genotype, mild cognitive impairment, episodic memory, longitudinal follow-up
1. Introduction
There is substantial evidence that a subset of older individuals who are cognitively normal have Alzheimer's disease (AD) pathology in their brains, based on both autopsy findings [1-3] and amyloid imaging studies [4-6]. It has been hypothesized that such individuals are at increased risk for developing cognitive decline over time, and that at some point during this ‘preclinical’ phase of disease, cognitive changes become evident [7], even though clinical symptoms have not yet been reported by the individual or his/her collateral source. Additionally, it has been proposed that Apolipoprotein E (ApoE) genotype may alter the rate of cognitive decline during the preclinical phase of AD (see below).
A small number of studies have followed cognitively normal individuals over time and retrospectively examined their cognitive performance to determine if changes in cognition precede the clinical diagnosis of mild cognitive impairment (MCI). These studies have shown that lower baseline test scores in episodic memory predict progression from normal cognition to diagnosis of MCI or AD [8-10]. Tests assessing psychomotor speed [11] have also been associated with time to progress from normal cognition to mild impairment. A number of studies have also examined the rate of cognitive decline during the preclinical phase of AD. They have found that 3 to 4 years before a diagnosis of MCI, performance on tests of episodic memory [12-15], visuospatial processing [12, 14], executive function [13, 14], and verbal fluency [12] change at a greater rate in cognitively normal individuals who subsequently progress to MCI or AD dementia compared to those who remain normal.
A number of studies have also sought to determine whether cognitive changes evident during the preclinical phase of AD are influenced by the effects of the E-4 allele of the ApoE gene, the major genetic risk factor for late onset AD (see [16] for a meta-analysis). Cross-sectional studies provide evidence for lower cognitive test scores among ApoE-4 carriers vs. non-carriers. For example, cross-sectional studies of cognitively normal middle-aged and older adults have reported lower performance among ApoE-4 carriers compared to non-carriers on tests of episodic memory [17], verbal fluency [18], and attention [19]. However, cross-sectional cognitive differences as a function of ApoE genotype are not always evident, particularly in studies that largely focus on middle-aged (45 - 65 years) individuals [20-23].
Rate of change in cognitive test scores in relation to ApoE genotype has also been examined. Short-term longitudinal studies suggest that episodic memory declines more rapidly among cognitively normal older individuals who are ApoE-4 carriers vs. non-carriers [20, 24-27], while performance on other cognitive tests shows smaller or no differences in the rate of decline in relation to ApoE-4 genotype [20, 24-27]. However, the follow-up period in these studies has been relatively short, and clinical diagnostic outcomes were not available.
The current study, due, in part, to its large sample size and unusually long follow-up period, can address several issues that remain unresolved by these studies. First, few studies have examined differences in cognitive performance at both ‘baseline’ (when subjects were first enrolled) as well as in rate of change over time. Second, the outcome has, in all instances, been the diagnosis of MCI (or its equivalent). Since onset of clinical symptoms typically precedes the diagnosis of MCI, it is unclear if cognitive changes are evident even earlier than what has been previously reported. Third, the impact of ApoE genotype on cognitive changes during preclinical AD should be elucidated. There appear to be at least two alternative explanations for prior findings related to ApoE-4 status: (1) it is possible that the lower cognitive scores among ApoE-4 carriers reflect the fact that a greater proportion of ApoE-4 carriers than non-carriers are in the preclinical phase of AD, or (2) cognitive changes during the preclinical phase of AD progress more rapidly in ApoE-4 carriers than non-carriers. If the former were true, one would expect that the degree of cognitive change preceding the emergence of the clinical symptoms of AD would be similar for ApoE-4 carriers and non-carriers. Whereas, if the latter were the case, one would expect to see a greater rate of decline on test scores among ApoE-4 carriers in the preclinical phase of AD vs. non-carriers. To determine which of these two alternatives is correct, it is necessary to examine prospective longitudinal cognitive trajectories in cognitively normal adults who later develop clinical symptoms of AD. This was the goal of the present study.
2. Subjects and Methods
2.1 Study Design
This study was designed to recruit and follow a cohort of cognitively normal individuals who were primarily in middle age. By design, approximately three quarters of the participants had a first degree relative with dementia of the Alzheimer type. The overarching goal of the study was to identify variables among cognitively normal individuals that could predict the subsequent development of mild to moderate symptoms of AD. Toward that end, subjects were administered a comprehensive neuropsychological battery annually. Magnetic resonance imaging (MRI) scans, cerebrospinal fluid (CSF), and blood specimens were obtained every two years. ApoE genotype was established for each participant after enrollment. The original study was initiated at the NIH in 1995, and was stopped in 2005 for administrative reasons. In 2008, the National Institute on Aging (NIA) and the National Institute of Mental Health (NIMH) decided to re-establish the study. In 2009, a research team at the Johns Hopkins School of Medicine was funded to re-establish the cohort, continue the annual clinical and cognitive assessments, collect blood, and evaluate the previously acquired MRI scans, CSF and blood specimens. All participants who agreed to continued follow-up signed consent forms approved by the Johns Hopkins Institutional Review Board. To our knowledge, this is the only study in participants who were primarily middle aged and cognitively normal at entry, with this set of measures, and with such a long duration of follow-up. The approximate timeline of the study and types of measurements collected each year are shown in Figure 1.
Figure 1.
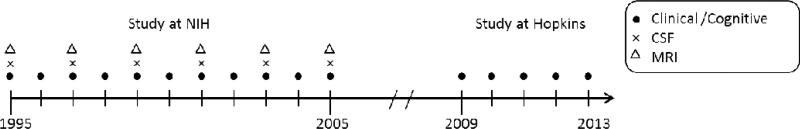
Approximate timeline of BIOCARD study indicating the types of measurements obtained each year when the study was at the NIH and since the study has been at Johns Hopkins.
2.2 Selection of Participants
A total of 354 individuals were initially enrolled in the study. Recruitment was conducted by the staff of the Geriatric Psychiatry Branch (GPB) of the intramural program of the NIMH, beginning in 1995 and ending in 2005. Subjects were recruited via printed advertisements, articles in local or national media, informational lectures, or word-of-mouth.
At enrollment, subjects in the study were admitted to the Clinical Center at the National Institutes of Health (NIH) for 3 days, after providing written informed consent. They received a detailed physical, neurological and psychiatric examination, an electrocardiogram, and standard laboratory studies (e.g., complete blood count, vitamin B12, thyroid function, etc). Mood was assessed with the Hamilton Depression Scale, the Beck Depression Inventory and the Spielberger Anxiety Scale. During the 3-day visit, a neuropsychological battery was administered, a magnetic resonance imaging (MRI) scan was obtained, blood was collected for genetic analysis, and a lumbar puncture was performed.
The GPB staff at the NIH reviewed the results of the clinical and cognitive assessments and excluded participants who were judged to be cognitively impaired, as determined by the cognitive testing or by evidence of clinical symptoms based on reports by collateral sources. Subjects were also excluded who had a history of significant medical problems such as severe cardiovascular disease (e.g., atrial fibrillation), chronic psychiatric disorders (e.g., schizophrenia, alcohol or drug abuse), chronic neurologic disorders (e.g., epilepsy, multiple sclerosis) or severe cerebrovascular disease (based on the MRI scan). Five subjects did not meet the entry criteria and were excluded at baseline, leaving a total 349 participants, who were followed over time. All but 5 of the participants were native English speakers.
2.3 Neuropsychological Assessment
All of the neuropsychological tests included in the battery administered by the Johns Hopkins research team were also administered when participants were examined at the NIH (with one exception, as described below). The battery covered a broad range of cognitive domains, including: memory, executive function, language, visuospatial ability, attention, speed of processing and psychomotor speed. The test battery was as follows: the Logical Memory (raw score and percent Retention) and Paired Associates Subtests of the Wechsler Memory Scale - Revised (WMS-R) [28], the California Verbal Learning Test (CVLT) [29], the Rey-Osterreith Complex Figure (copy and recall) [30], the Trail Making Test (TMT), Parts A and B [31], the Boston Naming Test (30-item version) (BNT) [32], Letter and Category Fluency [33], the Block Design (BD) subtest of the Wechsler Adult Intelligence Scale – Revised (WAIS-R) [34], Digit Span Forward and Backward from the WMS-R [28], the Digit Symbol Test from the WAIS-R [34], the Mini-Mental State Examination (MMSE) [35] and the Lafayette Grooved Pegboard test [36]. The Grooved Pegboard was the only test included in the battery at Johns Hopkins that had not been given previously. Additional neuropsychological tests were given at the NIH, which were not continued, primarily due to the time limitations of the study visit. These included: the complete Wechsler Adult Intelligence Scale- Revised, the complete Wechsler Memory Scale - Revised, a version of the Buschke Cued Selective Reminding test developed by the GPB staff, the Stroop Test, the Delis-Kaplan Executive Function System, and Clock drawing.
2.4 Clinical Assessment
Each of the participants also had an annual clinical examination at the NIH. Since the study has been conducted at Johns Hopkins, the annual examination has included the following: a physical and neurological examination, record of medication use, behavioral and mood assessments [37, 38], family history of dementia, history of symptom onset, and a Clinical Dementia Rating (CDR), based on a semi-structured interview [39, 40]. All measures are administered and coded by the evaluating clinician. The clinical assessments given at the NIH covered similar domains (as noted above).
2.5 Consensus Diagnoses
Each subject included in these analyses received a consensus diagnosis by the staff of the BIOCARD Clinical Core at Johns Hopkins. This research team included: neurologists, neuropsychologists, research nurses and research assistants. For the participants with evidence of clinical or cognitive dysfunction (i.e., individuals with a CDR score > 0 and/or evidence of decline on cognitive testing), a clinical summary was prepared that included information about demographics, family history of dementia, work history and past history of medical, psychiatric and neurologic disease, record of medication use and results from the neurologic and psychiatric evaluation. The reports of clinical symptoms from the subject and collateral sources were summarized, based on a version of the CDR that incorporates questions targeting the types of problems encountered by very mildly impaired individuals [41]. The results of the neuropsychological testing were also reviewed. The diagnostic process for each participant was handled in a comparable manner: (1) clinical data were examined pertaining to the medical, neurologic and psychiatric status of the subject, (2) reports of changes in cognition by the subject and by collateral sources were examined, and (3) decline in cognitive performance was established. All cognitive test scores were available during the consensus diagnosis. These data were used to: (1) determine whether the subject had become cognitively impaired, and (2) determine the likely etiology of such impairment. The age at which the clinical symptoms began was based primarily on the reports of clinical symptoms from the subject and from collateral sources reported during the semi-structured CDR interview. The version of the CDR used in the present study was specifically adapted to include questions about the types of problems that very mildly impaired patients experience (as noted above). To the extent possible, the clinical decision about symptom onset did not use the cognitive test scores. As such, our main outcome variable, the estimated age of onset of clinical symptoms, was largely independent of the cognitive test scores (i.e., our predictor variables). These diagnostic procedures are comparable to those implemented in the Alzheimer's Disease Centers program, supported by the NIA. It is acknowledged that, as this process is dependent on the clinical and cognitive data available at any one point in time, some subjects who are diagnosed as having MCI may subsequently be diagnosed as normal or as ‘Impaired not MCI’. This change in diagnosis occurred in 4 of the subjects whose data are presented here.
Diagnoses, cognitive testing, and clinical examinations were performed on an annual basis (see Figure 1) both at the NIH and since the study has been at Johns Hopkins. The age of symptom onset was established for the first visit at which the subject was deemed to be impaired and was reconfirmed on subsequent visits; thus there is a single age of symptom onset for each subject with a diagnosis of MCI or dementia.
2.6 APOE genotyping and coding
APOE genotype was established in all but one of the study participants (n = 348). Genotypes were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics, Worcester, MA). ApoE-4 carrier status was coded by creating an indicator variable, with ApoE-4 carriers coded as 1, if they had at least one ε4 allele, and non-carriers coded as 0.
2.7 Statistical Methods
The primary statistical analyses were designed to determine if performance on any of the cognitive tests administered when the subjects were first enrolled (i.e., at ‘baseline’) were related to time to onset of clinical symptoms. Exploratory analyses were first conducted based on data for two groups: (1) subjects who were cognitively normal at their last visit (n=208), and (2) subjects who received a diagnosis of MCI [42], or dementia of the Alzheimer type [43] (n=60) at their last visit (including subjects who are now deceased). A set of Cox regression analyses was then performed, using time to onset of clinical symptoms as the outcome variable. These analyses used baseline test scores and time-dependent-rate-of-change in scores as covariates; the censoring time was defined as the last date of diagnosis. These models tested whether each of the baseline neuropsychological test scores (adjusted by baseline age and education) were related to time to onset of cognitive impairment, and whether there was a differential rate of change over time in the test scores prior to the onset of clinical symptoms. A second set of analyses was completed, with ApoE-4 status included as a covariate. The interaction between ApoE-4 status and both the baseline test score and the rate of change in the test score was also examined. Note that analyses comparing those with a family history of dementia to those without will require longer follow-up, as only one-quarter of the cohort has no family history.
In Cox regression analysis (in which time-to-event’ is the outcome of interest), numerous types of ‘failure times’ can be selected as the outcome variable. The primary requirement is that the outcome measure must properly characterize the progression of the disease in question [44, 45]. In this study, we decided to use the age of onset of clinical symptoms as the 'failure time' of interest, specifically in those subjects who ultimately received a consensus diagnosis of MCI or AD dementia. The estimated age at which the clinical symptoms began was determined during the consensus review process, as described above. We chose to use the age of onset of symptoms as the ‘failure time’ instead of the date of diagnosis, specifically because of the 4-year gap in the study, during which the subjects were not examined. Using estimated age of onset of symptoms allowed us to generate a ‘failure time’ measure that was obtained in an identical manner for each subject, including those whose onset of symptoms occurred during the 4-year gap. This would not have been possible if we had used date of diagnosis, since no subjects could have received a consensus diagnosis during the 4-year gap. Additionally, this approach allowed us to exclude subjects whose symptoms were estimated to have begun at, or before, baseline (n=13).
In the Cox regression models, the rate of change was calculated as the slope of the individual changes in test score over time, which was then converted to a z-score, i.e., a ‘normed’ slope. The following procedure was used: (1) the cognitive test score at each follow-up time (time t) minus the cognitive test score at baseline was calculated, and divided by the difference in the time between these two measurements [(measurement at follow-up time t) -(measurement at baseline 0)] / t. (2) each of these individual slope values were then centered and standardized across subjects; thus, the rate of change had a mean of zero and a variance of 1. The standardized slope values were then used as time-dependent covariates in the Cox regression models, with each subject contributing a standardized slope value for each follow-up assessment that was available (e.g., 0-1, 0-2, 0-3, 0-4, etc). These measurements were not smoothed, but were instead treated as a ‘step function’, where the rate of change was not varied between the individual follow-up intervals. The baseline test scores were included as covariates in all rate-of change Cox models.
A multivariate model was also completed to determine whether a combination of baseline cognitive measures could be identified that significantly predicted time to progression from normal cognition to onset of clinical symptoms. Since individuals in the analyzed dataset were required to be symptom-free at baseline, we used statistical techniques to adjust for left-truncation in the data [46]. For these analyses, tests that were highly correlated with one another were excluded [e.g., of the three scores from the Logical Memory subtest (Immediate recall, Delayed recall and Retention score) only the Logical Memory Retention score was included in the model]. A least absolute shrinkage and selection operator (LASSO) method was also used to exclude variables, in advance, that did not contribute significantly to the model [47, 48].
Additionally, we calculated hazard ratios for each of the significant variables in the univariate baseline models and in the models examining rate of change over time. These analyses were completed with and without the inclusion of ApoE-4 status. Prior to these analyses, the test scores were converted to z scores (i.e., scores with a mean of 0 and a standard deviation of 1, averaged over the scores for the normal subjects) so that it would be possible to compare the hazard ratios for each test to one another along the same metric. The hazard ratio indicates the change in relative risk per one unit change in the predictor. For example, if the hazard ratio for the Paired Associates Immediate Recall is 0.53, the hazard of clinical symptom onset is reduced by a factor of 0.53 (i.e., by 47%) for each standard deviation increase in this test score. Likewise, a hazard ratio of 1.98 indicates that the hazard of clinical symptom onset is increased by a factor of 1.98 (i.e., 98%) for each standard deviation increase in the measure. All data analyses presented here used R, version 2.14.1.
3. Results
3.1 Subject Characteristics
The cognitive data presented here pertain to 268 of the 349 participants (mean duration of follow-up = 9.2 years, SD= 3.3). Their demographic characteristics are shown in Table 1, as are the characteristics of the cohort as a whole (n=349). The reasons that some subjects were excluded from the analyses were as follows: (1) The onset of symptoms was estimated to have occurred at or prior to baseline (N=13); (2) No follow-up data were available (N=8); (3) Subjects have withdrawn from the study (N=10) or are still considering whether to re-enroll (N=21); and (4) Subjects received a diagnosis of ‘Impaired-not-MCI’ (N=29). This diagnostic category includes individuals with no declines in cognitive testing but concerns of cognitive decline by the subject or informant, individuals with no concerns of cognitive decline by the subject or informant but evidence of decline on cognitive testing, and those with declines in cognition thought to be the result of non-AD pathology. Of note, when subjects with a diagnosis of ‘Impaired-not-MCI’ were included among the normal subjects comparable results were obtained.
Table 1.
Participant Characteristics at Baseline in Relation to Cohort as a Whole
| Variable | Cohort as a whole (N=349) | Subjects in analyses (N=268) |
|---|---|---|
| Age, mean years (SD) | 57.3 (10.4) | 56.9 (10.3) |
| Gender, females (%) | 57.6% | 61.6% |
| Education, mean years (SD) | 17.0 (2.4) | 17.1 (2.3) |
| Ethnicity, Caucasians (%) | 97.1% | 97.0% |
| ApoE-4 carriers (%) | 33.6% | 36.0% |
| MMSE, mean score (SD) | 29.5 (0.9) | 29.6 (0.8) |
| NART, mean score (SD) | 119.6 (7.9) | 120.4 (7.6) |
Abbreviations: ApoE-4, apolipoprotein E-4; MMSE, Mini-Mental State Exam; NART, National Adult Reading Test
Table 2 shows the characteristics at baseline of the subjects who remained normal at their last visit (n=208) vs. those who subsequently received a diagnosis of MCI (n=48) or dementia (n=12) (total n=60) (the primary differences between the two groups at baseline related to age). The percentage of subjects who were ApoE-4 positive was similar in the two groups (33.6% vs. 36.0%). The reasons for exclusion of specific groups of subjects in the analyses are summarized in Table 2. The data presented here exclude subjects with a classification of ‘Impaired Not MCI’ (N=26), but results were comparable when this group of individuals was included among the normal subjects (data not shown). The findings were also comparable when the one participant who became impaired, and also had a dominant mutation for AD, was excluded.
Table 2.
Participant Characteristics at Baseline
| Variable | Remained Normal (N=208) | Progressed to MCI or Dementia (N=60) |
|---|---|---|
| Age, mean years (SD) | 55.4 (9.6) | 62.4 (10.9)** |
| Gender, females (%) | 63.0 % | 56.7% |
| Education, mean years (SD) | 17.3 (2.3) | 16.6 (2.3) |
| Ethnicity, Caucasians (%) | 98.6% | 91.7%* |
| ApoE-4 carriers (%) | 33.2% | 45.8% |
| MMSE, mean score (SD) | 29.6 (0.7) | 29.4 (1.0) |
| NART, mean score (SD) | 121.5 (6.5) | 116.3 (10.0)* |
Abbreviations: ApoE-4, apolipoprotein E-4; MMSE, Mini-Mental State Exam; NART, National Adult Reading Test
p = .02
p = .001
3.2 Cox Regression Models of Test Scores at Baseline
A set of 17 variables from the cognitive battery were selected for this analysis. Selection of the variables was based on exploratory plots of the change pattern in each of the cognitive measures over time. Table 3 presents the means and standard deviations of these variables at baseline. The mean time from baseline to onset of clinical symptoms was 6.15 (+/− 3.4) years. Univariate Cox regression models were completed for each of the 17 variables at baseline (adjusted for age and education). For 9 of the 17 measures examined, there was a statistically significant association between the baseline measure and age-of-onset of impairment at the p < .001 level. The hazard ratios for these 9 variables and associated p-values are shown in Table 4 (Baseline Test Scores). For each of these cognitive variables, after accounting for baseline age and education, each standard deviation increase in the test score was associated with approximately a 40 – 60% reduction in the risk of symptom onset (all HR <=0.63).
Table 3.
Mean and Standard Deviation of Cognitive Test Scores at Baseline for Subjects who Remained Normal vs. Subjects who Developed Clinical Symptoms and were Diagnosed with MCI or AD Dementia
| Cognitive Test | Remained Normal | Progressed to MCI or AD Dementia |
|---|---|---|
| Logical Memory Immediate | 15.28 (2.78) | 13.48 (2.99) |
| Logical Memory Delayed | 13.43 (3.26) | 11.08 (3.70) |
| Logical Memory % Retention | 87.36 (11.33) | 80.63 (20.06) |
| Paired Associate Immediate | 21.03 (2.79) | 18.88 (3.12) |
| Paired Associate Delayed | 7.69 (0.63) | 7.31 (0.99) |
| CVLT Total Trials 1-5 | 53.93 (8.90) | 48.79 (10.43) |
| CVLT Short Delay Free Recall | 11.53 (2.92) | 10.68 (3.30) |
| CVLT Long Delay Free Recall | 12.19 (2.78) | 10.74 (3.30) |
| CVLT Short Delay % Retention | 86. 97 (18.18) | 85.19 (18.06) |
| Rey Figure Copy | 33.89 (2.22) | 32.83 (2.89) |
| Rey Figure Recall | 19.22 (6.10) | 14.66 (6.54) |
| Block Design Subtest | 34.48 (8.18) | 27.47 (8.72) |
| Boston Naming % Correct | 97.02 (4.51) | 93.67 (6.63) |
| Category Fluency Animals | 24.11 (4.86) | 21.25 (4.30) |
| Digit Symbol | 55.66 (11.33) | 45.78 (9.85) |
| Trail Making, Part A Time in sec | 30.22 (10.74) | 46.78 (28.45) |
| Trail Making, Part B Time in sec | 65.41 (19.92) | 104.00 (52.23) |
Abbreviations: CVLT, California Verbal Learning Test
Table 4.
Hazard Ratios for Univariate Analyses of Baseline and Rate of Change in Cognitive Test Scores in Relation to Onset of Clinical Symptoms
| Baseline Test Scores | Time-Dependent Rate of Change in Scores | |||
|---|---|---|---|---|
| Cognitive Test | Hazard Ratio* (95% CI) | p-value | Hazard Ratio* (95% CI) | p-value |
| Logical Memory Immediate | 0.59 (0.44-0.80) | 0.0005 | 0.58 (0.36-0.93) | 0.024 |
| Logical Memory Delayed | 0.48 (0.36-0.65) | 0.0001 | 0.54 (0.34-0.86) | 0.009 |
| Logical Memory % Retention | 0.56 0.44-0.71 | 0.0001 | 0.54 (0.38-0.76) | 0.001 |
| Paired Associate Immediate | 0.53 (0.41-0.70) | 0.0001 | 0.37 (0.24-0.57) | 0.001 |
| Paired Associate Delayed | 0.63 (0.49-0.82) | 0.0004 | 0.63 (0.43-0.92) | 0.016 |
| Rey Figure Recall | 0.62 (0.46-0.82) | 0.0008 | 0.49 (0.38-0.63) | 0.001 |
| Block Design Subtest | 0.53 (0.38-0.74) | 0.0001 | 0.40 (0.19-0.83) | 0.014 |
| Boston Naming Test | 0.57 (0.43-0.74) | 0.0001 | 0.69 (0.55-0.85) | 0.001 |
| Digit Symbol Test | 0.41 (0.29-0.58) | 0.0001 | 0.47 (0.35-0.64) | 0.001 |
The hazard ratios are for standardized scores (adjusted for age at baseline and education), and are presented per one standard deviation increase in each cognitive test.
Abbreviations: CI, confidence interval
A second set of Cox regression models were performed, additionally including ApoE-4 status as a predictor, as well as the interaction between ApoE-4 status and the baseline cognitive score. The interaction term was not significant for any of these 9 measures (all p > 0.1). but ApoE-4 status was significant in all models (all HR >=1.98, all p < 0.01, see Table 5, Baseline Test Scores). These results suggest that baseline cognitive performance and ApoE-4 status are independently associated with the time to onset of clinical symptoms, with ApoE-4 status increasing the risk of progressing from normal cognition to symptom onset by about 100% (i.e., a doubling of risk).
Table 5.
Hazard Ratios for Univariate Analyses of Baseline and Rate of Change in Cognitive Test Scores and ApoE-4 Status in Relation to Onset of Clinical Symptoms
| Baseline Test Scores | Time-Dependent Rate of Change in Scores | |||
|---|---|---|---|---|
| Cognitive Test | Hazard Ratio* (95% CI) | p-value | Hazard Ratio* (95% CI) | p-value |
| Logical Memory Immediate | 0.61 (0.47-0.81) | 0.001 | 0.56 (0.36-0.89) | 0.014 |
| ApoE-4 | 2.35 (1.32-4.17) | 0.004 | 2.25 (1.31-3.87) | 0.003 |
| Logical Memory Delayed | 0.51 (0.39-0.68) | 0.001 | 0.53 (0.33-0.83) | 0.006 |
| ApoE-4 | 2.18 (1.24-3.85) | 0.007 | 2.12 (1.23-3.64) | 0.007 |
| Logical Memory % Retention | 0.59 (0.46-0.75) | 0.001 | 0.55 (0.40-0.76) | 0.001 |
| ApoE-4 | 1.98 (1.14-3.45) | 0.015 | 1.78 (1.04-3.05) | 0.036 |
| Paired Associate Immediate | 0.55 (0.42-0.70) | 0.001 | 0.38 (0.25-0.60) | 0.001 |
| ApoE-4 | 2.39 (1.35-4.26) | 0.003 | 1.87 (1.05-3.32) | 0.033 |
| Paired Associate Delayed | 0.64 (0.52-0.79) | 0.001 | 0.64 (0.45-0.93) | 0.020 |
| ApoE-4 | 2.77 (1.56-4.91) | 0.001 | 2.31 (1.34-3.96) | 0.002 |
| Rey Figure Recall | 0.57 (0.43-0.75) | 0.001 | 0.46 (0.36-0.61) | 0.001 |
| ApoE-4 | 2.75 (1.56-4.84) | 0.001 | 2.74 (1.65-4.55) | 0.001 |
| Block Design Subtest | 0.54 (0.40-0.74) | 0.001 | 0.40 (0.19-0.82) | 0.013 |
| ApoE-4 | 2.35 (1.35-4.07) | 0.002 | 2.16 (1.28-3.65) | 0.004 |
| Boston Naming Test | 0.63 (0.51-0.78) | 0.001 | 0.68 (0.55-0.85) | 0.001 |
| ApoE-4 | 2.00 (1.16-3.43) | 0.013 | 1.80 (1.06-3.06) | 0.031 |
| Digit Symbol Test | 0.44 (0.31-0.61) | 0.001 | 0.47 (0.34-0.67) | 0.001 |
| ApoE-4 | 2.01 (1.17-3.47) | 0.012 | 1.94 (1.18-3.20) | 0.009 |
The first line for each cognitive test is the hazard ratio for the standardized scores of each cognitive test, adjusted by - baseline age, education and ApoE-4 status (i.e., ApoE-4 positive or ApoE-4 negative). The second line for each cognitive test is the hazard ratio, comparing those who were ApoE-4 positive to those who were ApoE-4 negative, adjusted by - the standardized scores of the cognitive test, baseline age and education.
Abbreviations: CI, confidence interval
3.3 Cox Regression Models of Rate of Change in Test Scores
For each of these 9 measures, univariate Cox regression models were also completed, with baseline test scores and time-dependent-rate-of-change in scores as covariates (adjusted for baseline age and education). As shown in Table 4, the time-dependent rate of change in scores was significant in all models (all HR <=0.069, all p < 0.025), indicating that there was a differential rate of change over time for participants who remained cognitively normal compared to those who progressed to MCI or AD for all 9 of these test scores, prior to the onset of clinical symptoms. For each one standard deviation increase in the rate of change over time (i.e., decrease in test score), the risk of progressing from normal cognition to symptom onset increased by 30 – 60%. ApoE-4 status was significant, when included in the models for each of these 9 measures (p < 0.03).
When ApoE-4 status and the interaction between ApoE-4 status and the rate of change were added to these models, ApoE-4 status was again significant in all models (all HR >1.78, all p < 0.04) and the interaction term was significant for 2 of the 9 measures [i.e., the Boston Naming Test (p=0.003) and the Logical Memory Retention Score (p ≤ 0.05)]. The hazard ratios for ApoE-4 and the cognitive variables are shown in Table 5 (Time-Dependent Rate of Change in Scores). These findings suggest that with the exception of two cognitive measures, the association between the rate of change in cognitive performance and the time to onset of clinical symptoms is comparable for ApoE-4 carriers and non-carriers. By comparison, for the Boston Naming Test and the Logical Memory Retention Score, there was evidence that ApoE-4 carriers declined at a greater rate relative to non-carriers prior to the onset of clinical symptoms.
3.4 Cox Regression Multivariate Model
Lastly, we completed a Cox multivariate model with baseline test scores as covariates (adjusted for age and education) in which the 9 variables mentioned above were included. Four of the 9 variables in the multivariate model were statistically significant (as well as age), as was the overall model (p < 0.0001). The significant cognitive tests were: the Digit Symbol Test (p = 0.0001), Paired Associates immediate recall (p= 0.008), Logical Memory Retention (p = 0.003), and the Boston Naming Test (p = .001). When ApoE-4 status was added to this model, the same 4 cognitive variables remained significant and ApoE-4 status was also significant (p < 0.005). The hazard ratios and p-values for significant variables in this model are shown in Table 6.
Table 6.
Hazard Ratios for Significant Variables in Multivariate Analysis of Baseline Test Scores in Relation to Onset of Clinical Symptoms
| Baseline Test Scores | ||
|---|---|---|
| Cognitive Test | Hazard Ratio* (95% CI) | p-value |
| Logical Memory % Retention | 0.75 (0.59-0.95) | 0.017 |
| Paired Associate Immediate | 0.67 (0.51-0.88) | 0.005 |
| Boston Naming Test | 0.70 (0.54-0.93) | 0.011 |
| Digit Symbol Test | 0.52 (0.37-0.72) | 0.001 |
| ApoE-4 | 2.43 (1.32-4.49) | 0.005 |
3.5 Cox Survival Curves
Survival curves from the Cox regression models were plotted to facilitate visualization of the results. These curves were derived from a Cox regression model for each variable, by categorizing the baseline variable into two groups (normal at follow-up vs. MCI or Dementia at follow-up), using the median score at baseline as a cut-off. As an example, Figure 2 presents the survival plot for the Digit Symbol test, showing the impact of ApoE-4 status.
Figure 2.

The survival plot for the Digit Symbol Test , based on the Cox regression model. This is one of the measures that was associated with time to onset of clinical symptoms (both with respect to the baseline score and the rate of change over time). The solid line represents scores below the median at baseline, and the dashed line represents scores above the median at baseline. The lines in black represent the scores of those who were ApoE-4 negative and the lines in red represent the scores of those who were ApoE-4 positive. The y axis represents the proportion of subjects who remained without symptoms. The plot starts at age 50 and is truncated at 75 years of age, since few participants remained unimpaired after this age, making the estimates unreliable after that age. Note that this survival curve is not age adjusted.
4. Discussion
In this study, longitudinal cognitive evaluations were conducted in a large group of normal individuals, some of whom subsequently were diagnosed with MCI or dementia (i.e. 17%). Approximately one-third of the subjects in both groups were carriers of the ApoE-4 allele, likely due to the over-recruitment of those with a family history of AD. Our data show that lower cognitive test scores at baseline and a greater rate of decline in test scores over time are significantly associated with time to onset of clinical symptoms, which were a harbinger of a diagnosis of MCI. Increased risk of progressing from normal cognition to onset of clinical symptoms was associated with lower baseline scores on several tests of episodic memory (i.e., Paired Associates immediate recall and Logical Memory Retention) as well as multifactorial tests of processing speed, where better memory ability leads to improved performance (e.g., Digit Symbol Test). Lower performance on tests in other cognitive domains was also associated with an increased risk of developing clinical symptoms among cognitively normal individuals, most notably performance on the Boston Naming Test (which was significant in both the univariate and multivariate models). Importantly, when both cognitive performance and ApoE-4 genotype were taken into account, the relative risk of progression from normal cognition to mild impairment was doubled (i.e., increased by 100%) for individuals with one or more e4 alleles. The rate of cognitive decline was higher in ApoE-4 carriers than in non-carriers for two of the nine tests.
These findings are notable for several reasons. First, given that lower baseline scores (obtained on average 6.5 years prior to symptom onset) were associated with time to symptom onset suggests that about 6.5 years prior to symptom onset, cognitive performance of individuals who progress is already lower than that of individuals who remain normal over the same time period. Since the onset of clinical symptoms typically precedes the diagnosis of MCI by several years, this extends previous findings that used the date of clinical diagnosis of MCI as the outcome. Second, the average age of the cohort was 56.9 years at baseline. Since previous studies have been conducted among individuals who were over the age of 65, this also extends the age at which cognitive changes may be observed among normal individuals who will subsequently develop MCI. Third, these findings suggest that the primary reason that test scores tend to be lower in cognitively normal individuals who are ApoE-4 carriers is that they are further along the preclinical trajectory of AD than non-carriers. That is, for an individual with a particular cognitive test score (i.e., at baseline or rate of change in score, adjusted by age and education) the risk of onset of symptoms is almost doubled for an individual who is ApoE-4 positive. Two cognitive tests had a differential rate of change in ApoE-4 carriers vs. non-carriers (i.e., the Boston Naming test and Logical Memory Retention), thus there is only modest evidence to support the hypothesis that ApoE-4 carriers progress at a more rapid rate than non-carriers during the preclinical phase of AD. These results may also be of relevance to clinical drug trials, as they highlight the importance of taking into account participants’ ApoE-4 genotype when evaluating drug efficacy in relation to cognitive change [see 49 for a discussion of this issue].
These findings are consistent with recent estimates of the onset of cognitive decline prior to diagnosis among individuals with a dominant AD mutation [50]. The strength of the findings is emphasized by the fact that increased risk of progression from normal cognition to onset of clinical symptoms were observed both with regard to baseline cognitive performance, and also in relation to rate of change over time in cognitive test scores. We cannot rule out the possibility that the lower baseline performance in our participants reflects life-long lower cognitive abilities, as has been suggested by others [51]. However, the fact that the rate of change was also predictive of time to develop clinical symptoms suggests that the baseline scores likely reflect an actual decline.
Our findings complement and extend those of prior studies. A number of prior studies have demonstrated differences in rate of change in cognitive test scores 4-6 years prior to diagnosis of MCI [12-15]. Likewise, baseline test scores obtained in normal individuals up to 6 years prior to a diagnosis of MCI have been significantly associated with outcome [8-11]. A number of cross-sectional and short-term longitudinal studies have reported lower cognitive performance [17-19] and a greater rate of cognitive decline in cognitively normal ApoE-4 carriers than non-carriers [20-24], particularly for tests of episodic memory.
It is also noteworthy that the relationships observed in this study are consistent with those that have examined cognitive change among individuals in the symptomatic phase of AD. For example, comparable findings have been reported in a number of longitudinal studies of non-demented individuals that have included both cognitively normal participants and subjects with MCI. These studies have found both lower baseline test scores [52-55] and greater rate of decline in test scores [56] among individuals who subsequently developed dementia, compared to those who remained non-demented. Such cognitive differences were evident 5 to 15 years prior to the diagnosis of dementia and encompassed several cognitive domains, including episodic memory.
Likewise, cross-sectional studies and short-term longitudinal studies of non-demented individuals (i.e., including both cognitively normal individuals and those with MCI) reported lower episodic memory scores and greater declines in episodic memory over time for ApoE-4 carriers vs. non-carriers [57-63]. The results of studies comparing performance on tests other than episodic memory in non-demented ApoE-4 carriers vs. non-carriers are more variable. Some have reported differences between the groups [22, 57, 61, 64] while others have not [58-60].
The multivariate analysis showed that in addition to two tests of episodic memory, the Boston Naming Test, which assesses semantic memory, and the multifactorial Digit Symbol test, represented the best combination of predictors of progression to cognitive impairment in this study. These findings are consistent with reports of differential brain atrophy in medial temporal lobe brain regions during the preclinical phase AD, particularly in the entorhinal cortex and hippocampus, which are involved in episodic memory performance [e.g., 65]. These findings further suggest that AD pathology may extend into other brain regions during this phase of the disease, such as the anterior and middle temporal cortex (which has been associated with Boston Naming performance [66, 67]. Performance on the Digit Symbol test benefits from episodic memory as well as a number of other cognitive skills, so it is unclear whether the association between performance on this test and risk of progression reflects pathology in the medial temporal lobe or in other regions.
The study results must be interpreted in the context of its limitations. First, the subjects are well educated, primarily Caucasian, with the majority having a family history of dementia, so the results may not generalize to the U.S. population at large. Second, the results may depend on the specific neuropsychological tests used. While many of the individual tests were significant in the univariate models, only a small set remained significant in the multivariate model, suggesting that a different set of initial tests might alter the findings in the multivariate model. Additionally, it remains unclear if other aspects of cognition not covered by our cognitive battery are also associated with the progression to clinical symptom onset and show differential rates of decline for ApoE-4 carriers and non-carriers. Third, the present work is limited to ApoE genotype and it remains unclear if the effects of ApoE single nucleotide polymorphisms (SNPs) influences cognitive change during preclinical AD [69]. Lastly, it is important to acknowledge the challenges of estimating age of symptom onset. While we used a semi-standardized instrument administered to both the subject and an informant, with specific targeted questions to assess onset of symptoms, the determination was ultimately based on the judgment of skilled clinicians. Additionally, although the test scores were not used to establish age of symptom onset, they were available to the clinician, thus making it unclear whether they influenced the decision making process in some way. It should be noted, however, that we have recently published data on CSF and MRI changes in relation to symptom onset in this cohort [65, 68], suggesting that this measure is biologically meaningful and related to variables above and beyond cognitive test scores.
Taken together, these findings are consistent with the hypothesis outlined in the working group report on ‘preclinical AD’ suggesting that decline in cognitive performance is detectable during the phase of AD when pathology is developing, but clinical symptoms have not yet been reported [7], and that genetic factors might influence the rate of decline. Interestingly, the working group report also hypothesized that more difficult or targeted cognitive tests might be more useful for demonstrating cognitive decline during the ‘pre-clinical’ phase of AD than the standard cognitive measures, particularly for the purposes of identification of individuals at risk. While our analyses do not include such comparisons, they nonetheless do demonstrate that standard cognitive tests are quite sensitive to declines in cognition during pre-clinical AD, and suggest that these tests might be useful indicators of which cognitively normal individuals will go on to subsequently develop clinical symptoms over a period of years.
Additionally, the hypotheses summarized in the working group report proposed that cognitive decline would follow alterations in both amyloid and tau levels in CSF, as well as volumetric changes on MRI scans. With additional follow-up of the participants it should soon be possible to address the question of the sequence in which changes in these biomarkers occur, and how our findings might compare with proposed hypothetical models [69].
Acknowledgements
This study is supported in part by grants from the National Institutes of Health: U01-AG03365, P50- AG005146 and P41-RR015241. The BIOCARD Study consists of 7 Cores with the following members: (1) the Administrative Core (Marilyn Albert, Barbara Rodzon, Richard Power), (2) the Clinical Core (Ola Selnes, Marilyn Albert, Rebecca Gottesman, Ned Sacktor, Guy McKhann, Scott Turner, Leonie Farrington, Maura Grega, Daniel D'Agostino, Sydney Feagen, David Dolan, Hillary Dolan), (3) the Imaging Core (Michael Miller, Susumu Mori, Tilak Ratnanather, Timothy Brown, Hayan Chi, Anthony Kolasny, Kenichi Oishi, Thomas Reigel, William Schneider, Laurent Younes), (4) the Biospecimen Core (Richard O'Brien, Abhay Moghekar, Richard Meehan), (5) the Informatics Core (Roberta Scherer, Curt Meinert, David Shade, Ann Ervin, Jennifer Jones, Matt Toepfner, Lauren Parlett, April Patterson, Lisa Lassiter), the (6) Biostatistics Core (Mei-Cheng Wang, Yi Lu, Qing Cai), and (7) the Neuropathology Core (Juan Troncoso, Barbara Crain, Olga Pletnikova, Gay Rudow, Karen Fisher).
We are grateful to the members of the BIOCARD Scientific Advisory Board who provide continued oversight and guidance regarding the conduct of the study including: Drs. John Csernansky, David Holtzman, David Knopman, Walter Kukull and John McArdle, as well as Drs. Neil Buckholtz, John Hsiao, Laurie Ryan and Jovier Evans, who provide oversight on behalf of the National Institute on Aging (NIA) and the National Institute of Mental Health (NIMH), respectively. We would also like to thank the members of the BIOCARD Resource Allocation Committee who provide ongoing guidance regarding the use of the biospecimens collected as part of the study, including: Drs. Constantine Lyketsos, Carlos Pardo, Gerard Schellenberg, Leslie Shaw, Madhav Thambisetty, and John Trojanowski.
We would like to acknowledge the contributions of the Geriatric Psychiatry Branch (GPB) of the intramural program of the NIMH who initiated the study (PI: Dr. Trey Sunderland). We are particularly indebted to Dr. Karen Putnam, who has provided ongoing documentation of the GPB study procedures and the data files received from NIMH.
References
- 1.Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM. Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol. 1998;57:1168–1174. doi: 10.1097/00005072-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 3.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 4.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blacker D, Lee H, Muzikansky A, Martin EC, Tanzi R, McArdle JJ, et al. Neuropsychological measures in normal individuals that predict subsequent cognitive decline. Arch Neurol. 2007;64:862–871. doi: 10.1001/archneur.64.6.862. [DOI] [PubMed] [Google Scholar]
- 9.Smith CD, Chebrolu H, Wekstein DR, Schmitt FA, Jicha GA, Cooper G, et al. Brain structural alterations before mild cognitive impairment. Neurology. 2007;68:1268–1273. doi: 10.1212/01.wnl.0000259542.54830.34. [DOI] [PubMed] [Google Scholar]
- 10.Rubin EH, Storandt M, Miller JP, Kinscherf DA, Grant EA, Morris JC, et al. A prospective study of cognitive function and onset of dementia in cognitively healthy elders. Arch Neurol. 1998;55:395–401. doi: 10.1001/archneur.55.3.395. [DOI] [PubMed] [Google Scholar]
- 11.Galvin JE, Powlishta KK, Wilkins K, McKeel DW, Jr., Xiong C, Grant E, et al. Predictors of preclinical Alzheimer disease and dementia: a clinicopathologic study. Arch Neurol. 2005;62:758–765. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- 12.Howieson DB, Carlson NE, Moore MM, Wasserman D, Abendroth CD, Payne-Murphy J, et al. Trajectory of mild cognitive impairment onset. J Int Neuropsychol Soc. 2008;14:192–198. doi: 10.1017/S1355617708080375. [DOI] [PubMed] [Google Scholar]
- 13.De Santi S, Pirraglia E, Barr W, Babb J, Williams S, Rogers K, et al. Robust and conventional neuropsychological norms: diagnosis and prediction of age-related cognitive decline. Neuropsychology. 2008;22:469–484. doi: 10.1037/0894-4105.22.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol. 2009;66:1254–1259. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol. 2011;68:351–356. doi: 10.1001/archneurol.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 17.Levy JA, Bergeson J, Putnam K, Rosen V, Cohen R, Lalonde F, et al. Context-specific memory and apolipoprotein E (ApoE) epsilon 4: cognitive evidence from the NIMH prospective study of risk for Alzheimer's disease. J Int Neuropsychol Soc. 2004;10:362–370. doi: 10.1017/S1355617704103044. [DOI] [PubMed] [Google Scholar]
- 18.Rosen VM, Sunderland T, Levy J, Harwell A, McGee L, Hammond C, et al. Apolipoprotein E and category fluency: evidence for reduced semantic access in healthy normal controls at risk for developing Alzheimer's disease. Neuropsychologia. 2005;43:647–658. doi: 10.1016/j.neuropsychologia.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 19.Greenwood PM, Lambert C, Sunderland T, Parasuraman R. Effects of apolipoprotein E genotype on spatial attention, working memory, and their interaction in healthy, middle-aged adults: results From the National Institute of Mental Health's BIOCARD study. Neuropsychology. 2005;19:199–211. doi: 10.1037/0894-4105.19.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caselli RJ, Reiman EM, Osborne D, Hentz JG, Baxter LC, Hernandez JL, et al. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology. 2004;62:1990–1995. doi: 10.1212/01.wnl.0000129533.26544.bf. [DOI] [PubMed] [Google Scholar]
- 21.Jorm AF, Mather KA, Butterworth P, Anstey KJ, Christensen H, Easteal S. APOE genotype and cognitive functioning in a large age-stratified population sample. Neuropsychology. 2007;21:1–8. doi: 10.1037/0894-4105.21.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer's disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer's Prevention. J Geriatr Psychiatry Neurol. 2005;18:245–249. doi: 10.1177/0891988705281882. [DOI] [PubMed] [Google Scholar]
- 23.Chen JG, Edwards CL, Vidyarthi S, Pitchumoni S, Tabrizi S, Barboriak D, et al. Learning and recall in subjects at genetic risk for Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 2002;14:58–63. doi: 10.1176/jnp.14.1.58. [DOI] [PubMed] [Google Scholar]
- 24.Caselli RJ, Dueck AC, Locke DE, Hoffman-Snyder CR, Woodruff BK, Rapcsak SZ, et al. Longitudinal modeling of frontal cognition in APOE epsilon4 homozygotes, heterozygotes, and noncarriers. Neurology. 2011;76:1383–1388. doi: 10.1212/WNL.0b013e3182167147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim YY, Ellis KA, Pietrzak RH, Ames D, Darby D, Harrington K, et al. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012;79:1645–1652. doi: 10.1212/WNL.0b013e31826e9ae6. [DOI] [PubMed] [Google Scholar]
- 26.Jochemsen HM, Muller M, van der Graaf Y, Geerlings MI. APOE epsilon4 differentially influences change in memory performance depending on age. The SMART-MR study. Neurobiol Aging. 2012;33:832, e815–822. doi: 10.1016/j.neurobiolaging.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 27.Mayeux R, Small SA, Tang M, Tycko B, Stern Y. Memory performance in healthy elderly without Alzheimer's disease: effects of time and apolipoprotein-E. Neurobiol Aging. 2001;22:683–689. doi: 10.1016/s0197-4580(01)00223-8. [DOI] [PubMed] [Google Scholar]
- 28.Wechsler D. Wechsler Memory Scale - Revised Manual. Psychological Corporation; San Antonio, TX: 1987. [Google Scholar]
- 29.Delis DC, Kramer JH, Kaplan E, Ober BA. California Verbal Learning Test. The Psychological Corporation; San Antonio, TX: 1987. [Google Scholar]
- 30.Rey A. L'examen psychologique dans les cas d'encephalopathie traumatique. Archives de Psychologie. 1941;28:286–340. [Google Scholar]
- 31.Reitan RM. Validity of the trail making test as an indicator of organic brain damage. Perceptual and Motor Skills. 1958;8:271–276. [Google Scholar]
- 32.Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Lee & Febiger; Philadelphia: 1983. [Google Scholar]
- 33.Benton AL, Hamsher KS. Multilingual Aphasia Examination. University of Iowa Press; Iowa City: 1976. [Google Scholar]
- 34.Wechsler D. Wechsler Adult Intelligence Scale - Revised Manual. The Psychological Corporation; New York: 1981. [Google Scholar]
- 35.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 36.Klove H. Grooved Pegboard. Lafayette. 1963 [Google Scholar]
- 37.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–2314. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 38.Yesavage JA, Brink TL, Rose TL, Lum O, Huang V, Adey M, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17:37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 39.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 40.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 41.Daly E, Zaitchik D, Copeland M, Schmahmann J, Gunther J, Albert M. Predicting conversion to Alzheimer disease using standardized clinical information. Arch Neurol. 2000;57:675–680. doi: 10.1001/archneur.57.5.675. [DOI] [PubMed] [Google Scholar]
- 42.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 43.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalbfleisch J, Prentice R. Statistical Analysis of Failure Time Data. Wiley & Sons; Hoboken, NJ: 2002. [Google Scholar]
- 45.Hosmer D, Lemeshow S, May S. Applied Survival Analysis. Wiley & Sons; Hoboken, NJ: 2008. [Google Scholar]
- 46.Wang MC, Brookmeyer R, Jewell NP. Statistical models for prevalent cohort data. Biometrics. 1993;49:1–11. [PubMed] [Google Scholar]
- 47.Tibshirani R. Regression shrinkage and selection via the Lasso. Journal of the Royal Statistical Society Series B-Methodological. 1996;58:267–288. [Google Scholar]
- 48.Tibshirani R. The lasso method for variable selection in the cox model. Statistics in Medicine. 1997;16:385–395. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Schneider LS, Lahiri DK. The perils of Alzheimer's drug development. Curr Alzheimer Res. 2009;6:77–78. doi: 10.2174/156720509787313871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer's Disease. N Engl J Med. 2012 doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riley KP, Snowdon DA, Desrosiers MF, Markesbery WR. Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiol Aging. 2005;26:341–347. doi: 10.1016/j.neurobiolaging.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 52.Kawas CH, Corrada MM, Brookmeyer R, Morrison A, Resnick SM, Zonderman AB, et al. Visual memory predicts Alzheimer's disease more than a decade before diagnosis. Neurology. 2003;60:1089–1093. doi: 10.1212/01.wnl.0000055813.36504.bf. [DOI] [PubMed] [Google Scholar]
- 53.Elias MF, Beiser A, Wolf PA, Au R, White RF, D'Agostino RB. The preclinical phase of alzheimer disease: A 22-year prospective study of the Framingham Cohort. Arch Neurol. 2000;57:808–813. doi: 10.1001/archneur.57.6.808. [DOI] [PubMed] [Google Scholar]
- 54.Saxton J, Lopez OL, Ratcliff G, Dulberg C, Fried LP, Carlson MC, et al. Preclinical Alzheimer disease: neuropsychological test performance 1.5 to 8 years prior to onset. Neurology. 2004;63:2341–2347. doi: 10.1212/01.wnl.0000147470.58328.50. [DOI] [PubMed] [Google Scholar]
- 55.Tierney MC, Yao C, Kiss A, McDowell I. Neuropsychological tests accurately predict incident Alzheimer disease after 5 and 10 years. Neurology. 2005;64:1853–1859. doi: 10.1212/01.WNL.0000163773.21794.0B. [DOI] [PubMed] [Google Scholar]
- 56.Amieva H, Le Goff M, Millet X, Orgogozo JM, Peres K, Barberger-Gateau P, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64:492–498. doi: 10.1002/ana.21509. [DOI] [PubMed] [Google Scholar]
- 57.Chey J, Kim JW, Cho HY. Effects of apolipoprotein E phenotypes on the neuropsychological functions of community-dwelling elderly individuals without dementia. Neurosci Lett. 2000;289:230–234. doi: 10.1016/s0304-3940(00)01288-x. [DOI] [PubMed] [Google Scholar]
- 58.Flory JD, Manuck SB, Ferrell RE, Ryan CM, Muldoon MF. Memory performance and the apolipoprotein E polymorphism in a community sample of middle-aged adults. Am J Med Genet. 2000;96:707–711. doi: 10.1002/1096-8628(20001204)96:6<707::aid-ajmg1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 59.Nilsson LG, Adolfsson R, Backman L, Cruts M, Nyberg L, Small BJ, et al. The influence of APOE status on episodic and semantic memory: data from a population-based study. Neuropsychology. 2006;20:645–657. doi: 10.1037/0894-4105.20.6.645. [DOI] [PubMed] [Google Scholar]
- 60.Reed T, Carmelli D, Swan GE, Breitner JC, Welsh KA, Jarvik GP, et al. Lower cognitive performance in normal older adult male twins carrying the apolipoprotein E epsilon 4 allele. Arch Neurol. 1994;51:1189–1192. doi: 10.1001/archneur.1994.00540240033012. [DOI] [PubMed] [Google Scholar]
- 61.Zehnder AE, Blasi S, Berres M, Monsch AU, Stahelin HB, Spiegel R. Impact of APOE status on cognitive maintenance in healthy elderly persons. Int J Geriatr Psychiatry. 2009;24:132–141. doi: 10.1002/gps.2080. [DOI] [PubMed] [Google Scholar]
- 62.Schiepers OJ, Harris SE, Gow AJ, Pattie A, Brett CE, Starr JM, et al. APOE E4 status predicts age-related cognitive decline in the ninth decade: longitudinal follow-up of the Lothian Birth Cohort 1921. Mol Psychiatry. 2012;17:315–324. doi: 10.1038/mp.2010.137. [DOI] [PubMed] [Google Scholar]
- 63.Caselli RJ, Dueck AC, Osborne D, Sabbagh MN, Connor DJ, Ahern GL, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med. 2009;361:255–263. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wetter SR, Delis DC, Houston WS, Jacobson MW, Lansing A, Cobell K, et al. Deficits in inhibition and flexibility are associated with the APOE-E4 allele in nondemented older adults. J Clin Exp Neuropsychol. 2005;27:943–952. doi: 10.1080/13803390490919001. [DOI] [PubMed] [Google Scholar]
- 65.Miller MI, Younes L, Ratnanather JT, Brown T, Trinh H, Postell E, et al. The diffeomorphometry of temporal lobe structures in preclinical Alzheimer's disease. Neuroimage Clin. 2013;3:352–360. doi: 10.1016/j.nicl.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baldo JV, Arevalo A, Patterson JP, Dronkers NF. Grey and white matter correlates of picture naming: evidence from a voxel-based lesion analysis of the Boston Naming Test. Cortex; a journal devoted to the study of the nervous system and behavior. 2013;49:658–667. doi: 10.1016/j.cortex.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Sachdev PS, Wen W, Kochan NA, Crawford JD, Brodaty H, et al. Grey matter correlates of three language tests in non-demented older adults. PLoS One. 2013;8:e80215. doi: 10.1371/journal.pone.0080215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moghekar A, Li S, Lu Y, Li M, Wang MC, Albert M, et al. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81:1753–1758. doi: 10.1212/01.wnl.0000435558.98447.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jack CR, Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maloney B, Ge YW, Petersen RC, Hardy J, Rogers JT, Perez-Tur J, Lahiri DK. Functional characterization of three single-nucleotide polymorphisms present in the human APOE promoter sequence: Differential effects in neuronal cells and on DNA-protein interations. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:185–201. doi: 10.1002/ajmg.b.30973. [DOI] [PMC free article] [PubMed] [Google Scholar]