

Abstract
Purpose
In this study, we investigated whether the phenotypic difference observed between two lattice corneal dystrophy type 1 (LCD type 1) cases caused by either a single A546D substitution or a A546D/P551Q double substitution in TGFBIp, can be ascribed to (I) a difference in the proteomes of corneal amyloid deposits, (II) altered proteolysis of TGFBIp or (III) structural changes of TGFBIp introduced by the P551Q amino acid substitution.
Experimental design
Amyloid deposits were isolated from the corneas of two siblings with LCD type 1 resulting from A546D/P551Q mutations in TGFBI using laser capture microdissection and a subsequently analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Proteolytic processing of TGFBIp was addressed by counting peptide spectra. Lastly, to study the possible effect of the P551Q substitution, recombinant FAS1-4 domain variants were subjected to in vitro stability assays.
Results
The amyloid proteomes and TGFBIp processing of the two A546D/P551Q LCD type 1 cases were similar to each other as well as to the A546D amyloid proteome previously reported by us. The stability assays revealed a minor destabilization of the FAS1-4 domain upon the addition of the P551Q mutation, moreover, it resulted in different accessibility to tryptic cleavage sites between the A546D and A546D/P551Q mutant FAS1-4 domain variants.
Conclusion
The difference in A546D and A546D/P551Q LCD type 1 phenotypes cannot be ascribed to altered corneal amyloid composition or altered in vivo proteolytic processing of TGFBIp. Instead, a small difference in thermodynamic stability introduced by the P551Q mutation most likely causes structural changes of TGFBIp.
Keywords: Amyloid deposits, cornea, laser capture microdissection, lattice corneal dystrophy, paraffin embedded tissue
1. Introduction
The transforming growth factor-beta induced protein (TGFBIp) (Uniprot: Q15582) is a 72 kDa extracellular matrix protein highly abundant in the human cornea [2]. TGFBIp consists of four consecutive, and highly conserved fasciclin 1 (FAS1) domains flanked by an N-terminal EMI domain and a C-terminal RGD integrin-binding motif. The role of TGFBIp is not fully understood, although the protein has been shown to be implicated in cell adhesion [3]. TGFBIp is associated with various genetically determined corneal disorders such as lattice corneal dystrophy type 1 (LCD type 1) characterized by amyloid deposits, granular corneal dystrophy type 1 (GCD type 1) leading to amorphous aggregates, granular corneal dystrophy type 2 (GCD type 2) resulting in a combination of amyloid and amorphous aggregates and Thiel-Behnke corneal dystrophy (TBCD), represented by the formation of curly fibers in the superficial corneal stroma [4]. Mutations within TGFBIp are believed to be responsible for the corneal deposits and, more than 50 different amino acid substitutions [5] have been linked to these disorders.
LCD type 1 leads to amyloid deposition throughout the corneal stroma [4]. Previously, we have identified the proteins present in deposits derived from unrelated individuals suffering from LCD type 1 with a V624M or A546D mutation within TGFBIp [6,7]. These studies indicated that different mutations in TGFBIp caused a similar protein composition of the amyloid deposits. In addition, one polypeptide region encompassing the tryptic peptide Y571-R588 was present in disproportionately large amounts, suggesting that a non-degradable fibril core is a major part of the corneal amyloid material in vivo. Furthermore, increased proteolysis was observed in the N-terminal region (F515-R533) of the fourth FAS1 (FAS1-4) domain in A546D deposited tissue compared to control tissue implying that altered TGFBIp proteolysis is involved in LCD type 1 progression.
In this study, we focus on the double A546D/P551Q substitution which has been shown to cause LCD type 1 [8]. The A546D and A546D/P551Q mutations both occur in the FAS1-4 domain of TGFBIp, but they have been associated with different phenotypes. Thus, the single A546D mutation has been found with bilateral multiple polymorphic, polygonal, refractile, gray and white opacities with a chipped ice appearance [9,10], whereas the A546D/P551Q double mutant has displayed refractile lattice-like corneal stromal deposits characterized by branching and nonbranching lattice figures resembling pipe stems [8]. The P551Q substitution alone has not been reported in association with any corneal disorder or in any normal cornea. We show that both the A546D TGFBIp mutation and the A546D/P551Q TGFBIp double mutation result in a similar corneal amyloid composition. The similarity is observed both in the protein composition of the corneal amyloid deposits and in the identity of proteolytic processing sites within deposited TGFBIp. Moreover, the two siblings with the A546D/P551Q TGFBIp mutations had nearly identical corneal amyloid proteomes. Using transverse urea gradient (TUG) gel electrophoresis, circular dichroism (CD) spectroscopy and limited proteolysis we show that introduction of the P551Q mutation in the FAS1-4 domain resulted in pronounced structural changes between the A546D the A546D/P551Q mutated FAS1-4 domain. Taken together, the P551Q mutation appears to alter the stability of the A546D TGFBIp genotype sufficient to cause structural changes of TGFBIp. These changes may influence the fibrillation rate and proteolytic turnover rate of TGFBIp resulting in the different morphological observations for the respective LCD genotypes.
2. Materials and methods
2.1. Corneal tissue
The corneas were obtained from two female siblings of a family with LCD type 1 previously documented with TGFBIp mutations as individuals III-1 and III-3 in a study by Afshari and colleagues [8]. Both siblings suffered from an A546D/P551Q double mutation within TGFBIp resulting in LCD type 1. Tissue was acquired with the approval of the Institutional Review Board of Duke University Medical Center and the Regional Committee for Scientific Medical Ethics, Denmark, according to the Declaration of Helsinki.
2.2. Tissue preparation, capture, and processing
Prior to laser capturing, 8 μm thick tissue sections of formaldehyde fixed and paraffin embedded corneas were cut perpendicular to the epithelium and placed centrally on non-charged glass sides. Sections were deparaffinized, stained with the amyloidophilic dye Congo red, and dehydrated using standard histological procedures.
Laser capture microdissection (LCM)
The Arcturus Autopix Laser Capture Microscope (Applied Biosystem) was used for tissue isolation. Deposits of amyloid areas stained by Congo red were isolated from each tissue section (Figure 1). Control stroma tissue was obtained from three healthy corneas. Tissue was collected on CapSure Macro LCM Caps (Applied Biosystem), subsequently placed in sterile 0.5 ml Eppendorf Biopur Microcentrifuge Tubes (VWR) and stored at −80°C until further processed.
Figure 1.
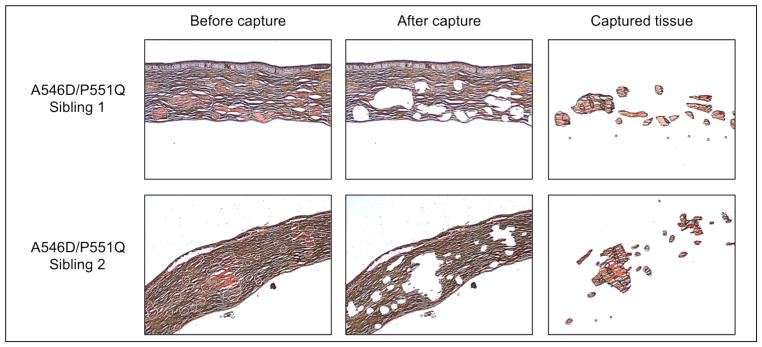
Corneal tissue section stained with Congo red. Upper row and lower row represent tissue sections of sibling 1 and 2, respectively. Left column; before laser capture, Middle column; after laser capture, and right column; corneal amyloid deposits isolated and used for further analysis. Light microscope, 4x magnification.
Tissue processing
Tissue from one or two caps was transferred to a 1.5 ml Protein LoBind Eppendorf Tube (VWR) and 20 μl of 10 mM Tris-HCl, pH 8, containing 0.1% RapiGest (Waters) was added for each cap. Samples were heated in a 95°C water bath for 90 min interrupted by centrifugation steps every 15 min. Samples were allowed to cool on ice, before the addition of 5 μg trypsin (Sequencing Grade Modified Porcine Trypsin, Promega, Madison, WI) and placed over night at 37°C. The produced peptides were subjected to C18 Stage Tips (Proxeon, Thermo Scientific) micro purification prior to LC-MS/MS analysis.
2.3. LC-MS/MS analysis
Samples were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an Easy-nLC II system (Thermo Scientific) coupled directly to a TripleTOF 5600 mass spectrometer (AB Sciex). Samples were loaded on a 0.1×21 mm C18 trap column and a 0.075×100 mm C18 analytical column (NanoSeparations). Peptides were eluted and directly electrosprayed directly into the mass spectrometer using a 50 min gradient from 5–40% acetonitrile in 0.1% acetic acid at a flow rate of 250 nL/min. Data were acquired using an ion spray voltage of 2.3 kV, curtain gas of 30, and an interface heater temperature of 150 °C. For information dependent acquisition (IDA) survey scans were acquired in 250 msec, and as many as 25 product ion scans were collected if exceeding a threshold of 150 counts per second (counts/s) and with a +2 to +5 charge-state. A sweeping collision energy setting of 35 ± 15 eV was applied to all precursor ions for collision-induced dissociation. Dynamic exclusion was set to 6 sec.
2.4. Data handling
Raw data were converted to mgf format using AB SCIEX MS Data Converter beta 1.1 (AB Sciex) and the “proteinpilot MGF” parameters and searched using an in-house Mascot search engine (Matrix Science, London, UK; version: 2.3.02) against the Swiss-Prot database (version: 2012_05). Enzyme was set to semi-trypsin, and missed cleavage was “1”. No fixed modifications were added. Instead oxidation of methionine and proline were included as variable modifications. The MS tolerance and MS/MS tolerance were 10 ppm and 0.4 Da, respectively and an ESI-QUAD-TOF system was set as the instrument used. All data sets were simultaneously searched against the decoy database to apply a false discovery rate (FDR) between 1.5 to 7% and a p-value < 0.01. Peptides with an ion score lower than 30 were rejected. Peptide spectra for protein hits identified by only one peptide were manually inspected, thereby reducing the FDR value. In these cases, only spectra with at least three consecutive y or b ions with high intensities were considered valid (supplementary material 4).
All cytokeratins were regarded as contaminants, based on recent findings in the proteome of the corneal stroma [2]. Furthermore, human tryptic peptides identical to porcine tryptic peptides were also regarded as contaminants as porcine trypsin was used for sample processing.
For each sample, four technical replicates were run. Rough relative protein quantification was performed using the exponentially modified Protein Abundance Index (emPAI). Average emPAI scores were calculated from the four technical replicates and the molar % of each protein was calculated by dividing average protein emPAI score by the total average emPAI score of the sample (supplementary material 3) [11].
TGFBIp processing
Spectral counts for all tryptic and semi-tryptic peptides observed for TGFBIp in all technical replicates for both sibling cases as well as the control case were performed using the processing software MS Data Miner v.1.2 [12]. Furthermore, positions and frequencies of non-trypsin P1 cleavage sites were counted and compared to our previous results [6,7].
2.5. Recombinant FAS1-4 domain expression and purification
WT and mutated FAS1-4 domain variants were expressed and purified as previously described [13]. Protein purity was assessed using SDS-PAGE [14] and samples were diluted to physiological NaCl concentration before CD spectroscopy, TUG gel electrophoresis and limited proteolysis assay.
2.6. Transverse urea gradient (TUG) gel electrophoresis
For TUG gel electrophoresis, 7% polyacrylamide gels (10 cm × 10 cm × 1.5 mm) were cast with a gradient mixer to give a linear 0–8 M urea gradient using a non-denaturing-PAGE buffer system [14]. The gels were rotated 90° and run at 23°C with a constant current of 15 mA until the front reached the end of the gel. Approximately, 100 μg of purified FAS1-4 domain was loaded evenly across the top of the gel and the proteins were visualized by staining with Coomassie Brilliant Blue. Delta changes between the most structured state and the fully unfolded state were measured and normalized to WT delta value.
2.7. Circular dichroism spectroscopy
The secondary structure elements and thermal unfolding of the WT and mutant FAS1-4 domains were assessed by circular dichroism (CD) on a Jasco J-800 spectropolarimeter using a quartz cuvette of 0.1 cm path length and a protein concentration of 0.15 mg/mL in PBS. Each domain variant was diluted to the desired concentration and 10 μL was analyzed by denaturing SDS-PAGE to verify comparable protein concentrations. The reported wavelength spectra were averages of three replicates recorded in the far-UV area from 205 to 250 nm at 20 °C and individually averaged over 10 scans. All spectra were baseline corrected by subtraction of a buffer spectrum and smoothed by the Savitzky-Golay method with a factor of 15 using the Jasco CD spectropolarimeter software. Thermal unfolding of each FAS1-4 domain variant was assessed by monitoring the change in signal at 222 nm over a temperature range from 10 to 95 °C with a scan rate of 90°C/h. The melting curves represented the mean of triplicate experiments performed for each domain variant. For WT and P551Q mutant FAS1-4 domains, the Kaleidagraph software (Synergy Software) was used to fit each thermal denaturation curve to a 2-state unfolding model and the second derivative was used to calculate the inflection point of the transition giving the indicated melting temperatures, Tm, with the standard deviation for the triplicate experiment.
2.8. Limited proteolysis
The recombinant FAS1-4 variants were subjected to trypsin limited proteolysis assay (Sequence grade, Promega) using a titration going from 1/500 to 1/50 trypsin/substrate (w/w) ratios. Reactions were incubated 1 hour at 37°C and subsequently incubated with 1mM PMSF for 15 min to stop the reaction. Samples were boiled 5 min in SDS sample buffer and applied to a 10–15% SDS-PAGE [14]. Protein bands were visualized using Coomassie Brilliant Blue. Densitometric analysis of the limited proteolysis was performed using ImageJ (v. 1.42q) using three digest replicates. All bands in the assay were normalized to the intensity of the control sample containing no trypsin.
3. Results
3.1. Microdissection of corneal tissues
Amyloid deposits within the stroma of corneal tissue with LCD type 1 were isolated from two adult female siblings suffering from A546D/P551Q double mutations within the FAS1-4 domain of TGFBIp (Figure 1). In addition, a pooled tissue sample consisting of corneal stroma from three non-related healthy individuals was included as a reference. The tissue samples were digested with trypsin, and four technical LC-MS/MS analysis replicates for each tissue sample were collected.
3.2. Protein composition of corneal deposits and healthy tissue
The LC-MS/MS analyses identified 139 and 159 proteins within the amyloid deposit derived from sibling 1 and 2, respectively, and 202 proteins in the healthy stroma control (supplementary material 3). The most abundant proteins identified in at least three out of the four technical replicates performed for sibling 1 and sibling 2 showed accumulation of TGFBIp, serum amyloid P-component (SAP), Apo A-I, Apo A-IV, Apo E, Apo D, clusterin, and HtrA1 when compared to control stroma tissue (Table 1). Furthermore, lysozyme C, dermcidin, complement component C9, protein S100-A8 and A9, and lactotransferrin were found in higher amounts in the amyloid deposits compared to the control. Among the 30 most abundant proteins identified for each sibling, the top 15 proteins were identified in the sibling counterpart except that serum albumin only was identified within top 15 in sibling 1 and lactotransferrin and thioredoxin (Trx) only were observed within top 15 in sibling 2.
Table 1.
The 30 most abundant proteins according to emPAI-based molar % identified in corneal amyloid deposits and healthy corneal tissue
| # | A546D/P551Q (Sibling 1) | A546D/P551Q (Sibling 2) | Healthy corneal stroma (3 individuals) | |||
|---|---|---|---|---|---|---|
| Name | Molar % | Name | Molar % | Name | Molar % | |
| 1 | TGFBIp | 69.5 ± 11.3 | TGFBIp | 47.0 ± 7.9 | Collagen alpha-1(I) chain | 36.5 ± 5.8 |
| 2 | Serum amyloid P-component | 3.0 ± 1.2 | Collagen alpha-1(I) chain | 4.1 ± 0.5 | Collagen alpha-2(I) chain | 17.3 ± 1.5 |
| 3 | Apolipoprotein A-I | 2.5 ± 0.6 | Serum amyloid P-component | 3.8 ± 1.0 | TGFBIp | 7.8 ± 2.0 |
| 4 | Collagen alpha-1(I) chain | 2.2 ± 0.4 | Lysozyme C | 3.4 ± 1.5 | Ig kappa chain C region | 5.0 ± 3.0 |
| 5 | Clusterin | 2.2 ± 0.7 | Dermcidin | 2.9 ± 1.9 | Serum albumin | 3.4 ± 0.3 |
| 6 | Collagen alpha-2(I) chain | 2.0 ± 1.0 | Collagen alpha-2(I) chain | 2.6 ± 0.3 | Keratocan | 1.8 ± 0.2 |
| 7 | Serine protease HTRA1 | 1.4 ± 0.8 | Clusterin | 2.5 ± 0.7 | Decorin | 1.6 ± 0.5 |
| 8 | Apolipoprotein A-IV | 1.3 ± 0.5 | Lactotransferrin | 2.0 ± 0.6 | Lumican | 1.4 ± 0.3 |
| 9 | Dermcidin | 1.1 ± 0.6 | Apolipoprotein A-IV | 1.7 ± 0.5 | Ig kappa chain V-III region SIE | 1.3 ± 0.4 |
| 10 | Apolipoprotein D | 0.8 ± 0.4 | Serine protease HTRA1 | 1.5 ± 0.6 | Collagen alpha-3(VI) chain | 1.2 ± 0.2 |
| 11 | Apolipoprotein E | 0.8 ± 0.1 | Thioredoxin | 1.1 ± 0.2 | Ig gamma-1 chain C region | 1.1 ± 0.2 |
| 12 | Protein S100-A8 | 0.7 ± 0.5 | Apolipoprotein E | 1.0 ± 0.3 | Ig gamma-3 chain C region | 1.1 ± 0.3 |
| 13 | Serum albumin | 0.6 ± 0.2 | Complement component C9 | 1.0 ± 0.2 | Collagen alpha-1(VI) chain | 0.9 ± 0.1 |
| 14 | Lysozyme C | 0.6 ± 0.4 | Apolipoprotein A-I | 1.0 ± 0.3 | Ig lambda-2 chain C regions | 0.8 ± 0.4 |
| 15 | Prostaglandin-H2 D-isomerase | 0.5 ± 0.3 | Apolipoprotein D | 0.9 ± 0.2 | Ig kappa chain V-III region NG9 | 0.8 ± 0.4 |
| 16 | Lumican | 0.5 ± 0.2 | Fatty acid-binding protein, epidermal | 0.8 ± 0.2 | Aldehyde dehydrogenase, dimeric NADP-preferring | 0.7 ± 0.1 |
| 17 | Protein S100-A9 | 0.4 ± 0.2 | Actin, cytoplasmic 1 | 0.8 ± 0.2 | Collagen alpha-2(VI) chain | 0.7 ± 0.2 |
| 18 | Ig gamma-1 chain C region | 0.4 ± 0.1 | Vimentin | 0.7 ± 0.2 | Vimentin | 0.7 ± 0.1 |
| 19 | Histone H4 | 0.4 ± 0.1 | Cystatin-A | 0.7 ± 0.4 | Collagen alpha-1(XII) chain | 0.6 ± 0.1 |
| 20 | Ubiquitin-40S ribosomal protein S27a | 0.4 ± 0.3 | Ig gamma-1 chain C region | 0.6 ± 0.2 | Collagen alpha-2(V) chain | 0.6 ± 0.2 |
| 21 | Complement component C9 | 0.4 ± 0.1 | Olfactomedin-like protein 3 | 0.6 ± 0.2 | Ig gamma-4 chain C region | 0.5 ± 0.1 |
| 22 | Olfactomedin-like protein 3 | 0.3 ± 0.1 | Protein S100-A6 | 0.6 ± 0.1 | Prostaglandin-H2 D-isomerase | 0.5 ± 0.2 |
| 23 | Angiopoietin-related protein 7 | 0.3 ± 0.2 | Cytokine-like protein 1 | 0.6 ± 0.2 | MAM domain-containing protein 2 | 0.4 ± 0.1 |
| 24 | Keratocan | 0.3 ± 0.1 | Ubiquitin-40S ribosomal protein S27a | 0.6 ± 0.3 | Histone H4 | 0.4 ± 0.1 |
| 25 | Decorin | 0.3 ± 0.2 | Histone H4 | 0.5 ± 0.1 | Dermcidin | 0.4 ± 0.1 |
| 26 | Ig alpha-1 chain C region | 0.3 ± 0.1 | Protein S100-A8 | 0.5 ± 0.1 | Collagen alpha-1(V) chain | 0.4 ± 0.1 |
| 27 | Phospholipase A2, membrane associated | 0.2 ± 0.1 | Ig kappa chain V-III region VG | 0.5 ± 0.1 | Protein S100-A6 | 0.4 ± 0.2 |
| 28 | Cytokine-like protein 1 | 0.2 ± 0.1 | Glyceraldehyde-3-phosphate dehydrogenase | 0.5 ± 0.3 | Ig alpha-1 chain C region | 0.4 ± 0.2 |
| 29 | Fatty acid-binding protein, epidermal | 0.2 ± 0.1 | Prostaglandin-H2 D-isomerase | 0.5 ± 0.2 | Alpha-enolase | 0.3 ± 0.1 |
| 30 | Actin, cytoplasmic 1 | 0.2 ± 0.1 | Metalloproteinase inhibitor 3 | 0.5 ± 0.2 | Collagen alpha-1(III) chain | 0.3 ± 0.1 |
We conclude that the LCD type 1 protein profiles of the amyloid deposits were similar in the two siblings analyzed in this study. In addition, we have previously described the proteome of corneal amyloid deposits of two other LCD type 1 (V624M and A546D) cases [6,7] revealing protein profiles very similar to the LCD type 1 cases described in this study.
3.3. In vivo proteolytic processing of TGFBIp
Aberrant proteolysis of TGFBIp by endogenous proteases is likely involved in the formation of corneal amyloid deposits [7]. To identify the specificity of these proteases, the in vivo proteolytic cleavage sites identified in the two A546D/P551Q mutant TGFBIp sibling cases were determined (Figure 2). A comparison of the cleavage sites found in TGFBIp in the amyloid deposits and in the control tissue revealed high frequencies of cleavages after Asn, Leu, Tyr, Thr, Phe, His and Ala residues of which Thr, Val, and Tyr were more pronounced in the corneal amyloid deposits (supplementary material 1).
Figure 2.

Spectral counting of TGFBIp tryptic (A) and semi-tryptic peptides (B) obtained from the MS analyses of corneal amyloid deposits from sibling 1 (gray), sibling 2 (white), and corneal stroma control tissue (black). Spectral counts from tryptic peptides indicate that the polypeptide region encompassed by the tryptic peptide Y571-R588 as well as peptides from the C-terminus of the FAS1-3 domain (Y488-R496) and the inter-domain region between the FAS1-3 and FAS1-4 domains (V497-K510) were accumulated in the amyloid deposits of both siblings, when compared to control tissue. Furthermore, according to the spectral counting of semi-tryptic peptides the same regions including peptide F515-R533 from the N-terminal part of the FAS1-4 domain appeared to be processed to a significantly higher extent than seen for the control tissue.
Similar to previous observations [6,7] the polypeptide region containing the tryptic TGFBIp peptide Y571-R588 was over-represented in the amyloid deposits (Figure 2A). Furthermore, the semi-tryptic data indicated that polypeptide regions corresponding to tryptic TGFBIp peptides V497-K510, F515-R533 and Y571-R588 were relatively more proteolytically processed in the amyloid deposits compared to control tissue (Figure 2B).
3.4. Stability comparison of FAS1-4 domain variants
As the proteomes of corneal amyloid deposits were similar for the A546D and A546D/P551Q LCD type 1 cases, we suggest that differences in phenotypes are independent of the surrounding extracellular matrix. Instead, a phenotypical difference may be attributed to differences in thermodynamic stability and/or proteolytic susceptibility caused by the P551Q mutation. We have previously shown that amino acid substitutions within the FAS1-4 domain of TGFBIp leads to altered thermodynamic stability and that these changes are mirrored by the FAS1-4 domain alone [13]. Therefore, A546D, A546D/P551Q, P551Q, and WT FAS1-4 domains were recombinantly expressed and the stabilities of the different FAS1-4 variants was compared using chaotropic denaturating TUG gel electrophoresis (Figure 3). The P551Q mutant migrated similar to the WT FAS1-4 domain and showed the same unfolding transition. Contrarily, both A546D and A546D/P551Q mutant FAS1-4 domains had a more extended or flexible structure at low urea concentrations and followed a more linear transition towards the unfolded state. The delta value measured as the distance between the highest and lowest mobility states were calculated and normalized to the WT domain delta value. The delta values indicate the degree of unfolding between native (0 M Urea) and unfolded (8 M urea) on the TUG gel. According to the delta values native domain structure were most preserved in the following order, WT > P551Q > A546D > A546D/P551Q.
Figure 3.

Transverse urea gradient gel electrophoresis of WT, A546D, P551Q, and A546D/P551Q recombinantly expressed FAS1-4 domains. Horizontal 0–8M urea gradient from left to right. Vertical separation based on the degree of native fold. WT and P551Q mutant FAS1-4 domains follow a classic two-state transition unfolding event whereas both the A546D and A546D/P551Q FAS1-4 domain variants follow a linear transition towards complete unfolding. Delta changes (Δ) are normalized to WT and indicates that the degree of unfolding at 0M urea is less for WT < P551Q < A546D < A546D/P551Q. The P551Q mutation alone does not contribute to FAS1-4 domain destabilization. However, in combination with the A546D mutations a destabilizing cooperative effecter is seen as the delta change is lower for A546D/P551Q than for A546D (n=2).
To support the TUG gel electrophoresis data, we subjected the FAS1-4 domains to heat denaturation monitored by circular dichroism spectroscopy (Figure 4). The absorbance spectra in the far-UV region at 20°C or 37°C indicated a similar content of secondary structure for all FAS1-4 variants (Figure 4A). Hence, the overall native structure appeared to be preserved in these conditions for A546D, A546D/P551Q, and P551Q mutants. Performing a thermal scan from 10 to 95°C for all variants showed a similar trend as observed for TUG gel electrophoresis (Figure 4B). The WT and P551Q FAS1-4 domains showed a classic two-state unfolding event with a clearly cooperative transition from the native to the denatured state, with melting temperatures Tm of 68.0 ± 0.1°C and 65.6 ± 0.2°C, respectively. Thus, introducing the P551Q mutation in the FAS1-4 domain caused a decrease in thermal stability of 2.4°C. In contrast, the A546D and A546D/P551Q mutant FAS1-4 domains showed no cooperative thermal transition, and the amount of residual structure at elevated temperatures was significantly higher than for WT and P551Q. This might be due to aggregation (stabilizing secondary structure) or simply altered conformational properties of the two mutants.
Figure 4.
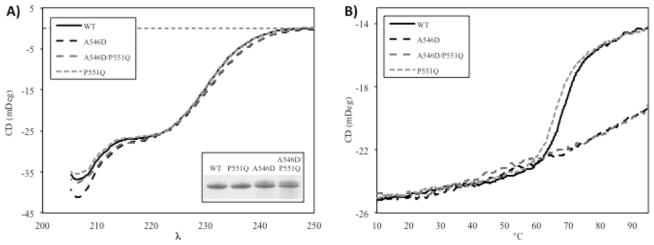
Circular dichroism spectroscopy revealed differences between WT and mutants of the FAS1-4 domain. (A) The overall structural fold of mixed α-helical and β-sheet was preserved at 20 and 37°C for all mutants (20°C shown here). The spectra of mutants A546D and A546D/P551Q only displayed small differences compared to WT and mutant P551Q. Insert represents SDS-PAGE of 10 μL of each sample used for CD to visualize purity and analyzed amounts. (B) Thermal scans at 222 nm from 10°C to 95°C showed significant differences in unfolding behavior. WT and P551Q follow a classical two-state unfolding event with Tm of 68.0 ± 0.1°C and 65.6 ± 0.2°C, respectively. A546D and A546D/P551Q mutant FAS1-4 domains undergo a linear thermal transition to a less unfolded state compared to WT.
We conclude that addition of the P551Q mutation contributes to a decrease in the FAS1-4 domain stability when compared to the WT FAS1-4 domain. However, in the context of the A546D background, the double mutant did not display any significant difference as a result of the P551Q introduction, suggesting that A546D is responsible for the most profound change in TGFBIp/FAS1-4 overall stability.
Neither TUG nor CD experiments revealed any large difference between the A546D mutant and the A546D/P551Q mutant FAS1-4 domains that could explain the different phenotypes. To further study any structural difference between WT, A546D and A546D/P551Q all four FAS1-4 domain variants were analyzed using limited proteolysis. All four FAS1-4 domain variants were titrated with trypsin going from 1/500 to 1/50 of enzyme to substrate ratio (Figure 5). The limited proteolysis showed that the WT and P551Q FAS1-4 domains were most resistant to proteolysis followed by the A546D/P551Q double mutated FAS1-4 domain. In contrast, the A546D FAS1-4 domain was prone to proteolysis even at the lowest concentration of trypsin. Hence, even though TUG gel electrophoresis and CD spectroscopy did not indicate significant thermodynamic stability differences between A546D and A546D/P551Q mutant FAS1-4 domains, the limited proteolysis assay indicated that the two FAS1-4 domain variants show conformational differences influencing the proteolytic susceptibility. Taken together, our results show that that addition of the P551Q mutation on its own maintains the overall fold and stability and contributes to increased resistance towards proteolytic susceptibility of the A546D mutation when combined.
Figure 5.
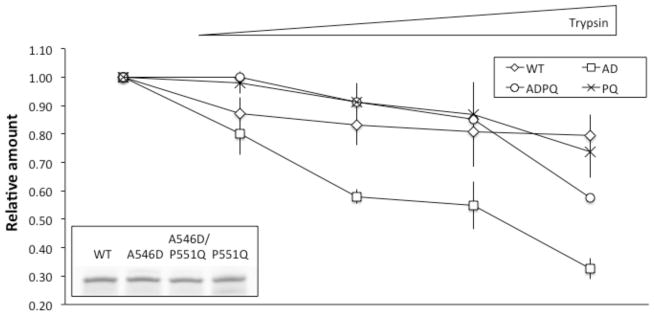
Limited proteolysis of FAS1-4 variants. WT, A546D, A546D/P551Q, and P551Q recombinant FAS1-4 were subjected limited to proteolysis using a titration of trypsin. Samples were subjected to SDS-PAGE and densitometric analysis was performed subsequently. A546D FAS1-4 was most sensitive toward trypsin proteolysis followed by A546D/P551Q, P551Q and WT FAS1-4 in the listed order. Insert in lower left corner shows protein amount for each FAS1-4 variant used in the assay (n=3).
4. Discussion
Previous reports have linked the A546D mutation within TGFBIp to atypical, and polymorphic LCD type 1 [9,10], whereas patients displaying the A546D/P551Q double mutation within TGFBIp have been reported to resemble the classic LCD type 1 phenotype normally observed for the more frequent R124C mutation within TGFBIp [8]. In this study, we investigated if the observed difference in phenotypes is caused by the P551Q mutation in addition to the A546D mutation and if it can be linked to (I), difference in proteomes of corneal amyloid deposits (II), different in vivo proteolytic processing of TGFBIp in the deposits or (III), changes in stability or structure of TGFBIp resulting in different fibrillation pathways giving rise to the various phenotypes.
4.1. LCD type 1 deposits caused by different mutations in the FAS1-4 domain share similar protein profiles
Using mass spectrometry we analyzed the protein composition of the amyloid deposits captured from two female siblings suffering from a LCD type 1 variant (A546D/P551Q). Proteins identified included TGFBIp, serum amyloid P-component, Apo A-I, Apo A-IV, Apo E, Apo D, clusterin, and HtrA1 (table 1). Remarkably, the proteomes of the two closely related cases analyzed in this study were similar as the 15 most abundant proteins identified were among the 30 most abundant proteins identified from their sibling counterpart. In addition, the sibling amyloid proteomes showed similarities to previous non-familiar studies of LCD type 1 cases caused by other TGFBIp mutations [6,7]. Altogether, this suggests that the molecular mechanism leading to LCD type 1 amyloid deposit formation is not the result of a random process but rather an orchestrated assembly of proteins most likely initiated by an initial misfolding event. The protein composition of the amyloid deposits is therefore not assumed to cause the phenotypic difference observed for the A546D and A546D/P551Q LCD type 1 cases. At this point it has not been clarified if the similarity in proteomes of corneal amyloid deposits include LCD type I caused by the more common R124C mutation in the first FAS1 domain. Thus, it cannot be out ruled that a different molecular mechanism is responsible for the R124C cases of LCD type 1 resulting in different proteomes of amyloid deposits when compared to the FAS1-4 related LCD type 1 cases presented here and described previously [6,7].
4.2. TGFBIp processing leads to accumulation of polypeptide regions from the FAS1-4 domain
Tryptic peptide counts observed by mass spectrometry analyses indicated accumulation of the internal polypeptide region Y571-R588 of the FAS1-4 domain in both siblings (Figure 2). Region Y571-R588 also accumulated in LCD type 1 caused by the A546D mutation, supporting our hypothesis that this may constitute the amyloidogenic part of TGFBIp.
Focusing on the non-tryptic P1 cleavage sites in TGFBIp in the deposits of the two siblings revealed Asn, Leu, Tyr, Thr, Phe, His and Ala residues as the preferred P1 cleavage sites of which Thr, Val, and Tyr were more profound in the amyloid deposits compared to stroma control tissue (supplementary material 1). This is in agreement with our previous observation for TGFBIp processing in LCD type 1 tissue [6,7] suggesting that clearance of misfolded TGFBIp involves the same proteases in all LCD type 1 variants caused by mutations in the FAS1-4 domain and therefore most likely does not lead to the different phenotypes observed for the A546D and the A546D/P551Q mutations. The observed P1 cleavage sites correlate well with the specificity of the serine protease HtrA1 [15] and we have previously proposed that the serine protease HtrA1 found to be associated with the amyloid deposits in LCD type 1 is responsible for processing of amyloidogenic and/or aggregated TGFBIp [7].
4.3. The P551Q mutation slightly decreases the thermodynamic stability of the FAS1-4 domain
Distinct TGFBI-linked corneal dystrophy phenotypes are known to correlate with different TGFBIp mutations [4]. Mutations within the FAS1-1 domain of TGFBIp associated with LCD type 1, GCD type 1, and GCD type 2 all show preserved stability compared to WT TGFBIp, whereas mutations in the FAS1-4 domain result in either a less stable or more stable TGFBIp for LCD type IIIA and GCD type 2, respectively [13]. Our stability measurements agreed with this finding as both A546D and A546D/P551Q mutant FAS1-4 domains showed a significant structural difference during chemical (Figure 3) or thermal denaturation (Figure 4) compared to the WT FAS1-4 domain. TUG gel signatures seen for both the A546D mutant FAS1-4 variants were similar to the one previously reported for the A546T mutant FAS1-4 domain (previously designated LCD type IIIA) [13] though the A546D mutant domains might be expected to be less stable than the A546T mutant FAS1-4 domain. The A546D mutation introduces a charged side chain pointing towards the hydrophobic core of the domain, as indicated by the NMR structure of human TGFBIp FAS1-4 domain (Protein Data Bank ID: 1X3B). In contrast, the P551Q mutation only contributed to a lesser extent to FAS1-4 domain destabilization. Inspection of the NMR structure reveals the P551 residue to be positioned in a loop region between two alpha helices. This loop region is rigid possibly due to two adjacent proline residues (P551 and P552). The change of one of these proline residues into a glutamic acid residue might increase mobility of the loop and surrounding α-helices and, thereby, locally destabilizes the FAS1-4 domain. Noteworthy, both the A546D and P551Q mutations are located in close proximity to the suggested amyloidogenic region, which thereby may enable inter FAS1-4 domain interactions to take place caused by local destabilization of the domain.
4.4. Limited proteolysis suggest structural difference between A546D and A546D/P551 mutant FAS1-4 domains
Limited proteolysis generally results in nicks in flexible polypeptide regions of the protein such as loops and domain linker regions [16]. The increased degradation observed for A546D and A546D/P551Q mutated FAS1-4 domains (Figure 5) is most likely caused by unfolding of the FAS1-4 domain on its path towards mature amyloid fibril as proteolysis of the WT FAS1-4 domain progresses much more slowly. Judged by the SDS-PAGE of the limited proteolysis assays no degradation products were seen for any of the four FAS1-4 domain variants (data not shown) indicating a global unfolding of the FAS1-4 domains. However, as the WT FAS1-4 domain also was processed to some extent the global unfolding may also rely on initial nicks in locally destabilized regions of the FAS1-4 domain consequently destabilizing the protein sufficiently to be completely degraded by trypsin afterwards. Addition of the P551Q mutation to the A546D mutated FAS1-4 domain only resulted in minor thermodynamic stability changes judged by TUG gel electrophoresis and CD spectroscopy. However, the limited proteolysis assay revealed a pronounced difference between A546D and A546D/P551Q mutations indicating profound structural changes that most likely accounts for the different phenotypically appearances of A546D and A564D/P551Q LCD type 1 cases. These changes could influence fibrillation rate and proteolytic turnover rate of TGFBIp resulting in the different morphological observations for the respective LCD genotypes.
In conclusion, the data support our previous findings [6,7] that the protein profile of amyloid deposits caused by different mutations within the fourth FAS1 domain of TGFBIp are alike. Furthermore, accumulations of the same polypeptide region of the FAS1-4 domain occur in the corneal amyloid deposits. This indicates that the proteolytic machinery involved in clearance of misfolded TGFBIp in the different genotypic variants of LCD type 1 rely on the same proteases. The stability data indicates that the P551Q mutant FAS1-4 domain resembles WT FAS1-4 domain in stability and the P551Q mutation on its own contributes only to a lesser extent to the destabilization of the molecule. However, in combination with the A546D mutation we observed structural changes when compared to the A546D mutant alone suggesting that the introduction of the P551Q mutations contributes to higher stability of the FAS1-4 domain. Hence, phenotypical variance in the A546D and A546D/P551Q LCD type 1 cases appears not to be correlated to different protein compositions associated with protein deposits but rather relies on stability differences associated with local dynamics and structure of TGFBIp that may affect fibrillation or turnover rates.
Supplementary Material
Clinical relevance.
Approximately 285 million people worldwide were in 2010 estimated to suffer from visual impairment of which corneal diseases accounted for 1% of the cases [1]. The corneal dystrophies linked to the transforming growth factor beta induced (TGFBI) gene are a group of poorly understood genetically determined corneal diseases. Most of them are characterized by an accumulation of mutant TGFBI gene product in the transparent corneal tissue, which hereby impairs vision. An evaluation of the protein profile of these deposits may provide valuable clues on the molecular mechanisms underlying the TGFBI corneal dystrophies. Understanding the cause and pathogenesis of the TGFBIp corneal dystrophies may ultimately lead to non-surgical therapeutic options for treating these genetically determined corneal disorders.
Acknowledgments
This study was supported by grants from the National Eye Institute (R01 EY012712), The Lundbeck Foundation, Denmark, The Danish National Research Foundation and The Danish Agency for Science Technology and Innovation, and Fight for Sight, Denmark. Lastly, we would like to thank the PRIDE team for assistance in data management in the ProteomeXchange Consortium.
Abbreviations
- CD
circular dichroism
- emPAI
exponentially modified Protein Abundance Index
- FAS1
fasciclin-1
- HtrA1
high temperature requirement A1
- LCM
laser capture microdissection
- LCD type 1
lattice corneal dystrophy type 1
- TGFBIp
transforming growth factor beta-induced protein
- TUG
transverse urea gradient
Footnotes
The authors have declared no conflict of interest
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [17] with the dataset identifier PXD000307 and DOI 10.6019/PXD000307.
References
- 1.Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012;96:614–618. doi: 10.1136/bjophthalmol-2011-300539. [DOI] [PubMed] [Google Scholar]
- 2.Dyrlund TF, Poulsen ET, Scavenius C, Nikolajsen CL, et al. Human cornea proteome: identification and quantitation of the proteins of the three main layers including epithelium, stroma, and endothelium. J Proteome Res. 2012;11:4231–4239. doi: 10.1021/pr300358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Runager K, Enghild JJ, Klintworth GK. Focus on molecules: Transforming growth factor beta induced protein (TGFBIp) Experimental Eye Research. 2008;87:298–299. doi: 10.1016/j.exer.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. doi: 10.1186/1750-1172-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Han X, Huang D, Yu L, et al. Analysis of TGFBI gene mutations in Chinese patients with corneal dystrophies and review of the literature. Mol Vis. 2010;16:1186–1193. [PMC free article] [PubMed] [Google Scholar]
- 6.Karring H, Runager K, Thøgersen IB, Klintworth GK, et al. Composition and proteolytic processing of corneal deposits associated with mutations in the TGFBI gene. Experimental Eye Research. 2012;96:163–170. doi: 10.1016/j.exer.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karring H, Poulsen ET, Runager K, Thøgersen IB, et al. Serine protease HtrA1 accumulates in corneal transforming growth factor beta induced protein (TGFBIp) amyloid deposits. Mol Vis. 2013;19:861–876. [PMC free article] [PubMed] [Google Scholar]
- 8.Klintworth GK, Bao W, Afshari NA. Two Mutations in the TGFBI (BIGH3) Gene Associated with Lattice Corneal Dystrophy in an Extensively Studied Family. Investigative Ophthalmology & Visual Science. 2004;45:1382–1388. doi: 10.1167/iovs.03-1228. [DOI] [PubMed] [Google Scholar]
- 9.Correa-Gomez V, Villalvazo-Cordero L, Zenteno JC. The TGFBI A546D mutation causes an atypical type of lattice corneal dystrophy. Mol Vis. 2007;13:1695–1700. [PubMed] [Google Scholar]
- 10.Eifrig DE, Afshari NA, Buchanan HW, Bowling BL, Klintworth GK. Polymorphic corneal amyloidosis: a disorder due to a novel mutation in the transforming growth factor beta-induced (BIGH3) gene. Ophthalmology. 2004;111:1108–1114. doi: 10.1016/j.ophtha.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 11.Ishihama Y, Oda Y, Tabata T, Sato T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Dyrlund TF, Poulsen ET, Scavenius C, Sanggaard KW, Enghild JJ. MS Data Miner: a web-based software tool to analyze, compare, and share mass spectrometry protein identifications. Proteomics. 2012;12:2792–2796. doi: 10.1002/pmic.201200109. [DOI] [PubMed] [Google Scholar]
- 13.Runager K, Basaiawmoit RV, Deva T, Andreasen M, et al. Human Phenotypically Distinct TGFBI Corneal Dystrophies Are Linked to the Stability of the Fourth FAS1 Domain of TGFBIp. Journal of Biological Chemistry. 2011;286:4951–4958. doi: 10.1074/jbc.M110.181099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bury AF. Analysis of protein and peptide mixtures: Evaluation of three sodium dodecyl sulphate-polyacrylamide gel electrophoresis buffer systems. Journal of Chromatography. 1981;213:491–500. [Google Scholar]
- 15.Truebestein L, Tennstaedt A, Mönig T, Krojer T, et al. Substrate-induced remodeling of the active site regulates human HTRA1 activity - Supp. Nature Publishing Group. 2011;18:386–388. doi: 10.1038/nsmb.2013. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 17.Vizcaíno JA, Côté RG, Csordas A, Dianes JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–9. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.