

Abstract
ERΔ3 transgenic mice expressing a dominant negative estrogen receptor α (ERα) variant lacking the second zinc finger in the DNA binding domain were developed to examine its potential to inhibit estrogen action in vivo. To investigate if ERΔ3 expression influences uterine carcinogenesis, ERΔ3 transgenic mice were exposed to diethylstilbestrol (DES) on post-natal days 1–5. Neonatal DES treatment induced uterine adenocarcinomas in 81% of 8-month-old ERΔ3 mice compared to 49% of wild-type females (p<0.016). ERΔ3 did not inhibit the expression of the estrogen-responsive progesterone receptor and lactoferrin genes in the presence of ERα or modify their expression in ERα knockout (αERKO) mice. Higher circulating 17β-estradiol levels and non-classical signaling by ERΔ3 may be related to the earlier incidence of uterine cancer. These findings indicate that expression of this ERα variant can influence determining events in uterine cancer development and its natural occurrence in the human uterus would unlikely be protective.
Keywords: diethylstilbestrol, dominant negative receptor, ERα variants, ERΔ3, lactoferrin, non-classical ER signaling, progesterone receptor, uterine cancer
1. INTRODUCTION
Estrogens are implicated in the initiation and promotion of carcinogenesis in the hormonally responsive tissues of the female reproductive tract [1, 2]. Estrogens stimulate cell proliferation and induce specific cellular responses through direct interaction with the estrogen receptors (ER), ERα and ERβ. Both ER subtypes are nuclear receptors that act as ligand-dependent transcription factors. In the presence of an estrogen agonist, ER dimers transactivate or repress estrogen-responsive genes containing one or more estrogen response elements (ERE) [3]. Alternative to this classical pathway, ER also acts through non-classical mechanisms by interacting with other transcription factors, such as the AP-1 family and Sp-1, to influence estrogen responses [4]. In the uterus, ERα is the predominant receptor; the requirement for ERα to elicit a uterotropic response to estrogens or epidermal growth factor (EGF) has been clearly demonstrated in ERα knockout mice (αERKO), which lack expression of wild-type (WT) ERα [5, 6]. Additionally, both ERα and ERβ act through rapid, nongenomic mechanisms [7]; however, a study with a nongenotropic-selective ligand suggests that ER genomic actions are required for uterine stimulation [8].
Exposure to synthetic estrogens, such as diethylstilbestrol (DES), during critical times of development is linked with an increased risk of reproductive tract cancers in women [9] and mice [10]. DES is a potent estrogen, which has high affinity for ERα and ERβ [11] and stimulates a uterotropic response at lower doses than the endogenous estrogen, 17β-estradiol (E2) [12]. In the late 1940’s, DES was approved for the treatment of pregnancy-related complications, including risk of abortion, premature labor, and diabetes. However, after puberty, daughters exposed to DES in utero have an increased risk for developing clear-cell adenocarcinoma of the vagina or cervix [9, 13]. The stages of reproductive tract differentiation that occur prenatally in humans include both prenatal and neonatal development in mice [13]. Correspondingly, like women exposed in utero, mice exposed prenatally or neonatally to DES also develop reproductive tract cancers, including uterine adenocarcinomas [10].
In women, reproductive tract tumors associated with in utero exposure to DES are detected after menarche, usually between the ages of 14–30 years [14, 15]. In mice, removal of the ovaries prior to puberty prevents the formation of DES-induced uterine tumors [16]. Therefore, DES-induced cancer is influenced by estrogens both at the time of treatment, from DES, as well after puberty, from endogenous estrogens. ERα is expressed in uterine epithelial and stromal cells during early stages of reproductive tract development in fetal and neonatal mice as well as in the uterus of sexually mature mice [17, 18]. αERKO mice are resistant to the effects of DES, demonstrating that ERα is required for DES-induced reproductive tract abnormalities and uterine cancer [19]. In contrast, elevated expression of ERα in MT-mER transgenic mice shortened the latency of uterine tumor development induced by neonatal DES treatment [20]. These data indicate that ERα expression levels and/or activity can influence susceptibility to DES-induced tumor formation. Therefore, neonatal DES treatment provides an effective model for investigating the effects of modified ER expression on hormonally-induced carcinogenesis, such as expression of an ERα variant with the potential to inhibit ER activity.
ERα variants were first detected in breast tumors and cell lines. ER variants arise by alternative splicing of the ERα transcript resulting in the deletion of one or more exons [21]. RNA expression is used to detect the presence of ERα variants in human tissues. A few studies have also verified that the variant RNA is translated into receptor proteins in human tissues and breast cancer cell lines [21–25]. Although the majority of the reports have focused on ER variant expression in breast cancer, ER variant expression has been found in other normal and neoplastic estrogen target tissues [21], including the uterus [26, 27]. The presence of ER variants in normal tissues suggests that these modified receptors may have a role in normal physiology, estrogen responsiveness, and, perhaps, tumor development.
The deletion of exon 3 (ERΔ3) from the human gene for ERα (ESR1) by alternative splicing was first detected in the T47D breast cancer cell line [28]. The message and protein for ERΔ3 also occur in MCF-7 cells [24, 25]. The in-frame deletion of exon 3 encodes a receptor protein missing the second zinc finger of the DNA binding domain (DBD). The second zinc finger contains the ligand-independent dimerization domain and may be responsible for discriminating the half-site spacing of DNA response elements [29]. The functional domains outside exon 3, including AF-1 and AF-2, first zinc finger, ligand binding, ligand-dependent dimerization, and nuclear localization domains, remain intact. Despite the loss of the dimerization domain within exon 3, dimerization with WT ERα occurs via its stronger, ligand-dependent dimerization domain [29–31]. In vitro, without the second zinc finger, human ERΔ3 does not bind to DNA containing the consensus estrogen response element (ERE) or activate transcription of an ERE-reporter gene[28]. However, in transfected HeLa cells, the ERΔ3 variant displays dominant negative activity; that is, coexpression of the ERΔ3 variant with WT ERα diminishes the ability of WT ERα to activate an ERE-reporter construct [28]. The postulated mechanism for its dominant negative activity is through the formation of ERΔ3:ERα and ERΔ3:ERβ heterodimers to prevent DNA binding and, thus, transactivation of ERE-regulated genes [30].
The in vivo activities of the ERΔ3 variant, such as inhibiting the activity of the WT ER, remain untested. Transgenic mouse models expressing other dominant negative receptors have been instructive for investigating the roles of the WT and repressor protein [32–35]. Therefore, our goals were to develop a transgenic mouse model expressing ERΔ3 and to investigate its actions in vivo and its effects on carcinogenesis in estrogen-responsive tissues. The resulting transgenic mice express the mouse ERα variant lacking the second zinc finger, which is encoded by exon 4 in the mouse Esr1 gene (third coding exon) and corresponds to the human variant lacking exon 3. The amino acid sequence for human exon 3 and mouse exon 4 is 100% conserved in the human and mouse ERα mRNAs, as are the splicing junctions for the message. Based on the reported absence of transactivation function and its ability to repress ERα activity in vitro, we speculated that expression of the ERΔ3 variant in transgenic mice may provide cancer protection to tissues in which abnormal proliferation has been associated with estrogen exposure. Therefore, in the present study, uterine tumors were induced by neonatal DES treatment in order to investigate the effects of ERΔ3 expression on the development of hormonally-induced cancer.
2. MATERIALS AND METHODS
2.1. ERΔ3 Construct and Generation of the Transgenic Mice
For constructing the transgenic mice expressing the mouse ERΔ3 variant, the sequences for exon 4 of mouse Esr1 cDNA encoding the second zinc finger were deleted. Due to the late discovery of the first exon in the human ESR1 gene [36], the numbering for mouse Esr1 and human ESR1 exons does not correspond. Exon 4 in the mouse Esr1 gene is equivalent to exon 3 in the human gene (with first and second exons in human ERα designated 1′ and 1, respectively). Therefore, for clarity and comparison with reports on ERα variants in humans, the transgenic model is named to reflect an equivalent deletion in the mouse gene as the naturally-occurring ERΔ3 variant in humans.
The ERΔ3 cDNA was generated by PCR to recreate the deletion of exon 3 in human ERα in the mouse ERα cDNA (exon 4 in Esr1). Primers P1 (forward), GCAAGCCCACTGTGTTCAAC, and P2 (reverse), GCGGATCCCTTGAATGCTTCTCTTAAAG, were used to amplify the region of the mouse ERα cDNA prior to the Not I site through the splice site of the third and fourth exons. At the junction of the third and fifth exons, a BamH I site was included in the primer sequences to aid in the cloning and verification of the variant ER. The PCR generated fragment was digested with Not I and BamH I enzymes and inserted into the Bluescript KS(−) plasmid. Primers P3 (forward), GTTGGATCCGCATACGGAAGACCGCCGA, and P4 (reverse), CATCAGAATCTCCAGCCAGG, were used to amplify the region from the splice site of the fourth and fifth exons to beyond the Xho I site in the mouse ER cDNA. This fragment was digested and inserted into the BamH I and Xho I sites of the vector containing the Not I/BamH I mouse ER fragment. The Not I/Xho I fragment from the mouse ER cDNA from the MOR-100 vector (kindly provided by M. Parker) [37] was replaced with PCR generated sequences containing the deletion. The ERΔ3 cDNA was removed with EcoR I for insertion into the final vector.
The BamH I-Sal I fragment from the MT-mER construct [38] containing the splicing and polyadenylation signals was inserted into the pUC18 plasmid. Since splicing has been shown to enhance expression of some cDNA transgenes [39, 40], this fragment, which contains the portion of the pKCR2 vector [41] with rabbit β-globin exons and one intron, was included to provide splicing signals for the ERΔ3 transgene. The β-globin sequences are present only in the untranslated sequences of the ERΔ3 transcript.
A murine viral enhancer was included in the vector to augment expression of the transgene. The EcoR I-BamH I fragment containing the Harvey murine sarcoma virus (HaMuSV) LTR (kindly provided by M. Ostrowski) [42] was inserted into the vector containing the β-globin sequences. The EcoR I site of the HaMuSV enhancer was converted to a Sal I site using linkers. The original intent of the ERΔ3 transgenic mice was to target expression of the variant to osteoblasts using the osteocalcin (Bglap) promoter. The promoter regions used for the rat osteocalcin promoter did not confer tissue specificity; therefore, in the ERΔ3 mice, this promoter region appears to act as a generic basal promoter element. The rat osteocalcin promoter, from sequences −194 to +26, was PCR amplified from DNA isolated from ROS 17/2.8 cells using primers P5 (forward), GCGGATCCGCAGCCTCTGATTGTGTCCT, and P6 (reverse), GCAGATCTCTAGGTCTGCACCGAGTTGC. The primers included the BamH I (5′) and Bgl II (3′) restriction sites for ligation of the digested PCR fragment into the BamH I site of the vector containing the β-globin and enhancer sequences. The ERΔ3 cDNA was then inserted into the EcoR I site within the second β-globin exon. The plasmid sequences were removed by Sal I digestion and purified prior to microinjection. The transgene DNA (Fig. 1), was microinjected into the pronuclei of fertilized eggs from FVB/N mice according to standard protocols [43]. This strain of mice has high fecundity as well as clear eggs with large, prominent pronuclei [44].
Figure 1. Diagram of the ERΔ3 construct used to generate the transgenic mice.
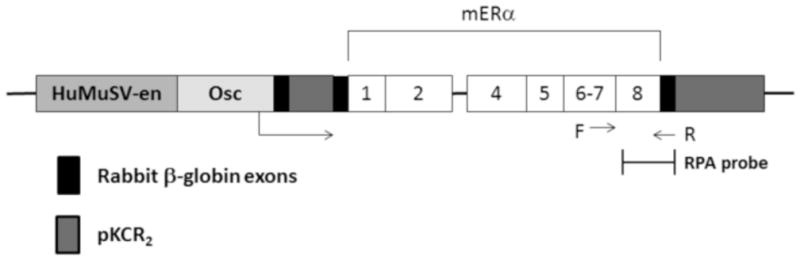
The exons encoding the mERα are numbered according to the coding exons only. The mouse ERα (mERα) cDNA sequences are designated with white bars and noting the loss of exon 3 (Δ3) sequences which correspond to exon 3 of human ERα. The sequences from the pKCR2 vector [38] include the two rabbit β-globin exons (black bars) and one intron (first dark gray bar between exons) as well as 3′ processing signals (second dark gray bar after mERα sequences). The mERα cDNA sequences were inserted into the second β-globin exon. The rat osteocalcin promoter sequences (Osc) are designated as a light gray bar. The enhancer sequences (en) from the U3 LTR of the Harvey murine sarcoma virus (HaMuSV) are indicated by the medium gray bar. The direction of transcription is noted from within the osteocalcin promoter by the arrow. The SV40 polyadenylation signals are located within the pKCR2 sequences. The region of the construct used for expression analysis by RPA is also indicated. The forward (F) and reverse (R) primers used for genotyping are noted. The thin bars at each end of the construct represent the plasmid sequences which are removed prior to microinjection into the FVB/N embryos.
Seven founders, 4 female and 3 male, were produced by the microinjections, but only 6 generated subsequent progeny. The highest levels of transgene expression were evident in lines D and F. Official designations for lines D and F are FVB/N-TgN(mERΔ3os)04Eme and FVB/N-TgN(mERΔ3os)06Eme, respectively.
2.2. Genotyping
Genomic DNA was isolated from tail biopsies [45] and analyzed using PCR [38] as previously described. Southern blots were also performed on the DNA from the founder mice as previously reported [38]. Lines D and F had copy numbers for the transgene of approximately 4 and 8, respectively (data not shown).
2.3. RNase Protection Assay Analyses
Total RNA was prepared using guanidine isothiocyanate-CsCl gradient procedure [46]. The RNase protection assay (RPA) was performed as previously described [38]. The presence of the vector sequences adjoining the ERα cDNA in the RPA probe (see Fig. 1) resulted in a smaller product for the ERα transcript compared to ERΔ3 mRNA for differentiating the two messages. The antisense cyclophilin probe used for the control was generated from the template pTRI-CYC (Ambion, Austin, TX). Quantitation of the RNA levels was determined with the Phosphoimager and ImageQuant software (Molecular Dynamics, Sunnyvale CA).
2.4. Real-time RT-PCR Analyses
RNA was prepared from mouse tissues using Absolutely RNA RT-PCR Miniprep Kit (Stratagene, La Jolla, CA) according to the kit instructions. The reverse transcriptase (RT) reaction was performed with qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD) prior to the PCR step. An aliquot of the RT reaction was amplified in an iCycler (Bio-Rad, Hercules, CA) with the BR SYBR Green SuperMix for iQ Systems (Quanta Biosciences, Gaithersburg, MD) and the specific primers for each gene using the following cycles: 1 cycle at 95°C for 90 sec followed by 50 cycles at 95°C for 15 sec and at 60°C for 45 sec. Primer sequences are listed in Table 1 for the mouse genes examined, including ERα, ERβ, ERΔ3, progesterone receptor (Pgr), and lactoferrin (Ltf). Relative mRNA levels were determined by the 2−ΔΔCt method by normalization to the cyclophilin A (Ppia) gene. Amplification of the mRNA was confirmed by comparison to the no RT control and by melting temperature determination. A subset of the RT-PCR samples were run on 2% NuSieve/0.7% agarose gel electrophoresis to verify the proper size product.
Table 1.
Primers for Real-Time RT-PCR Analysis
| Gene | Sequence |
|---|---|
| ERα (Esr1) | Forward: GTCCAGCTACAAACCAATGC Reverse: ATCTCTCTGACGCTTGTGCT |
| ERβ (Esr2) | Forward: AAATGTGCTATGGCCAACTTC Reverse: TTGGCGCTTGGACTAGTAAC |
| ERΔ3 | Forward: ATTCAAGGGATCCGCATAC Reverse: ACAAGGCAGGGCTATTCTTC |
| Lactoferrin (Ltf) | Forward: ACAATGCTGGAGATGTGGCT Reverse: TTGTCATTCGTGCTTCGGGA |
| PR (Pgr) | Forward: TGGGAGCTGCAAGGTCTTCT Reverse: TGCCAGCCTGACAACACTTT |
| Cyclophilin A (Ppia) | Forward: TATCTGCACTGCCAAGACTG Reverse: ACAGTCGGAAATGGTGATCT |
2.5. Western Blot Analysis
Total protein homogenates were prepared from uteri from individual WT, line F, and line D female mice at age 3 months in estrus; 10 ug of protein was loaded on a 10% NuPage Bis-Tris mini gel and MOPS buffer (Life Technologies, Grand Island, NY). The gel was run at 200 volts for 2 hours and transferred to nitrocellulose membrane using the iBlot transfer system (Life Technologies, Grand Island, NY). The membrane was incubated overnight with primary antibody for ERα (1:1000; MC-20 Santa Cruz Biotechnology, Santa Cruz, CA) and anti-rabbit IgG secondary (1:5000; Cell Signaling Technology, Danvers, MA). Signal was developed as directed with ECL Prime reagent (GE Healthcare Biosciences, Pittsburgh, PA).
2.6. Animal Care
All procedures involving the mice were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals with approved protocols by the NIEHS and Duquesne University Animal Care and Use Committees. All mice were housed with 12 h:12 h light:dark cycles in a temperature controlled room with diet and water provided ad libitum.
2.6.1. DES Studies
Wild-type FVB/N female mice (National Cancer Institute Animal Program, Bethesda, MD) were bred with hemizygous ERΔ3 males. Mice for this study were fed NIH 31 chow. The resulting WT and ERΔ3 progeny were treated with daily injections of DES (Sigma Chemical Co., St. Louis, MO) dissolved in corn oil at the dose of 2 μg/pup/day on days 1–5 after birth. Previous studies have shown this dose to be effective at inducing uterine abnormalities and tumors in CD-1 [16] and FVB/N mice [20]. Controls were left untreated. At 3 weeks of age the mice were weaned and genotyped using tail DNA. The mice were housed four or five females per cage. The mice were euthanized at 8 or 12 months of age and subjected to a complete necropsy. Tissues for histological examination were excised and fixed in 10% buffered formalin, embedded in paraffin, and sectioned at 6 μm. The sections were stained with hematoxylin and eosin and evaluated under a light microscope by the study pathologist (BCB). Some regions of pathological alteration noted initially were serially sectioned for further analysis.
2.6.2. ERΔ3 and WT (FVB/N) Mice for Examining Estrogen-responsive Gene Expression
ERΔ3 line D female and line F mice and FVB/N mice (Jackson Laboratories, Bar Harbor, ME) were euthanized at age 3 months in estrus. Stage of cycle was determined by vaginal smears stained with Dif-stain kit (IMEB Inc., San Marcos, CA) prior to necropsy. Uteri were frozen in liquid nitrogen for later RNA analyses for Pgr and Ltf RNA levels by real-time RT-PCR. Blood was collected by cardiac puncture in euthanized mice for the hormone assays.
2.6.3. ERΔ3 and αERKO Crossbred Mice
Dizygous ERΔ3 mice (FVB/N strain) were bred with heterozygous αERKO mates (C57BL/6 strain) to generate heterozygous αERKO/hemizygous ERΔ3 mice. These progeny were then mated with heterozygous αERKO mice to ensure all genotypes expressing ERΔ3 would be hemizygous. This breeding scheme generated littermates expressing ERα (WT), ERα and ERΔ3 (ERΔ3), no ERα (αERKO), and ERΔ3 without ERα (αERKO/ERΔ3) on a mixed background strain (FVB/N and C57BL/6), which were used for RNA analyses by real-time RT-PCR. Genotyping for ERΔ3 is described in section 2.2 and for the disruption of the ERα gene in the αERKO mice is previously reported [47]. Female mice with the desired genotypes were euthanized in estrus at age 3 months and the uteri quick frozen for later RNA analysis.
2.7. 17β-estradiol (E2) and Progesterone (P4) Serum Levels
Serum E2 and P4 were determined with the Double Antibody Estradiol and Coat-a-Count Progesterone kits (Siemens, Los Angeles, CA) on mice in estrus at necropsy.
2.8. Statistical Analyses
Statistics were performed using Graphpad Prism 5.0 software (San Diego, CA). Significance was designated for p values less than 0.05.
3. RESULTS
3.1. Generation of ERΔ3 transgenic mice
Two of the ERΔ3 transgenic lines, designated D and F, expressed the transgene in reproductive and non-reproductive tissues by the RNase protection assay (RPA) and real-time reverse transcriptase-polymerase chain reaction (RT-PCR) (Table 2). All organs thus far tested in both lines and genders expressed the transgene (Table 2). For female mice in both lines, similar expression was observed in the adrenal glands, bone, ovary, and uterus; but, line D had higher expression in the liver and mammary gland (Fig. 2A). In line F, the bone, brain, gonads, and liver had similar expression in male and female mice, but the male expressed the ERΔ3 transgene at higher levels in the kidney (Fig. 2B). As expected, the relative level of the ERΔ3 transgene expressed in the uterus was considerably less abundant than the endogenous ERα transcript, 1:7 (line D) to 1:9 (line F) ratio. In other tissues which typically express lower levels of ERα, such as the kidney and bone, the levels of the ERΔ3 transgene message exceeded the levels of the WT ERα. The ovary, which has high expression of ERβ [48], is the only organ in both lines in which the levels of ERΔ3 did not exceed ERβ. However, individual variations likely occur for these levels, as is observed with some tissues from the mice analyzed by RPA versus real-time RT-PCR (see Table 2). A receptor protein that corresponds to the expected size for ERΔ3 (approximately 61 kDa) was also detected in the uteri of line F and line D mice in addition to the 66 kDa WT ERα (Fig. 2C).
Table 2.
Relative Transcript Levels of ERΔ3 to ERα and ERβ in Tissues from Lines D and F Adult Transgenic Mice
| Line/Sex | Tissue | RNA Detection Method | Relative to ERα | Relative to ERβ |
|---|---|---|---|---|
| Line D ♀ | Adrenals | RT-PCR | ↑ (3:1) | ↑ (1400:1) |
| Bone (skull) | RT-PCR | ↑ (10:1) | ↑ (5200:1) | |
| Kidney | RPA | ↑ (9:1) | nd | |
| Liver | RT-PCR & RPA | ↑ (4:1 & 5:1) | ↑a | |
| Lung | RPA | ↑ (18:1) | nd | |
| Mammary gland | RT-PCR & RPA | ↑ (14:1 & 7:1) | ↑ (15800:1) | |
| Ovary | RT-PCR | = (0.7:1) | = (0.7:1) | |
| Uterus | RT-PCR & RPA | ↓ (0.1:1 & 0.4:1) | ↑ (100:1) | |
| Line F ♀ | Adipose | RT-PCR | ↓ (0.5:1) | ↑ (1500:1) |
| Adrenals | RT-PCR | ↑ (2:1) | ↑ (2400:1) | |
| Aorta | RT-PCR | = (1:1) | ↑a | |
| Bone (skull) | RT-PCR | ↑ (3:1) | ↑a | |
| Brain | RT-PCR | ↑ (23:1) | ↑ (110:1) | |
| Kidney | RT-PCR & RPA | ↑ (33:1 & 5:1) | ↑ (8500:1) | |
| Liver | RT-PCR & RPA | ↓ (0.2:1 & 0.2:1) | ↑a | |
| Lung | RPA | ↑ (6:1) | nd | |
| Mammary gland | RT-PCR & RPA | ↓ (0.6:1 & 0.6:1) | ↑ (4100:1) | |
| Ovary | RT-PCR | = (0.8:1) | = (1:1) | |
| Pituitary | RT-PCR | ↓ (0.1:1)b | nd | |
| Uterus | RT-PCR & RPA | ↓ (0.1:1c & 0.7:1) | ↑ (190:1) | |
| Line F ♂ | Bone (skull) | RT-PCR | ↑ (3:1) | ↑ (1500:1) |
| Brain | RT-PCR | ↑ (39:1) | ↑ (280:1) | |
| Kidney | RT-PCR | ↑ (23:1) | ↑ (30100:1) | |
| Liver | RT-PCR | = (0.9:1) | ↑ (2100:1) | |
| Testes | RT-PCR | ↑ (3:1) | ↑ (2300:1) |
ERβ not detected by real-time RT-PCR
n=3 versus n=1 for other tissues
n=6
RT-PCR: real-time RT-PCR (reverse transcriptase-polymerase chain reaction); for ERΔ3, ERα, and ERβ RPA: ribonuclease protection assay; for ERΔ3 and ERα on a different female mouse than for RT-PCR nd: not determined (tissue not examined for ERβ expression)
↑, ERΔ3 levels higher than ERα/ERβ; =, ERΔ3 levels are similar to ERα/ERβ; ↓, ERΔ3 levels lower than ERα
Figure 2. Comparison of transgene expression in multiple tissues of adult ERΔ3 male and female mice and detection of ERα protein in WT and ERΔ3 female mice.
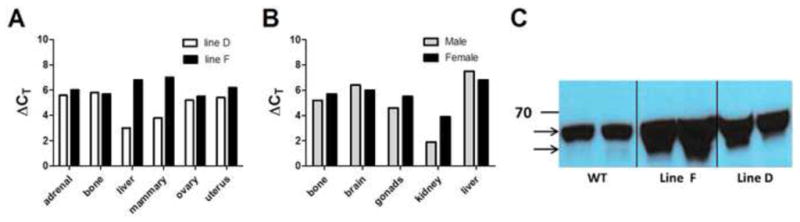
Total RNA was analyzed for ERΔ3 transgene transcripts by real-time RT-PCR from tissues dissected from a line D and line F female and line F male mouse. ΔCT value is shown for each tissue based on normalization to the cyclophilin A (Ppia) gene. Lower ΔCT values indicate higher expression levels. Expression relative to ERα and ERβ for these tissues is shown in Table 2. A. Comparisons between female mice for lines D and F are shown for ERΔ3 expression levels (n=1). B. Relative expression between male and female line F mice is indicated for the ERΔ3 transgene (n=1). Gonads represent the ovaries for the female and testes for the male mice. C. A protein in the appropriate size range for the ERΔ3 variant was detected by western blot analysis in the uteri of line D and F ERΔ3 mice in estrus using an antibody directed against the carboxy-terminus of ERα (MC-20). The images for the two uteri each from 3-month-old WT (FVB/N), line F, and line D mice were obtained from the same 10% gel and blot. The band for WT ERα is noted by the upper arrow at 66 kDa and the lower arrow corresponds to the smaller size expected for the ERΔ3 variant (approximately 61–62 kDa). The 70 kDa marker is indicated at the line marked with 70.
There is no evidence of infertility or diminished reproductive functions in the males or females in lines D and F. In the hemizygous mice, the only evident phenotype occurs in line F females, which develop spontaneous cataracts after puberty [49]. In dizygous mice, the growth of line D male and female mice is stunted, resulting in adult body weights that are less than half the weight of the WT (FVB/N), hemizygous line D and F, or dizygous line F mice (data not shown). It is unknown if the stunted growth in dizygous line D mice is related to transgene expression or due to the disruption of an unknown gene important for growth at the site of transgene insertion. Thus, only hemizygous mice were studied for DES-induced uterine cancer.
3.2. DES-Induced Uterine Cancer
To investigate the effects of ERΔ3 expression on DES-induced uterine cancer, DES (2 μg/pup) was administered to ERΔ3 and WT pups daily from birth through post-natal day 5. Both line D and F female mice were examined to ensure that the resulting outcomes would be due to the ERΔ3 transgene and not related to the site of transgene insertion, which would be random and, thus, unique for each line. The neonatal DES treatment induced strong cataracts in both male and female ERΔ3 mice from lines D and F, which were evident when the pups first opened their eyes [49]. In the reproductive tract, non-malignant abnormalities, which are common after neonatal DES treatment, were evident in the ERΔ3 and WT female mice (FVB/N strain). As with other strains [50], no corpora lutea were detected in the ovaries in the DES-treated ERΔ3 and WT mice, suggesting that normal cycling did not occur. In addition, 89% of the ERΔ3 females at 8 months of age and all of the ERΔ3 and WT females at 12 months of age had progressive proliferative lesions of the oviduct. All DES-exposed females also displayed excessive keratinization of the vagina (data not shown). The uteri of the treated mice for both genotypes were hypoplastic with minimal gland development. The glands that were observed were located at the cervical-uterine junction and were often hyperplastic. There were also “gland-like” structures in the cervix. Therefore, due to these similar effects in WT and ERΔ3 mice, the expression of the ERΔ3 transgene did not compound or diminish the previously reported effects of DES on reproductive tract development.
Besides the non-malignant phenotypes, neonatal DES exposure in hemizygous ERΔ3 (lines D and F) and WT littermates also resulted in the appearance of uterine adenocarcinomas. The histological appearance of the tumors in the WT FVB/N mice has been reported previously [20]. The malignant lesions usually arose at the junction of uterine and cervical epithelium in both WT and transgenic DES-treated females. Focal areas of squamous metaplasia were also evident in a few of the tumors. At age 8 months, a significantly higher number of ERΔ3 females developed uterine tumors compared with WT mice (p< 0.016, Fisher’s exact test; Table 3). No significant differences in uterine cancer incidence were noted between the two ERΔ3 lines, with 9/13 line D and 12/13 line F females having uterine tumors by 8 months of age, and the levels of transgene expression in the uteri of these two lines were comparable (Fig. 2).
Table 3.
Incidence of Uterine Adenocarcinomas in Wild-type and ERΔ3 Transgenic Mice with and without DES Exposure
| Age | UNTREATED
|
DES-TREATED
|
||
|---|---|---|---|---|
| WT | ERΔ3 | WT | ERΔ3 | |
| 8 months | 0% (0/21) | 0% (0/28) | 49% (17/35) | 81% (21/26)* |
| 12 months | 0% (0/17) | 10% (2/20) | 72% (13/18) | 86% (12/14) |
p<0.016, Fisher’s Exact Test
DES. Diethylstilbestrol; WT, wild-type (FVB/N strain)
At 12 months, the percentage of WT mice with neoplastic uterine lesions increased, but remained lower than the incidence in ERΔ3 mice at 8 and 12 months of age. However, the difference between the two genotypes was not significant at age 1 year. In addition to a higher incidence at younger ages, the uterine adenocarcinomas detected in the ERΔ3 females were more locally invasive and involved more of the uterine horn compared to tumors in WT females (data not shown). These data suggest that DES-induced tumor development is accelerated in the ERΔ3 transgenic mice. Unexpectedly, two untreated ERΔ3 females had uterine adenocarcinomas at 12 months of age (Table 3). The presence of this malignant lesion was not observed in the WT FVB/N females in our study or in those previously reported at ages 14 or 24 months [51]. These data indicate that the ERΔ3 female mice may have a slight predisposition for developing uterine adenocarcinomas, even in absence of DES exposure.
The unexpected higher incidence of uterine adenocarcinomas with neonatal DES treatment in ERΔ3 mice mimicked the incidence observed in transgenic mice overexpressing mouse ERα (mERα), MT-mER mice [20]. At 8 months of age, both ERΔ3 and MT-mER mice had significantly higher incidence of DES-induced uterine cancer compared to the WT group, which included WT mice from both studies (Fig. 3). Additionally, the tumor incidence was not significantly different between the two transgenic models. These results suggest ERΔ3 did not reduce estrogen activity in the uterus.
Figure 3. Similar increased uterine adenocarcinoma incidence in ERΔ3 and MT-mER transgenic mice compared to wild-type mice.
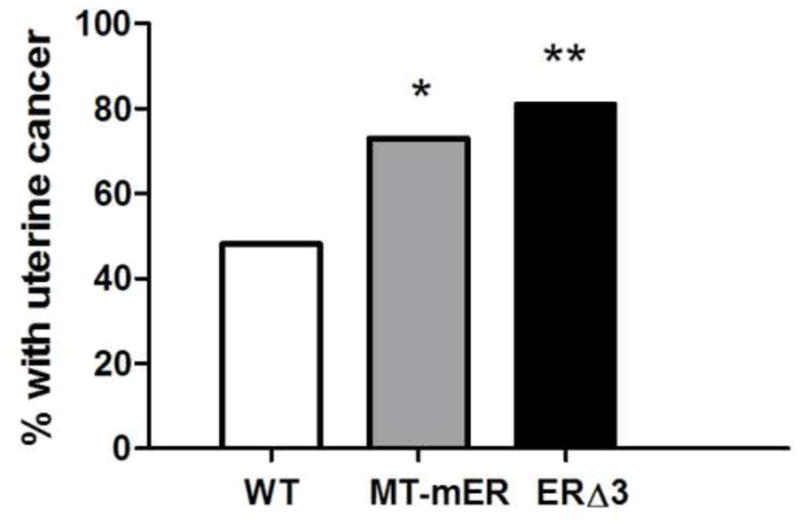
The percent of female mice with uterine adenocarcinoma detected by histopathology is depicted at age 8 months. The number of mice examined for the ERΔ3 mice (black bar) at 8 mo (n=26, from Table 3) and for the previously reported MT-mER mice (grey bar) at 8 mo (n=26) [20]. Wild-type (WT) mice (open bar) are shown with the combined incidence in WT mice (FVB/N strain) for both studies for 8 mo of age: 47.5% for the combined studies (n=59) compared to 49% for the ERΔ3 study (see Table 1) and 46% for the MT-mER study [20]. Chi-squared analysis demonstrated statistical significance for the 8 mo age groups (p=0.0054). Comparisons between the genotypes by the Fisher’s exact test demonstrated significant increases in uterine cancer incidence for MT-mER (*, p<0.035) and for ERΔ3 mice (**, p=0.0046) compared to the combined WT group; however, the difference in incidence for the ERΔ3 vs. MT-mER mice was not significant (p> 0.05, Fisher’s exact test).
3.3. Estrogen-Responsive Gene Expression in the Uterus
The accelerated onset of uterine cancer does not coincide with the predicted ability of ERΔ3 to inhibit ERα action. To test the potential of ERΔ3 to inhibit uterine estrogen responsive genes in the presence of WT ERα, the expression of progesterone receptor (Pgr) and lactoferrin (Ltf) was examined in line F ERΔ3 and WT uteri by real-time RT-PCR. No suppression was observed as their RNA levels were similar for WT and ERΔ3 mice (FVB/N strain) in estrus (when estrogen levels are high) for both the progesterone receptor (PR) (Fig. 4A) and lactoferrin genes (Fig. 4B).
Figure 4. Estrogen responsive genes in the uterus are not repressed by ERΔ3.
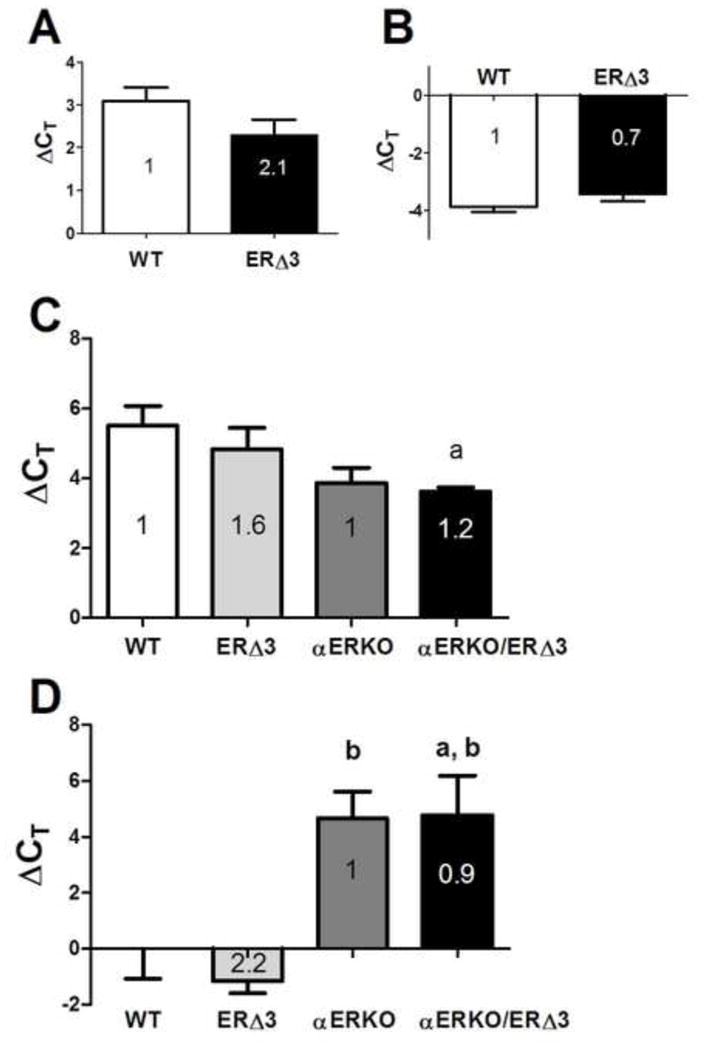
Uterine total RNA obtained from 3-month-old female mice in estrus were analyzed for each genotype. Relative mRNA expression was determined by real-time RT-PCR for the progesterone receptor (Pgr), panels A and C, and lactoferrin (Ltf) genes, panels B and D, with normalization to the cyclophilin A (Ppia) gene. In panels A and B, the average ΔCT values are displayed for WT (FVB/N, n=8) and ERΔ3 (line F, FVB/N strain) mice (n=8), with the fold difference determined by the 2−ΔΔCt method indicated within each bar: A. uterine progesterone receptor (Pgr) expression; B. uterine lactoferrin (Ltf) expression. Expression levels in the ERΔ3 mice were not significantly different than in WT uteri (p>0.05, Mann Whitney test). In panels C and D, uteri from intact female mice with a mixed background strain obtained by crossbreeding ERΔ3 (line F, FVB/N strain) and αERKO (C57BL/6 strain) were analyzed. The average ΔCT values for the 4 genotypes, including WT (n=4), ERΔ3 (hemizygous, +/−; n=4), αERKO (homozygous for the ERα disruption, −/−; n=4), and αERKO/ERΔ3 (−/− and +/−, respectively; n=6) are depicted. The fold difference of ERΔ3 relative to WT mice (1.0) and αERKO/ERΔ3 relative to αERKO determined by the 2−ΔΔCt method are indicated within each bar. C. Uterine progesterone receptor (Pgr) expression was significant (p=0.025, 1-way ANOVA) with a significant difference between WT and αERKO/ERΔ3 mice (Tukey’s test). Fold differences relative to WT (1) are 3.1 for αERKO and 3.7 for αERKO/ERΔ3 and relative to ERΔ3 (1) are 1.9 for αERKO and 2.3 for αERKO/ERΔ3. D. Uterine lactoferrin (Ltf) levels were significant (p=0.0042, 1-way ANOVA). The negative ΔCT levels reflect higher Ltf expression compared to the normalizing Ppia gene. Fold differences relative to WT (1) are 0.04 for both αERKO and αERKO/ERΔ3 and to ERΔ3 (1) are 0.2 for the two αERKO genotypes. Genotype comparisons for panels C and D were analyzed by Tukey’s test: a, p<0.05 compared to WT; b, p<0.05 compared to ERΔ3.
To determine how ERΔ3 influences the expression these estrogen-responsive genes in the absence of WT ERα, line F ERΔ3 mice were crossbred with αERKO mice (C57BL/6 strain). As observed above in the FVB/N strain (Fig. 4A–B), relative uterine expression of PR (Fig. 4C) and lactoferrin transcripts (Fig. 4D) also were not significantly different between the WT and ERΔ3 progeny on the mixed strain background (FVB/N and C57BL/6). In the absence of ERα, higher PR expression was detected in αERKO/ERΔ3 mice compared to WT mice (3.7 fold; p<0.05, Tukey’s test). In contrast, both αERKO and αERKO/ERΔ3 uteri had significantly lower lactoferrin expression compared to uteri from WT (0.04-fold) and/or ERΔ3 (0.02-fold) littermates (p<0.05, Tukey’s test; Fig. 4C–D). However, for both genes, no difference was observed between the αERKO and αERKO/ERΔ3 mice. Collectively, these data demonstrate that ERΔ3 does not modify uterine expression of these two estrogen-responsive genes compared to mice without ERΔ3, either in the presence (WT vs. ERΔ3) or absence of ERα (αERKO vs. αERKO/ERΔ3).
3.4 Circulating Estradiol and Progesterone Levels
A potential mechanism for enhanced estrogen action in ERΔ3 mice could be due to alterations in circulating hormone levels. ERΔ3 is expressed at equivalent levels as ERα and ERβ in the ovary of both lines and at decreased levels in the pituitary in line F female mice (7:1 ratio for ERα:ERΔ3). If ERΔ3 influences estrogen or progesterone synthesis through its expression in the ovaries and/or pituitary or other tissues, the resulting levels could influence tumor development in post-pubertal DES-treated mice. Although progesterone (P4) levels were similar in both genotypes, 17β-estradiol (E2) levels were significantly increased in ERΔ3 mice (p=0.023, Mann Whitney test) compared to WT mice in estrus (Fig. 5).
Figure 5. Serum 17β-estradiol levels are elevated in ERΔ3 mice.
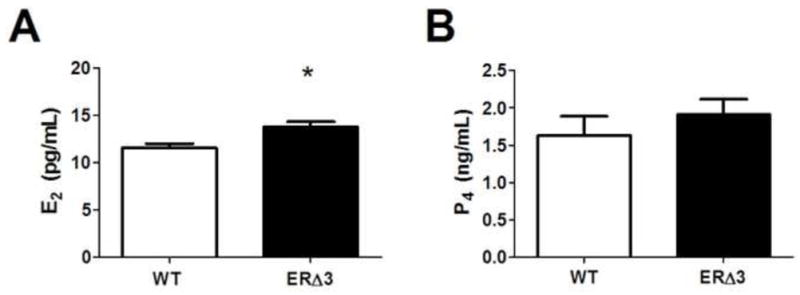
Serum hormone levels were measured in 3-month-old female mice in estrus. A. 17β-estradiol (E2) serum levels are increased in the ERΔ3 mice compared to wild-type (WT) mice (FVB/N strain). The 29 ERΔ3 mice examined includes female mice from lines F (n=16) and D (n=13); sera from 13 WT mice were analyzed. * p=0.023, Mann Whitney test. B. Progesterone (P4) serum levels analyzed in 14 WT FVB/N female mice and in 27 ERΔ3 mice (n=16 for line F and n=11 for line D) were not significant (p>0.05, Mann Whitney test).
4. DISCUSSION
Neonatal exposure to DES resulted in an increased incidence of uterine tumors in 8-month-old ERΔ3 females compared with WT mice. The similar effect in both lines D and F indicates that the increased tumor incidence is due to ERΔ3 expression versus model-specific effects from the site of transgene insertion. The lack of significance at 1 year indicates that DES-induced tumors appear at younger ages in ERΔ3 mice compared to WT mice. Therefore, contrary to our predicted results of providing cancer protection, these data indicate that expression of the ERΔ3 variant accelerates the development of hormonally-induced uterine cancer.
ERα is required for DES to induce the adverse effects on the female reproductive tract as evidenced by the lack of effects in neonatal-treated αERKO mice [19]. In MT-mER transgenic mice overexpressing ERα (which express both the WT mERα transgene plus the normal, endogenous ERα gene), neonatal DES treatment also induced an earlier onset of uterine adenocarcinomas [20]. The tumor results in MT-mER mice fit with the premise that estrogens acting through ERα are promoting tumor development; however, the similar tumor incidence in ERΔ3 females does not (Fig. 3). The paradox of both transgenic models accelerating DES-induced uterine tumor development despite expressing ERα receptors with opposite activities suggests that ERΔ3 expression resulted in increased versus decreased estrogen activity in the uterus.
Before the generation of the ERΔ3 transgenic mice, the ability of the ERΔ3 variant to inhibit WT ERα activity had only been tested in transfected mammalian cells. Transfecting a 1:10 ratio of WT ERα to ERΔ3 vectors into T47D breast cancer cells was found to inhibit approximately 80% of WT receptor activity [28]. Although the actual intracellular ratio of ERα:ERΔ3 receptors is unknown (since the 1:10 ratio reflects the relative levels of the transfected vectors and not the quantity of each receptor in an individual cell), higher or equal levels of dominant negative receptors are usually required to inhibit the activity of the WT receptor. With the inherently high levels of ERα in the uterus, ERΔ3 transcript levels do not exceed those of ERα in the uterus of lines D and F ERΔ3 mice (Table 2). A previous study in transfected breast cancer cells found that the ratio of ERΔ3:ERα transcripts also reflected their protein levels [52]. The high ratio of ERα to ERΔ3 transcripts (≥7:1) may be one reason that ERΔ3 did not provide protection against DES-induced uterine cancer and that the transcript levels of the PR and lactoferrin genes are not reduced in the ERΔ3 versus WT uterus, especially for lactoferrin which contains a palindromic ERE in its promoter [53].
The findings with the tested estrogen-responsive genes indicate ERΔ3 might not be expected to inhibit uterine cancer development, but would not explain the accelerated onset. Dominant negative activity for ERΔ3 has only been demonstrated with classical ERE-induced gene expression [30]. In contrast, ERα receptors with DBD mutations or deletions can activate transcription of estrogen-responsive genes by non-classical mechanisms through interactions with other transcription factors, such as the AP-1 family and Sp1 [4]. In transfected HeLa cells, ERΔ3 inhibits expression of an ERE-regulated reporter gene, but stimulates expression of a reporter construct regulated by an AP-1/ERE half site [30]. A study in cultured breast cancer cells also provides direct evidence that human ERα and mouse ERα with deletions in the second zinc finger stimulate non-classical pathways in an Sp1-regulated reporter gene [54]. These findings suggest ERΔ3 could stimulate versus inhibit genes regulated by these transcription factors. Thus, the lack of inhibition for the PR and lactoferrin genes in the ERΔ3 uteri in estrus may be related to their regulation at Sp1 and AP-1 sites, with and without half-ERE sites, which have been identified in the promoters of the PR [55–58] and lactoferrin genes [53]. Although ERΔ3 did not significantly increase expression of these two genes, other untested genes regulated by non-classical ER mechanisms, which are involved in promotion of the DES-induced uterine tumors, may be modified by ERΔ3. Additionally, PR and lactoferrin genes may be modified by ERΔ3 at other stages of the estrous cycle.
The lactoferrin, but not PR, gene contains a palindromic ERE [53, 58], which may explain why only its expression was significantly reduced in αERKO and αERKO/ERΔ3 mice. The expression of these genes in αERKO mice (Fig. 4C–D) are in accord with a previous study showing transcript levels for PR are unaffected, but lactoferrin mRNA is substantially repressed in the uteri of αERKO versus WT mice [59]. Additionally, treatment with estradiol did not modify the expression levels of either gene in ovariectomized αERKO mice, implicating ERα in regulating their expression [59]. ERΔ3 did not modify the constitutive levels of PR and lactoferrin transcripts in the uteri of αERKO/ERΔ3 mice compared to αERKO animals, suggesting that ERΔ3 expression is not sufficient to stimulate the preexisting levels of either gene through the AP-1 and Sp1 sites or that the genes may already be maximally stimulated in the αERKO mice to prevent further stimulation by ERΔ3. However, PR expression in the uteri of αERKO/ERΔ3 mice was significantly higher than their WT littermates (Fig. 4C), which may suggest ERΔ3 has a slight influence on PR expression.
The regulation of estrogen responses by non-classical mechanisms has been reported to be important in uterine epithelial proliferation. NERKI transgenic mice were developed that express ERα with a point mutation in first zinc finger of the DBD, which is unable to activate an ERE, does not have dominant negative activity, but retains non-classical ER signaling [60]. In NERKI mice lacking WT ERα (KIKO mice), estrogen did not induce a uterotropic response [61]. These findings demonstrate that non-classical signaling by the NERKI mutant in the absence of WT ERα is not sufficient for estrogen-induced uterine proliferation. However, in intact NERKI female mice expressing WT ERα, the uteri appear hypersensitive to estrogen since they are enlarged with cystic endometrial hyperplasia [60]. Although the NERKI and ERΔ3 receptors differ in action and structure and the ERΔ3 mice are fertile, these models have similarities since ERΔ3 transgenic mice also express WT ERα and a variant that can stimulate non-classical, but not classical, signaling. Therefore, in ERΔ3 mice, expression of the variant may augment WT ERα-induced endometrial proliferation, especially in the elevated E2 environment, which could ultimately lead to earlier tumor formation.
Since the ERΔ3 transgene is expressed in most tissues (Table 2), uterine tumor development may be influenced by ERΔ3 expression in other estrogen target tissues. This potential is supported by the higher circulating levels of E2, which would be due to ERΔ3 expression outside the uterus, such as in the pituitary gland and/or ovary (Table 2). Due to the known effects of excess estrogen on uterine cancer risk [1, 62], the higher E2 levels or the resulting imbalance in E2 to P4 levels may contribute to the earlier cancer development in the ERΔ3 mice.
DES-induced uterine cancer is influenced by both the neonatal and post-pubertal stages of development [16]. Consequently, the ERΔ3 transgene may influence either or both of these developmental stages: 1) during development and differentiation of the immature reproductive tract, when DES exposure occurs, and 2) after the onset of puberty and estrogen cycling. However, since latency is affected, these findings suggest a greater effect of the ERΔ3 variant on the mature uterus than during developmental DES exposure. Thus, in post-pubertal mice, elevated E2 levels and/or non-classical signaling may be promoting the growth of tumors initiated in the neonatal uterus to allow their detection at younger ages. Similar post-pubertal ERΔ3 actions are likely overstimulating uterine proliferation in the untreated adult female mice since two ERΔ3 females not treated with DES also developed uterine adenocarcinomas (Table 3).
In humans, transcripts for ERΔ3 [63] and other ERα splicing variants [26, 64, 65] have been detected in the normal human endometrium. No difference in ERΔ3 endometrial expression was detected between infertile and fertile women and patients with endometriosis. These findings suggest that the ERΔ3 variant does not influence fertility [63], which agrees with the findings in the ERΔ3 transgenic mice. Additionally, the ERΔ3 variant mRNA has been detected in human endometrial hyperplasia, but not in endometrial cancer [66]. Based on the results in the ERΔ3 transgenic mice, expression of ERΔ3 in the human uterus would not be expected to be protective. If sufficient levels of this variant were expressed in the human uterus, such as the 11–14% relative to ERα in the mouse uteri, a slight increase in uterine cancer risk may be possible, as was observed in the untreated mice (Table 3). Whether elevated circulating E2 levels would also be required to increase uterine cancer risk by ERΔ3 expression in other tissues, like the pituitary gland, is unknown; but, ERΔ3 variant transcripts have been reported in human pituitary adenomas and the normal rat pituitary gland [67, 68].
The tumor and gene expression results from this study do not provide direct evidence that ERΔ3 has dominant negative activity in vivo; however, dominant negative effects may be most evident in vivo in non-uterine tissues with lower ERα expression, such as the mammary gland. This premise is supported by the significant delay in mammary cancer in female ERΔ3 transgenic mice (line F) compared to mice not expressing the ERΔ3 transgene (Davis et al., unpublished results). Thus, ERΔ3 inhibits mammary tumor development despite the higher circulating estrogen levels. However, besides ERα levels, other tissue-specific characteristics may be related to the contrasting results on cancer onset in the uterus and mammary gland of ERΔ3 mice. For example, responses that differ between these two tissues include the contrasting actions of the non-classical signaling ERα mutant in NERKI mice, which have hyperplasia in the uterus and hypoplasia in the mammary gland [60], and the differing regulation of the PR promoter by various estrogenic ligands [55].
4.1. Conclusions
ER variants have been detected in many normal, premalignant, and cancerous tissues in humans and animals and speculated to have a role in normal physiology and cancer development, growth, endocrine responsiveness; however, studies in animal models are needed to understand their in vivo actions. Using a transgenic mouse model, this study demonstrates that expression of the ERΔ3 transgene can alter events important in normal uterine physiology that result in the earlier appearance of malignant lesions. Although ERΔ3 inhibits transcription from ERE-regulated genes in a dominant negative manner, it also stimulates expression of promoters with AP-1 and Sp1 sites [30, 54]. In addition, E2 levels were elevated due to ERΔ3 actions in other tissue(s). Therefore, the variant expression both in and outside the uterus may have a role in uterine cancer development in the ERΔ3 mice. In women expressing ERΔ3 mRNA in the uterus, the variant would be unlikely to protect the uterus from estrogen-induced uterine cancer due to its ability to stimulate non-classical signaling. The ERΔ3 transgenic mice provides a novel model system for future investigations into the roles of this ERα variant in cancer development, progression, and treatment as well as in the normal physiology of estrogen target tissues.
Highlights.
ERΔ3 transgenic mice express the mouse ERα variant lacking the second zinc finger
Many tissues express ERΔ3; a higher ratio of ERα to ERΔ3 occurs in the uterus
Neonatal diethylstilbestrol accelerated uterine cancer in ERΔ3 versus wild-type mice
Estrogen-responsive genes (Pgr, Ltf) are not modified by ERΔ3 in the uterus
17β-estradiol serum levels are higher in ERΔ3 than wild-type mice
Acknowledgments
We would like to thank William Tally and Mariana Molina for their skillful assistance with the mice and in processing the tissues for the histopathology. Support for the tumor study was provided through Z01ES70065 to KSK by the Division of Intramural Research of the NIEHS/NIH.
Abbreviations
- αERKO
estrogen receptor alpha knockout
- DES
diethylstilbestrol
- DBD
DNA binding domain
- ERα
estrogen receptor alpha
- ERβ
estrogen receptor beta
- ERE
estrogen response element
- PR
progesterone receptor
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tinelli A, Vergara D, Martignago R, Leo G, Malvasi A, Tinelli R. Hormonal carcinogenesis and socio-biological development factors in endometrial cancer: a clinical review. Acta Obstet Gynecol Scand. 2008;87:1101–13. doi: 10.1080/00016340802160079. [DOI] [PubMed] [Google Scholar]
- 2.Chen GG, Zeng Q, Tse GM. Estrogen and its receptors in cancer. Med Res Rev. 2008;28:954–74. doi: 10.1002/med.20131. [DOI] [PubMed] [Google Scholar]
- 3.Pettersson K, Grandien K, Kuiper GG, Gustafsson JA. Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol. 1997;11:1486–96. doi: 10.1210/mend.11.10.9989. [DOI] [PubMed] [Google Scholar]
- 4.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263–75. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–6. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, et al. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci U S A. 1996;93:12626–30. doi: 10.1073/pnas.93.22.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147:5557–63. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- 8.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–6. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 9.Holt LH, Herbst AL. DES-related female genital changes. Semin Oncol. 1982;9:341–8. [PubMed] [Google Scholar]
- 10.McLachlan JA, Newbold RR. Cellular and molecular mechanisms of cancers of the uterus in animals. Prog Clin Biol Res. 1996;394:175–82. [PubMed] [Google Scholar]
- 11.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 12.Shelby MD, Newbold RR, Tully DB, Chae K, Davis VL. Assessing environmental chemicals for estrogenicity using a combination of in vitro and in vivo assays. Environ Health Perspect. 1996;104:1296–300. doi: 10.1289/ehp.961041296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newbold RR, McLachlan JA. Transplacental hormonal carcinogenesis: Diethylstilbestrol as an example. In: Huff J, Boyd J, Barrett JC, editors. Cellular and molecular mechanisms of hormonal carcinogenesis: Environmental influences. New York: Wiley-Liss, Inc; 1996. pp. 131–47. [PubMed] [Google Scholar]
- 14.Bern HA. The fragile fetus. In: Colburn T, Clement C, editors. Chemically-induced alterations in sexual and functional development: The wildlife/human connection. Princeton, NJ: Princeton Scientific Publishing Co; 1992. pp. 9–15. [Google Scholar]
- 15.Horwitz RI, Viscoli CM, Merino M, Brennan TA, Flannery JT, Robboy SJ. Clear cell adenocarcinoma of the vagina and cervix: incidence, undetected disease, and diethylstilbestrol. J Clin Epidemiol. 1988;41:593–7. doi: 10.1016/0895-4356(88)90064-9. [DOI] [PubMed] [Google Scholar]
- 16.Newbold RR, Bullock BC, McLachlan JA. Uterine adenocarcinoma in mice following developmental treatment with estrogens: a model for hormonal carcinogenesis. Cancer Res. 1990;50:7677–81. [PubMed] [Google Scholar]
- 17.Korach KS, Horigome T, Tomooka Y, Yamashita S, Newbold RR, McLachlan JA. Immunodetection of estrogen receptor in epithelial and stromal tissues of neonatal mouse uterus. Proc Natl Acad Sci U S A. 1988;85:3334–7. doi: 10.1073/pnas.85.10.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamashita S, Newbold RR, McLachlan JA, Korach KS. Developmental pattern of estrogen receptor expression in female mouse genital tracts. Endocrinology. 1989;125:2888–96. doi: 10.1210/endo-125-6-2888. [DOI] [PubMed] [Google Scholar]
- 19.Couse JF, Dixon D, Yates M, Moore AB, Ma L, Maas R, et al. Estrogen receptor-alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol. 2001;238:224–38. doi: 10.1006/dbio.2001.0413. [DOI] [PubMed] [Google Scholar]
- 20.Couse JF, Davis VL, Hanson RB, Jefferson WN, McLachlan JA, Bullock BC, et al. Accelerated onset of uterine tumors in transgenic mice with aberrant expression of the estrogen receptor after neonatal exposure to diethylstilbestrol. Mol Carcinog. 1997;19:236–42. doi: 10.1002/(sici)1098-2744(199708)19:4<236::aid-mc4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 21.Herynk MH, Fuqua SAW. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–98. doi: 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- 22.Park W, Choi JJ, Hwang ES, Lee JH. Identification of a variant estrogen receptor lacking exon 4 and its coexpression with wild-type estrogen receptor in ovarian carcinomas. Clin Cancer Res. 1996;2:2029–35. [PubMed] [Google Scholar]
- 23.Desai AJ, Luqmani YA, Walters JE, Coope RC, Dagg B, Gomm JJ, et al. Presence of exon 5-deleted oestrogen receptor in human breast cancer: functional analysis and clinical significance. Br J Cancer. 1997;75:1173–84. doi: 10.1038/bjc.1997.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han F, Miksicek R, Clarke R, Conrad SE. Expression of an estrogen receptor variant lacking exon 3 in derivatives of MCF-7 cells with acquired estrogen independence or tamoxifen resistance. J Mol Endocrinol. 2004;32:935–45. doi: 10.1677/jme.0.0320935. [DOI] [PubMed] [Google Scholar]
- 25.Fasco MJ, Amin A, Pentecost BT, Yang Y, Gierthy JF. Phenotypic changes in MCF-7 cells during prolonged exposure to tamoxifen. Mol Cell Endocrinol. 2003;206:33–47. doi: 10.1016/s0303-7207(03)00256-9. [DOI] [PubMed] [Google Scholar]
- 26.Hirata S, Yamada-Mouri N, Nara M, Takizawa M, Ito H, Kato J. Presence of alternatively spliced-estrogen receptor mRNA variants in normal human uterine endometrium and endometrial cancer. Endocr J. 1995;42:289–93. doi: 10.1507/endocrj.42.289. [DOI] [PubMed] [Google Scholar]
- 27.Springwald A, Lattrich C, Skrzypczak M, Goerse R, Ortmann O, Treeck O. Identification of novel transcript variants of estrogen receptor alpha, beta and progesterone receptor gene in human endometrium. Endocrine. 2010;37:415–24. doi: 10.1007/s12020-010-9322-8. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Miksicek RJ. Identification of a dominant negative form of the human estrogen receptor. Mol Endocrinol. 1991;5:1707–15. doi: 10.1210/mend-5-11-1707. [DOI] [PubMed] [Google Scholar]
- 29.Leng X, Tsai SY, Tsai M-J. The nuclear hormone receptor superfamily: Structure and function. In: Vedeckis WV, editor. Hormones and Cancer. Boston: Birkhauser; 1996. pp. 91–126. [Google Scholar]
- 30.Bollig A, Miksicek RJ. An estrogen receptor-alpha splicing variant mediates both positive and negative effects on gene transcription. Mol Endocrinol. 2000;14:634–49. doi: 10.1210/mend.14.5.0460. [DOI] [PubMed] [Google Scholar]
- 31.Kumar V, Chambon P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell. 1988;55:145–56. doi: 10.1016/0092-8674(88)90017-7. [DOI] [PubMed] [Google Scholar]
- 32.Murillas R, Larcher F, Conti CJ, Santos M, Ullrich A, Jorcano JL. Expression of a dominant negative mutant of epidermal growth factor receptor in the epidermis of transgenic mice elicits striking alterations in hair follicle development and skin structure. EMBO J. 1995;14:5216–23. doi: 10.1002/j.1460-2075.1995.tb00206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. 1997;18:231–42. doi: 10.1016/s0896-6273(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 34.Sunaga S, Maki K, Lagasse E, Blanco JC, Ozato K, Miyazaki J, et al. Myeloid differentiation is impaired in transgenic mice with targeted expression of a dominant negative form of retinoid X receptor beta. Br J Haematol. 1997;96:19–30. doi: 10.1046/j.1365-2141.1997.8692483.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang XJ, Greenhalgh DA, Bickenbach JR, Jiang A, Bundman DS, Krieg T, et al. Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc Natl Acad Sci USA. 1997;94:2386–91. doi: 10.1073/pnas.94.6.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keaveney M, Klug J, Dawson MT, Nestor PV, Neilan JG, Forde RC, et al. Evidence for a previously unidentified upstream exon in the human oestrogen receptor gene. J Mol Endocrinol. 1991;6:111–5. doi: 10.1677/jme.0.0060111. [DOI] [PubMed] [Google Scholar]
- 37.White R, Lees JA, Needham M, Ham J, Parker M. Structural organization and expression of the mouse estrogen receptor. Mol Endocrinol. 1987;1:735–44. doi: 10.1210/mend-1-10-735. [DOI] [PubMed] [Google Scholar]
- 38.Davis VL, Couse JF, Goulding EH, Power SG, Eddy EM, Korach KS. Aberrant reproductive phenotypes evident in transgenic mice expressing the wild-type mouse estrogen receptor. Endocrinology. 1994;135:379–86. doi: 10.1210/endo.135.1.8013372. [DOI] [PubMed] [Google Scholar]
- 39.Palmiter RD, Sandgren EP, Avarbock MR, Allen DD, Brinster RL. Heterologous introns can enhance expression of transgenes in mice. Proc Natl Acad Sci U S A. 1991;88:478–82. doi: 10.1073/pnas.88.2.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991;11:3070–4. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breathnach R, Harris BA. Plasmids for the cloning and expression of full-length double-stranded cDNAs under control of the SV40 early or late gene promoter. Nucleic Acids Res. 1983;11:7119–36. doi: 10.1093/nar/11.20.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostrowski MC, Huang AL, Kessel M, Wolford RG, Hager GL. Modulation of enhancer activity by the hormone responsive regulatory element from mouse mammary tumor virus. EMBO J. 1984;3:1891–9. doi: 10.1002/j.1460-2075.1984.tb02064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hogan B, Constantini F, Lacy E. Manipulating the mouse embryo. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1986. [Google Scholar]
- 44.Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, et al. FVB/N: an inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci USA. 1991;88:2065–9. doi: 10.1073/pnas.88.6.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Couse JF, Davis VL, Tally WC, Korach KS. An improved method of genomic DNA extraction for screening transgenic mice. Biotechniques. 1994;17:1030–2. [PubMed] [Google Scholar]
- 46.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–9. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 47.Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) Null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol Endocrinol. 2003;17:1039–53. doi: 10.1210/me.2002-0398. [DOI] [PubMed] [Google Scholar]
- 48.Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. 1997;138:4613–21. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- 49.Davis VL, Chan C-C, Schoen TJ, Couse JF, Chader GJ, Korach KS. An estrogen receptor repressor induces cataract formation in transgenic mice. Proc Natl Acad Sci USA. 2002;99:9427–32. doi: 10.1073/pnas.132247999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forsberg J-G, Tenenbaum A, Rydberg C, Sernvi C. Ovarian structure and function in neonatally estrogen treated female mice. In: McLachlan JA, editor. Estrogens in the Environment. New York: Elsevier Science Publishing Co; 1985. pp. 327–46. [Google Scholar]
- 51.Mahler JF, Stokes W, Mann PC, Takaoka M, Maronpot RR. Spontaneous lesions in aging FVB/N mice. Toxicol Pathol. 1996;24:710–6. doi: 10.1177/019262339602400606. [DOI] [PubMed] [Google Scholar]
- 52.Erenburg I, Schachter B, Mira y Lopez R, Ossowski L. Loss of an estrogen receptor isoform (ER alpha delta 3) in breast cancer and the consequences of its reexpression: interference with estrogen-stimulated properties of malignant transformation. Mol Endocrinol. 1997;11:2004–15. doi: 10.1210/mend.11.13.0031. [DOI] [PubMed] [Google Scholar]
- 53.Teng CT. Lactoferrin gene expression and regulation: an overview. Biochem Cell Biol. 2002;80:7–16. doi: 10.1139/o01-215. [DOI] [PubMed] [Google Scholar]
- 54.Kim K, Thu N, Saville B, Safe S. Domains of estrogen receptor alpha (ERalpha) required for ERalpha/Sp1-mediated activation of GC-rich promoters by estrogens and antiestrogens in breast cancer cells. Mol Endocrinol. 2003;17:804–17. doi: 10.1210/me.2002-0406. [DOI] [PubMed] [Google Scholar]
- 55.Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors alpha and beta. J Biol Chem. 2005;280:347–54. doi: 10.1074/jbc.M407879200. [DOI] [PubMed] [Google Scholar]
- 56.Petz LN, Nardulli AM. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol Endocrinol. 2000;14:972–85. doi: 10.1210/mend.14.7.0493. [DOI] [PubMed] [Google Scholar]
- 57.Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143:4583–91. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- 58.Petz LN, Ziegler YS, Schultz JR, Nardulli AM. Fos and Jun inhibit estrogen-induced transcription of the human progesterone receptor gene through an activator protein-1 site. Mol Endocrinol. 2004;18:521–32. doi: 10.1210/me.2003-0105. [DOI] [PubMed] [Google Scholar]
- 59.Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, et al. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9:1441–54. doi: 10.1210/mend.9.11.8584021. [DOI] [PubMed] [Google Scholar]
- 60.Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002;16:2188–201. doi: 10.1210/me.2001-0174. [DOI] [PubMed] [Google Scholar]
- 61.Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285:2676–85. doi: 10.1074/jbc.M109.043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim JJ, Chapman-Davis E. Role of progesterone in endometrial cancer. Semin Reprod Med. 2010;28:81–90. doi: 10.1055/s-0029-1242998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rey JM, Pujol P, Dechaud H, Edouard E, Hedon B, Maudelonde T. Expression of oestrogen receptor-alpha splicing variants and oestrogen receptor-beta in endometrium of infertile patients. Mol Hum Reprod. 1998;4:641–7. doi: 10.1093/molehr/4.7.641. [DOI] [PubMed] [Google Scholar]
- 64.Marshburn PB, Zhang J, Bahrani-Mostafavi Z, Mostafavi BZ, Marroum M-C, Mougeot J-LC, et al. Estrogen receptor-alpha messenger RNA variants that lack exon 5 or exon 7 are coexpressed with wild-type form in human endometrium during all phases of the menstrual cycle. Am J Obstet Gynecol. 2004;191:626–33. doi: 10.1016/j.ajog.2004.05.082. [DOI] [PubMed] [Google Scholar]
- 65.Rice LW, Jazaeri AA, Shupnik MA. Estrogen receptor mRNA splice variants in pre- and postmenopausal human endometrium and endometrial carcinoma. Gynecol Oncol. 1997;65:149–57. doi: 10.1006/gyno.1997.4623. [DOI] [PubMed] [Google Scholar]
- 66.Horvath G, Leser G, Hahlin M, Henriksson M. Exon deletions and variants of human estrogen receptor mRNA in endometrial hyperplasia and adenocarcinoma. Int J Gynecol Cancer. 2000;10:128–36. doi: 10.1046/j.1525-1438.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- 67.Chaidarun SS, Klibanski A, Alexander JM. Tumor-specific expression of alternatively spliced estrogen receptor messenger ribonucleic acid variants in human pituitary adenomas. J Clin Endocrinol Metab. 1997;82:1058–65. doi: 10.1210/jcem.82.4.3864. [DOI] [PubMed] [Google Scholar]
- 68.Friend KE, Resnick EM, Ang LW, Shupnik MA. Specific modulation of estrogen receptor mRNA isoforms in rat pituitary throughout the estrous cycle and in response to steroid hormones. Mol Cell Endocrinol. 1997;131:147–55. doi: 10.1016/s0303-7207(97)00098-1. [DOI] [PubMed] [Google Scholar]