

Abstract
Background
Type 2 diabetes mellitus (T2DM) is the leading cause of chronic kidney disease and a major cause of cardiovascular disease (CVD) mortality. Inflammation is closely involved in the pathogenesis of T2DM, and reactive amyloidosis occurs in the presence of chronic inflammation. We hypothesized that patients with T2DM may have a higher prevalence of renal AA amyloidosis (RAAA) and that this could contribute to worse atherosclerosis and CVD.
Materials and Methods
We analyzed 330 autopsy kidneys from patients with a previous T2DM diagnosis. The kidney tissue was evaluated in order to determine the presence of diabetic nephropathy and RAAA, and systemic vessels were evaluated for the presence of atherosclerosis.
Results
RAAA was detected in 9% of our study population and was associated with an increased risk for nodular sclerosis [OR (95% CI)] [11 (2.04-59.16)], for chronic ischemic cardiomyopathy [4.59 (2.02-10.42)], for myocardial infarction [3.41 (1.52-7.64)] as well as for aortic [4.75 (1.09-20.69)], coronary [3.22 (1.47-7.04)], and intrarenal atherosclerosis [3.84 (1.46-10.09)].
Conclusions
RAAA is prevalent in T2DM and is associated with worse CVD and renal disease, likely because RAAA is a marker of severe chronic inflammation.
Key Words: Diabetic nephropathy, Advanced glycation end products, Atherosclerosis, Serum amyloid A
Introduction
Type 2 diabetes mellitus (T2DM) is the leading cause of chronic kidney disease and a major cause of cardiovascular disease (CVD) mortality [1]. There is clear evidence that inflammation is closely involved in the pathogenesis of T2DM and in the development of chronic complications such as atherosclerosis, coronary disease, retinopathy, neuropathy, and nephropathy [2,3,4,5,6,7]. Circulating levels of interleukin-6 (IL-6), interleukin-1 (IL-1), and tumor necrosis factor-α (TNF-α) are all elevated in diabetic patients and are associated with CVD [8,9,10,11].
Reactive amyloidosis (AAA) occurs in the presence of chronic inflammatory conditions such as rheumatoid arthritis and chronic infections [12,13,14,15,16,17]. Although AAA can affect many organs, renal involvement is one of the most common features in AAA. Renal AAA (RAAA) is diagnosed by renal biopsy, and amyloid deposition is found in different degrees in mesangium, capillary walls, tubules, and blood vessels.
T2DM is associated with the generation of advanced glycation end products (AGEs), which are closely associated with markers of inflammation such as IL-1, IL-6, and TNF-α [18], and play a role in the development of diabetic nephropathy and vasculopathy [19]. It has also been suggested that AGEs may be involved in either amyloidogenesis or complications related to amyloid [20,21]. Since RAAA is a disease that occurs under the influence of chronic inflammation and T2DM is considered per se as an inflammatory state, we thus hypothesized that patients with T2DM may have a higher prevalence of RAAA and that this could contribute to worse atherosclerosis and CVD. To our knowledge, there are no studies that have evaluated the association of T2DM and RAAA in a large number of autopsy cases.
Materials and Methods
Participants
The study included 330 autopsy kidneys from patients with a previous diagnosis of T2DM in the period from 2001 until 2011. Patients were included regardless of the cause of death or the presence of CVD. Patients were excluded if they had the clinical or postmortem diagnosis of tuberculosis, osteomyelitis, bronchiectasis, inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus and/or ankylosing spondylitis.
Variables
The primary outcome was RAAA, defined as the positivity of immunohistochemistry through indirect immunoperoxidase with the primary AA antibody. The postmortem registries were analyzed focusing on the description of cardiac and vascular tissues, assuming that affection in these structures would reflect inflammation and chronic disease. The variables included for the analyses were chronic ischemic cardiomyopathy (myocardial interstitial fibrosis with atrophy of myocardial fibers), myocardial infarction (myocardial coagulative necrosis) and aortic, coronary, and intrarenal atherosclerosis. The extension and severity of atherosclerosis were defined as follows: extension: (a) <25% of the vascular surface; (b) 26-50% of the vascular surface, and (c) >50% of the vascular surface; severity: (a) lipidic striae and nodules, (b) calcifications, and (c) ulcerations, fissures, and thrombosis.
Measurements
Kidney tissue was fixed in 10% formaldehyde solution and embedded in paraffin. Pieces of 3 μm were cut and stained with hematoxylin-eosin, Periodic Acid-Schiff, Masson's trichrome and silver methenamine. The tissue was evaluated by two different pathologists in order to determine the presence of diabetic nephropathy (according to the Pathologic Classification of Diabetic Nephropathy [22]). We intentionally analyzed the kidney for the presence of glomerular, interstitial, and vascular material suggestive of amyloidosis (extracellular, amorphous, and eosinophil material), which was confirmed by the characteristic apple-green birefringence under polarized light with Congo red stain [23] and by Thioflavin-T. In addition to the positivity by Congo red stain and Thioflavin-T, immunohistochemistry was performed through indirect immunoperoxidase. Pieces of 2 μm were obtained from the paraffin block and analyzed with the technique of avidin-biotin-peroxidase complex (ABC; StreptABComplex/HRP; DAKO Corp., Carpinteria, Calif., USA) previously described [24] with the primary AA antibody (FLEX Monoclonal Mouse Anti-Human Amyloid A, Code IR605) [25]. As a negative control, tissues were incubated at normal ram serum. As a positive test, we used a liver fragment with previously diagnosed AAA. Indirect immunoperoxidase was evaluated by two different pathologists. Demographic data was obtained from the medical records. In order to confirm the presence of RAAA, ten well-preserved samples obtained from the paraffin blocks were randomly selected for electronic microscopy.
Statistical Analysis
The variables are described as percentages and frequencies. The association measures were analyzed by χ2 and odds ratios using STATA version 8.
Results
The mean age of the population was 61 ± 13 years, and 47% were female. The main cause of death was sepsis (46%), followed by neoplasia (18%) and uremia-associated complications (15%) (table 1). Eighty percent (n = 257) of the population had diabetic nephropathy. RAAA was detected in 9% (n = 30) of our study population (table 1) but had only been diagnosed in 2% of the cohort when the autopsies were evaluated for the first time. As shown in table 1, we observed that patients with RAAA had a longer mean duration of T2DM (18 ± 13 vs. 11 ± 9 years, p = 0.01), and diabetic nephropathy was present in 93% of the RAAA patients versus 73% in the no RAAA (NRAAA) patients (p = 0.03). The data was analyzed by the type of diabetic nephropathy; nodular sclerosis (type III diabetic nephropathy) was more frequent in the RAAA patients (10 vs. 1%, p < 0.001). Types I (0 vs. 0.3%, p = 0.75), IIa (0 vs. 7%, p = 0.12), IIb (17 vs. 17%, p = 0.9), and IV (67 vs. 50%, p = 0.09) of the diabetic nephropathy classification were similar among RAAA and NRAAA patients.
Table 1.
General characteristics of the study population
| Total | RAAA | NRAAA | p | |
|---|---|---|---|---|
| Number of patients | 330 | 30 | 300 | |
| Mean age ± SD, years | 61.2 ± 13.1 | 63.7 ± 12.6 | 61 ± 13.1 | 0.27 |
| Male, % | 53 | 60 | 52 | 0.42 |
| Mean duration of T2DM, years | 12 ± 9.5 | 17.5 ± 13 | 11.4 ± 9 | 0.01 |
| Without antidiabetic drugs, % | 57 | 50 | 57 | 0.73 |
| Drugs, % | ||||
| Glibenclamide | 25 | 30 | 24 | 0.73 |
| Metformin | 18 | 33 | 16 | 0.04 |
| Insulin | 11 | 13 | 11 | 0.76 |
| Hypertension, % | 66 | 73 | 66 | 0.39 |
| Hypertension duration, years | 4.5 | 5.5 ± 8.8 | 4.4 ± 7.8 | 0.44 |
| Causes of death, n (%) | ||||
| Sepsis | 152 (46) | 11 (37) | 141 (47) | 0.29 |
| Neoplasia | 57 (17) | 0 | 57 (20) | 0.01 |
| Uremia-related complications | 49 (15) | 8 (27) | 41 (14) | 0.05 |
| Heart/cerebral ischemia | 26 (8) | 4 (13) | 22 (7) | 0.24 |
| Gastrointestinal bleeding | 13 (4) | 1 (3) | 12 (4) | 0.85 |
| Chronic obstructive pulmonary disease | 13 (4) | 3 (10) | 10 (3) | 0.07 |
| Valvular heart disease | 6 (2) | 1 (3) | 5 (2) | 0.73 |
| Hemorrhagic stroke | 6 (2) | 1 (3) | 5 (2) | 0.62 |
| Liver cirrhosis | 6 (2) | 0 | 6 (2) | 0.22 |
| Diabetic nephropathya | 257 (78) | 28 (93) | 229 (73) | 0.03 |
| Type I (mild or nonspecific LM changes) | 1 (0.3) | 0 | 1 (0.3) | 0.75 |
| Type IIa (mild mesangial expansion) | 22 (7) | 0 | 22 (7) | 0.12 |
| Type IIb (severe mesangial expansion) | 57 (17) | 5 (17) | 50 (17) | 0.9 |
| Type III (nodular sclerosis) | 6 (2) | 3 (10) | 3 (1) | <0.001 |
| Type IV (advanced diabetic glomerulosclerosis) | 171 (52) | 20 (67) | 151 (50) | 0.09 |
LM = Light microscopy.
According to the 2010 Pathologic Classification of Diabetic Nephropathy [22].
The cause of death was similar among RAAA and NRAAA patients, except for uremia complications, which were significantly higher in RAAA patients, and for neoplasia, which was only present in NRAAA patients. The percentages of death from sepsis heart/cerebral ischemia, gastrointestinal bleeding, valvular heart disease, hemorrhagic stroke, and liver cirrhosis were similar in the two groups. There was a trend towards more complications from chronic obstructive pulmonary disease in RAAA patients.
When systemic vascular tissues were analyzed, we found that chronic ischemic cardiomyopathy, myocardial infarction, aortic atherosclerosis, coronary atherosclerosis, and renal atherosclerosis were more frequent in the RAAA group (table 2). In addition, when we analyzed the data according to the severity score for aortic, coronary, and renal atherosclerosis, the differences between the RAAA and the NRAAA groups were observed only in the more severe categories (table 2).
Table 2.
Differences between atherosclerosis and CVD in RAAA and NRAAA patients
| RAAA |
NRAAA |
p | |||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Chronic ischemic cardiomyopathy | 20 | 67 | 91 | 30 | <0.001 |
| Myocardial infarction | 12 | 40 | 49 | 16 | 0.01 |
| Aortic atherosclerosis | 28 | 93 | 224 | 75 | 0.02 |
| IIa | 2 | 7 | 8 | 3 | 0.22 |
| IIb | 3 | 10 | 47 | 16 | 0.4 |
| IIc | 2 | 7 | 12 | 4 | 0.49 |
| IIIb | 1 | 3 | 28 | 9 | 0.26 |
| IIIc | 20 | 67 | 113 | 38 | 0.01 |
| Coronary atherosclerosis | 25 | 83 | 185 | 62 | 0.02 |
| Ia | 3 | 10 | 31 | 10 | 0.9 |
| IIa | 1 | 3 | 7 | 2 | 0.73 |
| IIb | 6 | 20 | 60 | 20 | 1 |
| IIc | 1 | 3 | 6 | 2 | 0.62 |
| IIIc | 14 | 47 | 64 | 21 | 0.01 |
| Renal atherosclerosis | 7 | 23 | 22 | 7 | 0.01 |
| IIb | 0 | 0 | 6 | 2 | 0.43 |
| IIIc | 7 | 23 | 13 | 4 | <0.001 |
Likewise, vascular disease was associated with the presence of RAAA and was more severe in the RAAA group for chronic ischemic cardiomyopathy [OR (95% CI)] [4.59 (2.02-10.42)], for myocardial infarction [3.41 (1.52-7.64)], for aortic atherosclerosis [4.75 (1.09-20.69)], and for intrarenal atherosclerosis [3.84 (1.46-10.09)] (table 3).
Table 3.
Factors associated with RAAA
| Risk factor | OR (95% CI) | p |
|---|---|---|
| Chronic ischemic cardiomyopathy | 4.59 (2.02 – 10.42) | <0.001 |
| Myocardial infarction | 3.41 (1.52 – 7.64) | 0.01 |
| Aortic atherosclerosis | 4.75 (1.09 – 20.69) | 0.02 |
| Aortic IIIc atherosclerosis | 3.30 (1.47 – 7.41) | 0.01 |
| Coronary IIIc atherosclerosis | 3.22 (1.47 – 7.04) | 0.01 |
| Renal atherosclerosis | 3.84 (1.46 – 10.09) | 0.01 |
| Renal IIIc atherosclerosis | 6.71 (2.37 – 19.03) | <0.001 |
| Diabetic nephropathya | 4.34 (0.99 – 18.9) | 0.01 |
| Nodular sclerosis | 11 (2.04 – 59.16) | <0.001 |
| Advanced diabetic glomerulosclerosis | 1.97 (0.88 – 4.37) | 0.08 |
Univariate analysis.
Kimmelstiel-Wilson lesion.
The arterioles and diabetic nodules were the most common areas for RAAA deposition in the kidneys (fig. 1). Diabetic glomerular lesions were also associated with RAAA for diabetic nephropathy [4.34 (0.99-18.9)] and for nodular sclerosis or Kimmelstiel-Wilson lesions [11 (2.04-59.16)] (table 3).
Fig. 1.
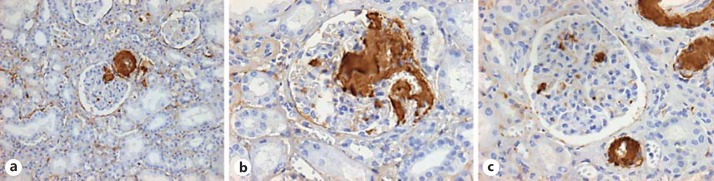
Distribution of RAAA. a Renal tissue photomicrography ×10 with diabetic nephropathy and RAAA by indirect immunoperoxidase with amyloid deposition in a preglomerular arteriole and glomerular nodule. b Photomicrography ×40 with extensive glomerular amyloid deposition. c Photomicrography ×40 with mild mesangial and extensive arteriolar amyloid deposition.
When samples were analyzed by electron microscopy, there was diffuse thickening of the glomerular basal basement membranes and mesangial expansion with fibrillary collagen deposits, findings consistent with diabetic nephropathy. In addition, irregular nonorganized mesangial fibrillary deposits were found in the glomerular basal basement membranes, interstitium, and some tubular membranes. These deposits were formed by 7-10-nm fibrils (fig. 2).
Fig. 2.
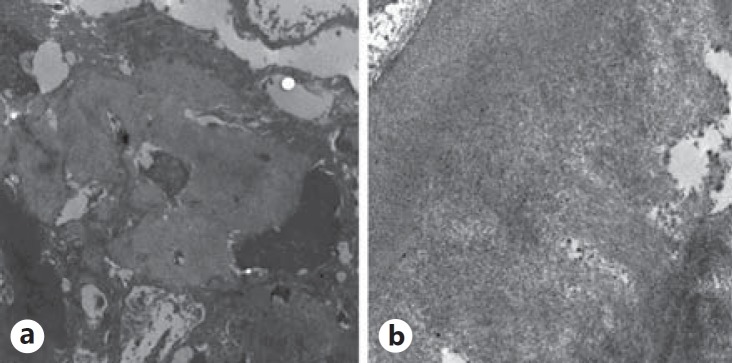
Electronic microscopy (a ×8,000 and b ×20,000) showing irregular nonorganized fibrillary deposits in the mesangium.
Discussion
In this large cohort of autopsies of patients with T2DM, we identified a 9% prevalence of RAAA. This prevalence is similar to that reported in other chronic inflammatory conditions such as ankylosing spondylitis, psoriatic arthritis, familial Mediterranean fever, and rheumatoid arthritis [26,27], in which AAA is more frequent with long-lasting and continuous activity [28]. The autopsies from patients with RAAA indicated a more severe chronic inflammatory disease as suggested by worse coronary, aortic, and renal vascular disease. In addition, diabetic nephropathy was more severe in the RAAA group.
The beginning and progression of atherosclerosis in T2DM patients is due, among other factors, to inflammation [29]. Hyperglycemia induces oxidative stress, which mediates inflammation through cytokines and transcription factors (IL-1, nuclear factor-κB) [30]. In hyperglycemia, there is increased production of AGEs, which damage target cells by modifying the intracellular proteins, extracellular matrix components, and by binding plasma proteins (modified by AGE precursors) to AGE receptors [31] on endothelial cells. The AGE receptor ligation activates nuclear factor-κB, leading to proinflammatory gene expression [32,33]. This inflammatory cascade may promote amyloidogenic molecules that result in AAA in parallel to severe atherosclerosis.
In addition, it has been suggested that inflammation plays an important role in the pathogenesis of diabetic nephropathy [34]. It is known that among inflammatory cytokines, IL-1, IL-6 and TNF-α are relevant for the development of diabetic nephropathy [35]. Studies in T2DM patients demonstrate a significant association between IL-6 and glomerular basement membrane thickening, a crucial lesion of diabetic nephropathy and a strong predictor of renal progression [36]. TNF-α is cytotoxic to renal cells and able to induce direct renal injury [37,38].
Serum amyloid A (SAA) levels in patients with T2DM are significantly higher than those in healthy subjects and are associated with an increase in urinary albumin excretion, suggesting that SAA levels are associated with the development of diabetic nephropathy [38]. Although the only clearly established adverse role of SAA has been in the pathogenesis of AAA, it is possible that SAA is involved in the pathogenesis of both atherosclerosis and diabetic nephropathy through the above-mentioned mechanisms. Nevertheless, our data does not prove that SAA is involved in the progression of these disorders, and it is possible that SAA is a marker of severe inflammation rather than playing a causative role in these diseases.
What are the implications of our results? First, we found that T2DM is associated with RAAA and that the diagnosis is often missed when tissue is routinely analyzed. Second, we found a clear association of RAAA with worse CVD and renal disease, likely because RAAA is a marker of severe chronic inflammation. Third, RAAA may explain the adverse renal outcomes observed in some patients with T2DM. We found that uremia complications were significantly higher in patients with RAAA. Finally, RAAA was treated with eprodisate, a drug that interferes with the interactions between amyloidogenic proteins and glycosaminoglycans and thereby inhibits polymerization of amyloid fibrils and deposition of the fibrils in tissues. In a study involving 183 patients with RAAA mostly related to arthritis and inflammatory autoimmune disease, eprodisate was associated with a slower decline in kidney function [39]. Whether this treatment would be beneficial for diabetic nephropathy with RAAA needs to be determined.
Our study has several strengths. First, this is the first large cohort of autopsies from T2DM to describe the prevalence of RAAA and to evaluate the association between the severity of CVD and renal disease with RAAA. Second, all autopsies were analyzed by two different pathologists blinded to the investigation. Finally, RAAA was analyzed by the gold standard method.
The main limitations of the study are the lack of serum or urinary markers of kidney disease or inflammation. In addition, only 40% of the patients had optimal treatment for diabetes, which could make the population less generalizable, especially for high-income countries. Finally, we did not have a control group without diabetes in which the prevalence of RAAA was determined for comparison.
In conclusion, RAAA is prevalent in T2DM and is associated with worse CVD and renal disease. Studies replicating our findings are warranted in order to confirm these associations. Whether treating RAAA in this population is beneficial needs to be determined in further studies.
Disclosure Statement
None of the authors has any conflicts of interest.
Acknowledgements
This paper was supported in part by grants No. 87236 and 138464 from the Mexican Council of Science and Technology to M.M. and G.G., respectively, and by a grant from the Fundación Miguel Alemán to M.M.
References
- 1.Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 2012;59(Suppl 1):A7, 21–1420. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 3.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 4.Barzilay JI, Abraham L, Heckbert SR, Cushman M, Kuller LH, Resnick HE, et al. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes. 2001;50:2384–2389. doi: 10.2337/diabetes.50.10.2384. [DOI] [PubMed] [Google Scholar]
- 5.Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 6.King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527–1534. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 7.Meleth AD, Agrón E, Chan CC, Reed GF, Arora K, Byrnes G, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:4295–4301. doi: 10.1167/iovs.04-1057. [DOI] [PubMed] [Google Scholar]
- 8.Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F Ciotola, et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002;106:2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae. [DOI] [PubMed] [Google Scholar]
- 9.Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumor necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000;67:291–300. doi: 10.1016/s0024-3205(00)00622-6. [DOI] [PubMed] [Google Scholar]
- 10.Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–1668. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welsh N, Cnop M, Kharroubi I, Bugliani M, Lupi R, Marchetti P, et al. Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes. 2005;54:3238–3244. doi: 10.2337/diabetes.54.11.3238. [DOI] [PubMed] [Google Scholar]
- 12.Merlini G, Bellotti V. Mechanisms of disease: molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 13.Brunt E, Tiniakos D. Metabolic storage diseases: amyloidosis. Clin Liver Dis. 2004;8:915–930. doi: 10.1016/j.cld.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Husby G. Amyloidosis and rheumatoid arthritis. Clin Exp Rheumatol. 1985;3:173–180. [PubMed] [Google Scholar]
- 15.Mladenovic J, Geisic L, Kerimovic D. Incidence of amyloidosis in rheumatoid arthritis and ankilosing spondylitis. Scand J Rheumatol. 1975;4(suppl 8):39–103. [Google Scholar]
- 16.El Mansoury TM, Hazenberg BP, El Badawy SA, Ahmed AH, Bijzet J, Limburg PC, et al. Screening for amyloid in subcutaneous fat tissue of Egyptian patients with rheumatoid arthritis: clinical and laboratory characteristics. Ann Rheum Dis. 2002;61:42–47. doi: 10.1136/ard.61.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koivuniemi R, Paimela L, Suomalainen R, Törnroth T, Leirisalo-Repo M. Amyloidosis is frequently undetected in patients with rheumatoid arthritis. Amyloid. 2008;15:262–268. doi: 10.1080/13506120802524676. [DOI] [PubMed] [Google Scholar]
- 18.Shoelson SE, Lee J, Goldfine AB. Inflammation and insuline resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruderman NB, Williamson JR, Brownlee M. Glucose and diabetic vascular disease. FASEB J. 1992;6:2905–2914. doi: 10.1096/fasebj.6.11.1644256. [DOI] [PubMed] [Google Scholar]
- 20.Foitschik T, Saeger W, Riebe M, Röcken C. Advanced glycation end products in intervertebral discs and hip joint capsules: correlation with senile amyloid? Amyloid. 2005;12:167–173. doi: 10.1080/13506120500222094. [DOI] [PubMed] [Google Scholar]
- 21.Matsunaga N, Anan I, Rosenberg P, Nagai R, Lundström O, Horiuchi S, et al. Advanced glycation end product is implicated in amyloid-related kidney complications. Scand J Clin Lab Invest. 2005;65:263–272. doi: 10.1080/00365510510013794. [DOI] [PubMed] [Google Scholar]
- 22.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 23.Röcken C, Sletten K. Amyloid in surgical pathology. Virchows Arch. 2003;443:3–16. doi: 10.1007/s00428-003-0834-y. [DOI] [PubMed] [Google Scholar]
- 24.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 25.FLEX Monoclonal Mouse Anti-Human Amyloid A Clone mc1 Ready-to-use. http://www.dako.com/dist/download.pdf?objectid=117411002 (accessed March 5, 2010).
- 26.Boers M, Croonen AM, Dijkmans BA, Breedveld FC, Eulderink F, Cats A, et al. Renal findings in rheumatoid arthritis: clinical aspects of 132 necropsies. Ann Rheum Dis. 1987;46:658–663. doi: 10.1136/ard.46.9.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gertz MA, Kyle RA. Secondary systemic amyloidosis: response and survival in 64 patients. Medicine (Baltimore) 1991;70:246. [PubMed] [Google Scholar]
- 28.Tiitinen S, Kaarela K, Helin H. Amyloidosis – incidence and early risk factors in patients with rheumatoid arthritis. Scand J Rheumatol. 1993;22:158–161. doi: 10.3109/03009749309099264. [DOI] [PubMed] [Google Scholar]
- 29.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 30.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease pathophysiology, clinical consequences, and medical therapy: part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84:489–497. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- 32.Yerneni K, Bai W, Khan B, Medford R, Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48:855–864. doi: 10.2337/diabetes.48.4.855. [DOI] [PubMed] [Google Scholar]
- 33.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 34.Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16:S78–S82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 35.Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, Ienaga K, et al. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991;40:1007–1012. doi: 10.1038/ki.1991.308. [DOI] [PubMed] [Google Scholar]
- 36.Nosadini R, Velussi M, Brocco E, Abaterusso C, Carraro A, Piarulli F, et al. Course of renal function in type 2 diabetic patients with abnormalities of albumin excretion rate. Diabetes. 2000;49:476–484. doi: 10.2337/diabetes.49.3.476. [DOI] [PubMed] [Google Scholar]
- 37.Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, et al. Tumor necrosis factor induces glomerular damage in rabbit. Am J Pathol. 1989;134:419–430. [PMC free article] [PubMed] [Google Scholar]
- 38.Kumon Y, Suehiro T, Itahara T, Ikeda Y, Hashimoto K. Serum amyloid A protein in patients with non-insulin-dependent diabetes mellitus. Clin Biochem. 1994;27:469–463. doi: 10.1016/0009-9120(94)00044-v. [DOI] [PubMed] [Google Scholar]
- 39.Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356:2349–2360. doi: 10.1056/NEJMoa065644. [DOI] [PubMed] [Google Scholar]