

Abstract
Background
The results of several clinical trials suggest that the integrase inhibitor dolutegravir may be less prone than other drugs to the emergence of HIV drug resistance mutations in treatment-naive patients. We have shown that the R263K mutation commonly emerged during tissue culture selection studies with dolutegravir and conferred low levels of resistance to this drug while simultaneously diminishing both HIV replication capacity and integrase enzymatic activity. E138K has been identified as a secondary mutation for dolutegravir in selection studies and has also been observed as a secondary mutation in the clinic for the integrase inhibitors raltegravir and elvitegravir.
Methods
We used biochemical cell-free strand-transfer assays and tissue culture assays to characterize the effects of the E138K/R263K combination of mutations on resistance to dolutegravir, integrase enzyme activity and HIV-1 replication capacity.
Results
We show here that the addition of the E138K substitution to R263K increased the resistance of HIV-1 to dolutegravir but failed to restore viral replication capacity, integrase strand-transfer activity and integration within cellular DNA. We also show that the addition of E138K to R263K did not increase the resistance to raltegravir or elvitegravir. The addition of the E138K substitution to R263K was also less detrimental to integrase strand-transfer activity and integration than a different secondary mutation at position H51Y that had also been selected in culture.
Conclusions
The E138K substitution failed to restore the defect in viral replication capacity that is associated with R263K, confirming previous selection studies that failed to identify compensatory mutation(s) for the latter primary mutation. This study suggests that the R263K resistance pathway may represent an evolutionary dead end for HIV in treatment-naive individuals who are treated with dolutegravir and will need to be confirmed by the long-term use of dolutegravir in the clinic.
Keywords: HIV-1, integrase inhibitors, antiviral
Introduction
The acquisition of drug resistance is an outcome of microbial evolution and is shared by multiple human pathogens. Clinical resistance of HIV has been shown to occur to inhibitors of each of viral reverse transcriptase, protease, integrase, entry and fusion.1 The only anti-HIV drug to date for which HIV resistance has not been demonstrated in treatment-naive patients is dolutegravir, which is also the most recently approved member of the integrase strand-transfer inhibitor (INSTI) family of drugs.2,3 In contrast, some treatment-naive patients treated with raltegravir or elvitegravir did fail therapy through the emergence of mutations in integrase at positions T66, E92, Y143, Q148 and N155, which also confer cross-resistance between these two drugs. Mutations at position Q148, in association with several secondary resistance mutations, can also decrease the likelihood of treatment success if patients are treated with dolutegravir after first failing either raltegravir or elvitegravir.4 Single mutations at position Q148 correlated with ∼80% treatment success with dolutegravir, whereas the addition of one and ≥2 secondary resistance mutations resulted in diminished levels of treatment success of ∼60% and ∼30%, respectively.5 Importantly, R263K was not one of the additional mutations that emerged in viruses isolated from patients who failed dolutegravir therapy after having previously failed raltegravir or elvitegravir.4
In tissue culture selections with dolutegravir, we have shown that the R263K substitution commonly emerged under drug pressure.6 R263K has also been detected after 24 weeks in several treatment-experienced patients who had received suboptimal regimens together with dolutegravir and who failed therapy,7 although no other mutations in integrase were observed at week 48, creating a contrast with raltegravir and elvitegravir, for which multiple mutations were observed at the time of treatment failure.1,3,7
These observations with dolutegravir are in agreement with two other findings: (i) none of the secondary mutations that were selected by dolutegravir in the presence of R263K led to the development of highly resistant viruses;8,9 and (ii) R263K decreased the ability of HIV-1 to become resistant to nevirapine and lamivudine.10 Since it is possible that some patients may eventually develop viruses that are both fit and resistant to dolutegravir, it is important to better understand how HIV can develop resistance to this drug and, in particular, which secondary mutations might emerge in the presence of R263K.
We previously characterized the H51Y/R263K combination and showed that the addition of H51Y to R263K increased resistance to dolutegravir while decreasing both integrase strand-transfer activity and viral fitness.8 Similar results have been observed with the M50I/R263K combination, which increased resistance to dolutegravir but failed to restore integration and viral replication capacity.9 Our tissue culture selection studies also revealed E138K as another secondary mutation that emerged following R263K under dolutegravir pressure.6 E138K was also selected together with G118R during selection studies with an experimental INSTI termed MK-2048, and we have shown that E138K when combined with G118R increased both resistance to this compound and viral replication capacity compared with G118R alone.11 E138K has also emerged in the presence of several primary resistance mutations that confer resistance to raltegravir and elvitegravir, including Q148R/H/K and Q148/G140, both in tissue culture and in patients.4,12–14 One individual enrolled in the VIKING clinical trial and who had previously failed raltegravir with the Q148H/G140S combination of mutations failed treatment with dolutegravir with a combination of T97A/T, E138E/K, G140S, Q148H and N155H mutations.4 In tissue culture, E138K and Q148K can synergize to confer ∼19-fold increased resistance to dolutegravir, although they minimally impacted the susceptibility of HIV to this drug when tested individually.15
We report here the characterization of the E138K substitution in the presence of R263K in regard to resistance, integrase strand-transfer activity and viral replication capacity. Our results show that E138K increased levels of resistance to dolutegravir above those conferred by R263K alone, but failed to restore integrase strand-transfer activity and HIV-1 DNA integration into host cells.
Methods
Cells and reagents
TZM-bl, 293T, and PM1 cells were cultured as described previously.6 Human primary peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque and stimulated for 72 h with 10 μg/mL of phytohaemagglutinin A and 20 U/mL of human interleukin-2. Merck & Co., Inc., Gilead Sciences, Inc. and ViiV Healthcare Ltd. kindly provided raltegravir, elvitegravir and dolutegravir, respectively.
Integrase strand-transfer activity assay
Site-directed mutagenesis was used to introduce the E138K mutation within the pET15b-integrase B plasmid for recombinant protein expression by using sense (5′-GGGCAGGTATCCAACAGAAATTTGGGATTCCCTAC-3′) and antisense (5′-GTAGGGAATCCCAAATTTCTGTTGGATACCTGCCC-3′) oligonucleotides. Site-directed mutagenesis was performed as previously described.8 The construction of the plasmids bearing the R263K mutation has previously been reported.6 Recombinant integrase proteins were expressed in the BL21 (DE3) bacterial strain and purified as published.6 Strand-transfer assays were performed using preprocessed LTR DNA as reported previously.8 Briefly, DNA-BIND 96-well plates (Corning) were coated with LTR DNA, blocked and washed before the addition of purified recombinant proteins that were either wild-type (WT) or carrying the E138K and R263K mutations. Biotinylated target DNA was added and strand-transfer reactions were allowed to occur for 1 h at 37°C. Integrated target DNA was detected after washes through the use of europium-labelled streptavidin molecules. The strand-transfer assay used in our laboratory has been described in previous publications.6,8,16
Generation of replication-competent genetically homogenous HIV-1
The generation of the pNL4.3IN(R263K) plasmid has previously been reported.6 Similar methods were used to generate the pNL4.3IN(E138K) and pNL4.3IN(E138K/R263K) plasmids through site-directed mutagenesis using the following primers: sense (5′-GGGCAGGTATCCAACAGAAATTTGGGATTCCCTAC-3′) and antisense (5′-GTAGGGAATCCCAAATTTCTGTTGGATACCTGCCC-3′). Genetically homogenous viral stocks were produced by transfecting 293T cells with the pNL4.3, pNL4.3IN(E138K), pNL4.3IN(R263K) and pNL4.3IN(E138K/R263K) plasmids using Lipofectamine 2000 according to the manufacturer's protocol (Life Technologies, Burlington, ON, Canada). Forty-eight hours after transfection, cell culture fluids were harvested, treated with Benzonase (Sigma-Aldrich, Oakville, ON, Canada) and filtered at 0.45 μm to remove plasmids and cell debris. Viruses were then aliquoted and stored at –80°C. Viral stocks were quantified by measuring both p24 content and cell-free reverse transcriptase activity.
HIV susceptibility to antiretroviral compounds
HIV susceptibilities to dolutegravir, raltegravir and elvitegravir were measured through the infection of 40 000 TZM-bl cells using a constant amount of WT and mutant viruses in the presence of 1 : 3 serial dilutions of drugs, as previously described.8 After 48 h, the cells were lysed and luciferase production was measured using the Luciferase Assay System (Promega, Madison, WI, USA). The results were analysed with GraphPad Prism 4.0 Software to calculate values for 50% inhibitory concentration (IC50) and 95% CI.
HIV infectivity and replication capacity
HIV-1 infectivity was measured through the infection of TZM-bl cells using serial 1: 3 dilutions of the NL4.3-WT, E138K, R263K and E138K/R263K viruses. Levels of infection were measured as described above. Replication capacity was measured by the quantification of reverse transcriptase activity in the cell culture fluids of PM1 cells infected with WT or mutant viruses over 27 days.
Determination of HIV integration in PBMCs
PBMCs were infected with WT and mutant viruses for 72 h, after which integrated DNA was quantified using an Alu-mediated quantitative PCR (qPCR) assay, as previously described.6,8,17 Briefly, cellular DNA was purified using the DNeasy Blood and Tissue Kit (Qiagen) and amplified by PCR using the following primers: sense (5′-GCCTCCCAAAGTGCTGGGATTACAG-3′) and antisense (5′-GTTCCTGCTATGTCACTTCC-3′).17 An additional reaction was performed in the absence of the sense primer to measure the background signal. A nested qPCR was then performed with the following primers and probe: sense (5′-TTAAGCCTCAATAAAGCTTGCC-3′), antisense (5′-GTTCGGGCGCCACTGCTAGA-3′) and probe (5′-FAM-CCAGAGTCACACAACAGAGGGGCACA-TAMRA-3′) on a Corbett Rotor-Gene 6000 apparatus (Corbett), as previously described.6,8,17 β-Globin was used for sample normalization.
Homology modelling
Homology models of HIV-1 WT and R263K integrase have previously been published.6 The homology structure of the E138K/R263K integrase was created by introducing the E138K substitution into the published R263K structure using the molecular visualization program PyMOL (http://pymol.org/, PyMOL Molecular Graphics System, Version 1.3; Schrödinger, LLC).18 All three structures were subsequently optimized at pH 6.9 in YASARA Structure.19 Briefly, the protein database (PDB) files containing the homology models were loaded into YASARA and corrected for any obvious structural errors. Semi-empirical quantum mechanics Molecular Orbital PACkage (MOPAC) was used to optimize the geometries of selected chains of residues (a maximum of 60 atoms/optimization): 62–67, 115–120, 134–141, 149–155 and 261–266.20 The structures were then corrected for structure errors and protein side chains optimized using combined rotamer sampling and steepest descent minimization.21 DNA and dolutegravir interaction hints were obtained by overlaying the HIV-1 homology models with the crystal structure of prototype foamy virus integrase co-crystallized with DNA and dolutegravir (PDB ID: 3S3M). The final structures were aligned, with global root mean square deviations of less than 1.5 Å indicating very good overall similarity. PyMOL Version 1.3 was used for structural visualization, analysis and image processing.
Results
Addition of E138K to R263K increases resistance to dolutegravir
We have previously shown that E138K can emerge in the presence of R263K under dolutegravir pressure.6 We now tested the effects of E138K on HIV-1 susceptibility to INSTIs using TZM-bl cell assays (Table 1). As previously reported,8 R263K conferred low-level resistance to dolutegravir [fold change (FC) = 2.3]. The addition of E138K to R263K modestly increased the resistance to dolutegravir (FC = 4.3). In agreement with previous studies, E138K alone did not confer resistance to raltegravir, elvitegravir or dolutegravir.15 The addition of E138K to R263K did not increase the resistance to elvitegravir (FC = 21.8 for R263K alone compared with FC = 16 for the E138K/R263K combination). We have previously shown and confirm here that R263K is innocuous to raltegravir susceptibility.8 Now, we also show that the addition of E138K to R263K did not confer resistance to raltegravir.
Table 1.
Effects of the E138K and R263K mutations on resistance to dolutegravir (DTG), raltegravir (RAL) and elvitegravir (EVG) in tissue culture and in cell-free strand-transfer assays, as well as on integrase enzyme activity
| Genotype | NL4.3 TZM-bl |
Cell-free strand-transfer assays |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DTG |
RAL |
EVG |
FC (Ki) |
enzyme activity |
|||||||||||
| IC50 (nM) | 95% CI (nM) | FC | IC50 (nM) | 95% CI (nM) | FC | IC50 (nM) | 95% CI (nM) | FC | DTG | RAL | EVG | FC (Km) | relative Vmax | enzyme efficiency | |
| WT | 8.6 | 6.4–11.7 | 1 | 9.9 | 6.6–14.7 | 1 | 0.5 | 0.4–0.7 | 1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.03 | 1 |
| E138K | 3.3 | 2.6–4.1 | 0.4 | 9.9 | 7.1–14.0 | 1 | 0.4 | 0.3–0.6 | 0.8 | 0.4 ± 0.1 | 0.8 ± 0.04 | 0.4 ± 0.05 | 0.5 ± 0.05 | 1.2 ± 0.04 | 2.4 |
| R263K | 20.2 | 15.8–26 | 2.3 | 11.5 | 7.1–18.7 | 1.2 | 10.9 | 6.2–19 | 21.8 | 3 ± 0.2 | 1.4 ± 0.3 | 4.5 ± 0.6 | 1.7 ± 0.3 | 0.8 ± 0.04 | 0.5 |
| E138K/R263K | 37 | 29.3–46.4 | 4.3 | 9.9 | 6.5–15.1 | 1 | 8 | 4.7–13.8 | 16 | 4.4 ± 0.2 | 1.7 ± 0.4 | 2.5 ± 0.5 | 1.3 ± 0.2 | 0.8 ± 0.04 | 0.6 |
We also measured the effects of E138K and R263K on integrase susceptibility to INSTIs in cell-free strand-transfer assays (Table 1). In agreement with the tissue culture experiments presented in Table 1, the addition of E138K to R263K increased the resistance to dolutegravir from FC = 3 to FC = 4.4. The addition of E138K to R263K decreased the resistance to elvitegravir in these cell-free assays (FC = 4.5 for R263K alone compared with FC = 2.5 for the E138K/R263K mutant enzyme). In agreement with previous studies8 and results from TZM-bl cell assays (Table 1), susceptibility to raltegravir remained unchanged for the R263K mutant enzyme compared with WT, and the E138K/R263K combination of mutations did not decrease susceptibility to raltegravir. The E138K mutant enzyme was associated with increased susceptibility to all the INSTIs tested.
Addition of E138K to R263K fails to restore integrase strand-transfer activity
We previously showed that the addition of H51Y to R263K decreases integrase strand-transfer activity.8 In order to determine whether E138K would have a similar effect, we measured the strand-transfer activity of recombinant purified integrase proteins bearing the E138K and R263K mutations alone or in combination. First, we showed that the optimal protein concentration for strand-transfer assays was 400 nM for each of the WT, E138K, R263K and E138K/R263K integrase enzymes (Figure 1). Next, we characterized the strand-transfer activity of these proteins in the presence of varying concentrations of target DNA (Table 1). In agreement with our previous results, the R263K substitution alone increased the integrase Michaelis constant (Km) by 1.7-fold compared with WT.6,22 The E138K mutant enzyme increased the target affinity (decreased the Km) as well as the maximal strand-transfer activity (Vmax). This finding is in agreement with the effect of E138K in the presence of the G118R mutation.16 However, the E138K/R263K combination failed to restore integrase strand-transfer activity to levels similar to those obtained with the WT enzyme (enzyme efficiency = 0.6 with E138K/R263K compared with 1 for WT).
Figure 1.
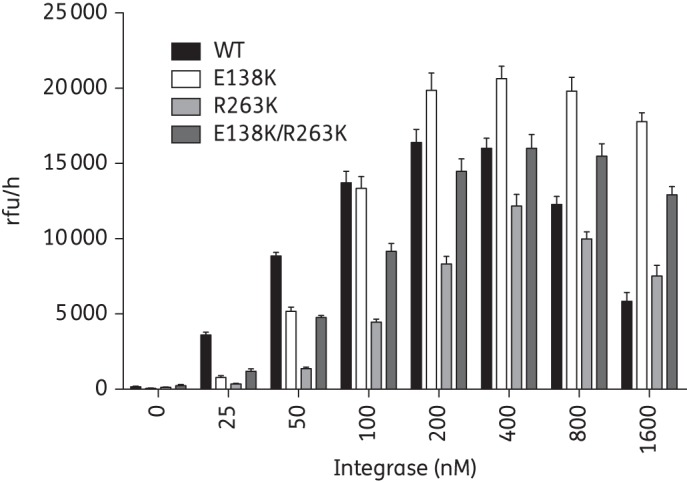
Strand-transfer activities of purified recombinant integrase proteins. Purified recombinant integrase proteins INWT, INE138K, INR263K and INE138K/R263K were used to measure strand-transfer activity in relative fluorescent units (rfu/h) in the presence of 18 nM target DNA and various concentrations of purified recombinant protein. Error bars indicate mean ± SEM.
Addition of E138K to R263K fails to restore HIV-1 infectivity and replication capacity
To determine whether the effects on integration observed with E138K in strand-transfer assays were significant, we measured the infectivity of WT virus and viruses bearing the E138K and R263K mutations in TZM-bl cells (Figure 2a). Our results show that infectivity was decreased by ∼2-fold for each of the mutant viruses, i.e. NL4.3IN(E138K), NL4.3IN(R263K) and NL4.3IN(E138K/R263K) (Figure 2a, inset).
Figure 2.

Effects of the E138K and R263K mutations on HIV infectivity and replicative fitness. (a) Luciferase produced by TZM-bl cells infected with increasing concentrations of the NL4.3, NL4.3IN(E138K), NL4.3IN(R263K) or NL4.3IN(E138K/R263K) virus was measured to assess infectivity after 48 h. The lines indicate the best fits. Error bars indicate mean ± SEM. The inset shows the relative EC50 ± SEM. (b) Reverse transcriptase (RT) activity in the culture fluids of PM1 cells infected with NL4.3, NL4.3IN(E138K), NL4.3IN(R263K) or NL4.3IN(E138K/R263K) virus was measured as cpm. Error bars indicate mean ± SEM.
We also monitored the effects of the E138K and R263K mutations on long-term viral replication capacity in PM1 cells (Figure 2b). In agreement with our previous results, R263K diminished HIV-1 replication capacity over 27 days of infection.8,9 Similar results were observed in the presence of the E138K mutation alone, while the addition of E138K to R263K did not restore the replication deficit observed with R263K. In agreement with the infectivity results, these experiments showed that all of the mutated viruses were impaired in terms of viral replication capacity. The addition of E138K to R263K did not improve either HIV-1 infectiousness or replication capacity.
Addition of E138K to R263K fails to restore HIV integration
To determine whether the effects of E138K on viral replication capacity also applied to integration in tissue culture, we used an Alu-mediated qPCR assay to measure the integration of HIV-1 DNA into the genome of primary human PBMCs that were infected with viruses bearing the E138K and R263K mutations (Figure 3). Our results confirmed the decrease in HIV DNA integration previously observed with R263K.6 The E138K mutation and the E138K/R263K combination of mutations did not increase levels of integration compared, respectively, with WT and R263K alone. Integration levels measured for the E138K/R263K virus remained significantly lower than those observed with WT virus.
Figure 3.
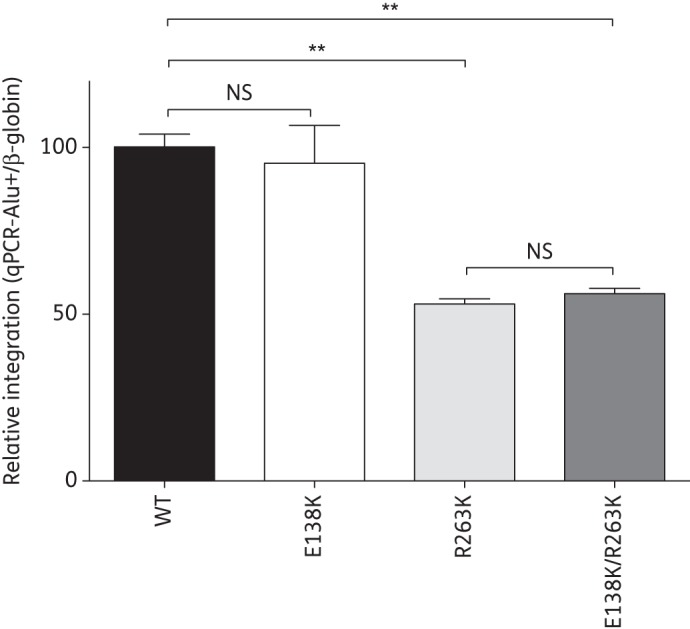
Effects of the E138K and R263K mutations on HIV integration. Alu-mediated qPCR quantification of integrated HIV DNA in primary human PBMCs infected with WT NL4.3 and with viruses containing the E138K, R263K and E138K/R263K mutations for 72 h. Results were normalized for β-globin gene content and expressed relative to the signal detected for WT, arbitrarily set at 100%. Error bars indicate mean ± SEM. NS, not significant. **Statistically significant differences (unpaired t-test, P < 0.001).
In silico studies of E138K mutant integrase
To gain insight into the effect of the E138K mutation on integrase catalytic activity, we performed a structural modelling of HIV integrase in the presence of E138K and R263K (Figure 4). In agreement with our previous results, our models predicted that the introduction of the R263K mutation changed the orientation of the H51 residue compared with WT structure.8 The addition of E138K was predicted to partially restore the orientation of H51 (Figure 4c). Additionally, the models predicted that the E138K and R263K mutations would alter the position and orientation of several other residues, including F139, Q146 and most notably Q148. Altogether, these in silico models suggest that the structure of the E138K/R263K mutant integrase differs from that of WT enzyme.
Figure 4.
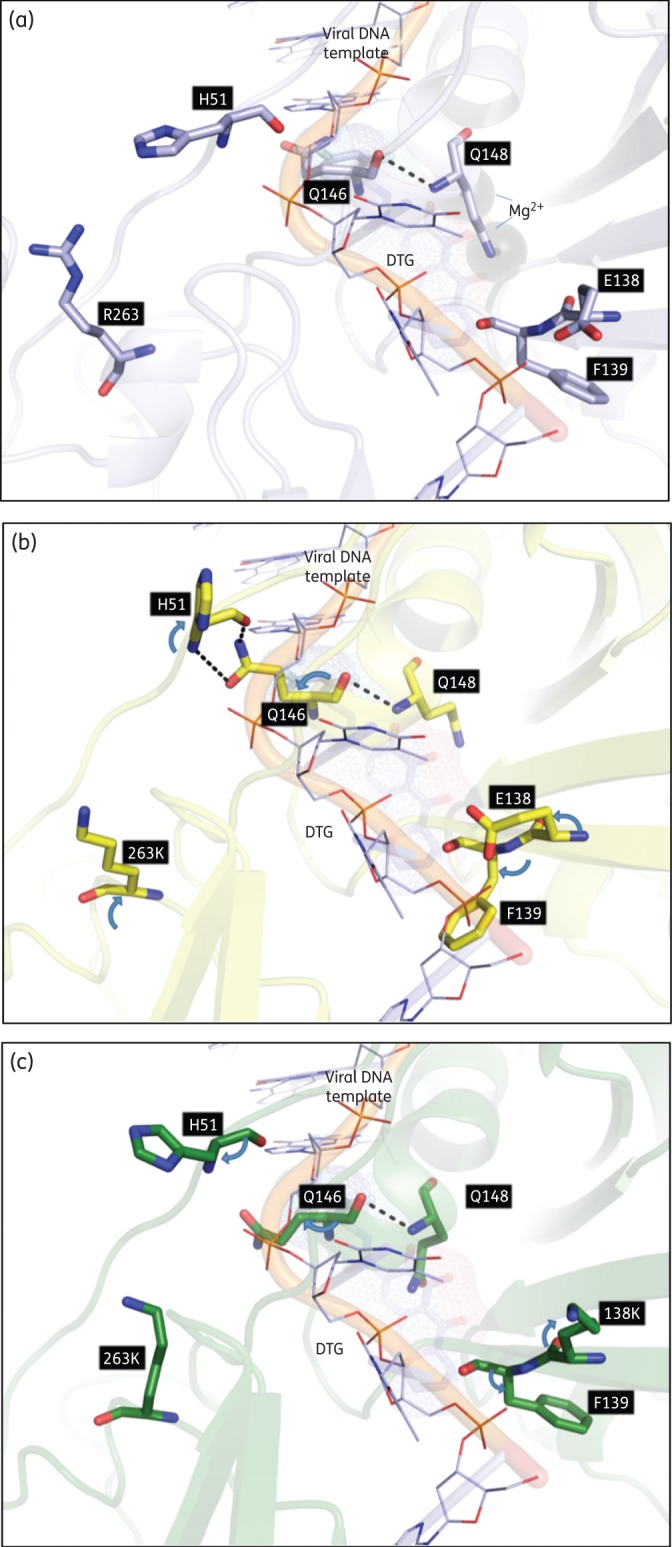
Visualization of putative interactions between HIV residues at positions 263 and 138. (a) WT model (light blue). (b) The R263K model showing altered orientations (blue arrows) and interactions with WT residues. (c) The E138K/R263K model showing an altered orientation from the R263K mutation alone (blue arrows). Key residues are labelled and shown as stick structures with standard atomic coloration. DTG, dolutegravir. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Discussion
Previous studies have shown that HIV-1 developed the R263K substitution under dolutegravir pressure.6 Several secondary mutations have also been selected following the emergence of R263K, including H51Y, E138K and the M50I polymorphism.6 However, none of these mutations resulted in viruses that possessed high levels of resistance to dolutegravir or high-level viral replication capacity.8,9 In fact, these selection studies have continued for more than 3 years and have not yet led to the emergence of highly fit and/or highly resistant viruses.8,10 Other investigators have also reported that HIV-1 did not develop high levels of resistance to dolutegravir in tissue culture selections.15
By way of comparison, raltegravir pressure led to the emergence of mutant viruses that were fully replication competent and highly resistant to this drug after only 42 days.15 More significant is that resistance mutations have not thus far been detected in treatment-naive patients who were enrolled in clinical trials with dolutegravir and who failed therapy.7,23–26 These results are different from those obtained during clinical trials with raltegravir and elvitegravir in which resistance mutations were detected in treatment-naive patients who failed treatment with these drugs.23,24,27–38
Here, we report the characterization of the E138K mutation in combination with R263K in regard to resistance, enzymatic activity and replication capacity. Mutations at position E138 have been observed with raltegravir and elvitegravir in tissue culture and in patients, often in association with primary resistance mutations including Q148.13 Several participants in the VIKING trial who had previously failed raltegravir subsequently developed additional E138K/A mutations upon dolutegravir failure.4 Previous studies have reported that the addition of E138K to Q148H did not increase resistance to raltegravir, elvitegravir and dolutegravir.15 In contrast, E138K and Q148K can synergize to confer 19-fold increased resistance to dolutegravir, whereas neither of these mutations alone conferred resistance to this drug.15 These observations underline the importance of E138 substitutions in pathways leading to resistance to dolutegravir.
We previously reported that the addition of both H51Y and M50I to R263K increased resistance to dolutegravir, from 2- to 6-fold with R263K alone to 16.5-fold and 15.5-fold for H51Y/R263K and M50I/R263K, respectively.8,9 Similarly, the H51Y/R263K and M50I/R263K combinations increased resistance to elvitegravir to 41.5-fold and 34-fold, respectively, compared with 3- to 24-fold with R263K alone.8,9 The H51Y/R263K combination of mutations did not alter raltegravir susceptibility whereas M50I/R263K modestly increased resistance to this drug.
We have previously demonstrated that levels of drug resistance can vary depending on differences in assays or target cells.8 Now, we report that the addition of E138K to R263K increased resistance to dolutegravir from 2.3-fold for R263K alone to 4.3-fold (Table 1). Direct comparisons showed that H51Y/R263K conferred the highest levels of resistance to dolutegravir, followed by M50I/R263K and E138K/R263K (T. Mesplède, unpublished observations). Similarly to H51Y and to a lesser extent M50I, the addition of E138K to R263K did not affect susceptibility to raltegravir (Table 1).8,9
In contrast with H51Y and M50I, the E138K/R263K combination decreased resistance to elvitegravir compared with R263K alone (Table 1).8,9 Our results also confirm previous reports that E138K alone does not affect HIV susceptibility to dolutegravir, raltegravir or elvitegravir (Table 1).15 Importantly, there was a direct correlation between resistance levels measured in tissue culture experiments and in biochemical cell-free assays (Table 1).
We have previously shown that R263K impaired HIV-1 replication capacity by diminishing integrase strand-transfer activity.6 The addition of H51Y to R263K further decreased viral fitness, integration and enzymatic activity.8 The M50I/R263K combination was less detrimental to these activities but failed to compensate for the deficit in viral replicative fitness observed with R263K.9 Here we show that the addition of E138K to R263K also failed to restore integrase strand-transfer activity (Table 1), integration into cellular DNA (Figure 3) and infectiousness and viral fitness (Figure 2). There was also an excellent correlation between the results of our tissue culture experiments and cell-free assays. However, E138K improved integrase maximal strand-transfer activity as well as the affinity of integrase for the target DNA (Table 1). In contrast, HIV genomic integration was not increased for E138K-containing virus, an observation that is consistent with the fact that glutamate is the natural amino acid at position 138 in the HIV-1 integrase. This discrepancy may be explained by the possibility that E138K diminishes integrase 3′-processing activity. Importantly, this may help to explain the common emergence of E138K in the presence of primary resistance mutations for raltegravir and elvitegravir as well as R263K.
Viruses that have lower 3′-processing activity and higher strand-transfer activity may be advantaged in the presence of INSTIs that specifically target the strand-transfer step of integration. However, E138K by itself does not confer resistance to dolutegravir, raltegravir or elvitegravir and is, therefore, unlikely to be selected in isolation (Table 1; Mesplede et al.8 and Wares et al.9). We have previously shown that the addition of E138K to G118R increased integrase strand-transfer activity through the enhancement of LTR binding.22 Whether E138K possesses similar activity when associated with R263K is currently under investigation in our laboratory.
In silico characterization of the E138K and R263K substitutions helps to explain the functional interaction between these two residues (Figure 4). We have previously reported that H51 is in close proximity to R263 and that its orientation is affected by the R263K substitution.8 The H51 and E138 residues lie on opposite sides of the viral DNA binding trough, next to the catalytic core domain and the disordered active site loop, respectively (Figure 4a).39 Both residues participate in viral DNA binding.8,22 Although E138 and R263 are separated by more than 10 Å (Figure 4a), our modelling suggests that these two residues might communicate through interactions involving H51 and Q146 in the active site loop (Figure 4b).
Based on these models, the presence of R263K might alter the H51, E138, F139 and Q146 main and side chain orientations and create hydrogen-bonding interactions between H51 and Q146. This could affect the conformation of the active site loop and help to explain the effect of the R263K mutation on integrase catalytic activity. The modelling simulation further suggested that the addition of E138K to R263K partially restores the orientation of H51, F139 and Q146 and abrogates hydrogen bonds between H51 and Q146 (Figure 4c). Superimposition of the viral DNA from the prototype foamy virus (PFV) IN structure of others also supports our model.40
A functional interplay between R263, H51 and E138 is further supported by tissue culture selection studies with lamivudine, which showed that H51Y/R263K viruses could rapidly acquire the E138K mutation in integrase.10 Importantly, the H51Y/E138K/R263K combination of mutations did not confer high-level resistance to dolutegravir.
Conclusions
Altogether, our tissue culture selection studies suggest that R263K is the signature resistance mutation for dolutegravir, but that none of the secondary mutations identified together with it in tissue culture selection studies can restore viral fitness. These results help to explain the absence of resistance mutations in individuals who have undergone treatment with dolutegravir.
Funding
This project was supported by the Canadian Institutes for Health Research (CIHR), the Canadian Foundation for AIDS Research (CANFAR), ISTP Canada and an unrestricted educational grant from ViiV Healthcare Inc. P. K. Q. is the recipient of a CIHR pre-doctoral fellowship. D. N. S. is the recipient of a CIHR doctoral scholarship.
Transparency declarations
None to declare.
References
- 1.Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance. N Engl J Med. 2011;365:637–46. doi: 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- 2.Wainberg MA, Mesplede T, Raffi F. What if HIV were unable to develop resistance against a new therapeutic agent? BMC Med. 2013;11:249. doi: 10.1186/1741-7015-11-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesplede T, Wainberg M. Integrase strand transfer inhibitors in HIV therapy. Infec Dis and Therapy. 2013;2:83–93. doi: 10.1007/s40121-013-0020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eron JJ, Clotet B, Durant J, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING study. J Infect Dis. 2013;207:740–8. doi: 10.1093/infdis/jis750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the Phase III VIKING-3 study. J Infect Dis. 2014 doi: 10.1093/infdis/jiu051. doi:10.1093/infdis/jiu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quashie PK, Mesplede T, Han YS, et al. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol. 2012;86:2696–705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382:700–8. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 8.Mesplede T, Quashie PK, Osman N, et al. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology. 2013;10:22. doi: 10.1186/1742-4690-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wares M, Mesplede T, Quashie PK, et al. The M50I polymorphic substitution in association with the R263K mutation in HIV-1 subtype B integrase increases drug resistance but does not restore viral replicative fitness. Retrovirology. 2014;11:7. doi: 10.1186/1742-4690-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliveira M, Mesplede T, Quashie PK, et al. Resistance mutations against dolutegravir in HIV integrase impair the emergence of resistance against reverse transcriptase inhibitors. Aids. 2014;28:813–9. doi: 10.1097/QAD.0000000000000199. [DOI] [PubMed] [Google Scholar]
- 11.Bar-Magen T, Sloan RD, Donahue DA, et al. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol. 2010;84:9210–6. doi: 10.1128/JVI.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimura K, Kodama E, Sakagami Y, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137) J Virol. 2008;82:764–74. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco JL, Varghese V, Rhee SY, et al. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011;203:1204–14. doi: 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki T, Kobayashi M, Wakasa-Morimoto C, et al. S/GSK1349572 is a potent next generation HIV integrase inhibitor and demonstrates a superior resistance profile substantiated with 60 integrase mutant molecular clones; Alexandria, VA, USA: Abstract 555. Foundation for Retrovirology and Human Health; Abstracts of the Seventeenth Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, 2010. [Google Scholar]
- 15.Kobayashi M, Yoshinaga T, Seki T, et al. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55:813–21. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quashie PK, Mesplede T, Han YS, et al. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother. 2013;57:6223–35. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu JJ, Wu TL, Liszewski MK, et al. A more precise HIV integration assay designed to detect small differences finds lower levels of integrated DNA in HAART treated patients. Virology. 2008;379:78–86. doi: 10.1016/j.virol.2008.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The PyMol Molecular Graphics System, Version 1.3. Schrodinger: LLC; [Google Scholar]
- 19.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 20.Dewar MJ. Some recent developments in quantum organic chemistry. Ciba Found Symp. 1978:107–29. doi: 10.1002/9780470720349.ch8. [DOI] [PubMed] [Google Scholar]
- 21.Canutescu AA, Dunbrack RL., Jr. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–72. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quashie PK, Mesplede T, Han YS, et al. Erratum: Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother. 2014;58:633. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raffi F, Jaeger H, Quiros-Roldan E, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13:927–35. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PubMed] [Google Scholar]
- 24.Raffi F, Rachlis A, Stellbrink HJ, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381:735–43. doi: 10.1016/S0140-6736(12)61853-4. [DOI] [PubMed] [Google Scholar]
- 25.van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect Dis. 2012;12:111–8. doi: 10.1016/S1473-3099(11)70290-0. [DOI] [PubMed] [Google Scholar]
- 26.Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807–18. doi: 10.1056/NEJMoa1215541. [DOI] [PubMed] [Google Scholar]
- 27.Eron JJ, Jr., Rockstroh JK, Reynes J, et al. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011;11:907–15. doi: 10.1016/S1473-3099(11)70196-7. [DOI] [PubMed] [Google Scholar]
- 28.Lennox JL, Dejesus E, Berger DS, et al. Raltegravir versus efavirenz regimens in treatment-naive HIV-1-infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr. 2010;55:39–48. doi: 10.1097/QAI.0b013e3181da1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lennox JL, DeJesus E, Lazzarin A, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet. 2009;374:796–806. doi: 10.1016/S0140-6736(09)60918-1. [DOI] [PubMed] [Google Scholar]
- 30.Markowitz M, Nguyen BY, Gotuzzo E, et al. Sustained antiretroviral effect of raltegravir after 96 weeks of combination therapy in treatment-naive patients with HIV-1 infection. J Acquir Immune Defic Syndr. 2009;52:350–6. doi: 10.1097/QAI.0b013e3181b064b0. [DOI] [PubMed] [Google Scholar]
- 31.Markowitz M, Nguyen BY, Gotuzzo E, et al. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46:125–33. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 32.Reynes J, Lawal A, Pulido F, et al. Examination of noninferiority, safety, and tolerability of lopinavir/ritonavir and raltegravir compared with lopinavir/ritonavir and tenofovir/ emtricitabine in antiretroviral-naive subjects: the progress study, 48-week results. HIV Clin Trials. 2011;12:255–67. doi: 10.1310/hct1205-255. [DOI] [PubMed] [Google Scholar]
- 33.Reynes J, Trinh R, Pulido F, et al. Lopinavir/ritonavir combined with raltegravir or tenofovir/emtricitabine in antiretroviral-naive subjects: 96-week results of the PROGRESS study. AIDS Res Hum Retroviruses. 2013;29:256–65. doi: 10.1089/aid.2011.0275. [DOI] [PubMed] [Google Scholar]
- 34.Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77–85. doi: 10.1097/QAI.0b013e31828ace69. [DOI] [PubMed] [Google Scholar]
- 35.DeJesus E, Rockstroh JK, Henry K, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379:2429–38. doi: 10.1016/S0140-6736(12)60918-0. [DOI] [PubMed] [Google Scholar]
- 36.Rockstroh JK, DeJesus E, Henry K, et al. A randomized, double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62:483–6. doi: 10.1097/QAI.0b013e318286415c. [DOI] [PubMed] [Google Scholar]
- 37.Sax PE, DeJesus E, Mills A, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379:2439–48. doi: 10.1016/S0140-6736(12)60917-9. [DOI] [PubMed] [Google Scholar]
- 38.Zolopa A, Sax PE, DeJesus E, et al. A randomized double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;63:96–100. doi: 10.1097/QAI.0b013e318289545c. [DOI] [PubMed] [Google Scholar]
- 39.Greenwald J, Le V, Butler SL, et al. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry. 1999;38:8892–8. doi: 10.1021/bi9907173. [DOI] [PubMed] [Google Scholar]
- 40.Hare S, Smith SJ, Metifiot M, et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol Pharmacol. 2011;80:565–72. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]