

CSF constituents are altered in multiple sclerosis, but whether this is a cause or a consequence of axonal degeneration is unclear. Vidaurre et al. identify two lipids that are enriched in the CSF of patients, and show that these induce bioenergetic dysfunction and oxidative damage in rat neuronal cultures.
Keywords: neurodegenerative mechanism, demyelinating disease, axonal degeneration, mitochondria, lipid metabolism
Abstract
Axonal damage is a prominent cause of disability and yet its pathogenesis is incompletely understood. Using a xenogeneic system, here we define the bioenergetic changes induced in rat neurons by exposure to cerebrospinal fluid samples from patients with multiple sclerosis compared to control subjects. A first discovery cohort of cerebrospinal fluid from 13 patients with multiple sclerosis and 10 control subjects showed that acute exposure to cerebrospinal fluid from patients with multiple sclerosis induced oxidative stress and decreased expression of neuroprotective genes, while increasing expression of genes involved in lipid signalling and in the response to oxidative stress. Protracted exposure of neurons to stress led to neurotoxicity and bioenergetics failure after cerebrospinal fluid exposure and positively correlated with the levels of neurofilament light chain. These findings were validated using a second independent cohort of cerebrospinal fluid samples (eight patients with multiple sclerosis and eight control subjects), collected at a different centre. The toxic effect of cerebrospinal fluid on neurons was not attributable to differences in IgG content, glucose, lactate or glutamate levels or differences in cytokine levels. A lipidomic profiling approach led to the identification of increased levels of ceramide C16:0 and C24:0 in the cerebrospinal fluid from patients with multiple sclerosis. Exposure of cultured neurons to micelles composed of these ceramide species was sufficient to recapitulate the bioenergetic dysfunction and oxidative damage induced by exposure to cerebrospinal fluid from patients with multiple sclerosis. Therefore, our data suggest that C16:0 and C24:0 ceramides are enriched in the cerebrospinal fluid of patients with multiple sclerosis and are sufficient to induce neuronal mitochondrial dysfunction and axonal damage.
Introduction
Multiple sclerosis is a chronic inflammatory demyelinating disease of the CNS and cause of neurological disability in young adults. Imaging studies and detailed neuropathological analyses of brain tissue have supported the notion that axonal loss is the cellular substrate of progressive disability (De Stefano et al., 1998; Bjartmar et al., 2000). Neurodegeneration occurs not only in lesions but also in normal-appearing white matter (Fu et al., 1998) and cortical grey matter in multiple sclerosis brains (Peterson et al., 2001). The presence of lesions around the ventricle (Swanton et al., 2007) or close to meningeal surfaces (Kutzelnigg et al., 2005) suggests that diffusible factors present in the CSF might contribute to multiple sclerosis pathology. Previous studies have attempted to identify components in the CSF as biomarkers, with the ability to predict disease course and to distinguish among patients with distinct clinical phenotypes and potential candidates [i.e. neurofilament light chain (NF-L)] have been suggested, although their predictive value remains to be thoroughly investigated (Malmestrom et al., 2003).
At a cellular level it is unclear whether components in the CSF contribute to mechanisms of axonal degeneration, or whether their presence in the CSF is the consequence of neurodegeneration. The most accepted theory is that axonal demise in multiple sclerosis is consequent to bioenergetic failure resulting from the inability of neurons to meet increased energetic demands after demyelination (Dutta et al., 2006). Redistribution of sodium channels along the axolemma increases ATP demands to maintain signal conduction. Mitochondrial dysfunction (Smith and Lassmann, 2002) leads to reversal of the function of the Na+/Ca2+ exchanger, resulting in the accumulation of cytosolic Ca2+, which is toxic to the cell (Waxman, 2006a; Trapp and Stys, 2009). This study addresses the effect of the CSF on neuronal mitochondrial function.
We used a xenogeneic system consisting of primary rat neuronal cultures incubated with CSF from patients with multiple sclerosis or controls and investigated the mitochondrial bioenergetic profile and axonal damage induced in these cells. We correlated these parameters with the concentration of several components present in the CSF. We then conducted genome-wide transcriptomic profiling of CSF-treated neurons to define the pathways leading to neurodegeneration. We also performed a targeted lipid analysis to detect whether the profile could distinguish patients with multiple sclerosis from control subjects.
Materials and methods
Reagents and antibodies
Oligomycin, FCCP [carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone], rotenone and antimycin A were obtained from Sigma-Aldrich; H2O2 from Fisher Scientific Co. Cell culture reagents were obtained from Invitrogen. Antibodies against nitrotyrosine, NF-L, NeuN and Olig2 were obtained from Millipore; anti-NF-L from Abcam; Iba1 from Wako Chemicals USA Inc.; and GFAP from Dako North America, Inc. All fluorophore-conjugated secondary antibodies were obtained from Invitrogen.
Cerebrospinal fluid sample collection
Patients in the discovery cohort were recruited at the Corinne Goldsmith Dickinson Centre for multiple sclerosis at Mount Sinai, New York, NY per an Institutional Review Board-approved protocol. All patients (ages 18–80 years) who had lumbar punctures performed for routine diagnostic evaluation at the multiple sclerosis centre, were eligible for the study. Patients who consented to participate had additional CSF withdrawn for the study. All subjects’ medical records were reviewed by a clinical multiple sclerosis specialist and categorized by final diagnosis after the lumbar puncture and additional diagnostics, including a follow-up period. Subjects classified as CIS or multiple sclerosis with a high level of certainty, comprised the ‘multiple sclerosis patient’ subset. Those classified as ‘not multiple sclerosis’ with a high level of certainty were included as controls. Subjects with probable diagnosis were excluded. Expanded Disability Status Scale scores were estimated from the neurological examination detailed in the chart. CSF samples were provided in a de-identified fashion that blinded the investigators to the clinical information until after the experiments were complete. Clinical investigators were similarly unaware of the experimental results until clinical classification was complete. CSF was kept at 4°C immediately after extraction, aliquoted and immediately frozen on dry ice and stored at −80°C. The CSF samples for the validation cohort were collected at the Johns Hopkins Multiple Sclerosis Centre, Baltimore, MD. As in the discovery cohort all subjects were reviewed by a clinical multiple sclerosis specialist. Control subject specimens were collected from the Department of Neurology at Johns Hopkins, outside the multiple sclerosis division. CSF was centrifuged before freezing.
Primary neuronal cultures
Hippocampal neuronal cultures derived from embryonic Day 18 Sprague-Dawley rat embryos were obtained as previously described (Kim et al., 2010). Cells were grown in Neurobasal® medium supplemented with B-27, GlutaMAX™ and penicillin/streptomycin and maintained at 37°C and 5% CO2 until the experiments were performed on in vitro Day 17. Antimitotics were added to the culture medium at in vitro Day 3 and removed after 24 h. Replacement of half of the culture medium was performed twice a week for 3 weeks. Only mature neurons kept in vitro for at least 17 days were used in the experiments.
Bioenergetic analysis
Oxygen consumption rate was measured using the Seahorse XF24 Bioanalyzer (Seahorse Bioscience) by following the manufacturer’s instructions. Neurons were seeded in Seahorse assay plates at a density of 8.4 × 104 cells per well. After 3 weeks in culture, they were incubated for 24 h with CSF from either multiple sclerosis or control patients. Mitochondrial bioenergetic function was then assessed by the sequential injection of 2 µM oligomycin, 4 µM FCCP, 0.5 µM rotenone and 4 µM antimycin A into the culture media. Results from at least three independent experiments are shown and data are expressed as a percentage of the basal oxygen consumption rate measured in cells incubated with control CSF.
Immunostaining
Immunocytochemistry was performed as previously described (Shen et al., 2008). Briefly, cells were washed once with PBS and fixed with 4% paraformaldehyde. After permeabilization in 0.1% Triton™ X-100 in PBGA buffer (0.1 M PB, 0.1% gelatin, 1% BSA, 0.002% azide) and blocking in 10% goat serum in PGBA, primary antibodies were incubated overnight followed by detection using fluorophore-conjugated secondary antibodies.
Neurofilament light chain measurement in cerebrospinal fluid
ELISA kit for NF-L detection from MyBiosource (cat no.: MBS705750) was used according to the manufacturer’s instructions.
Biochemical parameters in cerebrospinal fluid
IgG, glucose and cellularity in the CSF were measured by the clinical laboratory as part of routine diagnostic testing. Glutamate levels were measured using a glutamate colorimetric assay kit (BioVision Inc., cat no.: K629-100) following manufacturer’s instructions. Briefly, samples were incubated with a reaction mix in order to develop a colorimetric reaction that was quantified at λ = 450 nm in a microplate reader. Human cytokine arrays consisted of a sandwich-ELISA assay for detection of human cytokines in biological fluids (Panomics Inc., cat no: MA6150). The assays were performed according to the manufacturer’s instructions. Briefly, cytokine arrays were blocked with blocking buffer and incubated with CSF samples diluted 1:10 (v/v) in wash buffer. Diluted CSF was incubated with the membranes for 2 h before addition of a biotinylated cytokine antibody mix, and incubation with streptavidin-horseradish peroxidase. Membranes were developed using enhanced ECL solution (GE Healthcare). l-Lactate levels in the CSF were measured using Fluoro-Lactate kit from Cell Technology Inc. (cat. no: FLLACT100-2) following manufacturer’s instructions. Briefly, CSF samples were incubated with a reaction enzyme mix containing lactate oxidase in a 96-well plate, followed by addition of a detection reagent. Fluorescence at excitation/emmmision = 570–590 nm was detected using a fluorescence microplate reader.
RNA-Seq
Analysis of the RNA-Seq data was conducted as previously described (Huber et al., 2012). Briefly, reads were mapped to the reference genome (rn4) using the BWA alignment algorithm (Li and Durbin, 2009). For each transcript, we considered only the number of reads that were uniquely aligned to exon and splice-junction regions for each gene. This value was considered an estimated expression level and it was further subjected to log2 transformation and global median normalization, to compare transcript levels among distinct samples. Differentially expressed genes were identified with the software DESeq2 (Anders and Huber, 2010) using a false discovery rate (FDR) < 0.05 as a cut-off for statistical significance. The list of genes resulting from the study was subjected to Gene Ontology enrichment analysis using the software GOrilla (Eden et al., 2009).
RNA extraction and real-time reverse transcription polymerase chain reaction
RNA was extracted and purified with RNeasy® Micro kit (Qiagen) following the manufacturer’s protocol. RNA was reverse transcribed with qScript™ cDNA Supermix (Quanta BioSciences) and quantitative real-time PCR was performed using PerfeCTa® SYBR® Green FastMix® Rox 1250 (Quanta BioSciences) (primers listed in Table 1). After normalization to GAPDH transcripts, the average value for each transcript was calculated based on the values obtained in all the samples included for each experimental condition. Transcripts levels were also normalized for primer efficiency.
Table 1.
Primers for RNA extraction and real-time reverse transcription PCR
| Target gene | Accession No. | Forward (5’–3’) | Reverse (5’–3’) |
|---|---|---|---|
| Gapdh | NM_017008.4 | AGACAGCCGCATCTTCTTGT | CTTGCCGTGGGTAGAGTCAT |
| Hmox1 | NM_012580 | AGCATGTCCCAGGATTTGTC | GAGGCCATCACCAGCTTAAA |
| Nrg1 | NM_001271118.1 | TTCATCACAACCCTGCACAT | TGGGTCGTCTCGTACTCCTC |
| Mmp2 | NM_031054 | TTTGCTCGGGCCTTAAAAGTAT | CCATCAAACGGGTATCCATCTC |
| Vip | NM_053991 | CAGAAGCAAGCCTCAGTTCC | AGCCTGTCATCCAACCTCAC |
| Serpinb2 | NM_021696 | CACCACGCTTGGGAGATTAT | TTCATTGGCACATTCAAGGA |
| Adamts1 | NM_024400 | ACAGCCCAAGGTGGTAGATG | GATCACAGCCAGCTTTCACA |
| Grik1 | NM_017241 | ACAACCCTGGACTTGTTTGC | CAACCTCTCCATGGACCACT |
| Grin2b | NM_012574 | GCATGCCTACATGGGAAAGT | GTTGAGCACAGCTGCATCAT |
| Birc5 | NM_022274 | CTGATTTGGCCCAGTGTTTT | ACGGTCAGTTCTTCCACCTG |
| Ptprz1 | NM_001170685 | GGTTGGGAAAAACCATCCTT | TTTGCTTGCCTTGAAGACCT |
| Zeb1 | NM_013164 | ATGATCCCAACGTGGAAGAG | TGCATCTGGTGTTCCATTGT |
| Fabp7 | NM_030832 | CCAGCTGGGAGAAGAGTTTG | TTTCTTTGCCATCCCACTTC |
| Gsn | NM_001004080 | TGCAGCTGGATGACTACCTG | TTGGGTACCACGTGTTTGAA |
Quantitative lipidomic analysis
A crude lipid fraction was extracted from samples using a modified Bligh and Dyer procedure. Each sample was homogenized in 10 volumes of deionized water and three volumes of pure methanol were added containing 50 mM ammonium acetate, and the internal standards (C12:0 ceramide; Avanti Polar Lipids). Four volumes of chloroform were added and the mixture vortexed and centrifuged at 1000g for 10 min. The chloroform layer was removed and dried under a positive stream of nitrogen. Samples were resuspended in pure methanol for mass spectrometry using methods similar to those reported in our previous studies (Bandaru et al., 2013). Samples were injected using a PAL autosampler into a PerkinElmer HPLC equipped with a 2.6 µm C18 100 Å LC Column 50 × 2.1 mm (Kinetex) and a guard column (Phenomenex). For a typical run, the LC column was first pre-equilibrated for 0.5 min with the first mobile phase consisting of 85% methanol, 15% H2O, and 5 mM ammonium formate. The column was then eluted with the second mobile phase consisting of 99% methanol, 1% formic acid, and 5 mM ammonium formate at the flow rate of 400.0 µl/min. The eluted sample was injected into an electrospray ionization tandem mass spectrometer (ESI/MS/MS) API3000 (AB Sciex) and sample analysis was conducted by multiple reaction monitoring (MRM). Area under the curve for each ceramide molecular species (C16:0–C26:0 and C16:1–C26:1) was identified and quantified using Analyst 1.4.2 and MultiQuant (AB Sciex) softwares. Resulting data were normalized to the internal standard ceramide C12:0.
Unilamellar vesicle construction
Lipid micelles were constructed using a 1:1:2:2 ratio of phosphatidylethanolamine (C18:0), sphingomyelin (C18:0), cholesterol, and ceramide (one of C16:0, C22:0 or C24:0). In previous studies we found that this composition creates stable microparticles that are able to fuse with cellular membranes (Wheeler et al., 2009). Lipid emulsions in methanol were dried under a positive nitrogen gas flow and resuspended in PBS to 0.5 mg/ml total lipid content. Unilamellar vesicles were created at 37°C by extrusion using a polycarbonate membrane with a 0.08 µm pore size (Avanti Polar Lipids).
Magnetic resonance imaging analysis
The clinical MRI scans completed at the time of the diagnostic lumbar puncture by the patients diagnosed with multiple sclerosis in the discovery cohort were examined retrospectively and evaluated in an exploratory fashion. Axial 2D turbo spin-echo T2-weighted from the routine MRI scans, acquired at 3 T (Allegra, Siemens Medical Solutions), were used to measure the lesion volume. T2-hyperintense lesion volume was measured by a single experienced observer, blinded to the subject’s identity, as described in Inglese et al. (2010).
Data analysis and statistics
All data were analysed using GraphPad Prism 5.0 software. All data represent means ± standard error of the mean. All statistical tests were performed using either independent t-tests, or one-way ANOVA with Dunnett’s multiple comparison test to compare all groups to a given control, or Tukey’s test to compare all groups to each other. For correlation analysis, Spearman rank correlation method was used. All data were considered significant at P < 0.05.
Results
Cerebrospinal fluid from patients with multiple sclerosis induces oxidative damage in rat cultured neurons
To determine whether diffusible CSF factors in a multiple sclerosis patient might directly affect neuronal mitochondrial function, we assessed the bioenergetic profile of primary rat neurons cultured for 24 h in the presence of human CSF using the Seahorse Bioanalyzer. The CSF was obtained from 13 patients with multiple sclerosis and 10 non-multiple sclerosis controls. The multiple sclerosis group included only patients with a high level of certainty of diagnosis of multiple sclerosis, on the basis of clinical and imaging parameters. The control group included only subjects with a definite diagnosis of not having multiple sclerosis and comprised several unrelated disorders, such as amyotrophic lateral sclerosis, fibromyalgia, peripheral neuropathy, chronic relapsing inflammatory optic neuritis, lymphoma or chemotherapy-related sensory symptoms, steroid-dependent headache, chronic fatigue syndrome, non-specific cognitive complaints, and patients with non-specific sensory complaints and no objective evidence for multiple sclerosis. Because any potential inaccuracies resulting in a true ‘patient with multiple sclerosis’ being placed in the control group would tend to bias against detecting bioenergetic and biochemical differences between the groups, we excluded five potential controls for whom the diagnosis remained unclear at the time of data analysis. The demographic characteristics of the CSF donors in the two groups were similar (Table 2). Nearly all the patients with multiple sclerosis (12/13) had unmatched (i.e. present in the CSF but not in corresponding serum sample) oligoclonal bands in the CSF and most of them were also characterized by elevated IgG index and slightly elevated white blood cell count (Table 2). Measurement of the oxygen consumption rate in basal condition revealed disparities between the two groups, with greater consumption in neurons exposed to CSF samples from patients with multiple sclerosis compared to controls (Fig. 1A). A mitochondrial stress test was performed by the sequential addition to the culture medium of the mitochondrial respiratory chain inhibitor oligomycin (specific for complex V), the uncoupling agent FCCP, rotenone (specific for complex I) and antimycin A (specific for complex III). Subtraction of the baseline oxygen consumption rate from measurements after injection of the inhibitors allowed the calculation of additional parameters of mitochondrial function including proton leak, spare respiratory capacity, and coupling efficiency. Proton leak, a measurement of the proton flow from the inter-membrane space to the mitochondrial matrix and uncoupled from ATP synthase, was significantly greater (P < 0.05) in neurons exposed to CSF from patients with multiple sclerosis compared to non-multiple sclerosis control subjects (Fig. 1B). Because proton efflux is considered a mechanism that regulates mitochondrial membrane potential and that is activated to counteract the production of free radicals (Echtay et al., 2003; Mattiasson et al., 2003; Sullivan et al., 2003), these data suggested the possibility that the neurons exposed to CSF from patients with multiple sclerosis were subjected to increased oxidative stress. Indeed, immunostaining with antibodies specific for nitrotyrosine, a marker for oxidative damage (Fig. 1C), revealed greater immunoreactivity in neurons exposed to the patients’ CSF than controls (Fig. 1D). Characterization of the primary cultures by immunocytochemistry for neuronal and glial marked revealed a highly pure neuronal population (97 ± 1%), with only 3 ± 1 % of astrocytes and virtually no microglial cells or oligodendrocytes (Fig. 1E and F), suggesting that the oxidative radicals responsible for the protein nitration were produced by neurons. This interpretation was further supported by the detection of elevated transcript levels for Nos1, the neuronal form of the enzyme responsible for the synthesis of this free radical and by the significantly 1.2-fold (P < 0.001) increased expression of this enzyme in neurons treated with CSF from patients with multiple sclerosis compared to control CSF (Fig. 1G). Together, these data suggest that CSF-induced oxidative stress in neurons may contribute to the neurodegenerative changes detected in multiple sclerosis brains.
Table 2.
Patient and CSF characteristics of multiple sclerosis versus control subjects
| Multiple sclerosis cases, n = 13 | Controls, n = 10 | |
|---|---|---|
| n (%) | n (%) | |
| Age at time of lumbar puncture | ||
| Mean ± SE | 38.9 ± 2.9 | 42.6 ± 4.3 |
| Range | 21–56 | 18–56 |
| Sex | 5F, 8M | 8F, 2M |
| Final neurological diagnosis | ||
| CIS or RRMS | 8 (62%) | |
| PPMS | 4 (31%) | |
| PRMS | 1 (8%) | |
| Other | 0 | 10 (100%) |
| Presence of unmatched oligoclonal bands | ||
| Positive | 12 (92%) | 1 (10%) |
| Negative | 1 (8%) | 7 (70%) |
| Not done | 0 | 2 (20%) |
| IgG index | ||
| Elevated | 9 (69%) | 1 (10%) |
| Normal | 2 (15%) | 7 (70%) |
| Not done | 2 (15%) | 2 (20%) |
| CSF WBC (cells/µl) | 6 ± 1.7 | 2 ± 0.5 |
CIS = clinically isolated syndrome; CSF WBC = white blood cell count in CSF; PPMS = primary-progressive multiple sclerosis; PRMS = progressive-relapsing multiple sclerosis; RRMS = relapsing-remitting multiple sclerosis.
Figure 1.
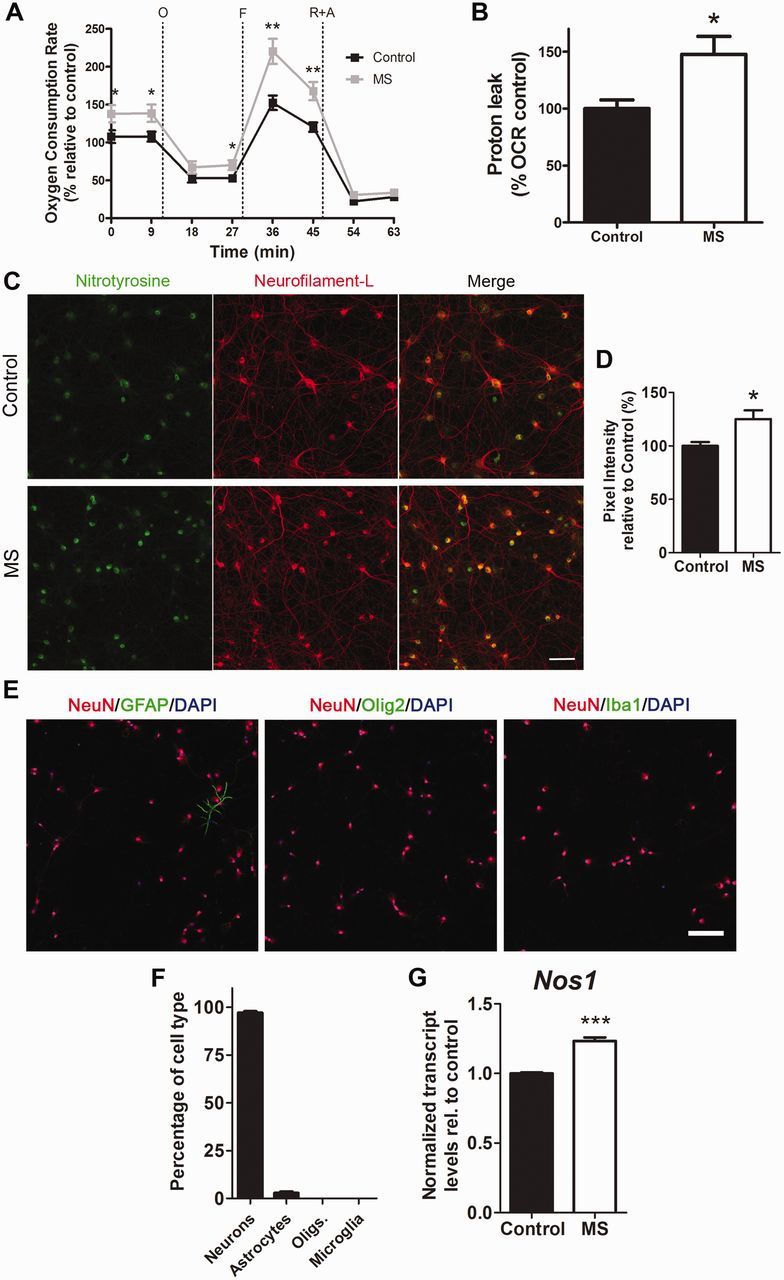
Oxidative damage induced in rat cultured neurons by exposure to the CSF of patients with multiple sclerosis. (A) Primary rat hippocampal neurons were incubated for 24 h with the CSF from patients with multiple sclerosis (MS) or from neurological controls and the neuronal profile was measured using a Seahorse Analyzer. Oxygen consumption rate (OCR) was measured as an indicator of mitochondrial respiratory function under basal conditions and during the sequential addition of 2 µM oligomycin (O), 4 µM FCCP (F), 0.5 µM rotenone and 4 µM antimycin (R+A). Data represent the mean ± SEM of the oxygen consumption rate levels relative to basal respiration levels from control CSF samples. (B) Proton leak levels were determined as readout of the bioenergetic analysis. Data are expressed as percentage relative to the oxygen consumption rate measured in cultured neurons exposed to CSF. (C) Confocal images of cultured neurons incubated for 24 h with CSF from control (n = 7) or patients with multiple sclerosis (n = 8) and stained with antibodies specific for nitrotyrosine (green) and NF-L (red) to assess oxidative damage. Scale bar = 50 µm. (D) Quantification of nitrotyrosine pixel intensity levels from three random regions (n = ∼80 cells per region) for each CSF sample and represented as the mean ± SEM relative to control CSF samples. (E) Immunocytochemistry of neuronal cultures with antibodies for NeuN (red) to label neurons, GFAP (green) for astrocytes, Olig2 (green) for oligodendrocytes and Iba1 (green) for microglia. DAPI (blue) was used as nuclear counterstain. Scale bar = 100 µm. (F) Quantification of the different cell types according to the immunostaining shown in E. (G) Transcripts levels of Nos1 in cultures treated with CSF from controls or patients with multiple sclerosis was assessed by RNA-Seq and data are shown relative to samples with control CSF after global median normalization. Statistical differences for A, B and D were determined using two-tailed independent t-test, *P < 0.05, **P < 0.01. Significance in G was determined by multiple comparison analysis, ***FDR < 0.001.
Cerebrospinal fluid-induced transcriptional changes unmask the activation of cellular response to stress in neurons
To define pathways uniquely activated in neurons exposed to multiple sclerosis CSF, we conducted an unbiased genome-wide transcriptomic deep sequencing analysis (RNA-Seq), followed by genome alignment and detection of differentially expressed transcripts (Fig. 2A). A principle component analysis revealed independent clustering of the data and clearly segregated the data into two transcriptional profiles corresponding to those detected in neurons exposed to the CSF from control subjects and patients with multiple sclerosis (Fig. 2B). A total of 13 021 transcripts (representing 12 331 genes) were detected, of these 1802 genes were significantly upregulated (FDR < 0.05) in the neurons exposed to CSF from patients with multiple sclerosis and could be further classified into functional categories related to response to stress (including neuronal injury or excitotoxicity), response to lipids, response to reactive oxygen species and regulation of cell death (Fig. 2C). Several genes from each category were further validated by quantitative real-time PCR and included genes activated in response to reactive oxygen species (e.g. Hmox1, Nrg1, Mmp2), response to neuronal injury (e.g. Vip, Serpinb2, Adamts1) or excitotoxicity (e.g. Grik1, Grin2b) (Fig. 2D). A total of 2147 genes were significantly downregulated in neurons after exposure to CSF from patients with multiple sclerosis, compared to the levels detected after exposure to control CSF. They could be classified into functional categories such as response to DNA damage, cellular response to stimulus, establishment of organelle organization, and nitrogen compound metabolic process (Fig. 2C). The majority of these genes were shown to have neuroprotective properties (e.g. Birc5, Ptprz1, Zeb1, Fabp7, Gsn) and were further validated by quantitative real-time PCR (Fig. 2E).
Figure 2.

Unbiased genome-wide transcriptomic analysis of neurons exposed to CSF reveals a signature of gene expression that is consistent with the impaired bioenergetic function. (A) Flowchart of the RNA-Seq analysis performed in neurons incubated with CSF from patients with multiple sclerosis or controls for 24 h. (B) Principle component analysis was used to determine data clustering. Note the segregation between the transcriptional changes induced in neurons by the CSF of patients with multiple sclerosis compared to those induced by the CSF from controls. (C) Gene ontology analysis revealed that the functional categories associated with elevated transcripts induced by multiple sclerosis CSF were related to response to stress, generation of reactive oxygen species (ROS) and cell death, whereas the downregulated ones included response to DNA damage and neuroprotection. (D and E) Validation of the transcriptional changes detected by RNA-Seq. RNA was isolated from cultured neurons incubated with CSF from controls or patients with multiple sclerosis and processed for quantitative real-time PCR. Values were normalized to GAPDH levels and referred as fold increase (D) or decrease (E) relative to the values measured in neurons treated with control CSF. Statistical differences for D and E were determined using two-tailed independent t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
Therefore, exposure to CSF from patients with multiple sclerosis increased the expression of genes involved in oxidative damage and neurotoxicity while decreasing the expression of genes involved in neuroprotection.
Cerebrospinal fluid-induced mitochondrial dysfunction in chronically stressed neurons correlates with the levels of neurofilament light chain
To better mimic the condition of chronic oxidative stress endured by neurons in the multiple sclerosis brain, we cultured neurons from the rat brain and subjected them to non-toxic oxidative stimuli (30 µM H2O2 added every other day to the medium) for 17 days. These stressed neurons were then challenged again with a 24 h exposure to the CSF from patients with multiple sclerosis or controls. Axonal damage was assessed by immunohistochemistry and scoring neurons for the presence of local enlargement, including varicosities and segmentations, which are considered to be one of the first signs of neurodegeneration (Trapp et al., 1998; Kim et al., 2010; Nikic et al., 2011) (Fig. 3A). The bioenergetic profile of these neurons further confirmed the functional impairment of these cells, as indicated by the decreased maximal respiratory rate and decreased spare respiratory capacity (Fig. 3B). Because this parameter provides a quantitative measure of the potential of neurons to adapt to increased energetic demand—and therefore considered an indicator of overall mitochondrial health—its decrease in chronically stressed neurons exposed to the CSF from patients with multiple sclerosis supported the notion that CSF components induced mitochondrial dysfunction and increased energetic demand in neurons, independently of demyelination (Fig. 3C).
Figure 3.
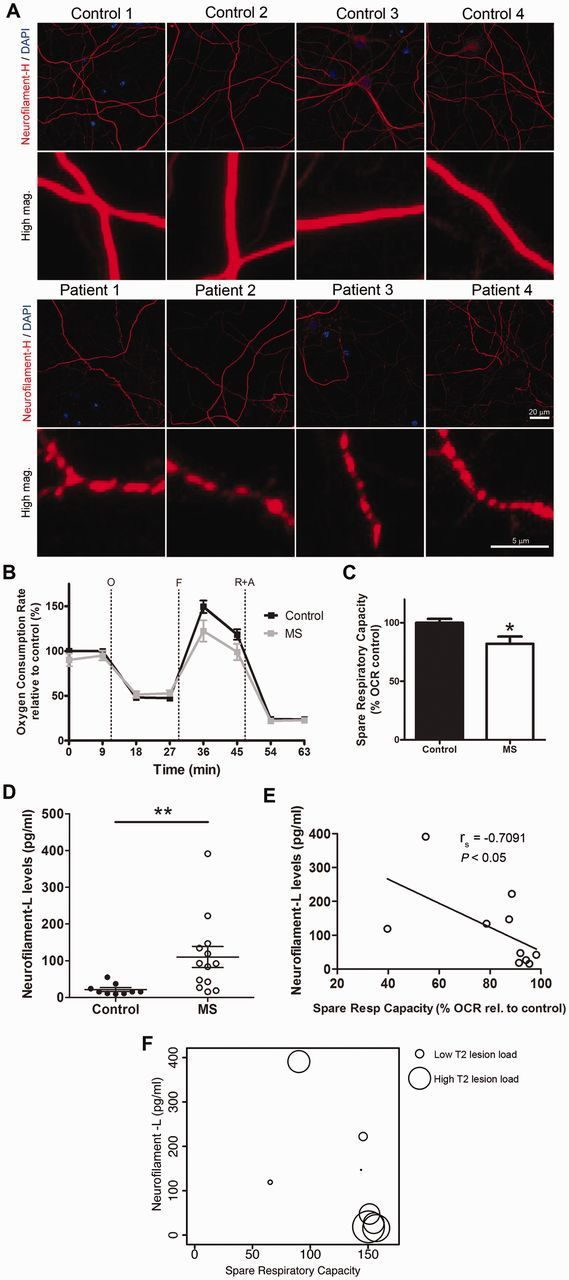
CSF from patients with multiple sclerosis (MS) induces axonal damage and mitochondrial dysfunction in chronically stressed neurons and correlates with NF-L levels. (A) Hippocampal neurons were cultured in mild oxidative stress conditions for 17 days and then incubated with CSF from control subjects or patients with multiple sclerosis for 24 h. Representative confocal images after immunocytochemical analysis with antibodies specific for neurofilament heavy chain (red) to visualize neurites and DAPI (blue) to label nuclei. Scale bar = 20 µm. High magnification shows axonal beading, a sign of axonal damage, in cultures incubated with CSF from patients with multiple sclerosis. Scale bar = 5 µm (B) Bioenergetic analysis was performed using a Seahorse Analyzer. Oxygen consumption rate (OCR) was measured as an indicator of mitochondrial respiratory function under basal conditions and during the sequential addition of 2 µM oligomycin (O), 4 µM FCCP (F), 0.5 µM rotenone and 4 µM antimycin A (R+A). Data represent the mean ± SEM of the oxygen consumption rate levels relative to basal respiration levels from control CSF samples. (C) Spare respiratory capacity levels of the cultures based on the results obtained from the bioenergetic analysis. Data are expressed as the percentage of the oxygen consumption rate from cultures incubated with control CSF samples. (D) NF-L levels were measured on the CSF samples from control and patients with multiple sclerosis using an ELISA method. The scatter plot shows the individual measurement of each sample and the mean ± SEM for each group. (E) Scatter plot indicating the correlation between the levels of NF-L in the CSF and the spare respiratory capacity measured in the neuronal cultures incubated with the same CSF. Correlation assessed by the Spearman's rank test; rho coefficient is shown. (F) Lesion volume on T2-weighted MRI (circle diameter) was determined from patients with multiple sclerosis and correlated with NF-L levels in CSF samples (y-axis) and spare respiratory capacity induced in the neuronal cultures (x-axis). Statistical differences for C and D were determined using two-tailed independent t-test, *P < 0.05, **P < 0.01.
To determine whether the detected impaired mitochondrial function in vitro could be correlated with any biomarker in the CSF (Lycke et al., 1998; Semra et al., 2002; Malmestrom et al., 2003) we measured the levels of NF-L by ELISA (Lycke et al., 1998; Semra et al., 2002; Malmestrom et al., 2003) and detected significantly higher levels in the CSF from patients with multiple sclerosis than controls (Fig. 3D). Only the CSF samples from patients with multiple sclerosis revealed an inverse significative correlation between NF-L levels and the bioenergetic parameter spare respiratory capacity, since the samples with the highest levels of NF-L were also the ones inducing the greatest decrease of spare respiratory capacity in neuronal cultures (Fig. 3E). Finally, we attempted to correlate these findings to imaging parameters, but we were unable to find any direct correlation as higher lesion volumes (T2 lesion volume = 26.13 ± 15.65 ml) were detected in patients whose CSF contained lower levels of NF-L (33.7 ± 5.3 pg/ml) and did not significantly impair mitochondrial function (Fig. 3F). The CSF with high levels of NF-L (134.0 ± 38.3 pg/ml) was the one inducing greater impairment of neuronal mitochondrial function (as measured by the smaller respiratory reserve capacity) and yet was characterized by lower lesion volume (T2 lesion volume = 1.87 ± 1.73 ml). Together these results suggest that the CSF from patients with multiple sclerosis induces mitochondrial dysfunction and axonal damage in chronically stressed rat neurons and the bioenergetic failure inversely correlates with NF-L levels present in the multiple sclerosis CSF samples. However, no significant correlation was detected for the T2 lesion volume.
Validation cohort of cerebrospinal fluid samples
These results were validated in an independent cohort (Table 3) of CSF, composed of eight controls and eight patients with multiple sclerosis recruited at Johns Hopkins Hospital. The controls included four subjects who underwent lumbar puncture to help differentiate normal pressure hydrocephalus from neurodegenerative disorders, a patient with congenital hydrocephalus, a patient with headaches, and a patient with idiopathic intracranial hypertension. For the patients with multiple sclerosis, five of eight subjects (62.5%) had unmatched oligoclonal bands in the CSF and an elevated IgG index (Table 3). The CSF samples from this validation cohort were used to conduct a similar analysis of bioenergetic profiling of chronically stressed neuronal cultures (Fig. 4A). Also in this case, exposure of rat neurons to the CSF from patients with multiple sclerosis decreased the maximal respiratory capacity and induced a dramatic decrease of spare respiratory capacity, compared to neurons exposed to control CSF (Fig. 4B). The NF-L levels in the CSF from patients with multiple sclerosis were significantly higher than those measured in controls (Fig. 4C) and they were inversely correlated with the spare respiratory capacity values measured in treated neurons (Fig. 4D). The validation of the findings in an independent cohort of CSF samples provided further support to the concept of direct toxicity of CSF on neurons, even in the absence of demyelination.
Table 3.
Patient and CSF characteristics of multiple sclerosis versus control subjects from the validation cohort
| Multiple sclerosis cases, n = 8 | Controls, n = 8 | |
|---|---|---|
| n (%) | n (%) | |
| Age at time of lumbar puncture | ||
| Mean ± SE | 48.4 ± 6.8 | 63.5 ± 8.3 |
| Range | 26–84 | 18–90 |
| Sex | 5F, 3M | 1F, 7M |
| Final neurological diagnosis | ||
| CIS or RRMS | 5 (62.5%) | |
| PPMS | 3 (37.5%) | |
| Other | 0 | 8 (100%) |
| Presence of unmatched oligoclonal bands | ||
| Positive | 3 (37.5%) | |
| Negative | 5 (62.5%) | |
| Not done | 0 | 8 (100%) |
| IgG index | ||
| Elevated | 5 (62.5%) | |
| Normal | 1 (12.5%) | |
| Not done | 2 (25%) | 8 (100%) |
| CSF WBC (cells/µl) | 2 ± 0.3 | 1 ± 0.5 |
CIS = clinically isolated syndrome; CSF WBC = white blood cell count in CSF; PPMS = primary-progressive multiple sclerosis; RRMS = relapsing-remitting multiple sclerosis.
Figure 4.

Independent validation of the effect of CSF from patients with multiple sclerosis (MS) on cultured neurons. (A) Hippocampal neurons were cultured in the presence of oxidative stimulus (30 µM H2O2) for 17 days in vitro and a bioenergetic analysis was performed using a Seahorse Analyzer after a 24 h incubation with CSF from control or patients with multiple sclerosis. Oxygen consumption rate (OCR) was measured as an indicator of mitochondrial respiratory function under basal conditions and during the sequential addition of 2 µM oligomycin (O), 4 µM FCCP (F), 0.5 µM rotenone and 4 µM antimycin A (R+A). Data represent the mean ± SEM of the oxygen consumption rate levels relative to basal respiration levels from control CSF samples. (B) Spare respiratory capacity levels of the cultures based on the results obtained from the bioenergetic analysis. Data are expressed as the percentage of the oxygen consumption rate from cultures incubated with control CSF samples. (C) NF-L levels were measured on the CSF samples from control and patients with multiple sclerosis using an ELISA method. The scatter plot shows the individual measure of each sample and the mean ± SEM of each group. (D) Scatter plot showing the correlation between NF-L in the CSF samples and the spare respiratory capacity induced in the neuronal cultures after incubation with CSF from patients with multiple sclerosis. Correlation was assessed by the Spearman’s rank test and rho coefficient is shown. Statistical differences for A–C were determined using two-tailed independent t-test, *P < 0.05, **P < 0.01. n.s. = not significant.
Elevated ceramide species distinguish the composition of cerebrospinal fluid samples from control subjects and patients with multiple sclerosis
We then reasoned that mitochondrial bioenergetics in neurons could be affected by the levels of glucose or lactate, as energy sources or by the levels of glutamate or inflammatory cytokines, as neurotoxins. Measurement of lactate or glucose revealed similar levels in the CSF from controls (glucose = 58.88 ± 3.96 mg/dl; lactate = 75.57 ± 1.45 µmol/l) and patients with multiple sclerosis (glucose = 55.31 ± 1.65 mg/dl; lactate = 76.80 ± 2.30 µmol/l) (Table 4). Furthermore, glutamate levels in multiple sclerosis CSF (8.71 ± 1.83 µmol/l) were not higher than in control CSF (13.58 ± 1.93 µmol/l) and a cytokine array able to detect levels of 30 different cytokines (Fig. 5A) did not reveal major differences between both groups (Fig. 5B). Thus, the bioenergetic changes induced in neurons exposed to the CSF collected from patients with multiple sclerosis could not be explained in terms of differential levels of energy sources, cytokines or glutamate. Given the previously described importance of antibodies directed against lipids in multiple sclerosis progression (Villar et al., 2005; Bosca et al., 2010) we then hypothesized that differences in lipid composition could distinguish the CSF of control and multiple sclerosis samples. Interestingly, several ceramide species were elevated in multiple sclerosis CSF samples compared to controls, including C16:0 and C24:0 ceramide and C16:0 monohexosyl ceramide (Fig. 5C). These data suggested that the sphingomyelin-ceramide pathway in multiple sclerosis might be relevant to the effect of the CSF on neurons.
Table 4.
Diffusible factors distinguishing the composition of CSF samples from multiple sclerosis subjects compared with controls
| Control |
Multiple sclerosis |
P-value | |||
|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | ||
| Glucose (mg/dl) | 58.88 | 3.96 | 55.31 | 1.65 | 0.426 n.s. |
| Lactate (µmol/l) | 75.57 | 1.45 | 76.80 | 1.87 | 0.611 n.s. |
| Glutamate (µmol/l) | 13.58 | 1.93 | 8.71 | 1.83 | 0.103 n.s. |
Glucose, lactate and glutamate levels were measured in the CSF obtained from multiple sclerosis and from control subjects. See Fig. 5.
n.s. = not significant.
Figure 5.

The search for diffusible factors distinguishing the composition of CSF samples from multiple sclerosis subjects compared with controls, revealed elevated ceramide species. (A) Levels of cytokines in CSF samples were assessed by ELISA. (B) Quantification of the cytokine arrays relative to control CSF samples. (C) Lipid profile on CSF samples from control subjects and patients with multiple sclerosis was performed using mass spectrometry and values are represented as relative to those measured in control samples. See also Table 4.
Exposure of cultured neurons to micelles containing the same ceramide species detected in the cerebrospinal fluid of patients with multiple sclerosis recapitulate the effect of cerebrospinal fluid on the neuronal bioenergetic profile
To determine whether the elevated ceramide species detected in the CSF from patients with multiple sclerosis were the mediators of the mitochondrial impairment observed in neuronal cultures, we conducted a reconstitution experiment. Lipid micelles containing the specific ceramide species elevated in the CSF of patients with multiple sclerosis (i.e. C16:0 and C24:0) were synthesized and assayed on cultured neurons for their ability to affect mitochondrial function. Micelles were synthesized in vitro by combining cholesterol, phosphatidylethanolamine, sphingomyelin and one of the ceramide species of interest in a 1:1:1:2 ratio. Two types of control micelles were also synthesized: one lacking any ceramide species and one containing a ceramide equally present in multiple sclerosis and controls (i.e. C22:0). Lipid incorporation into the cultured neurons after micelle treatment was confirmed by mass spectrometry and showed a clear dose-dependent enrichment (Fig. 6A). Exposure of neurons to the ceramide-containing micelles was sufficient to induce oxidative damage, as indicated by the positive immunoreactivity for nitrotyrosine (Fig. 6B). Remarkably, this effect was only seen when neurons were treated with micelles containing the ceramide species elevated in the CSF of multiple sclerosis (i.e. C16:0 and C24:0), whereas treatment with micelles either lacking ceramides or containing other ceramide species (i.e. C22:0), did not affect neurons (Fig. 6C). Treatment of chronically stressed neurons with micelle containing the ceramide species C16:0 and C24:0 was also sufficient to decrease neuronal maximal respiratory rate (Fig. 6D) and the spare respiratory capacity, whereas treatment with micelles either lacking ceramide or containing ceramide C22:0 did not affect the bioenergetic parameters (Fig. 6E). Therefore, only those ceramide species present at higher levels in the CSF of patients with multiple sclerosis compared with controls, were able to induce the functional bioenergetic changes and oxidative damage in treated neurons, recapitulating the effect of exposure to CSF. This reconstitution experiment underscores the importance of specific ceramide species present in the CSF in the induction of neuronal oxidative damage and defective bioenergetics in multiple sclerosis brains.
Figure 6.
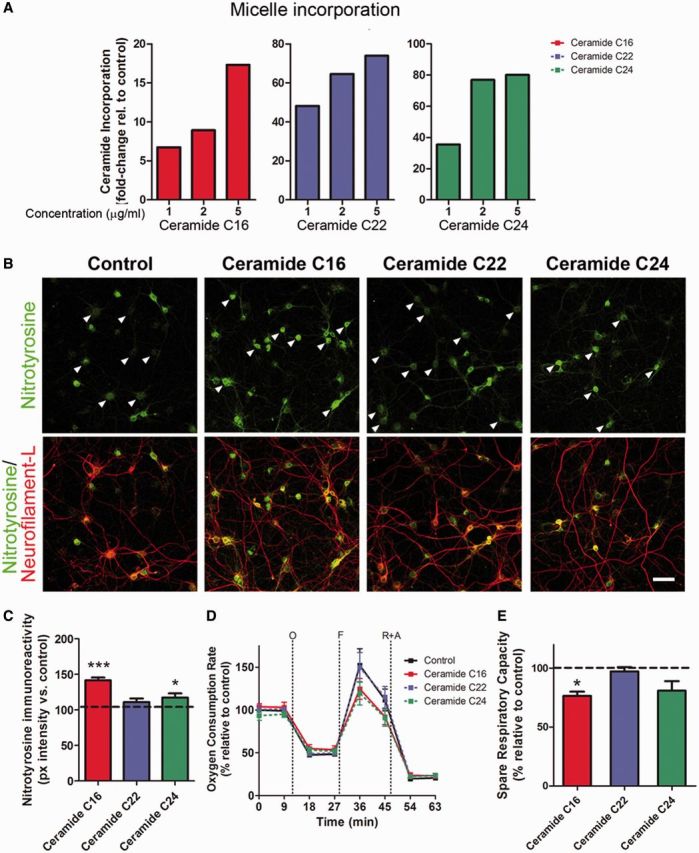
Incubation of neurons only with micelles containing ceramide species increased in the CSF of patients with multiple sclerosis affects mitochondrial bioenergetics and oxidative damage. (A) Lipid incorporation measured by mass spectrometry in cultured neurons that had been incubated for 24 h with micelles containing the same ceramide species detected in the CSF of patients with multiple sclerosis. (B) Immunocytochemistry of neuronal cultures after 24-h treatment with micelles composed of the indicated ceramide species and then stained with antibodies for nitrotyrosine (green) and NeuN (red) to assess oxidative damage. Arrowheads indicate differences in nitrotyrosine staining intensity in the different conditions. Scale bar = 50 µm. (C) Quantification of nitrotyrosine pixel intensity levels represented as the mean ± SEM relative to control levels. *P < 0.05, ***P < 0.001. (D) Bioenergetic analysis of neurons exposed to chronic oxidative stress and then treated with micelles for 24 h was performed using a Seahorse Analyzer. Data represent the mean ± SEM of the oxygen consumption rate levels relative to basal respiration levels measured in neurons treated with control micelles, containing ceramide species found in the CSF of controls. (E) Spare respiratory capacity levels calculated from the results of the bioenergetic analysis. Data are expressed as the percentage of the oxygen consumption rate from cultures incubated with control micelles sample. Statistical differences for C and E were determined using one-way ANOVA with Dunnett’s multiple comparison test. *P < 0.05, ***P < 0.001 versus control.
Discussion
One of the overarching hypotheses in the field of neurodegeneration in multiple sclerosis is that axonal damage in demyelinated axons results from the increased energetic demand consequent to the redistribution of Na+/K+ ATPase before increased intracellular Ca2+ levels (Waxman, 2006b). Axonal damage has also been attributed to defective metabolic or impaired trophic support, due to damaged oligodendrocytes (Funfschilling et al., 2012; Lee et al., 2012). However, it is conceivable that diffusible factors present in the CSF could also impact the ability of neurons to respond with adequate energy production, especially in the pathogenesis of cortical lesions. Decreased ability to meet energetic demand can be observed in the setting of mitochondrial impairment, as suggested by studies in cultured neurons (Kim et al., 2010), preclinical animal models of multiple sclerosis (Nikic et al., 2011) and in multiple sclerosis samples (Dutta et al., 2006; Fischer et al., 2013).
Based on the subpial (Kutzelnigg et al., 2005) location of grey matter lesions and to the frequent periventricular (Swanton et al., 2007) distribution of multiple sclerosis plaques, we designed this study to test the hypothesis that factors present in the CSF of controls and patients with multiple sclerosis could differentially modulate the bioenergetic profile of neurons, by a mechanism independent of demyelination. Therefore, we tested the effects of exposure to human CSF on the mitochondrial function of cultured rat neurons and show here that acute exposure to multiple sclerosis CSF induced a bioenergetic failure that was associated with increased mitochondrial proton leak, increased expression of genes involved in oxidative damage such as Hmox1, Mmp2 and Nrg1 (Marballi et al., 2010; Hill et al., 2012; Tronel et al., 2013), neuronal injury (Vip, Serpinb2, Adamts1) (Sasaki et al., 2001; Yamanaka et al., 2005; Waschek, 2013) and excitotoxicity (Grik1, Grik2b) (Micu et al., 2006; Newcombe et al., 2008), and decreased expression of neuroprotective genes (Birc5, Ptprz1, Zeb1, Fabp7, Gsn) (Qiao et al., 2005; Bui et al., 2009; Baratchi et al., 2011; Huang et al., 2012; Matsumata et al., 2012). These findings are consistent with a recent study in multiple sclerosis cortical lesions (Fischer et al., 2013), where neuronal oxidative damage was also found to be associated with increased expression of genes related to DNA damage and repair, excitotoxic cell death, oxidative stress and mitochondrial injury, compared with other inflammatory and non-inflammatory control brains. (Fischer et al., 2013).
Previous studies in similar xenogeneic models revealed that exposure to human CSF resulted in neurotoxicity in culture, although the molecular mechanisms remained elusive (Xiao et al., 1996; Alcazar et al., 2000). A potential explanation was the presence of pro-inflammatory cytokines in the CSF, leading to cytokine-induced synaptic hyperexcitability and consequent glutamate-dependent neurotoxicity (Rossi et al., 2012, 2013). This interpretation was supported by elevated glutamate levels detected in acute lesions and normal-appearing white matter in patients with multiple sclerosis (Srinivasan et al., 2005) and by studies reporting elevated levels of TNF-α, INF-γ, IL-1β, IL-6, IL-12 in patients with multiple sclerosis. Our study did not identify any significant difference in the levels of cytokines in the CSF of patients with multiple sclerosis compared to controls. There are several possible explanations for such a discrepancy, which include methodological differences, disease state and selection of the control population. The issue of variable methods of cytokine detection has been amply discussed as the main source of variability among different studies, and a careful and comprehensive discussion on the topic has been recently published (Malekzadeh et al., 2012). The variability of cytokine levels according to disease state and subtype has also been discussed in previous studies, although we believe that it was not an issue in our patient selection (Link, 1998; Matsushita et al., 2013; Rossi et al., 2014). A likely possibility for the similar cytokine levels in multiple sclerosis and non-multiple sclerosis samples in our study is the heterogeneity of our control population, as similarly elevated cytokine levels were also reported in the CSF of patients with other inflammatory diseases (Bielekova et al., 2012). The remarkable finding of our study is that—despite similar cytokine levels between multiple sclerosis and control CSF samples—only the CSF from patients with multiple sclerosis was capable of inducing oxidative damage and bioenergetic failure.
The bioenergetic dysfunction detected in multiple sclerosis brains has been often associated with aberrant production of reactive oxygen species and thought to contribute to spreading of the pathology. Indeed, neuronal damage consequent to reactive oxygen species has been reported in models of axonal injury, animal experimental autoimmune encephalomyelitis models and in human multiple sclerosis lesions (Smith and Lassmann, 2002; Dutta et al., 2006; Fischer et al., 2013; Persson et al., 2013). Our study supports these previous reports by detecting increased levels of nitrotyrosine, the product of peroxynitrite-mediated nitration of protein in neurons exposed to human CSF from patients with multiple sclerosis. In addition, our study indicates that oxidative damage in neurons is cell-autonomous and occurs as the consequence of the impaired bioenergetics state of the cell. This interpretation was supported by two lines of experimental evidence. First, oxidative damage occurred in highly pure neuronal cultures exposed to either CSF or to micelles containing the ceramide species C16:0 and C24:0. Second, the enzyme responsible for NO production, nitric oxide synthase Nos1 was expressed in cultured neurons in basal condition and its levels were increased by exposure to CSF from patients with multiple sclerosis. Thus, the high levels of nitrotyrosine detected in neurons treated with either CSF or ceramide-containing micelles were consistent with neurons as the source of the oxidative radicals responsible for the formation of peroxynitrites and subsequent protein nitration and possibly consequent to impaired mitochondrial function. The increased content of NF-L was detected in the most neurotoxic CSF samples thereby suggesting a correlation between this biochemical parameter and greater neuronal damage occurring in patients.
The mitochondrial changes induced by exposure to CSF from patients with multiple sclerosis and not by exposure to control CSF could not be explained in terms of differential cytokine expression, but correlated with the detection of elevated concentrations of specific ceramide species (i.e. C16:0 and C24:0). Because long-chain ceramides have been previously shown to suppress mitochondrial permeability transition pore activity and alter the bioenergetic profile (Novgorodov et al., 2008), the impaired mitochondrial function detected in neurons exposed to the CSF from patients with multiple sclerosis could be explained by a direct effect of specific ceramide species on the mitochondria. In this study we provide proof of concept for this hypothesis, as exposure to micelles composed of the same ceramides found in the CSF of patients with multiple sclerosis, was sufficient to impair mitochondrial function, decrease neuronal energy production and increase the production of reactive oxygen species and free radicals, with consequent neuronal damage. We hypothesize that a similar mechanism occurs in the brains of patients with multiple sclerosis as a consequence of the chronic exposure to cytotoxic ceramides that are present in the CSF. The dysfunctional mitochondria may attempt to compensate for the increased energetic demand, thereby generating free radicals that induce further damage and gradually exhaust the mitochondrial energy reserve capacity.
The importance of lipids in multiple sclerosis has been suggested by a series of previous studies relating the presence of antibodies against specific membrane lipids to disease progression (Villar et al., 2005; Kanter et al., 2006) and alterations in the lipid composition of normal-appearing white matter (Wheeler et al., 2008). These findings were interpreted within the context of an immune response to myelin debris but our study suggests the importance of ceramides as signalling molecules leading to impaired mitochondrial function. The concept of lipid signalling in multiple sclerosis is not unprecedented, as current therapies (e.g. fingolimod) have been targeted to specific sphingosine-phosphate receptors (Brinkmann et al., 2010). It is also not unique to multiple sclerosis, but shared with other neurological disorders characterized by neurodegeneration, as ceramide C16:0, ceramide C24:0 and C16:0 monohexosyl ceramide are also elevated in kainate-induced neuronal injury, Alzheimer’s disease (Guan et al., 2006; Mielke et al., 2012) and HIV-associated dementia (Haughey et al., 2004). Moreover, short-chain ceramides have been shown to induce mitochondrial alterations such as reactive oxygen species production and permeabilization of the mitochondrial membrane leading to neuronal death (Darios et al., 2003; Falluel-Morel et al., 2004). We hypothesize that ceramide species detected in the CSF of patients with multiple sclerosis were likely originated within the brain itself and possibly released into the CSF as lipoprotein complexes. We have not yet determined the exact mechanism for uptake but these ceramide-rich microparticles have been shown to readily cross endothelial and other plasma membranes and enter into cells (Tovar-Y-Romo et al., 2013).
Finally, it is important to mention that despite the relatively small size of our first discovery cohort of CSF samples, the major findings were validated in an independent cohort of samples obtained from a different multiple sclerosis centre, thereby supporting the reproducibility of the original findings and providing the rationale for the design of large longitudinal studies. Of note, when consolidating the two independent patient cohorts together and dividing them in two groups depending on the levels of spare respiratory capacity induced in neurons, we observed that six of seven patients with primary-progressive multiple sclerosis showed a greater decrease in spare respiratory capacity, whereas other parameters such as Expanded Disability Status Scale, oligoclonal bands, IgG index or white blood cell count did not discriminate patients with distinct multiple sclerosis type in these small cohorts (Table 5).
Table 5.
Patient and CSF characteristics in samples inducing high versus low spare respiratory capacity
| High spare respiratory capacity | Low spare respiratory capacity | |
|---|---|---|
| n = 9 | n = 9 | |
| Age at time of lumbar puncture | ||
| Mean ± SE | 35.7 ± 2.6 | 51.3 ± 5.5 |
| Range | 26.3–49.4 | 30.4–84.0 |
| Sex | 6F, 3M | 3F, 6M |
| Final neurological diagnosis | ||
| CIS or RRMS | 7 | 3 |
| PPMS | 1 | 6 |
| PRMS | 1 | 0 |
| Disease duration | ||
| Mean ± SE | 1.2 ± 0.4 | 6.0 ± 2.4 |
| Range | 0.1–3.7 | 0.2–21.1 |
| EDSS | ||
| Mean ± SE | 3.39 ± 0.7 | 4.17 ± 0.8 |
| Range | 0.0–6.5 | 0.0–7.0 |
| Presence of unmatched oligoclonal bands | ||
| Positive | 7 | 7 |
| Negative | 2 | 2 |
| IgG index | ||
| Elevated | 6 | 6 |
| Normal | 1 | 1 |
| Not done | 2 | 2 |
| CSF WBC (cells/µl) | 4 ± 1.7 | 4 ± 2 |
| NF-L (pg/ml) | 33.7 ± 5.3 | 134.0 ± 38.3 |
| Spare respiratory capacity on neuronal cultures (% OCR relative to control) | 81.2 ± 5.2 | 60.5 ± 6.5 |
CIS = clinically isolated syndrome; CSF WBC = white blood cell count in cerebrospinal fluid; EDSS = Expanded Disability Status Scale; PPMS = primary-progressive multiple sclerosis; PRMS = progressive-relapsing multiple sclerosis; RRMS = relapsing-remitting multiple sclerosis; OCR = oxygen consumption rate.
Future controlled studies in larger cohorts of patients will be necessary to define the potential use of bioenergetic profiling in cultured neurons as a tool for the definition of neurotoxic parameters in the CSF and to investigate the underlying brain tissue correlates of neurotoxicity with advanced neuroimaging measures.
Acknowledgements
We are grateful to Christine Hannigan, Colleen Farrell and Yadira Beconsme (MSSM) and Angela Vidal (JHH) for assistance with data collection, and Jacqueline Lovett (JHH) for her assistance with data analysis.
Glossary
Abbreviations
- FCCP
carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone
- NF-L
neurofilament light chain
Funding
These studies were performed with the support of grant R01 NS69835 to P.C. and funds from the Friedman Brain Institute and from the Multiple Sclerosis Research Foundation and from the Noto Foundation to P.C. and M.I. J.D.H. is the recipient of a postdoctoral fellowship from the Multiple Sclerosis Society of Canada and Fonds de la recherche en santé du Québec. N.J.H. receive support from the US Department of Defense (MS100151).
References
- Alcazar A, Regidor I, Masjuan J, Salinas M, Alvarez-Cermeno JC. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J Neuroimmunol. 2000;104:58–67. doi: 10.1016/s0165-5728(99)00225-8. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, Mielke MM, Sacktor N, McArthur JC, Grant I, Letendre S, et al. A lipid storage-like disorder contributes to cognitive decline in HIV-infected subjects. Neurology. 2013;81:1492–9. doi: 10.1212/WNL.0b013e3182a9565e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratchi S, Kanwar RK, Kanwar JR. Survivin mutant protects differentiated dopaminergic SK-N-SH cells against oxidative stress. PLoS One. 2011;6:e15865. doi: 10.1371/journal.pone.0015865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Komori M, Xu Q, Reich DS, Wu T. Cerebrospinal fluid IL-12p40, CXCL13 and IL-8 as a combinatorial biomarker of active intrathecal inflammation. PLoS One. 2012;7:e48370. doi: 10.1371/journal.pone.0048370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- Bosca I, Magraner MJ, Coret F, Alvarez-Cermeno JC, Simo-Castello M, Villar LM, et al. The risk of relapse after a clinically isolated syndrome is related to the pattern of oligoclonal bands. J Neuroimmunol. 2010;226:143–6. doi: 10.1016/j.jneuroim.2010.05.032. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–97. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Bui T, Sequeira J, Wen TC, Sola A, Higashi Y, Kondoh H, et al. ZEB1 links p63 and p73 in a novel neuronal survival pathway rapidly induced in response to cortical ischemia. PLoS One. 2009;4:e4373. doi: 10.1371/journal.pone.0004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Lambeng N, Troadec JD, Michel PP, Ruberg M. Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J Neurochem. 2003;84:643–54. doi: 10.1046/j.1471-4159.2003.01590.x. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121(Pt 8):1469–77. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–89. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–10. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falluel-Morel A, Aubert N, Vaudry D, Basille M, Fontaine M, Fournier A, et al. Opposite regulation of the mitochondrial apoptotic pathway by C2-ceramide and PACAP through a MAP-kinase-dependent mechanism in cerebellar granule cells. J Neurochem. 2004;91:1231–43. doi: 10.1111/j.1471-4159.2004.02810.x. [DOI] [PubMed] [Google Scholar]
- Fischer MT, Wimmer I, Hoftberger R, Gerlach S, Haider L, Zrzavy T, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(Pt 6):1799–815. doi: 10.1093/brain/awt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, et al. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain. 1998;121(Pt 1):103–13. doi: 10.1093/brain/121.1.103. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–21. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XL, He X, Ong WY, Yeo WK, Shui G, Wenk MR. Non-targeted profiling of lipids during kainate-induced neuronal injury. FASEB J. 2006;20:1152–61. doi: 10.1096/fj.05-5362com. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, et al. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–67. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- Hill JW, Poddar R, Thompson JF, Rosenberg GA, Yang Y. Intranuclear matrix metalloproteinases promote DNA damage and apoptosis induced by oxygen-glucose deprivation in neurons. Neuroscience. 2012;220:277–90. doi: 10.1016/j.neuroscience.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Ferrari CC, Monteiro de Castro G, Lafont D, Zhao C, Zaratin P, et al. Accelerated axonal loss following acute CNS demyelination in mice lacking protein tyrosine phosphatase receptor type Z. Am J Pathol. 2012;181:1518–23. doi: 10.1016/j.ajpath.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AK, Finkelman FD, Li CW, Concepcion E, Smith E, Jacobson E, et al. Genetically driven target tissue overexpression of CD40: a novel mechanism in autoimmune disease. J Immunol. 2012;189:3043–53. doi: 10.4049/jimmunol.1200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M, Madelin G, Oesingmann N, Babb JS, Wu W, Stoeckel B, et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain. 2010;133(Pt 3):847–57. doi: 10.1093/brain/awp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–43. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, et al. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci. 2010;13:180–9. doi: 10.1038/nn.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(Pt 11):2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link H. The cytokine storm in multiple sclerosis. Mult Scler. 1998;4:12–5. doi: 10.1177/135245859800400104. [DOI] [PubMed] [Google Scholar]
- Lycke JN, Karlsson JE, Andersen O, Rosengren LE. Neurofilament protein in cerebrospinal fluid: a potential marker of activity in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;64:402–4. doi: 10.1136/jnnp.64.3.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malekzadeh A, de Groot V, Beckerman H, van Oosten BW, Blankenstein MA, Teunissen C. Challenges in multi-plex and mono-plex platforms for the discovery of inflammatory profiles in neurodegenerative diseases. Methods. 2012;56:508–13. doi: 10.1016/j.ymeth.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Malmestrom C, Haghighi S, Rosengren L, Andersen O, Lycke J. Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology. 2003;61:1720–5. doi: 10.1212/01.wnl.0000098880.19793.b6. [DOI] [PubMed] [Google Scholar]
- Marballi K, Quinones MP, Jimenez F, Escamilla MA, Raventos H, Soto-Bernardini MC, et al. In vivo and in vitro genetic evidence of involvement of neuregulin 1 in immune system dysregulation. J Mol Med (Berl) 2010;88:1133–41. doi: 10.1007/s00109-010-0653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumata M, Sakayori N, Maekawa M, Owada Y, Yoshikawa T, Osumi N. The effects of Fabp7 and Fabp5 on postnatal hippocampal neurogenesis in the mouse. Stem Cells. 2012;30:1532–43. doi: 10.1002/stem.1124. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Matsuse D, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One. 2013;8:e61835. doi: 10.1371/journal.pone.0061835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–8. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–92. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Mielke MM, Bandaru VV, Haughey NJ, Xia J, Fried LP, Yasar S, et al. Serum ceramides increase the risk of Alzheimer disease: the Women's Health and Aging Study II. Neurology. 2012;79:633–41. doi: 10.1212/WNL.0b013e318264e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcombe J, Uddin A, Dove R, Patel B, Turski L, Nishizawa Y, et al. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol. 2008;18:52–61. doi: 10.1111/j.1750-3639.2007.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495–9. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- Novgorodov SA, Gudz TI, Obeid LM. Long-chain ceramide is a potent inhibitor of the mitochondrial permeability transition pore. J Biol Chem. 2008;283:24707–17. doi: 10.1074/jbc.M801810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AK, Kim I, Zhao P, Estacion M, Black JA, Waxman SG. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J Neurosci. 2013;33:19250–61. doi: 10.1523/JNEUROSCI.2148-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Qiao H, Koya RC, Nakagawa K, Tanaka H, Fujita H, Takimoto M, et al. Inhibition of Alzheimer's amyloid-beta peptide-induced reduction of mitochondrial membrane potential and neurotoxicity by gelsolin. Neurobiol Aging. 2005;26:849–55. doi: 10.1016/j.neurobiolaging.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, et al. Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol. 2012;71:76–83. doi: 10.1002/ana.22512. [DOI] [PubMed] [Google Scholar]
- Rossi S, Motta C, Studer V, Barbieri F, Buttari F, Bergami A, et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult Scler. 2013;20:304–12. doi: 10.1177/1352458513498128. [DOI] [PubMed] [Google Scholar]
- Rossi S, Studer V, Motta C, Germani G, Macchiarulo G, Buttari F, et al. Cerebrospinal fluid detection of interleukin-1beta in phase of remission predicts disease progression in multiple sclerosis. J Neuroinflammation. 2014;11:32. doi: 10.1186/1742-2094-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Seo-Kiryu S, Kato R, Kita S, Kiyama H. A disintegrin and metalloprotease with thrombospondin type1 motifs (ADAMTS-1) and IL-1 receptor type 1 mRNAs are simultaneously induced in nerve injured motor neurons. Brain Res Mol Brain Res. 2001;89:158–63. doi: 10.1016/s0169-328x(01)00046-8. [DOI] [PubMed] [Google Scholar]
- Semra YK, Seidi OA, Sharief MK. Heightened intrathecal release of axonal cytoskeletal proteins in multiple sclerosis is associated with progressive disease and clinical disability. J Neuroimmunol. 2002;122:132–9. doi: 10.1016/s0165-5728(01)00455-6. [DOI] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–34. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232–41. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128(Pt 5):1016–25. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–7. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton JK, Rovira A, Tintore M, Altmann DR, Barkhof F, Filippi M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol. 2007;6:677–86. doi: 10.1016/S1474-4422(07)70176-X. [DOI] [PubMed] [Google Scholar]
- Tovar-Y-Romo LV, Tabatadze N, Bandaru VV, Megra B, Williams D, Kanmonge M, et al. Microvesicles released from astrocytes regulate the peripheral immune response to CNS inflammation. Abstracts of the 12th International Symposium on NeuroVirology. October 29–November 2, 2013. Washington, DC, USA. J Neurovirol. 2013;19(Suppl 1):S1–101. doi: 10.1007/s13365-013-0211-9. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–91. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- Tronel C, Rochefort GY, Arlicot N, Bodard S, Chalon S, Antier D. Oxidative stress is related to the deleterious effects of heme oxygenase-1 in an in vivo neuroinflammatory rat model. Oxid Med Cell Longev. 2013;2013:264935. doi: 10.1155/2013/264935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar LM, Sadaba MC, Roldan E, Masjuan J, Gonzalez-Porque P, Villarrubia N, et al. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J Clin Invest. 2005;115:187–94. doi: 10.1172/JCI22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschek JA. VIP and PACAP: neuropeptide modulators of CNS inflammation, injury, and repair. Br J Pharmacol. 2013;169:512–23. doi: 10.1111/bph.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006a;7:932–41. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Ions, energy and axonal injury: towards a molecular neurology of multiple sclerosis. Trends Mol Med. 2006b;12:192–5. doi: 10.1016/j.molmed.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Wheeler D, Bandaru VV, Calabresi PA, Nath A, Haughey NJ. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain. 2008;131(Pt 11):3092–102. doi: 10.1093/brain/awn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, et al. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–49. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao BG, Zhang GX, Ma CG, Link H. The cerebrospinal fluid from patients with multiple sclerosis promotes neuronal and oligodendrocyte damage by delayed production of nitric oxide in vitro. J Neurol Sci. 1996;142:114–20. doi: 10.1016/0022-510x(96)00164-5. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Obata K, Fukuoka T, Dai Y, Kobayashi K, Tokunaga A, et al. Induction of plasminogen activator inhibitor-1 and -2 in dorsal root ganglion neurons after peripheral nerve injury. Neuroscience. 2005;132:183–91. doi: 10.1016/j.neuroscience.2004.12.003. [DOI] [PubMed] [Google Scholar]