

SUMMARY
In complex biological systems, small molecules often mediate microbe-microbe and microbe-host interactions. Using a systematic approach, we identified 3,118 small molecule biosynthetic gene clusters (BGCs) in genomes of human-associated bacteria and studied their representation in 752 metagenomic samples from the NIH Human Microbiome Project. Remarkably, we discovered that BGCs for a class of antibiotics in clinical trials, thiopeptides, are widely distributed in genomes and metagenomes of the human microbiota. We purified and solved the structure of a new thiopeptide antibiotic, lactocillin, from a prominent member of the vaginal microbiota. We demonstrate that lactocillin has potent antibacterial activity against a range of Gram-positive vaginal pathogens, and we show that lactocillin and other thiopeptide BGCs are expressed in vivo by analyzing human metatranscriptomic sequencing data. Our findings illustrate the widespread distribution of small-molecule-encoding BGCs in the human microbiome, and they demonstrate the bacterial production of drug-like molecules in humans.
INTRODUCTION
The human microbiome is composed of hundreds of bacterial species and thousands of strains, and its composition differs from person to person and between different body sites of the same individual (2012b). During the last decade, tremendous efforts have been made to sequence isolates of the human microbiota and metagenomic samples from various body sites (2012a; 2012b; Nelson et al., 2010; Qin et al., 2010). These studies have yielded a basic understanding of the “healthy” human microbiome and have correlated deviations from the healthy state to maladies such as obesity, diabetes, bacterial vaginosis, and Crohn’s disease (Gajer et al., 2012; Gevers et al., 2012; Gevers et al., 2014; Ravel et al., 2011; Turnbaugh et al., 2009). Several recent studies have begun to examine the human microbiome from a functional point of view, where direct molecular interactions between host and microbe are revealed (An et al., 2014; Hsiao et al., 2013; Mazmanian et al., 2005; Mazmanian et al., 2008; Nougayrede et al., 2006; Wieland Brown et al., 2013; Wyatt et al., 2010).
Diffusible and cell-associated small molecules often mediate host-microbe interactions in complex environments. Examples of small-molecule-mediated interactions have been revealed in symbioses between bacteria and insects (Oh et al., 2009), marine invertebrates (Kwan et al., 2012), nematodes (McInerney et al., 1991), and plants (Long, 2001). In addition, several studies have explored the role of small molecules in interactions between microbiota and the mammalian host. For example, Staphylococcus aureus pyrazinones were shown to be inducers of bacterial virulence (Wyatt et al., 2010), the Escherichia coli metabolite colibactin was found to contribute to colon cancer (Nougayrede et al., 2006), and polysaccharide A from Bacteroides fragilis has been shown to suppress the gut mucosal immune response (Mazmanian et al., 2005; Mazmanian et al., 2008). Recently, we and others showed that Bacteroides fragilis produces the canonical CD1d ligand α-galactosylceramide, revealing a specific mechanism by which the gut microbiota are capable of modulating host natural killer T cell function (An et al., 2014; Wieland Brown et al., 2013). Another recent study correlated the prevalence of hepatic cancer in mice to deoxycholic acid, a secondary bile acid produced by certain members of the gut microbiota (Yoshimoto et al., 2013). A recent metatranscriptomic study showed the expression of genes matching the COG category “secondary metabolites biosynthesis, transport, and catabolism”, which is consistent with the possibility of small molecule production but could also indicate the expression of catabolic and/or transport genes unrelated to biosynthesis (Leimena et al., 2013). These examples raise the question of whether there exists a much larger set of bacterially produced molecules that mediate microbiota-host interactions. Due to the complexity of the human microbiome and its vast coding potential, a more systematic approach is needed to explore small-molecule-mediated interactions between humans and their microbiota.
In this study, we explored the biosynthetic capacity of the human microbiome by performing the first systematic identification and analysis of its biosynthetic gene clusters. Unlike previous approaches that have focused on one compound or bacterial strain at a time, our approach allows the global analysis of biosynthetic gene clusters (BGCs) that encode small molecules in thousands of isolates of the human microbiota. By measuring the representation of these BGCs in human metagenomic samples, we can assess the small molecule coding capacity of a community, generating powerful hypotheses about which molecules might mediate microbe-host and microbe-microbe interactions in a particular community and how their prevalence differs among individuals. To illustrate the utility of this approach, we used a combination of chemistry, genetics, metagenomics, and metatranscriptomics to study a family of gene clusters that is widely distributed in the human microbiome, including the characterization of its small molecule product and the analysis of its prevalence among body sites and individuals.
RESULTS
A systematic approach identifies 3,118 biosynthetic gene clusters that are present in human metagenomic samples
The identification of biosynthetic gene clusters in bacterial genome sequences has become a powerful tool for natural product discovery. We began by using an algorithm we recently developed, ClusterFinder, to systematically analyze 2,430 reference genomes of the human microbiota from a range of body sites (Cimermancic et al., 2014). ClusterFinder detected >14,000 biosynthetic gene clusters (average of 6 gene clusters per genome) for a broad range of small molecule classes, including saccharides, nonribosomal peptides (NRPs), polyketides (PKs), ribosomally-encoded and posttranslationaly modified peptides (RiPPs), NRPS-independent siderophores and hybrids thereof (Figure 1 and see also Figure S1).
Figure 1. Overview of BGCs in the human microbiome.

A) Computational and experimental workflow for the identification of BGCs from human-associated bacteria. B) A bar graph showing the top 30 families by average BGC abundance from the human microbiome, and the average number and type of BGCs discovered in isolates of each genus using the workflow in A. See also Figure S1 and Supplemental Data File 1 for the full data set of predicted BGCs.
Reasoning that the small molecule products of BGCs that are widely distributed in the human population are most likely to mediate conserved microbe-host and microbe-microbe interactions, our next goal was to identify the subset of BGCs that is commonly found in healthy individuals. To achieve this goal, we examined the representation of these gene clusters in shotgun metagenomic sequencing data generated by the Human Microbiome Project (HMP, 752 samples collected from five main body sites of healthy subjects) (2012b). Briefly, we used mblastx (Davis et al., 2013) to detect metagenomic reads that match the >14,000 predicted BGCs (see Experimental Procedures for a more detailed description of how gene clusters were quantified in metagenomic samples). Using this method, we calculated that a subset of 3,118 biosynthetic gene clusters (22%) are present in the microbiomes of healthy individuals; all subsequent analyses focused on this subset (Figure 1 and see also Figure S1 and Supplemental Data File 1).
An overview of BGC types and distribution in the human microbiome
The identification of >3,000 biosynthetic gene clusters in the human microbiome was remarkable, given that almost nothing is known about their small molecule products or biological activities. We next examined the distribution and abundance of each of these BGCs in the 752 HMP samples.
The gut and oral cavity are by far the richest in BGCs, reflecting their increased microbial abundance relative to the other body sites (see also Figure S2-A, see Experimental Procedures) (2012b). A typical gut harbors 599 clusters, while a typical oral cavity harbors 1061 clusters; notably, the standard deviation of each of these values is large (152 and 143, respectively), pointing to a far greater degree of gene cluster diversity in some individuals than others. Typical skin, airway, and urogenital tract samples harbor many fewer BGCs on average (101, 31, and 41, respectively, see also Table S1). The smaller number of BGCs in the skin, airway, and urogenital tract communities could be a result of the lower microbial diversity in these communities, in comparison to the oral and gut microbiota (2012b). In addition, fewer samples were sequenced from skin, airways and urogenital tract communities (27, 65 and 94 samples, respectively) than from oral and gut communities (147 and 415, respectively), possibly affecting the total number of BGCs that were found to be present in these body sites.
The gene cluster classes in the human microbiota differ in important ways from those in non-human-associated bacteria (see also Figure S2-B). Even in light of the predominance of saccharides among environmental bacteria (Cimermancic et al., 2014), saccharides are significantly enriched in the microbiota (see also Figure S2-B), making them by far the most abundant BGC class in the gut and oral cavity (average of 443 and 662 saccharide clusters per sample, respectively). RiPPs, which are modestly enriched, are broadly distributed in every body site. Although nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) gene clusters are significantly depleted in the microbiota, they are still present at moderate levels (average of 57 PKS and 19 NRPS clusters in a gut sample and 129 PKS and 46 NRPS clusters in an oral cavity sample, see also Table S1) and notable exemplars are widely distributed in the healthy human population (see below). Most of the BGCs from non-human environmental isolates are harbored by members of the abundant genera Streptomyces, Bacillus, Pseudomonas, Burkholderia, and Myxococcus (Cimermancic et al., 2014); in contrast, most human-associated BGCs are harbored by members of the abundant human-associated genera Bacteroides, Parabacteroides, Corynebacterium, Rothia, and Ruminococcus (up to 7 BGCs per genome), pointing to undermined taxa that should be rich BGC sources for experimental mining efforts (Figure 1 and see also Figure S1). Many of these genomes harbor large BGCs despite being only 2–3 Mb, suggesting an important ecological role for their small molecule products. Some common genera of the human microbiota including Escherichia, Lactobacillus, Haemophilus, and Enterococcus harbor fewer than 2 BGCs per genome. Taken together, these findings indicate that the human microbiome harbors a rich and diverse array of BGCs that is mostly distinct in source and composition from that of non-human isolates.
The best-studied gene clusters are not widely distributed among healthy individuals. With the exception of the pyrazinone BGC from S. aureus (found in >9% of the airways samples), two of the most thoroughly studied BGCs from human-associated bacteria are found in <5% of the samples from their body site of origin (colibactin from E. coli, and polysaccharide A from Bacteroides fragilis (Mazmanian et al., 2005; Nougayrede et al., 2006; Wyatt et al., 2010). In contrast, nearly all of the BGCs from our data set that are widely distributed in healthy humans (present in >10% of the samples from the body site of origin) have never been studied or even described; a selected subset of these clusters is shown in Figure 2 (bgc01-bgc25). These results illustrate how little is known about the small molecule products of the most common BGCs in human-associated niches.
Figure 2. A selected subset of BGCs from the human microbiota.

A) 25 selected BGCs from the human microbiota, spanning each of the body sites (gut, vagina, airways and skin, and oral cavity), BGC types (PKS, NRPS, RiPPs, terpenes, NI siderophores and saccharides) and prevalent bacterial phyla (Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria). The label of each gene cluster indicates its source organism, body site of origin, and the percentage of HMP samples harboring this cluster in its body site of origin. All but two of these BGCs are present in more than 10% of the samples from their body site of origin, indicating that they are widely distributed among healthy subjects. B) Heat map showing the representation of BGCs from A in a subset of 60 selected HMP metagenomic samples from four body sites. The color of the cells in the heat map represents an abundance score ranging from 10 (blue) to 1000 (red) (key shown to the right, see Extended Experimental Procedures for calculation of abundance scores, and see also Figure S2 and Table S1).
Intriguingly, a smaller subset of 519 clusters is present in >50% of the gut samples; similar ‘common’ BGC subsets are present in the oral cavity (582 clusters), skin (65 clusters), urogenital tract (16 clusters) and airways (11 clusters) (see also Table S1). In addition, several widely distributed families of closely related BGCs were revealed by our analysis. Among these BGC families, four examples stood out because of their predominance in a body site, resemblance to a known BGC, or cosmopolitan distribution: a family of NRPS BGCs from the gut, a pair of PKS BGCs from the oral cavity, Bacteroidetes saccharides, and RiPPs.
A large family of nonribosomal peptide BGCs is widely distributed in the gut community
The first BGC family we identified is a set of NRPS clusters that are found exclusively in gut isolates (hereafter bgc26-bgc54). Surprisingly, members of this family are harbored by species of a wide variety of Gram-positive (Clostridium, Ruminococcus, Eubacterium, Lachnospiraceae, Holdemania) and Gram-negative genera (Bacteroides, Desulfovibrio). In addition to human gut isolates, this NRPS family is found in isolates from the chicken gut (Bacteroides barnesiae DSM 18169) and bovine rumen (Methanobrevibacter ruminantium M1) (Figure 3). Remarkably, 137/149 (92%) of the HMP stool samples contained at least one member of this family of NRPS clusters (see also Figure S3), while none or very few of the airways (0/94), oral (2/406), vaginal (2/65) and skin (1/27) metagenomic samples harbored a member of this family. The wide distribution and broad representation of this NRPS family suggests its importance for gut microbial inhabitants. Members of this BGC family fall into two main groups: the first consists of three NRPS modules while the second is composed of only two (Figure 3). The predicted substrate of the penultimate adenylation domain is an aliphatic amino acid (Leu, Ile, Val, or Ala) while the other adenylation domains have predicted substrates that vary widely among the clusters, suggesting that the small molecule products of these BGCs may be a family of closely related molecules.
Figure 3. An abundant family of NRPS BGCs is found exclusively in gut isolates and stool metagenomes.

Left, a phylogenetic tree (Maximum Parsimony, MEGA5) based on the main NRPS gene of 28 clusters from human bacterial gut isolates, two from bovine rumen archaeal isolates, and four from environmental isolates (see Experimental Procedures). The numbers next to the branches represent the percentage of replicate trees in which this topology was reached in a bootstrap test of 1000 replicates. Middle, schematic of each BGC (see also Figure S3 for a full heat map showing the prevalence and abundance of each member of this NRPS family in HMP stool samples). Right, domain organization of the NRPS genes of each cluster (A, adenylation domain; C, condensation domain; T, thiolation domain; R, terminal reductase domain).
A family of complex polyketides from oral Actinobacteria are widely distributed in the oral cavity
The most widely distributed multi-modular PKS in the oral cavity is a pair of closely related ~80 kb clusters of the trans-AT type (hereafter bgc54-bgc55). These clusters, which are found in two different species of oral Actinobacteria (Propionibacterium propionicum F0230a and Actinomyces timonensis DSM 23838), are strikingly similar in their domain architecture to a BGC from the marine isolate Streptomyces sp. A7248 that encodes the production of the homodimeric macrolide SIA7248 (Figure 4) (Zou et al., 2013). Despite this similarity, the enzymes encoded by the three clusters share only ~40% identity and no mobile elements were found in either of the human-associated clusters. SIA7248 is chemically similar to the marinomycins, which are known to have potent antibacterial and antitumor activity (Kwon et al., 2006). Our computational analysis of PKS architecture and AT domain selectivity predicts that the small molecule products of both PKS clusters will be nearly identical to the marinomycins and SIA7248, except for an alternative loading moiety activated by an acyl-CoA ligase domain that is absent in the SIA7248 cluster. Notably, bgc54 is widely distributed in healthy subjects (34% of the HMP supragingival plaque samples), indicating that a close relative of a complex marine bacterial metabolite might be common in the oral cavity.
Figure 4. A family of complex PKS BGCs is prevalent in the human oral cavity.

A) Related PKS BGCs in human oral actinobacteria P. propionicum F0230a and A. timonensis DSM 23838 and the marine actinobacterium Streptomyces sp. A7248. The label of each BGC indicates its source organism. B) Domain organization of the three BGCs shown in A (AT, acyltransferase domain; KS, ketosynthase domain; MT, methyltransferase domain; Ox, oxidation domain; ECH, enoyl-CoA hydratase domain; DH, dehydratase domain; KR, ketoreductase domain; ACL, acyl-CoA ligase domain; T, thiolation domain, HCS, HMG-CoA synthase domain; FKB, FkbH-like protein. Note that the domain architecture is remarkably conserved among the three pathways, despite low amino acid sequence identity (~40%). C) Structures of SIA7248, the product of the Streptomyces sp. A7248 BGC shown in A, and the related molecule marinomycin A.
Bacteroidetes saccharides are predominant and variable in the gut
Saccharide BGCs are the most abundant family in each of the five body sites, and are particularly predominant in the gut (74% of all BGCs in a typical HMP stool sample are saccharides, see also Table S1). The phylum Bacteroidetes harbors the largest number of saccharide BGCs (Cimermancic et al., 2014), two of which have been structurally and functionally characterized (polysaccharide A and B, (Baumann et al., 1992; Mazmanian et al., 2005; Mazmanian et al., 2008; Tzianabos et al., 1993)). Two observations were notable: First, among the most common saccharide BGCs were capsular polysaccharide loci from B. vulgatus and B. ovatus (see also Figure S4), which differ considerably from the better known examples in B. fragilis, suggesting that structurally distinct capsular polysaccharides might be remarkably common in the healthy human population. Second, 10 HMP stool samples that show similar taxonomic composition by MetaPhlAn profiling (Segata et al., 2012) harbor largely distinct sets of saccharide BGCs, with only a small number of BGCs common among the samples (see also Figure S4). Thus, knowledge about the taxonomic composition of gut communities from 16S rDNA analysis or metagenomic classification tools does not reveal the inherent diversity of BGCs in these communities.
Ribosomally synthesized, post-translationally modified peptides (RiPPs) are among the most widely distributed and variable BGCs encoded by the human microbiota
BGCs encoding modified ribosomal peptides (RiPPs) are widely distributed among the human microbiota. Three sub-classes of RiPPs were particularly notable: lantibiotics, which were numerous, variable, and broadly distributed throughout the Firmicutes and Actinobacteria from all of the major body sites; thiazole/oxazole-modified microcins (TOMMs), which were prevalent in the oral cavity; and thiopeptides. Although a small set of lantibiotics and TOMMs have been isolated from members of the human microbiota (Chikindas et al., 1995; Gonzalez et al., 2010; Lee et al., 2008), most are produced by pathogens or rare commensals.
Thiopeptides are highly modified RiPPs that have potent antibacterial activity against Gram-positive species. A semisynthetic member of this class, LFF571 (Novartis), is currently in Phase II clinical trials for treating Clostridium difficile infection (LaMarche et al., 2012). Although a thiopeptide BGC was previously predicted by our group and others in the genome of the skin isolate Propionibacterium acnes (Brzuszkiewicz et al., 2011; Wieland Brown et al., 2009), no thiopeptide BGC or small molecule product from the human microbiome has ever been characterized experimentally. Remarkably, in the analysis reported here, we discovered thiopeptide-like BGCs in isolates from every human body site (Lactobacillus gasseri, urogenital; Propionibacterium acnes, skin; Streptococcus downei and S. sobrinus, oral; and Enterococcus faecalis, gut). Additionally, we discovered a thiopeptide gene cluster in the porcine gut isolate Lactobacillus johnsonii PF01, indicating that thiopeptide BGCs are found in the microbiota of animals other than humans (Figure 5A).
Figure 5. Thiopeptide BGCs are widespread in isolates and metagenomes of all main human body sites.

A) Five thiopeptide BGCs from human isolates, eight thiopeptide BGCs from human metagenomes, and one thiopeptide BGC from a pig isolate are shown. The label of each BGC indicates its body site of origin. B) Precursor peptides corresponding to the thiopeptide BGCs shown in A. Note that the precursor peptides fall into six subgroups of nearly identical sequences. The structural portion of the precursor peptide is shown in red (see also Figure S5 for a phylogenetic analysis of thiopeptide BGCs). C) A heat map showing the representation and abundance of six oral thiopeptide BGCs in HMP metagenomic oral samples (see also Table S2 for the quantification of all thiopeptide BGCs in all HMP samples). Note that although each BGC shown in the heat map is well represented in the oral cavity, most samples harbor only one thiopeptide BGC.
We next turned to the question of whether we could discover additional thiopeptide gene clusters in metagenomic data that were not present in any sequenced bacterial genome. To address this question, we mined HMP metagenomic assemblies for the presence of additional thiopeptide gene clusters; remarkably, we discovered eight additional thiopeptide BGCs (7 complete, 1 partial) using this strategy, increasing the number of thiopeptide BGCs identified here to 13 (bgc56-bgc68) (Figure 5A) (see Experimental Procedures). Of these, we found one in human gut metagenomic samples (bgc61) and seven in human oral metagenomic data sets (bgc58, bgc59, bgc62, bgc64, bgc65, bgc67, and bgc68).
An analysis of the genes flanking these clusters suggests that bgc61 resides in a prominent member of the human gut microbiome (Eubacterium rectale), whereas two of the oral metagenomic thiopeptide gene clusters are found in Actinomyces and five others are harbored by Streptococcus (see also Table S2). Transposases and phage integrases are unusually prevalent among the thiopeptide BGCs (70%), and in at least one case (bgc66) we could show bioinformatically and experimentally that the cluster exists on a plasmid, suggesting a potential for mobility (see below, and note that the thiopeptide BGC from the porcine gut isolate Lactobacillus johnsonii PF01 (Lee et al., 2011) also exists on a plasmid). Four of the 13 thiopeptide BGCs are present in >20% of the HMP samples at one of the body sites, and 155/406 HMP oral samples (38%) harbor at least one thiopeptide BGC (Figure 5C and see also Table S2). For example, bgc61 is present in 11% of gut samples and bgc65 is present in 34% of oral samples, indicating that there exist widely distributed biosynthetic gene clusters in the healthy human population that are not found in the reference genome database.
In order to gain more insight into the evolution of these human-associated thiopeptide BGCs, we performed a detailed bioinformatic analysis of their biosynthetic genes and precursor peptides. Genomic and metagenomic thiopeptide BGCs fall into six sub-families in which members of each subfamily harbor similar precursor peptides (Figure 5A and 5B). Notably, the BGC precursor peptides and enzymes that post-translationally modify them cluster largely according to their body site of origin rather than the phylogeny of their host (Figure 5A and 5B and see also Figure S5). Moreover, the distribution of the oral thiopeptide clusters across the sample set indicates that most of the samples harbor only one thiopeptide BGC (Figure 5C). Taken together, these results are consistent with the possibility that thiopeptide gene clusters are mobile and that they undergo horizontal transfer among the microbiota within a body site (Smillie et al., 2011).
Discovery and characterization of a thiopeptide from the human microbiome
We next set out to purify and solve the structure of a thiopeptide from the human microbiome. Of the 14 thiopeptide BGCs described here, three resembled BGCs for known thiopeptides (Figure 5A). bgc56 is homologous to the berninamycin BGC from Streptomyces bernensis, and one of its two putative precursor peptides is identical to that of berninamycin (Malcolmson et al., 2013). Likewise, the genetic organization and precursor peptide sequences of bgc68 and bgc66 are similar to those of the TP-1161 (Nocardiopsis sp. TFS65-07) and thiocillin (Bacillus cereus ATCC 14579) BGCs, respectively (Engelhardt et al., 2010; Liao et al., 2009; Wieland Brown et al., 2009). Since there is more precedent for the genetic manipulation of Lactobacillus than Propionibacterium and bgc68 does not exist in any of the reference strains, we selected bgc66 for chemical and biological characterization.
The reference genome in which bgc66 resides is a vaginal isolate of Lactobacillus gasseri from a subject in Texas (L. gasseri JV-V03, HMP DACC); L. gasseri is one of the four Lactobacillus species that commonly dominate the vaginal community (Ravel et al., 2011). In order to determine whether the small molecule product of bgc66 is produced under conditions of laboratory culture, we constructed an insertional mutant of the gene harboring a YcaO domain (locus tag: HMPREF0514_11730) by single crossover mutagenesis (see Experimental Procedures). By comparing the HPLC and LC-MS profiles of organic extracts of the wild-type and bgc66::HMPREF0514_11730 mutant strains, we identified a single peak that is present in the wild-type but missing in the insertional mutant (Figure 6A). This peak had an absorption spectrum consistent with a compound that harbors a trithiazoylpyridine core (Figure 6A and see also Figure S6). High-resolution mass spectrometry enabled us to calculate the empirical formula C51H45N13O10S7 (observed [M+H]+ at m/z 1224.15184, calculated [M+H]+ 1224.15354, Δppm= 1.3). This mass and empirical formula deviated from the computationally predicted mass and empirical formula for the bgc66 product (1058.0667 and C42H34N12O8S7, respectively), suggesting that the bgc66 product harbored at least one unknown posttranslational modification.
Figure 6. Characterization of a new antibiotic, lactocillin, from a human vaginal isolate.
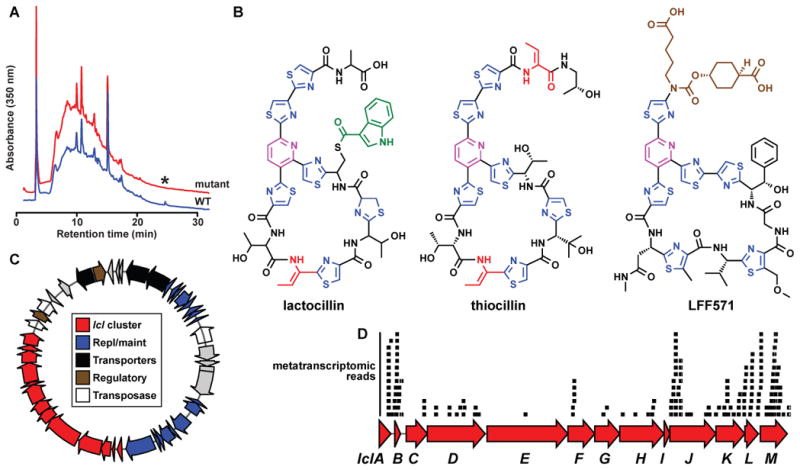
A) HPLC analysis of organic extracts of cell pellets from wild-type (red) and lclD insertional mutant (blue) strains of L. gasseri JV-V03, monitored at 350 nm. An asterisk indicates the HPLC peak corresponding to lactocillin, B) Planar structure of lactocillin (see Experimental Procedures and see also Figure S6 and Supplemental Data File 2 for details about its purification and structural elucidation), the Bacillus cereus antibiotic thiocillin, and the clinical candidate LFF571 (Phase II, Novartis). Note the structural similarities among the three thiopeptides. C) Plasmid harboring the lactocillin BGC (see Experimental Procedures for details of the experimental closure of the circular plasmid). The lactocillin gene cluster (red) occupies ~30% of the plasmid; other elements on the plasmid include plasmid replication and maintenance genes (blue), transporters (black), transcription regulators (brown) and transposases and phage integrases (white). D) Raw oral metatranscriptomic reads were recruited to the lcl cluster using blastn and aligned using Geneious. Each black bar represents one read. Note that the precursor peptide lclB is amongst the most deeply covered genes in the lcl cluster, as anticipated for a RiPP pathway (see Experimental Procedures and see also Figure S7 for metatranscriptomic analysis of the whole lcl plasmid, and of bgc65).
To obtain multi-milligram quantities of the bgc66 product for structural characterization, we performed a 50 L cultivation of L. gasseri JV-V03. Our initial attempts to perform NMR experiments on the isolated product failed due to its low solubility in NMR solvents and apparent instability. Hypothesizing that the bgc66 product harbored a free carboxylic acid that was partly responsible for its insolubility and instability, we treated the product extracted from a second 60 L cultivation with TMS-diazomethane to convert any free carboxylic acids to methyl esters. The resulting product had a mass consistent with the addition of a single methyl group, and exhibited greatly improved solubility and stability. After extensive purification by HPLC, the purified product (0.5 mg) was characterized using a series of 1D and 2D NMR experiments, high-resolution MS/MS and MSn, and isotope labeling experiments. Collectively, these data enabled us to determine the structure of the bgc66 product, to which we assign the name lactocillin (see Experimental Procedures, Figure 6B and see also Figures S6 and Supplemental Data File 2).
Lactocillin harbors a canonical 26-membered thiopeptide scaffold, with a trithiazolylpyridine core and a macrocycle with four cysteine-derived heterocycles and a single dehydrobutyrine residue. Three structural features set it apart from thiocillin: 1) The presence of an indolyl-S-cysteine residue at position 8, a feature that is structurally and regiochemically reminiscent of the 3-methylindolyl and quinaldic acid moieties of nosiheptide and thiostrepton, respectively (Just-Baringo et al., 2014; Kelly et al., 2009; Liao et al., 2009; Yu et al., 2009). Interestingly, the enzymes predicted to install the indolyl-S-cysteine in lactocillin (lclI, a thiolation domain, lclJ, an adenylation domain, and lclK, an alpha-beta hydrolase) are unrelated to any genes in the nosiheptide or thiostrepton BGCs, indicating that it may have arisen by convergent evolution. Lactocillin also 2) harbors a free carboxylic acid at the C-terminus, a rare feature among thiopeptides (Just-Baringo et al., 2014); and 3) lacks any oxygen-requiring posttranslational modifications, reflecting the anaerobic conditions under which L. gasseri thrives in the vaginal community (Figure 6B).
Next, we set out to determine whether lactocillin has a spectrum of activity similar to other thiopeptides, which are potent antibiotics against Gram-positive but not Gram-negative bacteria. To answer this question, we tested purified lactocillin against commonly observed pathogens and commensals from the vaginal community. Lactocillin was active against Staphylococus aureus, Enterococcus faecalis, Gardnerella vaginalis, and Corynebacterium aurimucosum but not Escherichia coli, an activity spectrum similar to that of other thiopeptides and suggestive of a potential role in protecting the vaginal microbiota against pathogen invasion. Interestingly, lactocillin was inactive against the vaginal commensals Lactobacillus jensenii JV-V16, Lactobacillus crispatus JV-V01, and L. gasseri SV-16A-US (a different strain from the lactocillin producer) at 425 nM, raising the possibility that the vaginal microbiota have evolved resistance against a compound they commonly encounter (see Experimental Procedures).
Metatranscriptomic data analysis reveals that the lcl cluster is expressed in humans
We next examined the distribution pattern of the lcl gene cluster in the human microbiota. In contrast to other human-associated thiopeptide BGCs (Figure 5A and 5C), the lcl gene cluster appears to have a limited distribution in genomic and metagenomic data sets: L. gasseri JV-V03 is the only one of the 10 sequenced L. gasseri genomes that harbors the cluster (see Extended Experimental Procedures), and none of the six vaginal metagenomic samples in which L. gasseri is the dominant strain contain reads that match the lcl cluster. However, building on the finding that lcl resides on a plasmid (Figure 6C) and that some BGCs are present in more than one body site (see also Figure S2), we broadened our search to publicly available metatranscriptomic data sets from the oral cavity (see Experimental Procedures). To our surprise, we found that two thiopeptide gene clusters – lcl and bgc65 – were covered by oral metatranscriptomic reads in multiple samples (in total, reads matching lcl and bgc65 were found in 3/38 and 11/38 of the supragingival plaque metatranscriptomic samples, respectively) (see Experimental Procedures, Figure 6D and see also Figure S7). In addition, the rest of genes on the plasmid in which the lcl cluster resides were also present in oral metatranscriptomic samples, further emphasizing its mobility (see also Figure S7). These results show that thiopeptide gene clusters are actively transcribed in human samples, suggesting a potential role in mediating microbe-microbe interactions in the communities in which they are expressed.
DISCUSSION
Here, we systematically identify BGCs in the genomes of human isolates, examine their representation in metagenomic and metatranscriptomic samples, identify widely distributed BGC families, and characterize the structure and activity of their small molecule products. Our approach introduces a new method for identifying and prioritizing BGCs for experimental characterization, and can easily be generalized to other BGC families. The resulting database of BGCs that were detected in human samples (see also Supplemental Data File 1) will serve as a resource for future studies that aim to discover small-molecule mediated interactions in the human microbiota.
Our approach has three important limitations. First, we rely on sequenced isolates of the human microbiota to predict the initial set of BGCs that we later use to recruit reads from whole-genome shotgun metagenomic samples. Although the ~2,400 bacterial genomes that we analyze here span all of the human-associated bacterial phyla and represent each major body site, they are biased toward, e.g., gut and oral residents and easily cultivated species.
Second, in examining the representation of BGCs in human metagenomic samples, we relied on the HMP whole-genome shotgun metagenomic data sets, which were sampled from fewer body sites and sub-sites than the larger 16S data set. Fewer samples were sequenced from skin than other body sites (27 versus at least 60 for all other body sites), and these samples were all obtained from one skin sub-site (retroauricular crease). We anticipate that the number of skin-associated BGCs will rise as the number and diversity of whole-genome shotgun metagenomic data sets increases.
Third, we show that metatranscriptomic reads matching the lcl cluster exist in RNA-seq data from the human oral cavity, suggesting that the lcl cluster is expressed in humans. To prove conclusively that lactocillin is produced in humans, it would need to be directly detected in human vaginal or oral samples using sensitive analytical techniques. The activity of lactocillin in its native context will need to be further explored in colonization experiments in which a lactocillin producer is compared to an isogenic strain deficient in lactocillin production.
Landmark studies of gene clusters encoding catabolic pathways from the microbiota have shown the power of connecting genes to microbiome-related functions. Sonnenburg (Sonnenburg et al., 2010) and Brumer and Martens (Larsbrink et al., 2014) have dissected the function of gene clusters from Bacteroides that catabolize fructans and xyloglucans, respectively. Both studies lend important insights into the role of specialized catabolic modules in competition for nutrient niches in the gut community, and they demonstrate that a mechanistic understanding of gene cluster function yields predictive power into how members of the gut community will respond to a change in diet. The current state of knowledge of how BGCs in the human microbiota function is comparatively less well developed, but holds great promise in yielding similar insights into microbe-host and microbe-microbe interactions.
The small molecule products of BGCs are widely used in the clinic, and they constitute much of the chemical language of interspecies interactions. Our data highlight the fact that there exist hundreds of widely distributed BGCs of unknown function in the human microbiome, and they provide a template for future experimental efforts to discover biologically active small molecules from the microbiota (see also Supplemental Data File 1 for a full dataset of human-associated BGCs). These molecules represent a promising starting point for studying microbe-host interactions at the level of molecular mechanism, and potentially a rich source of new therapeutics.
Experimental Procedures
Computational analysis of BGCs from the human microbiome
Genome sequences of 2,430 bacterial strains isolated from humans were obtained from JGI-IMG and BGCs were predicted using ClusterFinder (Cimermancic et al., 2014). The initial set of 44,000 putative BGCs was then analyzed by antiSMASH (Medema et al., 2011), which classified a subset of 14,000 BGCs into a known category. A database containing the amino acid sequence of each gene in these 14,000 BGCs was constructed and queried against the processed (post-QC) reads of 752 HMP metagenomic samples using mblastx (Davis et al., 2013). Abundance scores of genes and gene clusters were calculated and 3,118 BGCs were found to be present in at least one metagenomic sample (see also Extended Experimental Procedures and Supplemental Data File 3 for details about abundance score calculations, thresholds, and additional computational analyses).
Identification of thiopeptide BGCs from HMP metagenomic data
A database containing the amino acid sequence of 24 homologs of TclM (the enzyme responsible for pyridine ring formation in thiocillin) was constructed. This dataset was then used as a query for tblastn searches against metagenomic assemblies of the 752 HMP samples. Contigs that generated hits to multiple thiopeptide genes were analyzed using antiSMASH and results were verified manually. A thiopeptide BGC was called only when genes for all the essential posttranslational modifications were identified in addition to the precursor peptide (see also Extended Experimental Procedures).
Generation of an insertional mutant in L. gasseri JV-V03 and verification of lactocillin production
L. gasseri JV-V03 was cultivated in an anaerobic chamber (80% N2, 10% CO2 and 10% H2) in MRS broth (BD) at 37 °C. A 1000 bp fragment of lclD was PCR-amplified and cloned into pKM082, a suicide vector harboring an erythromycin resistance gene. L. gasseri JV-V03 was transformed with this vector by electroporation, and transformants were selected on erythromycin and verified using PCR (see also Extended Experimental Procedures and Table S3). Cell pellets from the wild-type strain and insertional mutants were grown in 1 L MRS broth, extracted with methanol, and analyzed using HPLC and LC-MS monitoring at 350 and 220 nm.
Large scale production and derivatization of lactocillin
A 60 L culture of L. gasseri JV-V03 was grown for two days at 37 °C. Cell pellets were harvested and extracted with 3 L methanol. Organic extracts were dried using rotary evaporation and fractionated on a C18 Sep Pak column using a step gradient of 20, 40, 60, 80 and 100% methanol in water. Lactocillin eluted in the 80 and 100% fractions, as determined by LC-MS. Semi-purified lactocillin from the 80% fraction was methylated using TMS-diazomethane, and the methylated lactocillin product was purified by preparative HPLC (see also Extended Experimental Procedures).
Structural elucidation of lactocillin methyl ester
The structure of purified lactocillin methyl ester was solved using a combination of mass spectrometry experiments (HRMS, HRMS/MS and HRMSn) and 1D and 2D NMR spectroscopy experiments (1D 1HNMR, gCOSY, HSQC, HMBC, and ROESY). In addition, heavy isotope feeding experiments were performed to confirm the identity of the indolyl acyl group in the structure (see also Extended Experimental Procedures).
Purification and MIC determination of lactocillin
Lactocillin was purified by preparative HPLC from the 100% Sep Pak fraction and quantified using HPLC. The minimal inhibitory concentration of purified lactocillin against a suite of commensal and pathogenic vaginal and oral isolates was tested using a range of concentrations from 42 to 425 nM. A vehicle-alone control was used in each experiment (see also Extended Experimental Procedures).
Metatranscriptomic analysis of thiopeptide BGCs
A database containing the nucleotide sequences of all human-associated thiopeptide BGCs was generated. Raw Illumina reads from 38 oral metatranscriptomic samples were compared to this database using blastn, and hits with expectation values <1e−10 were recruited to the clusters using Geneious.
Supplementary Material
1H, gCOSY, TOCSY, HSQC, HMBC, and ROESY spectra are shown. B. MS fragmentation analysis of lactocillin methyl ester. MS fragmentation was performed on an HR Orbitrap Velos mass spectrometer, and selected fragments are shown here. Fragment ion types are indicated next to the fragmentation site, and key fragments that were crucial for determining the position of the indolyl-S-cysteine (position 8) and thiazoline (position 7) moieties are shown in red boxes. Note the subsequent loss of one residue at a time, enabling their relative position to be determined. C. Chemical shifts of lactocillin in DMSO-d6 (600 MHz)
Selected read tiling maps of three BGCs in nine HMP metagenomic samples. Labels indicate the BGC number, the HMP sample identifier, and the abundance score computed using the mblastx method described in Extended Experimental Procedures. Note that the increase in the score computed using mblastx represents, to a great extent, the increase in the depth of coverage of a given BGC.
Counts of BGCs that are “commonly found” (detected in >50% of the HMP samples at the body site of origin), and counts of BGCs that are “typically found” at a certain body site (average number of BGCs per HMP samples at the body site of origin).
A) Phylogenetic tree representing 2,430 analyzed genomes, with the corresponding number and class of predicted BGCs shown as bars (total of ~14,000 BGCs). The tree was generated from 16S rDNA sequences downloaded from JGI-IMG and the final figure was generated using iTOL (Letunic and Bork, 2007). B) The same tree showing only the 3,118 BGCs detected in at least one HMP metagenomic sample. C) A network view of all BGCs predicted from human-associated bacteria. Edges were calculated based on similarities of Pfam compositions between a pair of BGCs, with a cutoff of 0.4 (Cimermancic et al., 2014). The network was generated using Cytoscape (Smoot et al., 2011). D) Average number and class of BGCs detected in HMP samples by genus. The 40 genera with the highest average number of BGCs are shown.
A) Venn diagrams representing BGCs present in >20% of the HMP samples at multiple body sites. B) Percentages of certain classes of BGCs in human-associated bacterial genomes (red) in comparison to non-human-associated bacterial genomes (blue), showing the respective enrichments or depletions. To assess the statistical significance of the distribution differences between human and non-human organisms, we bootstrapped the two samples by randomly drawing and analyzing 20% of BGCs 1000 times. C) A comparison between the fraction of BGCs predicted per source organism phylum in human-associated bacterial genomes (green) and non-human-associated bacterial genomes (red). Note: only the 3,118 BGCs present in HMP metagenomic samples were included in the analyses.
A heat map showing the representation of nine members of the gut NRPS family in HMP stool samples (samples shown here represent 92% of the HMP stool samples).
A) Ten HMP stool samples that have similar Bacteroidetes compositions based on their MetaPhlAn (Segata et al., 2012) classifications were selected (SRS015369, SRS012902, SRS023176, SRS013158, SRS024388, SRS016989, SRS016267, SRS013215, SRS058723, and SRS049900). Only members of the phylum Bacteroidetes are shown in the heat map displayed here. B) A heat map showing the representation of saccharide BGCs predicted from members of the phylum Bacteroidetes and detected in the same ten samples shown in A. Although some of the BGCs displayed here show similar patterns of abundance among samples, a large subset varies substantially across the sample set.
A maximum likelihood phylogenetic tree of the lantibiotic dehydratase enzyme of all thiopeptide BGCs described here, constructed using MEGA5. The numbers on the branches indicate the percentage of times this topology was reached in a bootstrap test of 1000 replicates. A similar tree was obtained using TclM homologs (responsible for pyridine ring formation).
A) UV-Vis absorbance profiles of lactocillin and thiocillin I (note the similarity between profiles). B) (+) HR-FTMS of lactocillin, consistent with the empirical formula C51H45N13O10S7 (observed [M+H]+ at m/z 1224.15184, calculated [M+H]+ 1224.15354, Δ ppm = 1.3) C) Time course of lactocillin production by L. gasseri JV-V03 in MRS broth under anaerobic conditions at 37 °C. D) HPLC-MS chromatogram of the trimethylsilyldiazomethane derivatization reaction (red) in comparison to the fraction containing native lactocillin (blue). E) (+) HR-ESI Orbitrap data of the resulting reaction showing a mass shift of 14 amu corresponding to the addition of one methyl group to the parent lactocillin and consistent with the empirical formula C52H47N13O10S7 (observed [M+H]+ at m/z 1238.16916, calculated for [M+H]+ at m/z 1238.16916, Δ ppm = − 0.02) F) Selected HMBC and COSY correlations observed and used to determine the structure of lactocilin methyl ester (see also Supplemental Data File 2 for NMR spectra and table of chemical shifts) G) HPLC-MS analysis of L. gasseri feeding experiments with 20 mM L-tryptophan (Top), 20 mM L-tryptophan D5 (middle), 20 mM L-alanine D3 (bottom). Mass spectra of the lactocillin peaks are shown in all three cases, where the +5 and +3 amu mass shifts are apparent in the case of L-tryptophan D5 and L-alanine D3, respectively. H) Selected MS/MS and MSn spectra of lactocillin methyl ester (See also Supplemental Data File 2 for an illustration of the detected fragments).
A) Mapping of metatranscriptomic reads from a human supragingival plaque sample (HMP DACC) to bgc65 (colors indicate the same key as in Figure 5). B) Mapping of metatranscriptomic reads from a human supragingival plaque sample (HMP DACC) to the L. gasseri JV-V03 plasmid harboring lcl (colors indicate the same key as in Figure 6).
The complete matrix of the 3,118 BGC predicted here (as columns) and 752 HMP metagenomic samples (as rows). Values given are abundance scores ranging from 10–1000. Matrices of body site and class of BGC are included as separate tabs. Locus tags of all BGCs are given in a tab containing all 3,118 classified BGCs. Pfams eliminated from the ClusterFinder output prior to computing the abundance of BGCs in HMP samples are also given in a separate tab.
Table 1.
Minimum inhibitory concentration (MIC) of lactocillin against vaginal and oral pathogens and commensals (in nM). See Experimental Procedures for details.
| Strain | Phylum | Description | Lactocillin MIC (nM) |
|---|---|---|---|
| Staphylococcus aureus | Firmicutes | Pathogen | 42a |
| Escherichia coli | Proteobacteria | Commensal | NAb |
| Enterococcus faecalis | Firmicutes | Pathogen | 425 |
| Lactobacillus jensenii | Firmicutes | Vaginal commensal | NAb |
| Lactobacillus gasseric | Firmicutes | Vaginal commensal | NAb |
| Lactobacillus crispatus | Firmicutes | Vaginal commensal | NAb |
| Corynebacterium aurimucosum | Actinobacteria | Vaginal pathogen | 42a |
| Finegoldia magna | Firmicutes | Vaginal pathogen | NAb |
| Gardnerella vaginalis | Actinobacteria | Vaginal pathogen | 212 |
| Streptococcus sanguinis | Firmicutes | Oral commensal | 212 |
| Streptococcus sobrinus | Firmicutes | Oral commensal/pathogen | 85 |
| Streptococcus mutans | Firmicutes | Oral commensal/pathogen | 425 |
Lowest concentration tested was 42 nM.
Highest concentration tested was 425 nM.
Note that the strain of L. gasseri tested was SV-16A-US, which is a different strain from the lactocillin producer (JV-V03).
- Streptococcus sanguinis and Gardnerella vaginalis were partially inhibited at 80 nM lactocillin, but not completely until the next concentration tested (212 nM).
- Streptococcus mutans was partially inhibited at 212 nM, but not completely until the next concentration tested (425nM).
HIGHLIGHTS.
3,118 biosynthetic gene clusters were identified in the human microbiome.
All families, including complex polyketides and nonribosomal peptides, are present.
Thiopeptide antibiotic gene clusters are widespread among the human microbiota.
A new antibiotic, lactocillin, has been discovered from a vaginal commensal.
Acknowledgments
We are indebted to Marnix Medema (Max Planck Institute for Marine Microbiology), Amrita Pati (JGI), and members of the Fischbach Group for helpful advice. We thank Krishna Parsawar and Chad Nelson (University of Utah) and Jeff Johnson (UCSF) for help with mass spectrometry experiments and Mark Kelly (UCSF) for help with NMR experiments. This work was supported by a Howard Hughes Medical Institute Predoctoral Fellowship (P.C.), NIH grant TW006634 (R.G.L.), a Medical Research Program Grant from the W.M. Keck Foundation (M.A.F.), a Fellowship for Science and Engineering from the David and Lucile Packard Foundation (M.A.F.), DARPA award HR0011-12-C-0067 (M.A.F.), and NIH grants OD007290, AI101018, AI101722 and GM081879 (M.A.F.). M.A.F. is on the scientific advisory boards of NGM Biopharmaceuticals and Warp Drive Bio.
Footnotes
Author Contributions
M.S.D., C.J.S., R.G.L., and M.A.F. designed the research and analyzed the data, with substantial input from P.C., M.M., and J.C. M.S.D., C.J.S. and L.C.W.B. performed the experimental research. M.S.D., P.C. and S.A. performed the computational research. M.S.D. and M.A.F. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- A framework for human microbiome research. Nature. 2012a;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structure, function and diversity of the healthy human microbiome. Nature. 2012b;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, Lu X, Zeissig S, Blumberg RS, Kasper DL. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156:123–133. doi: 10.1016/j.cell.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann H, Tzianabos AO, Brisson JR, Kasper DL, Jennings HJ. Structural elucidation of two capsular polysaccharides from one strain of Bacteroides fragilis using high-resolution NMR spectroscopy. Biochemistry. 1992;31:4081–4089. doi: 10.1021/bi00131a026. [DOI] [PubMed] [Google Scholar]
- Brzuszkiewicz E, Weiner J, Wollherr A, Thurmer A, Hupeden J, Lomholt HB, Kilian M, Gottschalk G, Daniel R, Mollenkopf HJ, et al. Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS One. 2011;6:e21581. doi: 10.1371/journal.pone.0021581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikindas ML, Novak J, Driessen AJ, Konings WN, Schilling KM, Caufield PW. Mutacin II, a bactericidal antibiotic from Streptococcus mutans. Antimicrobial agents and chemotherapy. 1995;39:2656–2660. doi: 10.1128/aac.39.12.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. 2014. Sumbitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C, Kota K, Baldhandapani V, Gong W, Abubucker S, Becker E, Martin J, Wylie KM, Khetani R, Hudson ME, et al. mBLAST: Keeping up with the sequencing explosion for (meta)genome analysis. Journal of Data Mining in Genomics and Proteomics. 2013;4:3. doi: 10.4172/2153-0602.1000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt K, Degnes KF, Zotchev SB. Isolation and characterization of the gene cluster for biosynthesis of the thiopeptide antibiotic TP-1161. Applied and environmental microbiology. 2010;76:7093–7101. doi: 10.1128/AEM.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajer P, Brotman RM, Bai G, Sakamoto J, Schutte UM, Zhong X, Koenig SS, Fu L, Ma ZS, Zhou X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra152. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Knight R, Petrosino JF, Huang K, McGuire AL, Birren BW, Nelson KE, White O, Methe BA, Huttenhower C. The Human Microbiome Project: a community resource for the healthy human microbiome. PLoS Biol. 2012;10:e1001377. doi: 10.1371/journal.pbio.1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DJ, Lee SW, Hensler ME, Markley AL, Dahesh S, Mitchell DA, Bandeira N, Nizet V, Dixon JE, Dorrestein PC. Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. The Journal of biological chemistry. 2010;285:28220–28228. doi: 10.1074/jbc.M110.118554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just-Baringo X, Albericio F, Alvarez M. Thiopeptide antibiotics: retrospective and recent advances. Mar Drugs. 2014;12:317–351. doi: 10.3390/md12010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WL, Pan L, Li C. Thiostrepton biosynthesis: prototype for a new family of bacteriocins. J Am Chem Soc. 2009;131:4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, Schmidt EW. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci U S A. 2012;109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HC, Kauffman CA, Jensen PR, Fenical W. Marinomycins A-D, antitumor-antibiotics of a new structure class from a marine actinomycete of the recently discovered genus “marinispora”. J Am Chem Soc. 2006;128:1622–1632. doi: 10.1021/ja0558948. [DOI] [PubMed] [Google Scholar]
- LaMarche MJ, Leeds JA, Amaral A, Brewer JT, Bushell SM, Deng G, Dewhurst JM, Ding J, Dzink-Fox J, Gamber G, et al. Discovery of LFF571: an investigational agent for Clostridium difficile infection. J Med Chem. 2012;55:2376–2387. doi: 10.1021/jm201685h. [DOI] [PubMed] [Google Scholar]
- Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Chae JP, Lee JY, Lim JS, Kim GB, Ham JS, Chun J, Kang DK. Genome sequence of Lactobacillus johnsonii PF01, isolated from piglet feces. Journal of bacteriology. 2011;193:5030–5031. doi: 10.1128/JB.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimena MM, Ramiro-Garcia J, Davids M, van den Bogert B, Smidt H, Smid EJ, Boekhorst J, Zoetendal EG, Schaap PJ, Kleerebezem M. A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets. BMC genomics. 2013;14:530. doi: 10.1186/1471-2164-14-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W. Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chemistry & biology. 2009;16:141–147. doi: 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SR. Genes and signals in the rhizobium-legume symbiosis. Plant physiology. 2001;125:69–72. doi: 10.1104/pp.125.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolmson SJ, Young TS, Ruby JG, Skewes-Cox P, Walsh CT. The posttranslational modification cascade to the thiopeptide berninamycin generates linear forms and altered macrocyclic scaffolds. Proc Natl Acad Sci U S A. 2013;110:8483–8488. doi: 10.1073/pnas.1307111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- McInerney BV, Gregson RP, Lacey MJ, Akhurst RJ, Lyons GR, Rhodes SH, Smith DR, Engelhardt LM, White AH. Biologically active metabolites from Xenorhabdus spp., Part 1. Dithiolopyrrolone derivatives with antibiotic activity. J Nat Prod. 1991;54:774–784. doi: 10.1021/np50075a005. [DOI] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic acids research. 2011;39:W339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- Oh DC, Poulsen M, Currie CR, Clardy J. Dentigerumycin: a bacterial mediator of an ant-fungus symbiosis. Nat Chem Biol. 2009;5:391–393. doi: 10.1038/nchembio.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzianabos AO, Onderdonk AB, Rosner B, Cisneros RL, Kasper DL. Structural features of polysaccharides that induce intra-abdominal abscesses. Science. 1993;262:416–419. doi: 10.1126/science.8211161. [DOI] [PubMed] [Google Scholar]
- Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci U S A. 2009;106:2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, Sonnenburg JL, Comstock LE, Bluestone JA, Fischbach MA. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol. 2013;11:e1001610. doi: 10.1371/journal.pbio.1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt MA, Wang W, Roux CM, Beasley FC, Heinrichs DE, Dunman PM, Magarvey NA. Staphylococcus aureus nonribosomal peptide secondary metabolites regulate virulence. Science. 2010;329:294–296. doi: 10.1126/science.1188888. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS chemical biology. 2009;4:855–864. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Yin H, Kong D, Deng Z, Lin S. A trans-acting ketoreductase in biosynthesis of a symmetric polyketide dimer SIA7248. Chembiochem. 2013;14:679–683. doi: 10.1002/cbic.201300068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H, gCOSY, TOCSY, HSQC, HMBC, and ROESY spectra are shown. B. MS fragmentation analysis of lactocillin methyl ester. MS fragmentation was performed on an HR Orbitrap Velos mass spectrometer, and selected fragments are shown here. Fragment ion types are indicated next to the fragmentation site, and key fragments that were crucial for determining the position of the indolyl-S-cysteine (position 8) and thiazoline (position 7) moieties are shown in red boxes. Note the subsequent loss of one residue at a time, enabling their relative position to be determined. C. Chemical shifts of lactocillin in DMSO-d6 (600 MHz)
Selected read tiling maps of three BGCs in nine HMP metagenomic samples. Labels indicate the BGC number, the HMP sample identifier, and the abundance score computed using the mblastx method described in Extended Experimental Procedures. Note that the increase in the score computed using mblastx represents, to a great extent, the increase in the depth of coverage of a given BGC.
Counts of BGCs that are “commonly found” (detected in >50% of the HMP samples at the body site of origin), and counts of BGCs that are “typically found” at a certain body site (average number of BGCs per HMP samples at the body site of origin).
A) Phylogenetic tree representing 2,430 analyzed genomes, with the corresponding number and class of predicted BGCs shown as bars (total of ~14,000 BGCs). The tree was generated from 16S rDNA sequences downloaded from JGI-IMG and the final figure was generated using iTOL (Letunic and Bork, 2007). B) The same tree showing only the 3,118 BGCs detected in at least one HMP metagenomic sample. C) A network view of all BGCs predicted from human-associated bacteria. Edges were calculated based on similarities of Pfam compositions between a pair of BGCs, with a cutoff of 0.4 (Cimermancic et al., 2014). The network was generated using Cytoscape (Smoot et al., 2011). D) Average number and class of BGCs detected in HMP samples by genus. The 40 genera with the highest average number of BGCs are shown.
A) Venn diagrams representing BGCs present in >20% of the HMP samples at multiple body sites. B) Percentages of certain classes of BGCs in human-associated bacterial genomes (red) in comparison to non-human-associated bacterial genomes (blue), showing the respective enrichments or depletions. To assess the statistical significance of the distribution differences between human and non-human organisms, we bootstrapped the two samples by randomly drawing and analyzing 20% of BGCs 1000 times. C) A comparison between the fraction of BGCs predicted per source organism phylum in human-associated bacterial genomes (green) and non-human-associated bacterial genomes (red). Note: only the 3,118 BGCs present in HMP metagenomic samples were included in the analyses.
A heat map showing the representation of nine members of the gut NRPS family in HMP stool samples (samples shown here represent 92% of the HMP stool samples).
A) Ten HMP stool samples that have similar Bacteroidetes compositions based on their MetaPhlAn (Segata et al., 2012) classifications were selected (SRS015369, SRS012902, SRS023176, SRS013158, SRS024388, SRS016989, SRS016267, SRS013215, SRS058723, and SRS049900). Only members of the phylum Bacteroidetes are shown in the heat map displayed here. B) A heat map showing the representation of saccharide BGCs predicted from members of the phylum Bacteroidetes and detected in the same ten samples shown in A. Although some of the BGCs displayed here show similar patterns of abundance among samples, a large subset varies substantially across the sample set.
A maximum likelihood phylogenetic tree of the lantibiotic dehydratase enzyme of all thiopeptide BGCs described here, constructed using MEGA5. The numbers on the branches indicate the percentage of times this topology was reached in a bootstrap test of 1000 replicates. A similar tree was obtained using TclM homologs (responsible for pyridine ring formation).
A) UV-Vis absorbance profiles of lactocillin and thiocillin I (note the similarity between profiles). B) (+) HR-FTMS of lactocillin, consistent with the empirical formula C51H45N13O10S7 (observed [M+H]+ at m/z 1224.15184, calculated [M+H]+ 1224.15354, Δ ppm = 1.3) C) Time course of lactocillin production by L. gasseri JV-V03 in MRS broth under anaerobic conditions at 37 °C. D) HPLC-MS chromatogram of the trimethylsilyldiazomethane derivatization reaction (red) in comparison to the fraction containing native lactocillin (blue). E) (+) HR-ESI Orbitrap data of the resulting reaction showing a mass shift of 14 amu corresponding to the addition of one methyl group to the parent lactocillin and consistent with the empirical formula C52H47N13O10S7 (observed [M+H]+ at m/z 1238.16916, calculated for [M+H]+ at m/z 1238.16916, Δ ppm = − 0.02) F) Selected HMBC and COSY correlations observed and used to determine the structure of lactocilin methyl ester (see also Supplemental Data File 2 for NMR spectra and table of chemical shifts) G) HPLC-MS analysis of L. gasseri feeding experiments with 20 mM L-tryptophan (Top), 20 mM L-tryptophan D5 (middle), 20 mM L-alanine D3 (bottom). Mass spectra of the lactocillin peaks are shown in all three cases, where the +5 and +3 amu mass shifts are apparent in the case of L-tryptophan D5 and L-alanine D3, respectively. H) Selected MS/MS and MSn spectra of lactocillin methyl ester (See also Supplemental Data File 2 for an illustration of the detected fragments).
A) Mapping of metatranscriptomic reads from a human supragingival plaque sample (HMP DACC) to bgc65 (colors indicate the same key as in Figure 5). B) Mapping of metatranscriptomic reads from a human supragingival plaque sample (HMP DACC) to the L. gasseri JV-V03 plasmid harboring lcl (colors indicate the same key as in Figure 6).
The complete matrix of the 3,118 BGC predicted here (as columns) and 752 HMP metagenomic samples (as rows). Values given are abundance scores ranging from 10–1000. Matrices of body site and class of BGC are included as separate tabs. Locus tags of all BGCs are given in a tab containing all 3,118 classified BGCs. Pfams eliminated from the ClusterFinder output prior to computing the abundance of BGCs in HMP samples are also given in a separate tab.