

Abstract
BACKGROUND
Previous studies have demonstrated that antidepressant medication and electroconvulsive therapy increase occipital cortical GABA in major depressive disorder (MDD), but, a small pilot study failed to show a similar effect of cognitive behavioral therapy (CBT) on occipital GABA. In light of these findings we sought to determine if baseline GABA levels predict treatment response and to broaden the analysis to other metabolites and neurotransmitters in this larger study.
METHODS
40 MDD outpatients received baseline 1H-MRS, and 30 subjects completed both pre- and post-CBT 1H-MRS. Nine CBT non-responders completed an open-label medication phase followed by an additional/third 1H-MRS. The magnitude of treatment response was correlated with occipital amino acid neurotransmitter levels.
RESULTS
Baseline GABA did not predict treatment outcome. Furthermore, there was no significant effect of CBT on GABA levels. However, we found a significant group x time interaction (F(1,28) = 6.30, p = 0.02), demonstrating reduced glutamate in CBT responders with no significant glutamate change in CBT non-responders.
CONCLUSIONS
These findings corroborate the lack of effect of successful CBT on occipital cortical GABA levels in a larger sample. A reduction in glutamate levels following treatment, on the other hand, correlated with successful CBT and antidepressant medication response. Based on this finding and other reports, decreased occipital glutamate may be an antidepressant response biomarker. Healthy control comparator and non-intervention groups may shed light on the sensitivity and specificity of these results.
Keywords: cognitive-behavioral therapy, antidepressant, selective serotonin reuptake inhibitor, GABA, glutamine, glutamate
INTRODUCTION
Major depressive disorder (MDD) has the highest worldwide morbidity among neuropsychiatric disorders across all socioeconomic strata[1–3]. Standard treatment for unipolar depression includes antidepressant medications, device-based therapies and/or psychotherapy. At present, the selection of treatment modality for an individual patient is based primarily on patient preference and/or therapist orientation instead of a more personalized pathophysiological approach. Due to the heterogeneous nature of MDD, there remains a clinical need for trait and/or state-specific biomarkers to assist in antidepressant treatment prediction and response assessment.
The two manualized psychotherapies with the largest evidence base in MDD are interpersonal psychotherapy (IPT) and cognitive-behavioral therapy (CBT). Both psychotherapies were recommended as first-line treatments in the 2010 iteration of the American Psychiatric Association’s (APA) practice guideline for MDD[4]. Additionally, CBT is associated with lower risk of relapse after remission compared to antidepressant medications [5–8]. Although a multi-site study initially suggested that CBT might be less effective in patients with severe depression[9], a subsequent meta-analysis did not support this claim[10]. As illness severity, therefore, is not a reliable predictor of response, several studies have stratified subjects into depressive subtypes as a putative enrichment strategy. Stewart et al.[11] reported that atypical depression responded more robustly to cognitive therapy than other depressive subtypes. In contrast, hypothalamic-pituitary-adrenal (HPA) axis dysfunction (a biometric of melancholic depression) predicted a poorer treatment response to CBT and other psychosocial interventions[12, 13]. Yet, due to discrepancies in the literature[14], it remains inconclusive if depressive subtype reliably predicts antidepressant outcomes.
As a result of the discrepancies in the literature with demographic and clinical predictors, several investigators have instead focused on neurobiological measures that might predict and/or correlate with antidepressant treatment outcomes. Several studies have demonstrated that currently depressed outpatients have lower levels of the inhibitory amino acid neurotransmitter γ-aminobutyric acid (GABA) in plasma, cerebrospinal fluid, and occipital and prefrontal cortices[15–19]. We previously reported that a subgroup of MDD subjects exhibited markedly reduced occipital GABA concentrations[18]. Nearly 50% of MDD subjects had occipital GABA levels below the level found in greater than 95% of healthy subjects. The reduced GABA content was especially prominent in patients diagnosed with melancholic depression [18]. Other studies have reported a normalization of occipital cortex GABA in response to somatic interventions [selective serotonin reuptake inhibitors (SSRIs)[20] and ECT[21]]. In contrast, a preliminary study from our group[22] showed no significant cortical GABA change following a successful 12-week course of once-weekly CBT. However, this study was small (n=8 completers) and did not investigate other amino acid neurotransmitters and their precursors, e.g. glutamate and glutamine.
Alterations in the glutamate system in MDD have been described by PET, protein biochemistry and MRS. Examples of such alterations include decreased metabotropic glutamate receptor type 5 (mGluR5) expression in MDD brain based on both PET (with the mGluR5-selective radioligand [11C]ABP688) and immunoblotting in postmortem samples[23], as well as reduced prefrontal cortical glutamate receptor subunit NR2A and NR2B expression and the excitatory postsynaptic density protein, PSD-95, relative to matched psychiatrically healthy subjects[24]. Prior MRS studies, have shown brain region specific alterations of glutamate and/or glutamine levels, i.e. increased glutamate/glutamine in the occipital cortex [18, 25] but decreased glutamate/glutamine levels in the prefrontal cortex[19, 26], anterior cingulate cortex[27, 28] and left amgydala[29].
Considering our preliminary evidence that successful CBT did not affect occipital GABA, and that a selective subgroup of MDD patients appears to have markedly reduced GABA levels, we sought to determine if baseline occipital GABA content could serve as a biomarker for CBT treatment efficacy. Addressing the limitations of our previous study, i.e. the small sample size and the lack of data on other neurotransmitters and metabolites, we increased our sample size of currently depressed MDD outpatients and expanded the panel beyond GABA to other 1H-MRS detectable metabolites, glutamate and glutamine.
Based on these preliminary results[22], our primary hypotheses were that lower baseline GABA levels would predict a poorer antidepressant response to CBT and that a successful 12-week course of CBT would not alter occipital GABA levels. Pilot 1H MRS studies suggesting a relationship between glutamate changes (post-treatment – pre-treatment) and successful antidepressant therapies (either ECT[26, 28–30] or ECT + pharmacotherapy) led to our secondary hypothesis that a successful course of CBT will correlate with cortical glutamate changes.
MATERIALS AND METHODS
All study procedures were conducted at the Yale Depression Research Program (YDRP) and the Yale Magnetic Resonance Research Center (MRRC). Subjects enrolled in this study provided written informed consent prior to all research-related procedures. The institutional review board/human investigations committee at the Yale University School of Medicine approved all portions of this protocol before initiation. The study was initiated prior to 2007 and was therefore not registered with ClinicalTrials.gov.
Study Design
This longitudinal study enrolled medication-free (≥ 2 weeks for typical monoaminergic antidepressants with the exception of ≥ 4 weeks for fluoxetine), currently depressed outpatients who presented to the clinic for initial screening and evaluation. After obtaining informed consent, subjects received baseline clinical assessments from licensed psychiatric clinicians. All rating scales were conducted by trained research staff. Proton magnetic resonance spectroscopy (1H-MRS) was obtained for baseline occipital cortical amino acid measurements. Then, participants received structured CBT as outlined in Beck et al.[31]. The psychotherapy consisted of 50-minute individual sessions on a once-weekly basis for 12 weeks. Two therapists (L.F. and M.F., who were trained and certified at the Beck Institute) provided the manualized psychotherapy. The Cognitive Therapy Rating Scale[32] was used to assess audiotaped copies of the sessions to ensure treatment integrity, evaluate therapist competency and provide quality control. After the end of the 12-week therapy course, all clinical assessments and 1H-MRS were repeated (Fig. 1A). CBT non-responders were then offered an additional six weeks of a SSRI. At the end of this brief pharmacotherapy phase, a third 1H-MRS was obtained (Fig. 1A).
Figure 1. Study Design and Subject Flow.

A. Study Design – Unipolar unmedicated depressed subjects in a current major depressive episode meeting inclusion/exclusion criteria (Phase I) received a baseline 1H-MRS before starting 12 weeks of once-weekly structured CBT with clinician-administered psychiatric ratings every four weeks (Phase IIa). A second 1H-MRS was performed for all subjects retained at the end of Phase IIa (or after at least eight weeks if the subject dropped-out). CBT non-responders were then offered a 6-week course of an SSRI (Phase IIb). All subjects retained at the end of Phase Ib received a third and final 1H-MRS at study exit. B. Subject Flow – All available data were used in the analysis. Last Observation Carried Forward (LOCF) was used for patients with one post-treatment follow-up visit (at least 4 weeks of CBT). aA total of 8 subjects did not have a baseline 1H-MRS for the following reasons: contraindication to MRI (N=4), poor quality spectra (N=2), excessive motion artifact (N=1), poor water suppression (N=1). bMR contraindication (N=4), poor quality spectra (N=2), exited the study (N=11; 5 started medications, 1 poor adherence to study procedures, 1 no time for appointments, 1 had knee surgery, 1 moved to a different city, and 2 were lost to follow-up). cSubjects who completed at least 8 weeks of CBT received the second MRS. Eight subjects exited the study before completing 4 weeks of CBT (4 started medications, 1 moved to a different city, 1 had knee surgery, and 2 were lost to follow-up before receiving any CBT), 3 exited before 8 weeks of CBT (1 started medications, 1 no time for appointments, and 1 poor adherence to study procedures), and 5 exited before 12 weeks of CBT (2 started medications, 1 moved to another city, 1 got a new job, and 1 was lost to follow-up).
Abbreviations: N: number of subjects; 1H-MRS: proton magnetic resonance spectroscopy; CBT: cognitive-behavioral therapy.
Subjects
All subjects were 18–65 years old and met DSM-IV-TR criteria for MDD in a current major depressive episode. Diagnosis was determined by face-to-face psychiatric evaluation and confirmed by the Structured Clinical Interview for DSM-IV Disorders (SCID)[33]. Exclusion criteria were as follows: a current or past diagnosis of MDD with psychotic features, active suicidal ideation, a history of suicidal behavior in the preceding two years, current use of psychotropic medications, a history of or current uncontrolled medical or neurological problems, an alcohol or illicit substance use disorder within the preceding six months, current illicit substance use (as confirmed by urine toxicology), a history of psychiatric illness due to confirmed general medical condition(s), a history of primary personality disorder, and a history of psychotic spectrum illness. Predefined exit criteria for clinical deterioration were defined as 1.) a 25% increase over baseline BDI score at any time over the course of the trial; or 2.) an increase in passive suicidal ideation or the onset of active suicidal ideation at any time over the course of the trial. No subjects were terminated for clinical deterioration, and the reasons for in-protocol termination are presented in Fig. 1B.
Clinical Assessments and Rating Scales
Clinical assessments included comprehensive medical, neurological and psychiatric evaluations by experienced psychiatric research clinicians. Baseline blood and urine testing included the following: complete blood count (CBC), glucose, blood urea nitrogen (BUN), creatinine, electrolytes, liver and thyroid function tests, rapid plasma reagin (RPR), urinalysis, urine toxicology and urine pregnancy test (if applicable). Within three days prior to the first 1H-MRS scan, a battery of clinician-administered psychiatric assessments was completed including the 24-item Hamilton Depression Rating Scale (HDRS24)[34] and Hamilton Anxiety Rating Scale (HAM-A)[35] and other measures of maladaptive cognition/rumination – Dysfunctional Attitude Survey (DAS), Beck Hopelessness Scale (BHS) and Ruminative Responses Scale (RRS) (Table 2). Thereafter, the clinician-administered HDRS24 was repeated every 4 weeks and the self-reported Beck Depression Inventory (BDI)[36] was repeated on a weekly basis to monitor clinical response. HAM-A was repeated at the end of both the psychotherapy and pharmacotherapy phases.
Table 2.
Effect of CBT and SSRIs on Clinical Outcome Measures
| PRE-CBT | POST-CBT | df | Ta | Sig. | |
|---|---|---|---|---|---|
| HDRS24 | 28.1 ±1.2 | 16.5 ±1.5 | 41 | − 4.35 | < 0.001 |
| BDI | 25.6 ±1.2 | 11.6 ±1.4 | 41 | − 5.30 | < 0.001 |
| HAM-A | 14.7 ±0.8 | 9.0 ±0.9 | 37 | − 4.00 | < 0.001 |
| DAS | 152 ±5.1 | 129 ±5.5 | 34 | − 3.10 | 0.002 |
| HS | 13.6 ±0.7 | 7.5 ±0.9 | 34 | − 4.31 | < 0.001 |
| RRS | 25.9 ±0.9 | 25.3 ±0.7 | 34 | − 0.46 | 0.64 |
|
| |||||
| PRE-SSRI | POST-SSRI | ||||
|
| |||||
| HDRS24 | 24.5 ±1.7 | 16.9 ±2.9 | 12 | − 2.24 | 0.02 |
| BDI | 17.6 ±2.1 | 12.4 ±2.5 | 12 | − 1.91 | 0.06 |
| HAM-A | 12.5 ±1.5 | 11.4 ±2.2 | 10 | − 0.36 | 0.72 |
| DAS | 149 ±9.9 | 124 ±9.9 | 8 | − 2.26 | 0.02 |
| HS | 11.2 ±1.8 | 7.3 ±1.9 | 8 | − 2.17 | 0.03 |
| RRS | 24.3 ±1.2 | 22.2 ±1.5 | 8 | − 1.50 | 0.13 |
Data are presented as mean±SEM.
Test statistics from related-samples Wilcoxon Signed Rank test (significance set at p ≤ .05).
Abbreviations: SEM: Standard error of mean; HDRS24: Hamilton Depression Rating Scale; BDI: Beck Depression Inventory; HAM-A: Hamilton Anxiety Rating Scale; DAS: Dysfunctional Attitude Survey; HS: Hopelessness Scale; RRS: Ruminative Responses Scale (RRS).
1H-MRS Acquisition and Processing
The occipital cortex was selected in our initial MRS protocols due to technical limitations for the detection of amino acid neurotransmitters in other brain areas that have been more extensively studied in depression, e.g. the prefrontal cortex and amygdala. Since those initial studies, our group has consistently demonstrated decreased GABA[17, 18] and increased glutamate[18] levels with differential effects from successful antidepressant therapy[20–22, 37] (but not full restoration[25, 38]) in the MDD occipital cortex. Therefore, we continued to utilize the occipital cortex as our region of interest (ROI) in this study. 1H-MRS was acquired with a slight modification of the J-edited method previously described[18] (two editing pulses were inserted into the sequence instead of one). Briefly, metabolite levels were measured in an occipital cortex ROI – primarily in V1 in a 13.5 cc voxel (3.0 x 1.5 x 3.0 cm) placed across the midline of the brain centered 1.5 cm from the dura. Cortical GABA content was determined using J-editing, where sub-spectra were acquired with 8,192 data points over a 410 millsecond acquisition, a 2.5 second repetition time, and a TE of 68 millisecond with a 4-Tesla (4T) Bruker spectrometer, averaging data in 20 second blocks for a total of 20 minutes. Glutamate and glutamine were measured simultaneously using the unedited subspectra of the J-editing acquisition.
The unedited and the J-edited difference subspectra were fitted simultaneously using a basis set of metabolite spectra measured with the J-editing acquisition sequence. The metabolites fitted were aspartate, GABA, glutamate, glutamine, N-acetylaspartate (NAA), N-acetylaspartylglutamate (NAAG), creatine, phosphocreatine, myoinositol, scylloinositol, choline, phosphorylcholine, and glycerophosphorylcholine. The subspectrum obtained without the editing pulse was fitted simultaneously with the J-edited difference spectrum of GABA. Because of limited resolution in vivo, the results for NAA and NAAG were combined and recorded as NAA; creatine and phosphocreatine were combined and recorded as creatine (Cr); and the three choline-containing compounds were combined and recorded as choline (Cho). This implementation had no macromolecular contamination of GABA [37], so the basis set for fitting did not include a macromolecular signal. The level of aspartate, though present in the spectra, was poorly determined at the echo time of 68 milliseconds and, therefore, was not used. Uncertainties of individual measurements were determined using a Monte-Carlo analysis as previously described [37, 39]. The means of percent solid tissue in the acquired voxel were not different between groups or assessment time points (p > 0.1). Thus, no co-variance for tissue content was included. The means of Cr/water were not different between groups or assessment time points (p > 0.1). As such, the concentration of brain metabolites was estimated assuming a Cr concentration of 9 mmol/kg.
Statistical Analysis
Prior to conducting each analysis, the distribution of outcome measures was examined using probability plots and Kolmogorov-Smirnov test statistics. Response status was determined by 50% or more reduction in last observed HDRS24 scores. Paired t-test and related-samples Wilcoxon Signed Rank tests were used to examine pre- and post-treatment changes. General linear model (GLM) repeated-measures analyses were constructed as needed. Age and sex were considered as covariates in all models. Spearman’s Rank Order was used for correlational analyses. All tests were two-tailed, with a significance level set at p ≤ 0.05. False Discovery Rate correction for multiple comparisons was used when appropriate (padjusted).
RESULTS
A total of 50 subjects were enrolled [30 women, mean (±SD) age 42.6±11.4]. Of these, 42 subjects had a successful baseline 1H-MRS scan and had at least one treatment follow-up visit (≥ 4 weeks of CBT). 33 subjects had a successful post-CBT 1H-MRS scan, and 30 subjects had both baseline and post-CBT 1H-MRS scans (Fig 1B). 14 CBT non-responders then entered the open-label pharmacotherapy phase, of which nine completed the 2nd and 3rd 1H-MRS and all clinical assessments (Fig. 1B). The demographic and clinical features of the study sample entering and completing both the CBT and SSRI phases are provided in Table 1.
Table 1.
Demographic and Clinical Characteristics of Study Sample.
| CBTa | CBT-MRSb | SSRI-MRSc | |
|---|---|---|---|
|
| |||
| Median (range) | Median (range) | Median (range) | |
| Age (y) | 45.4 (21–62) | 42.4 (21–62) | 45.2 (24–58) |
| Duration of Illness (y) | 20.0 (0.2–45) | 15.8 (0.2–45) | 29.2 (6–45) |
| Age of Onset (y) | 18.0 (6–52) | 18.0 (6–52) | 13.0 (7–40) |
| Number of MDEs | 3 (1–12) | 3 (1–7) | 5 (1–6) |
| Number of ADTs | 1 (0–6) | 1 (0–6) | 1 (0–3) |
| Current MDE (y) | 2.0 (0.1–27) | 2.0 (0.2–19) | 0.2 (0.2–16) |
|
|
|||
| Percent | Percent | Percent | |
|
|
|||
| Female | 60% | 63% | 22% |
| Medication Naïve | 38% | 40% | 25% |
| Substance Abuse History | 24% | 25% | 25% |
Participants treated with CBT, have pre-CBT MRS and presented for at least one follow-up assessment (n = 42).
Participants treated with CBT and completed pre- and post-CBT MRS (n = 30).
Participants treated with SSRI and completed pre- and post-SSRI MRS (n = 9).
Abbreviations: CBT: Cognitive Behavioral Therapy; MRS: Magnetic Resonance Spectroscopy; SSRI: Selective Serotonin Reuptake Inhibitor; SEM: Standard Error of (the) Mean; y: years; MDEs: Major Depressive Episodes; ADTs: Antidepressant Treatment (Trials).
As shown in Table 2, CBT exerted a significant antidepressant effect (p < 0.001), resulting in an average 41% reduction in HDRS24 scores and 38% response rate (≥ 50% reduction from baseline HDRS24). CBT also had a significant effect on self-rated depressive symptoms (BDI) and anxiety symptoms (HAM-A), dysfunctional attitude (DAS), and hopelessness (BHS). CBT, however, did not affect total scores on the Ruminative Responses Scale (RRS) (Table 2). In CBT non-responders who entered the pharmacotherapy phase, SSRIs exerted a significant antidepressant effect (p = 0.02), leading to an average 31% reduction in HDRS24 scores and 36% response rate (Table 2).
Association of baseline amino acid neurotransmitter levels with treatment response
Baseline GABA did not correlate with delta HDRS24 (post-treatment minus pre-treatment) over the CBT period (rs = − 0.08, p = 0.66). There was also no association between baseline glutamate and delta HDRS24 (p = 0.59). However, baseline glutamine levels were negatively correlated with delta HDRS24, such that patients with higher baseline glutamine exhibited a better antidepressant response to CBT (rs = − 0.38, p = 0.03), although this correlation did not survive correction for multiple comparisons (padjusted = 0.06). In addition, baseline GABA, glutamate, and glutamine did not differentiate CBT responders and non-responders (p > 0.05). Pre-SSRI GABA, glutamate and glutamine were also not significantly correlated with HDRS24 change over the pharmacotherapy phase (p > 0.1).
Relationship between amino acid neurotransmitter changes and the efficacy of CBT
A repeated-measure GLM with GABA as the dependent variable, time of scans (pre-CBT vs. post-CBT) as the within-subject factor, and group (responders vs. non-responders) as the between-subjects factor showed no time (p = 0.39), group (p = 0.79), or group-by-time interactive effects (p = 0.96). A repeated-measure GLM with glutamate as the dependent variable, time of scans (pre-CBT vs. post-CBT) as the within-subject factor, and group (responders vs. non-responders) as the between-subjects factor found a significant group x time interaction (F(1,28) = 6.30, p = 0.02), demonstrating reduced glutamate in CBT responders with no significant change in CBT non-responders (Fig. 2A). The time effect was significant (F(1,28) = 5.42, p = 0.03) but there was no group effect (p = 0.98). Secondary analyses including gender and age, as covariates in the same repeated-measure GLM, showed no gender effects, no time x gender interaction, no age effect, and no time x age interaction (all p > 0.05). Over the 12-week CBT course, there was a significant correlation between delta glutamate and delta HDRS24 (rs = 0.43, padjusted = 0.02), delta BDI (rs = 0.46, padjusted = 0.02; Fig. 2B) and delta HAM-A (rs = 0.48, padjusted = 0.02; Fig. 2C). Secondary partial correlation analyses included gender and age as covariates. Controlling for gender, delta glutamate correlated with delta HDRS24 (rs = 0.42, p = 0.02), delta BDI (rs = 0.46, p = 0.01) and delta HAM-A (rs = 0.48, p = 0.01). Controlling for age, delta glutamate correlated with delta BDI (rs = 0.37, p = 0.046) and delta HAM-A (rs = 0.37, p = 0.047) but not delta HDRS24 (rs = 0.30, p = 0.11).
Figure 2. Successful CBT for MDD Is Associated With Decreased Glutamate Levels in the Occipital Cortex.
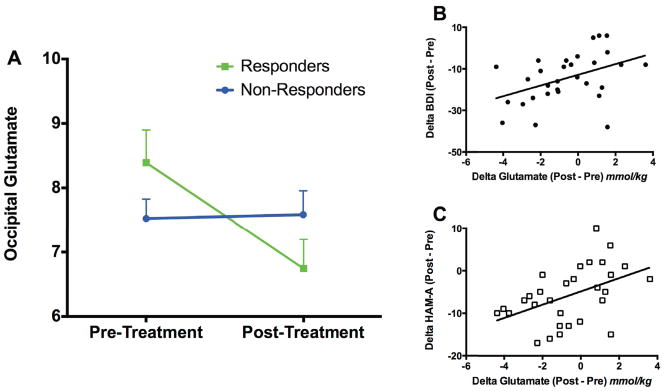
Unipolar unmedicated depressed subjects in a current major depressive episode received 12 weeks of once-weekly structured CBT with 30 completing both pre- and post-CBT MRS. A. Treatment responders (≥ 50% reduction in HDRS24) showed a significant reduction in occipital glutamate levels [group-by-time interaction (F(1,28) = 6.30, p = 0.02)]. B & C. Similarly, reductions in the secondary depression and anxiety outcome measures, BDI (rs = 0.46, p = 0.01) and HAM-A (rs = 0.48, p = 0.01), were positively correlated with decreased occipital glutamate.
Given the small sample completing the pharmacotherapy phase (n = 9), no GLM analyses were performed. To assess the relationship between delta glutamate and delta HDRS24, we conducted a complementary correlational analysis. During the pharmacotherapy phase, there was a correlation between delta glutamate (post-SSRI – pre-SSRI) and delta BDI (rs = 0.69, p = 0.04; Fig. 3A). There was a trend with delta glutamate and HDRS24 (rs = 0.63, p = 0.07; Fig. 3B), but, unlike CBT, the change in glutamate levels did not correlate with delta HAM-A (p = 0.23). Given the small sample size and the exploratory nature of the pharmacotherapy phase, no correction for multiple comparisons was performed.
Figure 3. In CBT Non-Responders, Positive SSRI Response Is Associated With Decreased Glutamate in the Occipital Cortex.

Unipolar unmedicated depressed subjects in a current major depressive episode received 12 weeks of once-weekly structured CBT. Of the 14 subjects who entered phase 2 due to CBT non-response, nine completed all neuroimaging and clinical assessments. In the medicated CBT non-responders, reductions in BDI and HDRS were positively correlated with reductions in occipital cortical glutamate.
DISCUSSION
This study investigated amino acid neurotransmitters at baseline, after a 12-week course of CBT, and, in CBT non-responders, a six-week course of antidepressant medication, in a well-characterized sample of currently depressed unipolar major depression subjects. Although several patients had an lengthy course of illness and index episode, our depressed outpatient sample specifically contacted our research clinic based on advertisements for evidence-based psychotherapy in lieu of antidepressant medications. This sample was also relatively treatment-naïve and had lifetime rates of substance misuse consistent with community rates seen in large epidemiological surveys[1, 40]. Therefore, unlike depression effectiveness trials with larger, more heterogeneous samples, e.g Sequenced Treatment Alternatives to Relieve Depression (STAR*D)[41] and Combining Medications to Enhance Depression Outcomes (CO-MED)[42], our research sample was generally homogenous, which maximizes effect size in depression treatment trials[43, 44].
Consistent with our preliminary report (n = 8 completers)[22], successful CBT had no effect on occipital cortical GABA levels in this larger sample. Yet, contrary to our a priori hypothesis, we did not find a significant relationship between baseline GABA levels and treatment outcome. The lack of CBT effects on cortical GABA measures contrasts with the findings of increased occipital GABA levels following treatment with other evidence-based therapies for depression such as SSRIs[20] and ECT[21]. Disparate effects of psychotherapy and antidepressant medications have previously been observed in the frontal cortex, cingulate cortex, and hippocampus, i.e. CBT relative to the SSRI paroxetine as detected by 18FDG-glucose positron emission tomography (PET) [45]. These results suggest that distinct neurotransmitter and/or neurocircuitry effects may be responsible for somatic and psychotherapeutic antidepressant response. However, it is also possible that the changes in occipital GABA associated with medications and ECT are not directly related to the antidepressant effects of the treatments. It should also be noted as a potential limitation of this study, that although pre-treatment HDRS24 scores resembled those observed in the earlier SSRI study, the current CBT study did not include subjects with melancholic, psychotic or treatment-resistant depression, which typically have been associated with the lowest levels of GABA. Therefore, the subjects in this study may have less room for improvement in GABA levels than the subjects with the more severe depressive subtypes. Nevertheless, in contrast to this hypothesis, healthy subjects (who presumably have normal baseline GABA levels) exhibited increased occipital cortical GABA in response to a single intravenous administration of the SSRI citalopram [46].
We did however observe a robust association between changes in occipital cortex glutamate levels with CBT and treatment response. More specifically, we found the subjects with the greatest reductions in glutamate levels during the CBT treatment period had the largest reductions in depression and anxiety symptom severity, as exemplified by decreased HDRS24/BDI and HAM-A. This finding is of interest in light of several previous studies showing evidence of decreased prefrontal Glx content (a mixture of MRS-detectable glutamine and glutamate at lower magnetic field strengths) that subsequently normalizes following successful antidepressant treatment of unipolar depression, i.e. either ECT[26, 28–30] or ECT + antidepressant medications[29]. Similarly, the rapid but transient antidepressant effect of total sleep deprivation has also been associated with increased Glx in the dorsolateral prefrontal cortex of male depressed patients with neurovegetative symptoms consistent with melancholia[47]. Our findings, showing decreased levels of occipital cortex glutamate following CBT to be associated with treatment response, appear in contrast to the existing literature. However, studies measuring glutamate or GLX in the occipital cortex have either reported no difference or increased metabolite levels in MDD subjects [18, 25, 48]. Considering the current findings in this light, the decreased glutamate content observed following CBT might reflect a regional normalization similar to what has been reported in the PFC regions.
Although the small number of subjects going on to participate in the pharmacotherapy phase of the study severely limits our ability to draw any firm conclusions, we did find suggestive evidence of a similar correlation between the change in glutamate following 6 weeks of SSRI treatment and clinical response as assessed by measures of depression severity. This data suggests that the change in glutamate occipital cortex content may be associated with clinical improvement regardless of the treatment modality employed. Obviously, this correlative finding will need to be replicated before any more definitive statements can be made.
Although our study has many strengths, including a relatively large and clinically homogenous sample initially requesting evidence-based psychotherapy for the treatment of major depression, longitudinal design, long treatment period (4–6 months total), two evidence-based treatment modalities (depression-focused CBT and antidepressant pharmacotherapy) with no overlap between treatment periods, the use of neurobiological measures[49, 50], and an MRS sequence at 4T that permitted specific quantification of glutamate, there are several limitations to consider. First, as with all proton MRS studies, the measures reflect total tissue content of the metabolites and does not provide any direct information related to amino acid neurotransmission. Second, our selection of a V1 occipital cortical ROI may be viewed by some as a limitation due to the occipital cortex’s primary function in vision (as opposed to other brain regions that have been more widely associated with depression, e.g. prefrontal cortex, anterior cingulate cortex and amygdala). However, as cited above, our group and others have demonstrated differential levels of amino acid neurotransmitters with response to successful antidepressant therapies in the occipital cortex. Additionally, there are several recent studies demonstrating abnormal function of the visual cortex in depression that changes [51, 52] and possibly predicts treatment outcome [53]. A third major limitation of our study is the absence of a non-intervention (“placebo”) group in both the CBT and pharmacotherapy phases to control for non-specific clinical and neuroimaging effects. The lack of a healthy control population does not allow us to directly make metabolite comparisons with non-depressed brains. Additionally, the fact that we excluded psychotic depression and had a limited number of subjects with melancholic or treatment-resistant depression (the subtypes that have been reported to have both the lowest GABA levels and poorest response to CBT[14, 48, 54]) limits our ability to generalize the findings to all MDD subtypes. It is unclear if CBT success would also correlate with baseline GABA levels or decreased glutamate in these typically more severely depressed patients. Moreover, it is plausible that there was some level of selection bias because the inclusion criteria required individuals who were not currently taking psychotropic medications. Therefore, it is possible that we had a higher percentage of patients that for a variety of reasons were opposed or hesitant to seeking medication therapy [51, 52]. Finally, the SSRI exploratory analysis is hampered by the small sample size (n=9) and data variability.
In conclusion, our observations provide exciting evidence of concordant glutamatergic effects from both evidence-based psychotherapy and antidepressant medication. Mechanistically, the relationship between the changes in occipital cortex glutamate content and successful treatment of depression may reflect several non-exclusive processes including a recovery from an underlying pathology affecting neuronal-glial cell amino acid metabolism [55–57], changes in mitochondrial energy metabolism [58], or a change in the relative cellular composition of the tissue. Future studies are required to confirm these observations and to identify their underlying neurophysiological bases.
Acknowledgments
This work was supported by the Patrick and Catherine Weldon Donaghue Medical Research Foundation (GS), the National Alliance for Research on Schizophrenia and Depression (GS, LRF), National Institute of Mental Health Grant R01 MH071676-05 and K02 MH076222-04 (GS), R21 AA018210 (GFM), and R01 DA021785 (GFM), The State of Connecticut, Department of Mental Health and Addiction Services (through its support of the Connecticut Mental Health Center and Clinical Neuroscience Research Unit), Veterans Affairs (VA) National Center for PTSD, and VA Alcohol Research Center (GFM). Salary support was provided by NIMH T32 MH19961 (MJN) and NIDA T32-DA022975 (CGA). We would also like to thank Ms. Lisa Roach, M.S., and June Watzl, Ph.D., for their research and administrative assistance.
Footnotes
Conflict of interest statement: Dr. Sanacora has received consulting fees from AstraZeneca, Avanier Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly & Co., Hoffman La-Roche, Naurex and Noven Pharmaceuticals over the last 36 months. He has also received additional grant support from AstraZeneca, Bristol-Myers Squibb, Hoffman La-Roche, Eli Lilly & Co., Janssen, Merck & Co. and Naurex over the last 36 months. In addition he is a co-inventor on filed patent application by Yale University (PCTWO06108055A1) and holds shares in BioHaven Pharmaceuticals Holding Company. Dr. Abdallah has received consulting fees from Genentech. None of the other authors report any potential conflict of interests.
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–392. doi: 10.1192/bjp.184.5.386. [DOI] [PubMed] [Google Scholar]
- 3.Ormel J, Petukhova M, Chatterji S, Aguilar-Gaxiola S, Alonso J, Angermeyer MC, Bromet EJ, Burger H, Demyttenaere K, de Girolamo G, Haro JM, Hwang I, Karam E, Kawakami N, Lepine JP, Medina-Mora ME, Posada-Villa J, Sampson N, Scott K, Ustun TB, Von Korff M, Williams DR, Zhang M, Kessler RC. Disability and treatment of specific mental and physical disorders across the world. Br J Psychiatry. 2008;192:368–375. doi: 10.1192/bjp.bp.107.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Psychiatric Association. Practice Guideline for the Treatment of Patients with Major Depressive Disorder. 2. Washington, D.C: American Psychiatric Association; 2000. [PubMed] [Google Scholar]
- 5.Simons AD, Murphy GE, Levine JL, Wetzel RD. Cognitive therapy and pharmacotherapy for depression. Sustained improvement over one year. Arch Gen Psychiatry. 1986;43:43–48. doi: 10.1001/archpsyc.1986.01800010045006. [DOI] [PubMed] [Google Scholar]
- 6.Evans MD, Hollon SD, DeRubeis RJ, Piasecki JM, Grove WM, Garvey MJ, Tuason VB. Differential relapse following cognitive therapy and pharmacotherapy for depression. Arch Gen Psychiatry. 1992;49:802–808. doi: 10.1001/archpsyc.1992.01820100046009. [DOI] [PubMed] [Google Scholar]
- 7.Thase ME, Simons AD, McGeary J, Cahalane JF, Hughes C, Harden T, Friedman E. Relapse after cognitive behavior therapy of depression: Potential implications for longer courses of treatment. Am J Psychiatry. 1992;149:1046–1052. doi: 10.1176/ajp.149.8.1046. [DOI] [PubMed] [Google Scholar]
- 8.Gloaguen V, Cottraux J, Cucherat M, Blackburn IM. A meta-analysis of the effects of cognitive therapy in depressed patients. J Affect Disord. 1998;49:59–72. doi: 10.1016/s0165-0327(97)00199-7. [DOI] [PubMed] [Google Scholar]
- 9.Elkin I, Shea MT, Watkins JT, Imber SD, Sotsky SM, Collins JF, Glass DR, Pilkonis PA, Leber WR, Docherty JP, Fiester SJ, Parloff MB. National institute of mental health treatment of depression collaborative research program. General effectiveness of treatments. Arch Gen Psychiatry. 1989;46:971–982. doi: 10.1001/archpsyc.1989.01810110013002. discussion 983. [DOI] [PubMed] [Google Scholar]
- 10.DeRubeis RJ, Gelfand LA, Tang TZ, Simons AD. Medications versus cognitive behavior therapy for severely depressed outpatients: Mega-analysis of four randomized comparisons. Am J Psychiatry. 1999;156:1007–1013. doi: 10.1176/ajp.156.7.1007. [DOI] [PubMed] [Google Scholar]
- 11.Stewart JW, Garfinkel R, Nunes EV, Donovan S, Klein DF. Atypical features and treatment response in the national institute of mental health treatment of depression collaborative research program. J Clin Psychopharmacol. 1998;18:429–434. doi: 10.1097/00004714-199812000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Thase ME, Dube S, Bowler K, Howland RH, Myers JE, Friedman E, Jarrett DB. Hypothalamic-pituitary-adrenocortical activity and response to cognitive behavior therapy in unmedicated, hospitalized depressed patients. Am J Psychiatry. 1996;153:886–891. doi: 10.1176/ajp.153.7.886. [DOI] [PubMed] [Google Scholar]
- 13.Robbins DR, Alessi NE, Colfer MV. Treatment of adolescents with major depression: Implications of the DST and the melancholic clinical subtype. J Affect Disord. 1989;17:99–104. doi: 10.1016/0165-0327(89)90031-1. [DOI] [PubMed] [Google Scholar]
- 14.Thase ME, Friedman ES. Is psychotherapy an effective treatment for melancholia and other severe depressive states? J Affect Disord. 1999;54:1–19. doi: 10.1016/s0165-0327(99)00033-6. [DOI] [PubMed] [Google Scholar]
- 15.Brambilla P, Perez J, Barale F, Schettini G, Soares JC. GABAergic dysfunction in mood disorders. Mol Psychiatry. 2003;8:721–737. 715. doi: 10.1038/sj.mp.4001362. [DOI] [PubMed] [Google Scholar]
- 16.Petty F. GABA and mood disorders: A brief review and hypothesis. J Affect Disord. 1995;34:275–281. doi: 10.1016/0165-0327(95)00025-i. [DOI] [PubMed] [Google Scholar]
- 17.Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56:1043–1047. doi: 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- 18.Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 19.Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- 20.Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex gaba concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159:663–665. doi: 10.1176/appi.ajp.159.4.663. [DOI] [PubMed] [Google Scholar]
- 21.Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, Berman RM, Krystal JH. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160:577–579. doi: 10.1176/appi.ajp.160.3.577. [DOI] [PubMed] [Google Scholar]
- 22.Sanacora G, Fenton LR, Fasula MK, Rothman DL, Levin Y, Krystal JH, Mason GF. Cortical gamma-aminobutyric acid concentrations in depressed patients receiving cognitive behavioral therapy. Biol Psychiatry. 2006;59:284–286. doi: 10.1016/j.biopsych.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Deschwanden A, Karolewicz B, Feyissa AM, Treyer V, Ametamey SM, Johayem A, Burger C, Auberson YP, Sovago J, Stockmeier CA, Buck A, Hasler G. Reduced metabotropic glutamate receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study. Am J Psychiatry. 2011;168:727–734. doi: 10.1176/appi.ajp.2011.09111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:70–75. doi: 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Boorman E, PMM, PJC Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int J Neuropsychopharmacol. 2008;11:255–260. doi: 10.1017/S1461145707007924. [DOI] [PubMed] [Google Scholar]
- 26.Michael N, Erfurth A, Ohrmann P, Arolt V, Heindel W, Pfleiderer B. Metabolic changes within the left dorsolateral prefrontal cortex occurring with electroconvulsive therapy in patients with treatment resistant unipolar depression. Psychol Med. 2003;33:1277–1284. doi: 10.1017/s0033291703007931. [DOI] [PubMed] [Google Scholar]
- 27.Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: An in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. 2000;47:305–313. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Narr KL, Woods RP, Phillips OR, Alger JR, Espinoza RT. Glutamate normalization with ECT treatment response in major depression. Mol Psychiatry. 2013;18:268–270. doi: 10.1038/mp.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michael N, Erfurth A, Ohrmann P, Arolt V, Heindel W, Pfleiderer B. Neurotrophic effects of electroconvulsive therapy: A proton magnetic resonance study of the left amygdalar region in patients with treatment-resistant depression. Neuropsychopharmacology. 2003;28:720–725. doi: 10.1038/sj.npp.1300085. [DOI] [PubMed] [Google Scholar]
- 30.Pfleiderer B, Michael N, Erfurth A, Ohrmann P, Hohmann U, Wolgast M, Fiebich M, Arolt V, Heindel W. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. 2003;122:185–192. doi: 10.1016/s0925-4927(03)00003-9. [DOI] [PubMed] [Google Scholar]
- 31.Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive Therapy of Depression. 2. The Guilford Press; 1987. [Google Scholar]
- 32.Young J, Beck AT. Cognitive therapy scale: Rating manual. 1980. [Google Scholar]
- 33.First M, Spitzer R, Gibbon M, Williams J. Biometric Research. New York State Psychiatric Institute; 1995. Structured Clinical Interview for DSM-IV Axis I Disorders: Patient Edition (SCIDI/P. Version 2.0) [Google Scholar]
- 34.Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967;6:278–296. doi: 10.1111/j.2044-8260.1967.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 35.Hamilton M. Diagnosis and rating of anxiety. Br J Psychiatry. 1969;3:76–79. [Google Scholar]
- 36.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 37.Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, Krystal JH, Sanacora G. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. 2011;191:122–127. doi: 10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Ashworth F, Sule A, Matthews PM, Cowen PJ. Reduction in occipital cortex gamma-aminobutyric acid concentrations in medication-free recovered unipolar depressed and bipolar subjects. Biol Psychiatry. 2007;61:806–812. doi: 10.1016/j.biopsych.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 39.Morgan PT, Pace-Schott EF, Mason GF, Forselius E, Fasula M, Valentine GW, Sanacora G. Cortical GABA levels in primary insomnia. Sleep. 2012;35:807–814. doi: 10.5665/sleep.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grant BF, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W, Pickering RP, Kaplan K. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61:807–816. doi: 10.1001/archpsyc.61.8.807. [DOI] [PubMed] [Google Scholar]
- 41.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 42.Rush AJ, Trivedi MH, Stewart JW, Nierenberg AA, Fava M, Kurian BT, Warden D, Morris DW, Luther JF, Husain MM, Cook IA, Shelton RC, Lesser IM, Kornstein SG, Wisniewski SR. Combining Medications to Enhance Depression Outcomes (CO-MED): Acute and long-term outcomes of a single-blind randomized study. Am J Psychiatry. 2011;168:689–701. doi: 10.1176/appi.ajp.2011.10111645. [DOI] [PubMed] [Google Scholar]
- 43.Tomba E. Nowhere patients. Psychother Psychosom. 2012;81:69–72. doi: 10.1159/000334112. [DOI] [PubMed] [Google Scholar]
- 44.Fava M, Mischoulon D, Iosifescu D, Witte J, Pencina M, Flynn M, Harper L, Levy M, Rickels K, Pollack M. A double-blind, placebo-controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPTA-A study) Psychother Psychosom. 2012;81:87–97. doi: 10.1159/000332050. [DOI] [PubMed] [Google Scholar]
- 45.Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, Mayberg H. Modulation of cortical-limbic pathways in major depression: Treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 46.Bhagwagar Z, Wylezinska M, Taylor M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. Am J Psychiatry. 2004;161:368–370. doi: 10.1176/appi.ajp.161.2.368. [DOI] [PubMed] [Google Scholar]
- 47.Murck H, Schubert MI, Schmid D, Schussler P, Steiger A, Auer DP. The glutamatergic system and its relation to the clinical effect of therapeutic-sleep deprivation in depression - an MR spectroscopy study. J Psychiatr Res. 2009;43:175–180. doi: 10.1016/j.jpsychires.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 48.Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, Murrough JW, Charney DS, Mathew SJ. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: Relationship to treatment resistance in major depressive disorder. Biol Psychiatry. 2009;65:792–800. doi: 10.1016/j.biopsych.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fava GA. Modern psychiatric treatment: A tribute to Thomas Detre, MD (1924–2011) Psychother Psychosom. 2013;82:1–7. doi: 10.1159/000343002. [DOI] [PubMed] [Google Scholar]
- 50.Zantvoord JB, Diehle J, Lindauer RJ. Using neurobiological measures to predict and assess treatment outcome of psychotherapy in posttraumatic stress disorder: Systematic review. Psychother Psychosom. 2013;82:142–151. doi: 10.1159/000343258. [DOI] [PubMed] [Google Scholar]
- 51.Keedwell P, Drapier D, Surguladze S, Giampietro V, Brammer M, Phillips M. Neural markers of symptomatic improvement during antidepressant therapy in severe depression: Subgenual cingulate and visual cortical responses to sad, but not happy, facial stimuli are correlated with changes in symptom score. J Psychopharmacol. 2009;23:775–788. doi: 10.1177/0269881108093589. [DOI] [PubMed] [Google Scholar]
- 52.Keedwell PA, Drapier D, Surguladze S, Giampietro V, Brammer M, Phillips M. Subgenual cingulate and visual cortex responses to sad faces predict clinical outcome during antidepressant treatment for depression. J Affect Disord. 2010;120:120–125. doi: 10.1016/j.jad.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 53.Furey ML, Drevets WC, Hoffman EM, Frankel E, Speer AM, Zarate CA., Jr Potential of pretreatment neural activity in the visual cortex during emotional processing to predict treatment response to scopolamine in major depressive disorder. JAMA Psychiatry. 2013;70:280–290. doi: 10.1001/2013.jamapsychiatry.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, Alonso CM, Shungu DC. Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: Relationship to anhedonia. Arch Gen Psychiatry. 2012;69:139–149. doi: 10.1001/archgenpsychiatry.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: Glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73:1172–1179. doi: 10.1016/j.biopsych.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao X, Li LP, Wang Q, Wu Q, Hu HH, Zhang M, Fang YY, Zhang J, Li SJ, Xiong WC, Yan HC, Gao YB, Liu JH, Li XW, Sun LR, Zeng YN, Zhu XH, Gao TM. Astrocyte-derived ATP modulates depressive-like behaviors. Nat Med. 2013;19:773–777. doi: 10.1038/nm.3162. [DOI] [PubMed] [Google Scholar]
- 58.Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]