

Abstract
The prospects for successful clinical trials of neuroprotective and neurorestorative interventions for patients with acute and chronic myelopathies depend on preclinical animal models of injury and repair that reflect the human condition. Remarkable progress continues in the attempt to promote connections between the brain and the sensory and motor neurons below a spinal cord lesion. Recent experiments demonstrate the potential for biological therapies to regenerate or remyelinate axons and to incorporate new neural cells into the milieu of a traumatic spinal cord injury. The computational flexibility and plasticity of the sensorimotor systems of the brain, spinal cord, and motor unit make functional use of new circuitry feasible in patients. To incorporate residual and new pathways, neural repair strategies must be coupled to rehabilitation therapies that drive activity-dependent plasticity for walking, for reaching and grasping, and for bowel and bladder control. Prevention of pain and dysautonomia are also clinical targets. Research aims to define the temporal windows of opportunity for interventions, test the safety and efficacy of delivery systems of agents and cells, and provide a better understanding of the cascades of gene expression and cell interactions both acutely and chronically after injury. These bench-to-bedside studies are defining the neurobiology of spinal cord injury rehabilitation.
Keywords: myelopathy, neuroprotection, rehabilitation, neural regeneration, activity-dependent plasticity, functional neuroimaging
Background
Investigators have learned valuable lessons from developmental and experimental neurobiology about the cues that enable axons to grow and stem cells to differentiate and migrate. As a result, the potential to reduce the neurological impairments and functional disabilities of people with traumatic spinal cord injury (SCI) has grown. Greater knowledge about how to manipulate activity-dependent plasticity to retrain sensorimotor skills complements strategies for neural repair (1). At the same time, patients and their families have become forceful advocates for a SCI cure. The possibilities for partial neurorestoration must be balanced against risky, premature clinical trials that employ poorly understood interventions drawn from limited pilot data in rodents. We review the promising biological, engineering, and rehabilitative strategies that may lessen disability and enhance quality of life for patients with acute and chronic myelopathies, primarily due to traumatic SCI.
Traumatic SCI causes disability in 8000–10,000 Americans yearly. Causes include penetrating bullet wounds and other forms of violence (26%) and nonpenetrating lesions from vehicular accidents (38%), and sports accidents (7%), as well as falls (22%), especially in elderly persons (2). Males 15 to 35 years old are most often injured, but another peak occurs in people over age 60 who have an underlying cervical spinal stenosis from degenerative disk disease. Approximately 50% of victims present with complete sensorimotor impairment below the lesion. Fewer than 5% of these patients will regain the ability to walk. When some movement is present, motor gains may continue for many months. Patients who present with even modest voluntary movement below the lesion by the time they begin inpatient rehabilitation usually recover some useful movement in the arms or legs and up to 60% will walk, though often with assistive devices. Table 1 shows the incidence and yearly costs of care for patients with tetraplegia and paraplegia. The American Spinal Cord Injury Association (ASIA) Impairment Scale defines the degree of neurological loss as A (complete sensorimotor loss below the lesion including absent sacral sensation), B (sensory but no motor function below the lesion level), C (some motor preservation, but the majority of muscles are less than 3/5), and D (muscle grade is 3 or greater in the majority of groups below the lesion). By the time of admission for inpatient rehabilitation, ~50% of cervical lesions and 75% of thoracic lesions are graded A. About 8% of lesions at all levels are ASIA B. About 32% of cervical lesions are ASIA D and 65% of L-1–L-5 lesions are ASIA D.
Table 1. Severity and costs beyond the first year after traumatic spinal cord injury.
| Severity of injury | Incidence | Yearly health expenses (in 1998 dollars) |
|---|---|---|
| Tetraplegia | ||
| Complete | 19% | |
| C-1–C-4 | $95,000 | |
| C-5-C-8 | $39,000 | |
| Incomplete | 30% | $12,000 |
| Paraplegia | ||
| Complete | 28% | $20,000 |
| Incomplete | 21% | $11,000 |
Source: National Spinal Cord Injury Statistical Center (http://www.spinalcord.uab.edu).
Causes of nontraumatic SCI include cervical myelopathy associated with cervical spondylosis or a spinal stenosis; epidural metastatic and primary cord tumors; cord ischemia from atherothrombosis, emboli, and venous anomalies; infection; and viral or immune-mediated transverse myelitis. Multiple sclerosis may present with spastic paraparesis. Thoracoabdominal aneurysm repair causes paraparesis or paralysis in up to 10% of surgeries, often after a postoperative delay (3). This review emphasizes traumatic SCI, but most of the interventions discussed for rehabilitation and neural repair can be applied to any cause of an acute or chronic myelopathy.
Experimental Acute Spinal Cord Injury
Pathogenesis
Traumatic SCI may include torsional and tearing mechanical forces on the cord, axonal tears and demyelination, ionic fluxes, ischemia, loss of vascular autoregulation, neurotoxicity from glutamate and products of cell membrane breakdown such as free radicals, central gray neuronal death, delayed cell death by apoptosis, and an early and a delayed inflammatory response. Debris cleared by macrophages leaves a cystic matrix with trabeculae of new vascular structures, basal lamina, fibroblasts, sprouting axons tacked to the wall, and a rim of preserved white matter with some spared axons surrounding the central damage. Thus, gray matter of the dorsal and ventral horns is destroyed over one or more levels of the cord, ascending and descending white matter tracts are partially to completely disconnected, and the dorsal and ventral roots at the level of the lesion may be avulsed or torn.
Models of traumatic SCI in rodents (4) and in transgenic mice (5) reproduce some aspects of human SCI, but because of neurobiological differences among strains and between rodents and humans, such models are most valuable for studies of particular biological processes. Each species and injury model has unique characteristics. Models include complete transection of the cord by surgical incision, incomplete transection that may include 1–3 quadrants of the cord, circumferential compression using a surgical clip, and an impact contusion using a weight or electromechanical device dropped over the cord after removing the dorsal vertebral bone. The impact contusion is reproducible in a graded fashion to create a focal injury similar to human lesions. Of course, a single blow at one angle does not reenact all of the torsional forces and prolonged compression that occur with nonpenetrating traumatic SCI from accidents.
Therapies
Experimental studies of acute SCI target partially characterized components of the pathophysiological cascades that follow injury within hours to days. The cellular and molecular mechanisms of injury resemble those associated with acute stroke and traumatic brain injury, including ischemia, hypoxia, mechanical injury, changes in gene expression, inflammation, and necrotic and apoptotic cell death. Microarray technology is revealing the upregulation and downregulation of gene expression associated with injury over the minutes, hours, and days after SCI (6), which may better focus future neuroprotective and repair therapies (7). Recent murine models of SCI permit genetic manipulations to better determine the effects of a single molecular component of injury.
Results from experimental models of neuroprotection in rodents have suggested interventions for human clinical trials. Highly inbred rodent species, usually of one sex, are typically injured under controlled experimental conditions and studied with outcome measures that do not fully reflect the behavioral and pathophysiological complexity of humans who suffer a SCI (2). Indeed, success in an animal model has led to only one proven, though controversial, human intervention, namely methylprednisolone. Potential targets for clinical trials of neuroprotection within hours to several days after acute SCI are similar to those derived from models of stroke and traumatic brain injury. Interventions that have been or are being studied include the following: ion channel and alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic (AMPA) receptor antagonists to protect axons; glutamate receptor antagonists or other forms of inhibition of the N-methyl-D-aspartate (NMDA) receptor to protect neurons; antioxidants and cell membrane stabilizers (8); caspase inhibitors to prevent apoptosis; neurotrophins, such as brain-derived neurotrophic factor (BDNF), or nitric oxide synthetase inhibition to prevent cell death; inhibition of cytokines and proteinases that contribute to inflammation and disruption of the blood-brain barrier, such as matrix metalloproteinase-9 and minocycline (8a); and modulation of macrophage activity to limit demyelination and secondary injury from inflammatory responses (9). Interventions for neuroprotection overlap with potential biological and pharmacological approaches that inhibit or accelerate mechanisms of spontaneous neural repair. For example, several families of metalloproteases activate or inhibit axon elongation and guidance signaling under differing conditions (10). Activated microglia and macro-phages at the wound site appear to augment secondary tissue damage within the first several days after injury, then provide both neurotrophins and other substrates to increase peri-wound axonal sprouting toward the microglia along the edge of the injury. Thus, the timing of an anti-inflammatory or proinflammatory cell or immune-mediated approach may be important for strategies of protection and repair.
Acute Spinal Cord Injury in Humans
Pathogenesis
Human traumatic SCI often produces a central hematoma, which is visible by magnetic resonance imaging in the most severe cases. Sparing of ascending and descending axons at the periphery of the central lesion may account for later sensorimotor gains. An autopsy study revealed that 28% of 130 patients who survived a complete SCI had some intact axons (11). Patients had been able to move against gravity, but not resistance, if 4%–10% of the axons at the fourth thoracic level were intact. Voluntary foot movement was associated with the presence of only 3000 of the usual 41,000 fibers in the corticospinal tract. Intact touch and vibration sensation required approximately 117,000 of the 452,000 sensory fibers. Sparing of >25% of the lateral or ventral white matter in studies of nonhuman primates permitted walking. Thus, rather modest incremental sparing of sensorimotor pathways from an acute therapeutic intervention, or modest regeneration elicited by subsequent biological intervention, may dramatically improve motor control. Many patients who present with ASIA A or B impairments improve 1–2 levels in motor function and 2–3 levels in sensory function below the lesion. Some of these gains may be related to physiological and structural improvements in the dorsal and ventral roots at the level of SCI and locally in gray and white matter.
Therapies
Traumatic SCI is managed with surgery within one week of onset in 40%–70% of patients. Importantly, however, no clinical trials have been carried out to demonstrate the efficacy of spinal decompression, realignment, or stabilization for lessening sensorimotor or bowel and bladder impairments (12). Various procedures probably help prevent late complications such as cord tethering, syringomyelia, recurrent cord compression, and perhaps some forms of neuropathic pain. Surgical approaches for cervical myelopathy due to spondylosis also lack support from formal trials but may prevent progression of spastic paraparesis (13).
Clinical trials of interventions for ameliorating the pathologic and behavioral effects of acute SCI suffer from problems in translating a single intervention in a rodent model to the complex, multifactorial disorder that exists outside the laboratory. Differences between humans and rodents in size, therapeutic window, dose-response relationships, drug penetration, and gene expression must be taken into account; differences in age and sex may also be important. In addition, interventions may aim at behavioral compensation or at innate restitutive or substitutive capacities. (Restitutive capacity allows the same neural pathways to be used after injury; substitutive capacity entails adapting a defective or partially spared network, usually via external stimulation such as rehabilitation of motor skills.) Most important, the designs of randomized human trials for neuroprotection have been less than optimal for showing clinically meaningful improvements, as opposed to statistically significant differences, due to an intervention.
Acute randomized clinical trials for neuroprotection have been carried out using methylprednisolone, tirilizad, naloxone, and GM-1 ganglioside (Table 2). The results of these trials have been rather disappointing and controversial (14). The culmination of three large trials from the National Acute Spinal Cord Injury Study (NASCIS I, II, and III) led the investigators to recommend that patients with acute SCI receive methylprednisolone (30 mg/kg bolus, then 5.4 mg/kg/h infusion) for 23 h if started within 3 h of injury. If the drug is started 3–8 h after injury, patients should stay on the regimen for 48 h. This recommendation is based on modest gains in motor scores, but not functional gains. The outcome measures of the NASCIS studies aimed rather low in looking for a benefit; the GM-1 studies may have statistically powered their trial based on an effect size that was too high to reveal efficacy.
Table 2. Clinical trials to enhance outcomes after spinal cord injury (SCI).
| Intervention RCTs* | Outcome | Confounds |
|---|---|---|
| Methylprednisolone (95) | Motor gains small; side effects greater with 48-h infusion | Design and statistical issues; limited impact of preventing lipid peroxidation |
| GM-1 ganglioside (96) | No better than steroid | Design, control, and statistical issues |
| 4-aminopyridine (fampridine) (92) | Improves axonal conduction to modestly increase strength or lessen fatigability with repeated movements | Plugs potassium channels in partially demyelinated fibers; in Phase 3 trial with 150 subjects for spasticity, bladder, and motor outcomes (Acorda Therapeutics, Hawthorne, NY) |
| BWSTT* for walking | Nonrandomized trials show value for poor walkers | High manpower and experience needed by therapists |
| Robotic- and FES*-assisted BWSTT | Robot-assistive stepper may be more practical than manual training for patients with no motor function; design ought to include proprioceptive/torque feedback | |
| SAFETY TRIALS FOR CHRONIC SCI (http://carecure.atinfopop.com; no peer-reviewed reports available) | ||
| Fetal tissue in cord syrinx | Cell plug persists without adverse reaction; no clear functional gains | Not sociopolitically viable in United States |
| Human fetal stem cells injected into lesion | Oral reports from Beijing and Moscow suggest no efficacy | No formal report of Phase 1/2 trials |
| Fetal OEGs* injected into cord | Report from Beijing in first 150 subjects and from Lisbon pending | Clinical assessments and outcome measures unclear |
| Fetal pig embryo cells cultured to become oligodendrocytes | Phase 1 trial not yet reported (Diacrin Inc., Charlestown, MA) | No cells found in one autopsy |
| Autologous monocytes activated against skin and injected into cord below lesion | Some sensorimotor gains in Phase 1 trial by oral report | Phase 2 trial of immune strategy pending (Proneuron Biotechnologies, Rehovat, Israel) |
| Peripheral nerve bridge with neurotrophins | Oral reports from Taiwan (H Cheng, http://www.nature.com/nsu/nsu_pf/030512-12.html) | Technical difficulty and could reinjure cord |
RCT, randomized clinical trial; BWSTT, body weight–supported treadmill training; FES, functional electrical stimulation; OEG, olfactory ensheathing glial cells.
An Israeli research group has taken rodent studies into a Phase 1 human trial of activated macrophages (15), which is leading to a Phase 2 trial that includes American sites. A subject's peripheral blood monocytes are activated with epidermal cells and placed into the injury site about two weeks after a traumatic SCI, where they presumably counteract the negative impact of inflammatory responses and tie up myelin products that may inhibit axonal regeneration. The mechanism of efficacy is still unclear. Risks include an exacerbation of injury by chronic macrophage activation and autoimmune responses (16).
Medical complications (17) in the first weeks after a SCI may decrease the benefits of rehabilitation and of future biological interventions for neural repair. Unsuspected fractures or local trauma and inflammation outside the spinal column may produce heterotopic ossification and limit joint range of motion and weight bearing on a limb (2). Low-level, focal irradiation may reduce this risk, along with disodium etidronate. Cerebral trauma may accompany up to 50% of traumatic SCIs, which may alter behavior, cognitive abilities, and ability to cooperate with rehabilitation if not managed. Even minor pain, or pain signals not appreciated from below the lesion, such as from a rectal fissure, bladder infection, thrombophlebitis, or traumatized joint, may induce spasms and dysautonomia with associated hypertension, sweating, and the potential for a cardiac arrythmia. With increasing time after the injury, the initial flaccid paralysis may be superseded by spacticity, manifested as abnormal postures, spasms, hyperreflexia, and, in some instances, contractures. Weakness, impaired motor control for skilled movements, and fatigability are even more disabling residua of an upper motoneuron lesion. Dystonic postures that interfere with hygiene or self-image and spasms that cause pain or make wheelchair activities dangerous require first-order oral antispasticity agents such as baclofen or tizanidine, local injection of botulinum toxin, or intrathecal injection of baclofen. Motor control for walking, reaching, and grasping is rarely improved by these drugs (2).
Neurologic Rehabilitation and Neuroplasticity
Much of rehabilitation for highly impaired patients involves behavioral compensation. Patients learn to use whatever sensorimotor function they possess, along with assistive devices, to become more independent in their daily mobility, self care, and community roles (2). Task-oriented practice, development of problem-solving skills, management of skin, bowel, and bladder care, and use of a wheelchair for those who cannot walk are priorities during inpatient therapy. Additional strategies take advantage of the growing understanding of the neurobiology of rehabilitation.
Successful rehabilitative techniques must induce activity in residual spinal pathways that interact with cortical, subcortical, and brainstem nodes for motor control to drive the plasticity required at all CNS levels for relearning motor skills (2). The spinal cord itself serves far more physiological functions than its appearance as a simple conduit for ascending and descending axons may suggest. The dorsal and ventral neurons of the gray matter integrate and project sensory information related to proprioception and cutaneous inputs related to posture, skilled actions, and automatic flexor and extensor movements such as stepping and reaching. This information drives learning and representational plasticity for movements along the neuroaxis. Interactions among spinal cord columns of rostrocaudal motor pools and oscillating circuits increase the flexibility of supraspinal projections for motor control.
Locomotor Training
Walking over flat ground depends, at minimum, on segmental sensory feedback to spinal and supraspinal networks and on output from cortical, brainstem, and lumbosacral locomotor regions, all of which interact with biomechanical and postural mechanisms for balance and motor control. Segmental proprioceptive and cutaneous sensory inputs associated with walking had powerful modulatory effects on spinal motor pools after complete spinal cord transection at a low thoracic level in the cat and rat (18, 19). These animals could be trained to walk on a moving treadmill when given truncal support. During training on the treadmill, they incorporated signals for stance that emphasized limb loading and for swing that emphasized hip extension at the end of the stance phase (20). Spinal locomotion requires oscillating flexor and extensor motor pools in the lumbar cord called central pattern generators. Studies in spinalized rodents and cats reveal that training to step reduces spinal levels of inhibitory neurotransmitter-associated substances such as glutamic acid decarboxylase (GAD-67) and glycine, whereas injury-induced paralysis raises these levels (21). The exercise also increases neurotrophin levels in muscle and cord. These biochemical changes, then, are activity-dependent and can perhaps be manipulated by physical therapies in patients.
Lessons learned from training spinal-transected animals to step have led to a technique called body weight–supported treadmill training (BWSTT) in human subjects with acute and chronic SCI, to improve walking over ground. In this training, subjects are placed on a treadmill with their weight partially supported by a climbing harness attached to a lift that provides 0%–50% unloading of the paretic legs (22). Therapists manually assist the legs to step with the joint angles and timing of stance and swing typical of normal gait. Hip kinematics and load bearing are critical elements in eliciting rhythmical electromyographic activity in the flexor and extensor muscles of the legs in patients with complete and incomplete spinal cord lesions (23, 24). This form of task-oriented massed practice enhances motorskills learning in the presence of spared pathways for locomotor control (22, 25).
Quasiexperimental studies suggested efficacy of BWSTT for walking in highly impaired subjects, especially with chronic SCI (25–27). However, a large multicenter randomized clinical trial of patients with recent SCI graded ASIA B, C, or D did not find significant differences for its primary outcome measures. In this study, 140 patients received either conventional mobility training or BWSTT for 12 weeks. Interventions started during initial inpatient rehabilitation, within 8 weeks of an incomplete SCI that spared sensation or some sensorimotor function below the lesion (28). At a 6-month follow-up, no significant differences were found in the percentage of subjects who recovered the ability to walk with minimal assistance or less help, and no differences were found in walking speed for those who could walk (29). It remains possible that BWSTT may be valuable for patients beyond the first 3–6 months after an incomplete SCI or other causes of a myelopathy, especially in subjects who have some ability to flex the hip and extend the knee. Treadmill training using robotic-assistive stepping devices is also coming into use to entrain sensorimotor integration for walking without physically taxing therapists (30).
Cortical representational plasticity within the sensorimotor network has been demonstrated after the deafferentation and deefferentation caused by SCI. Functional neuroimaging studies, for example, have shown that the primary sensorimotor cortex representation for the hand may expand into the representation of the trunk that no longer receives sensory input from below a thoracic SCI (31). Functional magnetic resonance imaging performed during passive dorsiflexion of an affected ankle reveals intact inputs to the cortex in some patients who were thought to have a complete lesion, and step training using BWSTT in patients who walk poorly over ground can induce cortical reorganization in the leg representation associated with improved walking ability (32). Such reorganization may occur primarily within the cortical representation or associated with collateral sprouting within the cord, which was demonstrated after a complete experimental lesion (33).
Interventions to Reverse Muscle Atrophy
Chronic nonuse, nonloading, and inactive biomechanical stress on skeletal muscle causes a drop in protein synthesis, an increase in protein degradation, and a preferential reduction in the myosin heavy-chain contractile proteins that are found in slow-twitch, fatigue-resistant muscles (34). Limb immobilization alone will cause atrophy at a rate of 1% to 5% a day for several weeks and a 40% loss of strength by 6 weeks. An upper motoneuron lesion that reduces selective motor control and strength may be magnified when patients do not perform resistance exercises. For muscles that can contract, selective isometric exercises against 60% of the maximal force of a muscle group may be the safest form of exertion for paretic groups. Neurotrophic factors such as insulin growth factor-1, neurotrophin-4, and BDNF are among the substances that have been used exogeneously in rodent experiments to enhance muscle function (35). Muscle normally produces these factors in response to stretch. Indeed, the neurotrophins help link mechanical stimuli to protein synthesis for muscle mass, motoneuron health, and synaptic efficacy. Beta-2 agonists, anabolic steroids, growth hormone, and angiotensin-converting enzyme inhibitors are among the other substances shown to exert an anabolic effect on muscle fibers in animals and humans (36). Clinical studies will be needed to reveal how to best employ such drugs to reverse atrophy and augment the strengthening effect of resistance exercises.
Functional electrical stimulation (FES) can increase muscle mass in the legs and improve cardiovascular conditioning in patients with paraplegia (37). Surface electrodes over the gluteal, hamstring, and quadriceps muscles are stimulated to sequentially contract against the resistance of bicycle pedals connected to an ergometer. However, more than half of patients who start a pedaling program drop out within 6 months and few continue beyond one year. The expense of the equipment, the assistance needed for set-up, the time taken from other daily activities, and the meager visible effects of FES exercise deter patients.
Neuroaugmentation Devices
FES has also been used to stimulate or potentiate functional movements. For example, it has been used to stimulate standing and simple stepping, usually with some bracing and upper-extremity aides (ParaStep from Sigmedics, Northfield, IL). Embedded muscle electrode stimulators have also been commercialized for finger pinching and grasping (the FreeHand from Neurocontrol, Cleveland, OH) and for emptying the bowel and bladder (Vocare from Neurocontrol, Cleveland, OH) (38, 39). The necessity for surgical laminectomy, implantation of electrodes into the S-2 to S-4 ventral roots, and destruction of several dorsal roots has dissuaded some patients with upper motoneuron neurogenic bladder from employing the bladder stimulator, even though its positive economic and quality-of-life consequences have been demonstrated (39). FES products have not been financially successful, which may constrain future investments in sophisticated consumer aids. Newer nerve and neuromuscular stimulation systems using injectable bionic neurons (BIONsTM) are approaching large-scale clinical trials. BIONs are wireless electronic stimulators (2 mm × 15 mm) with a circuit chip and antenna coil that have been safely implanted by hypodermic needle injection in patients (39a). Control paradigms are being developed for a variety of applications.
Microstimulation of the spinal cord is being studied to externally activate an alternating pattern of flexor and extensor muscle groups for stepping. The stimuli may drive small modules for synergistic movements called motor primitives (40) or central pattern generators for locomotion (41). Extradural stimulation at about L-1 near the dorsal roots elicits oscillating electromyographic activity in paraplegic patients (42). A patient who walked poorly after an incomplete SCI improved modestly with the combination of epidural electrical stimulation and treadmill training until walking ability was the same with or without stimulation (43).
Brain-computer interfaces are in development to use biofeedback-trained cortical signals to drive an exoskeleton or FES device that translates the thought of a movement into a stimulated action (44). A variety of robotic exoskeletons and upper- and lower-extremity assistive devices are being tested (45). Passive step training or upper-extremity movement induced by FES or robotic assists may temporarily alter some of the membrane properties of motoneurons and motorcircuit physiology or augment proliferation of neural progenitors and produce neurotrophins in the spinal cord to enhance a strategy for neural repair. Some investigators speculate that the modest sensorimotor gains by Christopher Reeve five years after his cervical SCI can be attributed, in part, to automatically evoked movements during FES bicycling. The notion is that FES signals are a form of patterned neural activity that may alter central activity-dependent plasticity, leading to regeneration and recovery of function (46). However, sensory feedback induced by FES is not likely to lead to persistent central sensorimotor activation or motor skills learning (47). At present, passive and FES-driven repetitive movement cannot be justified as medically necessary.
Neural Repair in Animal Models
Studies of pathological specimens and animal models of injury demonstrate that injured neurons and axons are capable of turning on genes that lead to attempted axonal regeneration, but the milieu for extension of axons inhibits such growth (48, 49). Regeneration-associated genes include those that make (a) cytoskeletal proteins, such as the tubulins, that extend the axon growth cone's protrusions or filipodia; (b) growth cone proteins, including GAP-43, that mediate signal transduction; and (c) cell adhesion molecules (NCAMS) that guide the growth cone, such as L1. Many of these genes require constant expression and interaction of their proteins for an axon to reach a target. The axon-elongation machinery interacts with the growth cone's assessment of signals in its immediate environment. An environment of obstacles, such as scar tissue that contains both physical and chemical barriers and other inhibitory molecules, ordinarily shuts these genes down soon after SCI.
Table 3 lists some specific approaches to neural repair that have led to axonal regrowth or sprouting of axons for 5–20 mm in a modest percentage of descending or ascending fibers beyond the site of injury. The efficacy of any intervention or combination of interventions depends on the extent of injury, the particular behavioral goal sought from regeneration, and the time from onset of injury. For example, most biological interventions have been attempted within one day to two weeks of the experimental SCI. The same approach that encouraged axonal regeneration within that period tends to fail when undertaken more than four weeks after injury (49a).
Table 3. Rodent studies of promising human interventions for spinal cord injury (SCI) repair.
| Intervention | Action | Delivery/Confounds |
|---|---|---|
| Bridges | ||
| Polymers, conduits | Fill cavity; may contain growth substances and cells | Injection of alginate or smart biodegradable fibers may release cells and factors, but timing of release and integration with cord uncertain |
| OEGs* | Inject just above and below lesion or within a matrix into a cystic area | |
| Schwann cell graft | Align axons, migrate, produce trophins | Quantity and quality of cells, associated matrix, ability of axon to travel beyond a bridge are uncertain; could induce scar, tumor |
| Bone marrow stromal/stem cells | Differentiate into matrix cells, neurons, and oligodendroglia; serve as neuronal relays within the bridge, repopulate gray and white matter, provide trophins | Inject |
| Stem/progenitor cells | Cell type needed may not differentiate or integrate; ethical issues for human studies with fetal tissues | |
| Fetal spinal cord | ||
| Injury-induced neurogenesis | ||
| Peripheral nerve micrografts | White-to-gray and gray-to-white matter connections | Technically complex, risky surgery |
| Nogo myelin inhibitor | ||
| Nogo-A antibody | Binds Nogo to block inhibition of axon growth | Injected locally by osmotic CSF pump; immunization with CNS myelin component (e.g., Nogo-66) or injection of activated macrophages |
| Nogo peptide antagonist | Bind to Nogo receptor, blocking inhibition of axon growth by Nogo, MAG,* and OMGP* | Potential for oral administration CSF pump; possible intravenous route |
| Nogo receptor antibody | Bind receptor | |
| Proteoglycan inhibitor | ||
| Chondroitinase | Digests inhibitors to foster axon growth in white matter | Infuse locally above and below edge of injury site |
| Growth cone signaling | ||
| cAMP | Overcomes growth cone inhibitors | Must be taken up by neuron (potential for oral administration) |
| cGMP* | Higher ratio of cAMP/cGMP for axon extension | |
| Rho GTPases | Block inhibitory effect of Mag, OMGP, Nogo A | Provide soon after injury for brief time by local infusion near injury site |
| Neurotrophic factors BDNF,* NT-3 | Limit neuronal apoptosis, aid axonal regeneration and guidance to targets; aid dendritic sprouting and learning mechanisms, such as LTP* | Inject engineered fibroblasts that secrete a trophin; inject or pump factor into CSF |
| Other trophins* | ||
| GDNF, FGF, VEGF, IGF-1 | ||
| Immunophilin | Aid axonal growth and transport | Provide orally or infuse near injury site |
| Inosine (93) | Neurite outgrowth | Oral or intravenous route |
| Electrical stimulation | Activates axonal growth cone | Methods and efficacy in humans uncertain |
| X-irradiation | Decrease number of cells making inhibitory molecules; lessen negative inflammatory cells | Safety and potential negative impact in humans |
| Remyelination | ||
| Neural stem cells | Stimulate to become oligodendrocytes | Must proliferate, differentiate, and migrate to where needed; only travel short distances |
| Oligodendrocyte precursors | Abundant in adult brain | Activated in situ precursors may inhibit growth cone |
| OEGs | Migrate to surround axons | Inject into cord |
| Ventral horn | ||
| Prevent apoptosis of injured or axotomized cells | Neurotrophins, antiapoptotic proteins, caspase inhibitors | CSF infusion |
| Implant neuronal precursors | Replace lower motoneurons | Inject near injury site |
| Reimplant ventral roots from below the lesion into cord above lesion | Regenerate axons into peripheral nerve to muscle, sphincters, bladder | Can reinnervate muscle within 1 month of cervical plexus avulsion in humans |
| Dorsal horn | ||
| Neurotrophins | Prevent small-fiber sprouting that leads to pain; increase large to small fiber ratio | Local infusion or implant cells that secrete agent |
| OEGs | Regenerate sensory axons | Inject at dorsal horn entry zone |
| Combinations of above (Figure 1) | Bridge lesion, aid axons to grow beyond the bridge, and target spinal neurons | Graded timing of interventions; more manipulations increase risk of tissue damage and adverse interactions to substances given |
OEG, olfactory ensheathing glial cells; MAG, myelin-associated glycoprotein; OMGP, oligodendrocyte myelin glycoprotein; AMP, adenosine monophosphate; GMP, guanosine monophosphate; BDNF, brain-derived neurotrophic factor; GDNF, glial cell-derived neurotrophic factors; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; IGF-1, insulin growth factor-1; LTP, long-term potentiation
Many behavioral studies have related the growth of axons to modest improvements in a few rodent behaviors, such as forelimb grasping and hindlimb movements (50, 51). Studies in some animal models of repair are compelling. Often, however, the tissue evidence for regenerating axons does not prove that the cut axons are regrowing. The apparent axons may actually be spared fibers or collateral branches from an uninjured axon (52). Sometimes, the rapidity of behavioral gains does not bear any plausible temporal relationship to the time needed for axons to regenerate past the lesion. Thus, evidence is still wanting for morphologic, physiologic, and behavioral proof of the influence of new inputs on their targets.
Figure 1 shows the potential effects of one or more neural repair strategies when combined with training. An initial goal might be to regenerate 10%–20% of one or more descending tracts for 3–4 cm, which would be about two levels in the human cervical cord. Figure 2 makes it clear that 10 mm of axonal growth in a rodent cord covers many more segments than in the human cord; the distance from a high thoracic injury to the lumbar motor pools in the human can exceed 20 cm.
Figure 1.
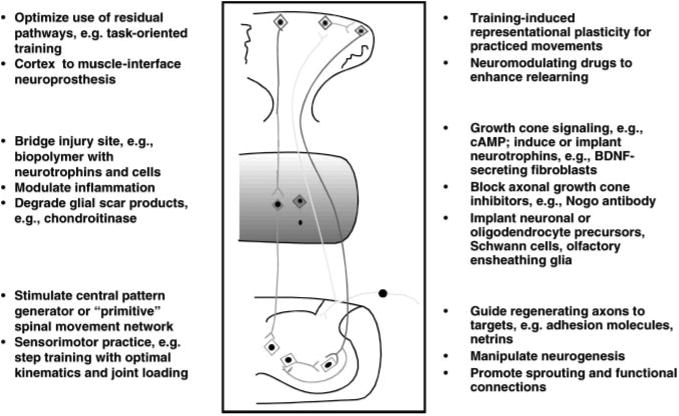
Experimental strategies for SCI repair. At the site of this hypothetical cystic lesion, a cellular matrix or biopolymer bridge is injected to fill the cavity. The matrix contains neuronal or oligodendroglial precursors and neurotrophins. Chondroitinase is injected into the white matter above and below the cavity to lessen inhibitory proteoglycans. An inhibitor to the Nogo receptor is injected around white matter tracts near the cavity to prevent growth-cone collapse. Fibroblasts modified to secrete brain-derived neurotrophic factor (BDNF) or neurotrophin-3 (NT-3) are placed intermittently along the white matter tracts below the injury site to help signal the growth cones of regenerating axons. Neurotrophins or other diffusible attractive cueing molecules are injected to lead regenerating axons into the ventral gray matter of the cord at the levels of targeted motoneuron pools. Task-oriented sensorimotor training promotes regeneration and specific neuronal targeting, incorporates the sensorimotor pools into functional units, and enhances cortical representational plasticity for the motor control needed to relearn functional skills.
Figure 2.
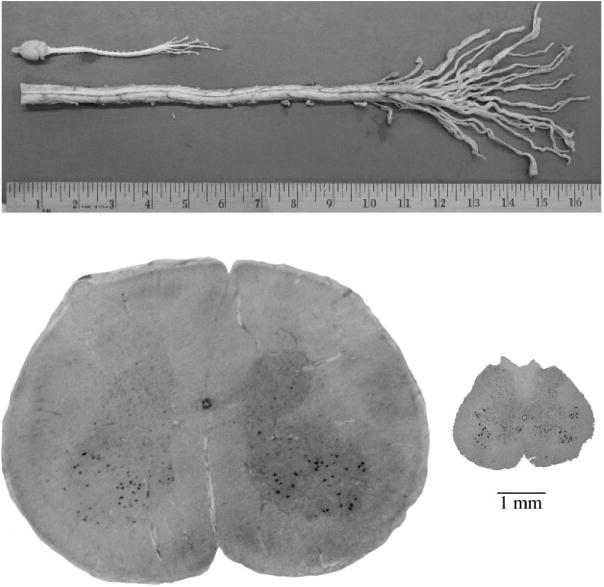
Physical comparison of human and rodent spinal cord anatomy. The overall length of a rat brain and spinal cord, including the cauda equina, measure about 15 cm. The human cord alone, up to the cauda equina averages about 65 cm in a person who is 68 inches tall. The splay of roots of the cauda equina of the human cord reveals that 2–3 lumbar roots equal the diameter of the rat spinal cord. The differences in diameter of the cord sectioned at the lumbar enlargement are shown in the axial sections. Approximately 20 axial sections from the rat would fit within the human cord at this level. Relative distances necessary for targeting the cervical or lumbar motor pools for axonal regeneration from any level is at least four times greater in humans.
Preventing Growth-Cone Inhibition
Gliotic scarring and the traumatic cavity represent a physical barrier to regeneration of axons. The glial scar includes many cell types, including glial progenitors, injured oligodendrocytes, reactive astrocytes, microglia, fibroblasts, macrophages, and meningeal cells. Most of these cells produce molecules that can inhibit axonal growth, including proteoglycans, collagen, growth cone–collapsing substances, and repulsive guidance molecules. Their identification has led to therapeutic strategies. For example, enzymatic digestion of a glycosaminoglycan chain from chondroitin sulfate proteoglycans has made the environs more permissive of axonal regeneration (53).
Recent studies point to the therapeutic potential for targeting myelin-associated inhibitory substances produced by oligodendrocytes—myelin-associated glycoprotein (MAG), oligodendrocyte-myelin glycoprotein (OMGP), and Nogo-A. Growth inhibition in neurites is caused by their Nogo-66 and amino-Nogo domains when oligodendrocytes, periaxonal CNS myelin, and myelin debris are exposed by an injury. The normal role of Nogo-A may be to prevent collateral sprouting of axons after development. MAG, OMGP, and Nogo bind to the same region of the Nogo receptor at the growth cone. When the receptor complex is signaled by one of these inhibitory substances, a small guanosine triphosphatase (GTPase) called Rho and other cascades of intracellular activity stop support for the growth cone. Partial reversal of this inhibition has been accomplished using an intrathecal infusion of the small peptide NEP1-40, which inhibits binding of Nogo-66 to the Nogo receptor (54). The inhibition of Rho using small molecules that cross the blood-brain barrier may eventually accomplish the same effect in patients (55). Infusion of a substance that irreversibly inhibits Rho or the Nogo receptor may best be carried out in the first 3–10 days after SCI and continued for several weeks. Monoclonal antibodies raised against Nogo-A permit spinal axons to regenerate modest distances below a lesion (56). Immunization with myelin also promotes axonal regeneration in rodents after transection of the corticospinal tracts by blocking myelin-associated inhibitors (56a). Another approach is to increase the amount of the second messenger cyclic nucleotide adenosine monophosphate (cAMP), which induces genes to activate protein kinase A and to synthesize polyamines (57). The cAMP also inactivates Rho to make the growth cone less sensitive to MAG, OMGP, and Nogo. Indeed, a high ratio of intracellular cAMP to cGMP may stimulate growth-cone motility, whereas a low ratio or just a low level of cAMP may lead to collapse of the growth cone. More information about the Nogo receptor's binding sites and the mechanisms that signal genes to make actin and other components of an extending axon growth cone may lead to practical ways to regenerate axons in an otherwise hostile milieu (57a).
Augmenting Axonal Regeneration
The neurotrophins are polypeptide growth factors that include nerve growth factor (NGF), neurotrophins 3 and 4 (NT-3 and NT-4), and brain-derived neurotrophic factor (BDNF). They act specifically on one of three Trk receptor tyrosine kinases and the p75 neurotrophin receptor. In many ways, the receptors act as omnipresent neural sensors for extracellular and intracellular signals. The p75 receptor increases its expression after SCI, axotomy, or ischemia in motoneurons and oligodendrocytes. With the Nogo receptor, it forms a complex that inhibits neurite outgrowth by MAG and OMGP (48). The neurotrophins induce axonal extension and dendritic arborization by acting on cytoskeletal proteins. In addition, they guide axons over distances, aid neuronal cell survival, and participate in the regulation of synapse formation, synaptic plasticity, long-term potentiation for learning, and the release of neurotransmitters (58). BDNF seems better at protecting corticospinal neurons than eliciting axonal growth, whereas NT-3 elicits axonal regeneration in the spinal cord (59). BDNF has reversed the atrophy of rubrospinal neurons and promoted the regeneration of spinal axons into a peripheral nerve graft, even when given one year after SCI (60). Thus, trophic support may rescue chronically injured neurons, probably by triggering regeneration-associated genes. Other growth factors, including insulin-related growth factor, cytokine family members such as leukemia inhibitory factor, and ciliary neurotrophic factor also have trophic effects on particular types of central axons, as well as on peripheral nerve axons. Trophic factors are diffusible and could be manipulated to make a gradient in their concentration for better axonal signaling over long distances. They can be delivered by a CSF pump or by perilesional implantation of fibroblasts, Schwann cells, or neural progenitor cells that have been genetically modified to express neurotrophins. The temporal pattern of providing neurotrophins may be important. For example, axotomized rubrospinal neurons showed greater survival and axonal regeneration after a delay, rather than after acute treatment with BDNF or NT-3 (50, 61).
Guiding Regenerating Axons
The demonstration that CNS axons may be enticed to regenerate is an important breakthrough. It is unclear, however, which axons among the descending tracts are most critical for restoring voluntary movement and which targets in the spinal gray matter below the lesion will enable restitution of function. In addition, strategies that induce axonal regeneration may result in aberrant sprouting of axons into both sides of the cord and into dorsal and ventral horns. Thus, strategies to guide the regenerating axons in SCI lesions are also needed.
The growth cone can be steered by local forces of attraction and repulsion. These include variations in the cone's adhesion to different surfaces in the milieu, the mechanical forces generated by the cone, and transduction of signals in the milieu. Growth cones may be guided by direct-current electrical fields, as well as by chemotropic gradients of attractive (e.g., netrin-1) and repulsive (e.g., MAG) cues (62). Physical, chemical, and electrical cues, then, may aid axonal guidance through the spinal cord white matter and into gray matter targets.
Extracellular axon guidance molecules present during development include the netrins, semaphorins, slits, and ephrins. Transmembrane proteins on the surface of the growth cone, such as plexin, initiate a signal transduction cascade for collapse, repulsion, or turning of the growth cone when they bind to one of these families of developmental guidance molecules (63). In general, guidance molecules affect actin-based motility by directing and stabilizing the assembly of microtubules. These steering molecules signal different receptors at a growth cone to attract or repel fingers of actin webs and bundles, as well as microtubule filaments within the stretching and shrinking protrusions of the growth cone. Interactions are complex. A guidance cue may act on a navigating growth cone differently over the course of the axon's journey, as occurs during development, by attracting, repelling, or modulating internal and external factors (64). Although investigators can only speculate on how to best control the distribution of cues over space and time for rewiring the spinal cord, manipulations have shown some success. For example, the Roundabout (Robo) family of slit receptors can be inhibited to promote axon sprouting. Cyclic AMP and cGMP can reverse growth-cone repulsion. Neurotrophins also have neurotropic actions that steer the growth cone toward them.
Mechanical guidance of regenerating axons in damaged peripheral nerves depends heavily on the integrity of the nerve's endoneurial tubes. An experimental SCI by surgical transection leaves much of the structure above and below the cut intact for axonal regeneration, compared, for example, to the focal contusion model, which disrupts this structure. Mechanical guidance by the intact central myelin or by implanted guidance channels such as peripheral nerve grafts may be a useful repair strategy (64a).
Implanting Cells or Stimulating Neurogenesis
Exogenous and endogenous stem cells, or more differentiated precursors of neurons and oligodendrocytes, offer an exciting but challenging approach for neural repair (59, 65). The cellular and extracellular milieu of the injury and the genetic makeup of the implants may alter the survival, differentiation, migration, and integration of these cells. The injury itself leads to signaling of progenitors of neurons and especially of glia to stream out from the central canal of the cord or proliferate from within the white matter of the brain and spinal cord (66). The fate and function of these endogenous multipotential cells is uncertain. However, exogenous multipotent stem cells and neural progenitor cells may be programmed in cell culture and by environmental signals to serve as potential tools for repair. A few examples of experimental manipulations follow (Table 2).
After a complete cord transection in adult rodents, fetal spinal cord tissue placed into the lesion produced greater descending axonal regeneration when combined with BDNF or NT-3 (50). Because fetal tissue poses ethical constraints, other cell types have been tried in rodent SCI. Progenitor cells used for spinal cord implantation include neural progenitors derived from subventricular and olfactory regions of embryonic and adult rodent and human brain tissue, and nonneural progenitors from marrow stromal cells.
Embryonic stem cells in rodents have been differentiated into specific neuronal cell types, such as motoneurons of the ventral horn of the cord (67), by exposure to signals drawn from normal development, such as neurotrophins. The motoneurons integrated within ventral horns of the cord and sent out axons that made cholinergic synapses on muscle. In one study, embryonic stem cells from mice were transplanted into a spinal contusion in rats after being manipulated into a neural lineage. Although most cells died, the majority of the survivors became oligodendrocytes that remyelinated adjacent axons, and 10% became neurons (68). Other studies have had less success, finding only a small percentage of surviving cells that did not become astrocytes. The milieu of a SCI may inhibit neuronal precursor differentiation and require interventions beyond transplantation (69), such as adding a neurotrophin. Some lines of neural stem cells produce their own neurotropic factors, which leads to host axonal regeneration (69a). Newer techniques may generate pure populations of neurons from human fetal stem cells that are capable of producing, for example, acetylcholine, regardless of the milieu into which they are grafted (70).
Schwann cells from peripheral nerve and cells from olfactory mucosa have special characteristics that make them candidates for use after SCI in humans. Cultured Schwann cells injected into a rat spinal cord contusion one week after injury were somewhat superior to cultured olfactory ensheathing glial (OEG) cells, grown from cells of the olfactory bulb, in enabling axonal growth, myelination, and partial hindlimb stepping (71). Cultured OEGs have promoted axonal regeneration over distances of 10 mm, repressed axonal branching, aligned growing axons, and improved forepaw and hindlimb movement when injected into a focal corticospinal tract lesion (72) or into a transected cord (73). The combination of olfactory fibroblasts and ensheathing cells embedded in a matrix improved phrenic nerve and climbing skills after a high cervical cord hemisection associated with possible dorsal and ventral tract repair (74). However, these models of injury may produce a different level of inflammatory and local signaling molecule response compared to a traumatic contusion. In addition, most regenerating axons in these models seem to end just below the lesion, suggesting that if they are making effective synapses 1–2 weeks after the transplantation, any restitution of function must be related to spared fibers or to propriospinal or other local polysynaptic connections that were engaged, and not to restitution of a pathway.
Other types of cells have been implanted to try to restore a particular descending pathway. For example, brainstem raphe cells that produce serotonin have restored treadmill locomotion after implantation into a T-11 injury in the rat. Engineered fibroblasts that produce BDNF or NT-3 injected below the lesion can signal axons from above a lesion to extend toward them (59), protect the cell bodies, and regenerate specific pathways such as dorsal root, serotonergic, and rubrospinal fibers (75). These trophins may also induce oligodendrocytes to myelinate fibers in a contusion (76).
Certain neural precursor cells derived from the human brain or from marrow have been induced to myelinate fibers to improve central conduction (77, 78). In a mouse model, cultured adult neural precursors drawn from the subventicular zone reversed much of the demyelination caused by experimental multiple sclerosis (79). Interventions for focal demyelination may be especially useful for patients with a transverse myelitis, focal infarct, or spinal multiple sclerosis (80). The success of implants will depend, in part, on the local cellular milieu produced by each disease and the timing of an implant in relation to onset of the injury.
Transplantation studies for SCI in humans have started in North America, Europe, Australia, South America, China, and Russia (Table 3). In the United States and Europe, patients with multiple sclerosis have received autologous Schwann cells and OEGs placed into plaques in safety trials. Oligodendrocyte precursors from pigs were implanted into the lesion bed of six subjects after SCI in another safety trial (Diacrin Corp). Results are pending. Reports from American physicians who visited Beijing describe implantation of OEGs in patients six or more months after a traumatic SCI (http://carecure.atinfopop.com). The cells were expanded from olfactory tissue of one fetus and injected just above and below the lesion during open visualization of the cord. Sensation supposedly improved within days of the injection over a few levels and motor function increased for up to two levels below the lesion. A study using embryonic stem cells in China was halted, apparently because no recovery was seen in 30 cases. One hopes that meaningful data will be published by these and other investigators.
Reimplanting Nerve Roots
Nerve roots may be torn or avulsed by traumatic SCI. This injury may occur at any segmental level but is perhaps a more critical cause of disability in patients who suffer a burst fracture, vertebral subluxation, or penetrating injury below the T-10 vertebral body, which accounts for 20% of traumatic SCIs. Here, the tip of the cord or conus medullaris and the nearby floating nerve roots of the cauda equina may be sheared, torn, crushed, or avulsed. Clinically, this lower motoneuron lesion causes flaccid paralysis, muscle atrophy, and loss of stretch reflexes from ventral root and anterior horn injury; flaccid bladder and sphincters from preganglionic parasympathetic and autonomic nerve injury; and at-level and below-level sensory loss and, often, central neuropathic pain from dorsal horn and ganglion injury. The motor and autonomic neurons of these roots start to die by apoptosis within one week after being disconnected at the interface between the central and peripheral nervous system (80a). Neurotrophic factors may be able to rescue these cell bodies. Remarkably, reimplantation of avulsed ventral roots, both experimentally and in humans after brachial plexus avulsions (81), can lead to at least partial recovery of motor function (82). The motoneurons and preganglionic parasympathetic neurons, at least in animal models, regenerate toward the reimplanted ventral root, probably lured by substances such as neurotrophins within the nerve's Schwann cells. This axonal regeneration is associated with remyelination of nerve root fibers (80a). Thus, at the time of surgical management of acute conus/cauda SCI, identification and reimplantation of ventral roots into the cord at the level of a tear or avulsion or into the ventrolateral cord at a level above the gray matter injury could restore motor and bladder function when combined with rehabilitation training.
Maintaining Dorsal Horn Function
Dorsal root and dorsal horn injury may cause sensory loss and pain at the segmental levels of the SCI. Dorsal root ganglion cells do not ordinarily regenerate into the dorsal horn after an avulsion. Strategies to promote spinal ingrowth of subpopulations of these neurons are similar to strategies for ventral root and central axonal regeneration. Interventions include placing fibroblast-secreting cells or a local intrathecal infusion of neurotrophins such as BDNF, NGF, or NT-3 into the dorsal horn, upregulation of cAMP, transplantation of embryonic spinal tissue or OEGs into the transition zone (83), and implantation of a peripheral nerve conduit from the entry zone to the dorsal root. These manipulations have restored sensation and bladder function after an experimental dorsal rhizotomy in rodents (84). Attempts to restore sensory inputs within and near the injured dorsal horn may, however, activate pathways for pain, autonomic dysreflexia, and flexor or extensor spasms. This potential negative aspect of exogenous neurotrophins derives from their role in pain-associated neuroplasticity, both neurite outgrowth and physiologic reorganization (85, 86). Several therapeutic strategies have prevented these complications in rodents. Antibodies to NGF prevented small fiber afferents from sprouting in the dorsal horn, which reduced autonomic responses to pain. Other neurotrophins, such as GDNF, enhance large-fiber proliferation and offset the balance toward pain induced by small-fiber proliferation. Neurotrophins may also increase the regeneration of ascending axons from the dorsal horn.
Bridges to Span SCI-Induced Scar and Cavities
The cavity and scar tissue that form after SCI may have to be filled or bridged to allow regenerating axons to descend and ascend across a cystic cord to reach their targets (Figure 1). Schwann cells, OEGs, embryonic spinal cord tissue, and tiny peripheral nerve segments have allowed at least some types of axons to cross from white matter into white or gray matter below the lesion. The reproducibility of successful experiments in other laboratories has been poor, however. Engineered substrates may include synthetic channels or biopolymers laced with guidance channels, Schwann cells or OEGs, a permissive substrate such as laminin, and neurotrophic factors. A bridge may release substances on a schedule that attracts axons into and then out of the bridge. Many forms of “smart” (biodegradable) bridges for central and peripheral axons have shown some efficacy in animal models (87, 88).
Pharmacologic Interventions
After a complete SCI in cats and rodents, inhibitory neurotransmitters are expressed in the motor pools below the lesion (89). Other neurotransmitters, such as serotonin and norepinephrine, tend to excite the pools related to locomotion (90). Thus, a strategy to augment rehabilitation and neural repair strategies may include medications that act as antagonists or agonists for the control of excitation and inhibition, as well as routine avoidance of drugs that may have a negative impact on excitation and inhibition of motor and bowel and bladder autonomic activity. The spinal cord is also capable of learning through training (91). Drugs that affect membrane properties and synaptic efficacy are being studied to determine if they may augment motor skills learning by affecting spinal motor pools or mechanisms of cortical representational plasticity for the practiced movements.
Future Directions
Animal models of injury and repair allow the researcher to study or manipulate specific biological changes and responses to interventions. Robust changes in preclinical studies should include anatomical, physiological, and behavioral correlative evidence of the effects of a biological and training strategy (94). Positive outcomes for limiting injury or inducing greater repair in a transgenic mouse or inbred rodent species cannot predict a similar outcome in human subjects. In this decade, interventions for neural repair that produce robust gains in rodents may have to be tested in small studies with larger mammals or nonhuman primates to evaluate safety and dose-response relationships.
Clinicians, including neurologists, physiatrists, neurosurgeons, and orthopedic surgeons, as well as rehabilitation therapists, must develop ethical designs to test these approaches and measure important and relevant outcomes in patients with recent motor-complete SCI. Clinical trials with chronically injured subjects may run in parallel, especially for safety trials of invasive interventions. Primary outcomes for trials could include, for example, a robust increase in the likelihood that patients with high quadriplegia breathe without a ventilator, gain better use of an upper extremity by improving two or more motor levels, or have fewer complications of dysautonomia. For subjects with paraplegia from a mid- to low-thoracic lesion, reciprocal stepping at a low energy cost, control of sphincters and voluntary bowel and bladder emptying, and less pain at and below the level of injury will be clinically meaningful outcome measures. The latter endpoints will also be appropriate for studies in patients with conus or cauda equina injuries.
The potential pool of participants eligible for clinical trials of neural repair interventions who have incurred a recent, profound SCI is rather small compared to other common neurological diseases. If only subjects classified as ASIA A are included, and 10%–20% of patients meet entry criteria (as is typical of such studies), as few as 600 subjects a year would be available across the United States and 800 in Europe. ASIA B subjects may also be reasonable to enter into early interventional studies if the repair strategy does not surgically disrupt the cord, as well as studies of chronically paraplegic subjects. However, experimental interventions in patients with chronic SCI, which perhaps means >3 months after trauma for certain approaches, probably should not include patients with debilitating pain, dysautonomia or spasms, or a symptomatic tethered cord or syrinx. These complications could worsen with biological manipulation and would interfere with interpretation of any adverse responses to the intervention. Thus, multicenter clinical trials will require great cooperation between institutions and adequate funding to identify and randomize just 50–100 subjects with a cervical or a thoracic injury of interest and to monitor outcomes for at least two years.
Present care and optimal deployment of biological interventions demand well-defined physical rehabilitation and pharmacological therapies. Clinicians must continue to develop interventions that drive mechanisms of activity-dependent plasticity to best incorporate spared and new circuitry into a neural matrix for important behavioral gains.
Acknowledgments
We thank the National Institutes of Health for funding through HD37439, HD39629, T32HD07416, NS42719, NS16333, and T32NS07449; the Nathan Shapell Foundation; the State of California Roman Reed Bill; and the Larry L. Hillblom Foundation.
Literature Cited
- 1.Raineteau O, Schwab M. Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci. 2001;2:263–73. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- 2.Dobkin BH. The Clinical Science of Neurologic Rehabilitation. New York: Oxford Univ Press; 2003. [Google Scholar]
- 3.Maniar H, Sundt T, Prasad S, et al. Delayed paraplegia after thoracic and thoracoabdominal aneurysm repair: a continuing risk. Ann Thorac Surg. 2003;75:113–20. doi: 10.1016/s0003-4975(02)04494-6. [DOI] [PubMed] [Google Scholar]
- 4.Kwon B, Oxland T, Tetzlaff W. Animal models used in spinal cord regeneration research. Spine. 2002;27:1504–10. doi: 10.1097/00007632-200207150-00005. [DOI] [PubMed] [Google Scholar]
- 5.Inman D, Guth L, Steward O. Genetic influences on secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol. 2002;451:225–35. doi: 10.1002/cne.10340. [DOI] [PubMed] [Google Scholar]
- 6.Di Giavanni S, Knoblach S, Brandoli C, et al. Gene profiling in spinal cord injury shows role of cell cycle neuronal death. Ann Neurol. 2003;53:454–68. doi: 10.1002/ana.10472. [DOI] [PubMed] [Google Scholar]
- 7.Nesic O, Svrakic N, Xu G, et al. DNA microarray analysis of the contused spinal cord: effect of NMDA inhibition. J Neurosci Res. 2002;68:406–23. doi: 10.1002/jnr.10171. [DOI] [PubMed] [Google Scholar]
- 8.Gilgun-Sherki Y, Rosenbaum Z, Melamed E, et al. Antioxidant therapy in acute central nervous system injury. Pharmacol Rev. 2002;54:271–84. doi: 10.1124/pr.54.2.271. [DOI] [PubMed] [Google Scholar]
- 8a.Wells J, Hurlbert RJ, Fehlings M, Yong V. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628–37. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- 9.Jones T, Basso D, Sodhi A, et al. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J Neurosci. 2002;22:2690–2700. doi: 10.1523/JNEUROSCI.22-07-02690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McFarlane S. Metalloproteases: carving out a role in axon guidance. Neuron. 2003;37:559–62. doi: 10.1016/s0896-6273(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 11.Kakulas B. A review of the neuropathology of human spinal cord injury with emphasis on special features. J Spinal Cord Med. 1999;22:119–24. doi: 10.1080/10790268.1999.11719557. [DOI] [PubMed] [Google Scholar]
- 12.Fehlings M, Tator C. An evidence-based review of decompressive surgery in acute spinal cord injury: rationale, indications, and timing based on experimental and clinical studies. J Neurosurg. 1999;91(Spine 1):1–11. doi: 10.3171/spi.1999.91.1.0001. [DOI] [PubMed] [Google Scholar]
- 13.Edwards C, Riew K, Anderson P, et al. Cervical myelopathy: current diagnostic and treatment strategies. Spine J. 2003;3:68–81. doi: 10.1016/s1529-9430(02)00566-1. [DOI] [PubMed] [Google Scholar]
- 14.Hadley M. Pharmacological therapy after acute cervical spinal cord injury. Neurosurgery. 2002;50:S63–S72. doi: 10.1097/00006123-200203001-00013. [DOI] [PubMed] [Google Scholar]
- 15.Hauben E, Schwartz M. Therapeutic vaccination for spinal cord injury: helping the body cure itself. Trends Pharmacol Sci. 2002;24:7–12. doi: 10.1016/s0165-6147(02)00013-5. [DOI] [PubMed] [Google Scholar]
- 16.Popovich P, Jones T. Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends Pharmacol Sci. 2002;24:13–17. doi: 10.1016/s0165-6147(02)00006-8. [DOI] [PubMed] [Google Scholar]
- 17.Consortium for Spinal Cord Medicine. Outcomes following traumatic spinal cord injury: clinical practices guidelines for health-care professionals. Washington DC: Paralyzed Veterans of America; 1999. [DOI] [PubMed] [Google Scholar]
- 18.de Leon R, Tamaki H, Hodgson J, et al. Hindlimb locomotor and postural training modulates glycinergic inhibition in the spinal cord of the adult spinal cat. J Neurophysiol. 1999;82:359–69. doi: 10.1152/jn.1999.82.1.359. [DOI] [PubMed] [Google Scholar]
- 19.Barbeau H, McCrea D, O'Donovan M, et al. Tapping into spinal circuits to restore motor function. Brain Res Rev. 1999;30:27–51. doi: 10.1016/s0165-0173(99)00008-9. [DOI] [PubMed] [Google Scholar]
- 20.Duysens J, Van de Crommert H. Neural control of locomotion: the central pattern generator from cats to humans. Gait Posture. 1998;7:131–41. doi: 10.1016/s0966-6362(97)00042-8. [DOI] [PubMed] [Google Scholar]
- 21.Edgerton V, de Leon R, Harkema S, et al. Retraining the injured spinal cord. J Physiol. 2001;533:15–22. doi: 10.1111/j.1469-7793.2001.0015b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobkin B. Overview of treadmill locomotor training with partial body weight support: a neurophysiologically sound approach whose time has come for randomized clinical trials. Neurorehabil Neural Repair. 1999;13:157–65. [Google Scholar]
- 23.Harkema S, Hurley S, Patel U, et al. Human lumbosacral spinal cord interprets loading during stepping. J Neurophysiol. 1997;77:797–811. doi: 10.1152/jn.1997.77.2.797. [DOI] [PubMed] [Google Scholar]
- 24.Dietz V, Muller R, Colombo G. Locomotor activity in spinal man: significance of afferent input from joint and load receptors. Brain. 2002;125:2626–34. doi: 10.1093/brain/awf273. [DOI] [PubMed] [Google Scholar]
- 25.Barbeau H. Locomotor training in neurorehabilitation: emerging rehabilitation concepts. Neurorehabil Neural Repair. 2003;17:3–11. doi: 10.1177/0888439002250442. [DOI] [PubMed] [Google Scholar]
- 26.Dietz V. Spinal cord pattern generators for locomotion. Clin Neurophysiol. 2003;114:1379–89. doi: 10.1016/s1388-2457(03)00120-2. [DOI] [PubMed] [Google Scholar]
- 27.Wernig A, Nanassy A, Miller S. Maintenance of locomotor abilities following Laufband (treadmill) therapy in para- and tetraplegic persons: follow-up studies. Spinal Cord. 1998;36:744–49. doi: 10.1038/sj.sc.3100670. [DOI] [PubMed] [Google Scholar]
- 28.Dobkin B, Apple D, Barbeau H, et al. Methods for a randomized trial of weight-supported treadmill training versus conventional training for walking during inpatient rehabilitation after incomplete traumatic spinal cord injury. Neurorehabil Neural Repair. 2003;17:153–67. doi: 10.1177/0888439003255508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobkin B, Apple D, Barbeau H, et al. Results of the Spinal Cord Injury Locomotor Trial (SCILT). Presented at annual joint scientific meeting of Am. Soc. Neurorehabil. and Am. Congr. Rehabil. Med; Tucson, AZ. Oct. 23.2003. [Google Scholar]
- 30.Colombo G, Joerg M, Schreier R, et al. Treadmill training of paraplegic patients using a robotic orthosis. J Rehabil Res Dev. 2000;37:693–700. [PubMed] [Google Scholar]
- 31.Bruehlmeier M, Dietz V, Leenders K, et al. How does the human brain deal with a spinal cord injury? Eur J Neurosci. 1998;10:3918–22. doi: 10.1046/j.1460-9568.1998.00454.x. [DOI] [PubMed] [Google Scholar]
- 32.Dobkin B. Spinal and supraspinal plasticity after incomplete spinal cord injury: correlations between functional magnetic resonance imaging and engaged locomotor networks. In: Seil F, editor. Progress in Brain Research. Vol. 128. Amsterdam: Elsevier; 2000. pp. 99–111. [DOI] [PubMed] [Google Scholar]
- 33.Fouad K, Pedersen V, Schwab M, et al. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Biol. 2001;11:1766–70. doi: 10.1016/s0960-9822(01)00535-8. [DOI] [PubMed] [Google Scholar]
- 34.Caiozzo VJ. Plasticity of skeletal muscle phenotype: mechanical consequences. Muscle Nerve. 2002;26:740–68. doi: 10.1002/mus.10271. [DOI] [PubMed] [Google Scholar]
- 35.Singleton J, Feldman E. Insulin-like growth factor-1inmuscle metabolism and myotherapies. Neurobiol Dis. 2001;8:541–54. doi: 10.1006/nbdi.2001.0416. [DOI] [PubMed] [Google Scholar]
- 36.Onder G, Penninx B, Balkrishnan R, et al. Relation between use of angiotensin-converting enzyme inhibitors and muscle strength and physical function in older women: an observational study. Lancet. 2002;359:926–30. doi: 10.1016/s0140-6736(02)08024-8. [DOI] [PubMed] [Google Scholar]
- 37.Scremin A, Kurta L, Gentili A, et al. Increasing muscle mass in spinal cord injured persons with a functional electrical stimulation exercise program. Arch Phys Med Rehabil. 2000;80:1531–36. doi: 10.1016/s0003-9993(99)90326-x. [DOI] [PubMed] [Google Scholar]
- 38.Peckham P, Keith M, Kilgore K, et al. Efficacy of animplanted neuroprosthesis for restoring grasp in tetraplegia: a multicenter study. Arch Phys Med Rehabil. 2001;82:1380–88. doi: 10.1053/apmr.2001.25910. [DOI] [PubMed] [Google Scholar]
- 39.Creasey G, Grill J, Korsten J, et al. An implantable neuroprosthesis for restoring bladder and bowel control to patients with spinal cord injuries: a multicenter trial. Arch Phys Med Rehabil. 2001;82:1512–19. doi: 10.1053/apmr.2001.25911. [DOI] [PubMed] [Google Scholar]
- 39a.Loeb GE, Peck RA, Moore W, Hood K. BIONTM system for distributed neural prosthetic interfaces. Med Eng Phys. 2001;23:9–18. doi: 10.1016/s1350-4533(01)00011-x. [DOI] [PubMed] [Google Scholar]
- 40.Thoroughman K, Shadmehr R. Learning of action through adaptive combination of motor primitives. Nature. 2000;407:742–47. doi: 10.1038/35037588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearson K. Neural adaptation in the generation of rhythmic behavior. Annu Rev Physiol. 2000;62:723–53. doi: 10.1146/annurev.physiol.62.1.723. [DOI] [PubMed] [Google Scholar]
- 42.Dimitrijevic M, Gerasimenko Y, Pinter M. Evidence for a spinal central pattern generator in humans. Ann NY Acad Sci. 1998;860:360–76. doi: 10.1111/j.1749-6632.1998.tb09062.x. [DOI] [PubMed] [Google Scholar]
- 43.Herman R, He J, D'Luzansky S, et al. Spinal cord stimulation facilitates functional walking in a chronic incomplete spinal cord injury. Spinal Cord. 2002;40:65–68. doi: 10.1038/sj.sc.3101263. [DOI] [PubMed] [Google Scholar]
- 44.Wolpaw J, Birbaumer N, McFarland D, et al. Brain-computer interfaces for communication and control. Clin Neurophysiol. 2002;113:767–91. doi: 10.1016/s1388-2457(02)00057-3. [DOI] [PubMed] [Google Scholar]
- 45.Reinkensmeyer D, Lum P, Winters J. Emerging technologies for improving access to movement therapy following neurologic injury. In: Winters J, Robinson C, Simpson R, Vanderheiden G, editors. Emerging and Accessible Telecommunications, Information and Healthcare Technologies. New York: IEEE Press; 2002. pp. 123–38. [Google Scholar]
- 46.McDonald J, Becker D, Sadowsky C, et al. Late recovery following spinal cord injury Case report and review of the literature. J Neurosurg. 2002;97:252–65. doi: 10.3171/spi.2002.97.2.0252. [DOI] [PubMed] [Google Scholar]
- 47.Dobkin B. Do electrically stimulated sensory inputs and movements lead to long-term plasticity and rehabilitation gains? Curr Opin Neurol. 2003;16 doi: 10.1097/01.wco.0000102622.38669.ac. In press. [DOI] [PubMed] [Google Scholar]
- 48.David S, Lacroix S. Molecular approaches to spinal cord repair. Annu Rev Neurosci. 2003;26:411–40. doi: 10.1146/annurev.neuro.26.043002.094946. [DOI] [PubMed] [Google Scholar]
- 49.Selzer M. Promotion of axonal regeneration in the injured CNS. Lancet Neurol. 2003;2:157–66. doi: 10.1016/s1474-4422(03)00322-3. [DOI] [PubMed] [Google Scholar]
- 49a.Houle JD, Tessler A. Repair of chronic spinal cord injury. Exp Neurol. 2003;182:247–60. doi: 10.1016/s0014-4886(03)00029-3. [DOI] [PubMed] [Google Scholar]
- 50.Coumans J, Lin T, Dai H, et al. Axonal regeneration and functional recoveryafter complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci. 2001;21:9334–44. doi: 10.1523/JNEUROSCI.21-23-09334.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murray M, Kim D, Liu Y, et al. Transplantation of genetically modified cells contributes to repair from spinal injury. Brain Res Rev. 2002;40:292–300. doi: 10.1016/s0165-0173(02)00211-4. [DOI] [PubMed] [Google Scholar]
- 52.Steward O, Zheng B, Tessier-Levigne M. False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. J Comp Neurol. 2003;459:1–8. doi: 10.1002/cne.10593. [DOI] [PubMed] [Google Scholar]
- 53.Bradbury E, Moon L, Popat R, et al. Chrondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–40. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 54.GrandPre T, Li S, Strittmatter S. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–51. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- 55.Dergham P, Ellezam B, Essagian C, et al. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–77. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bareyre F, Haudenschild B, Schwab M. Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci. 2002;22:7097–110. doi: 10.1523/JNEUROSCI.22-16-07097.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56a.Sicotte M, Tsatas O, Jeong S, et al. Immunization with myelin or recombinant Nogo-66/MAG in alum promotes axon regeneration and sprouting after corticospinal tract lesions in the spinal cord. Mol Cell Neurosci. 2003;23:251–63. doi: 10.1016/s1044-7431(03)00053-8. [DOI] [PubMed] [Google Scholar]
- 57.Cai D, Qiu J, Cao Z, et al. Neuronal cyclic AMP controls the developmentalloss in ability of axons to regenerate. J Neurosci. 2001;21:4731–39. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57a.Woolf CJ. No Nogo: Now where to go? Neuron. 2003;38:153–56. doi: 10.1016/s0896-6273(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 58.Chao M. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 59.Blesch A, Lu P, Tuszynski M. Neurotrophic factors, gene therapy, and neural stem cells for spinal cord repair. Brain Res Bull. 2002;57:833–38. doi: 10.1016/s0361-9230(01)00774-2. [DOI] [PubMed] [Google Scholar]
- 60.Plunet W, Kwon B, Tetzlaff W. Promoting axonal regeneration in the central nervous system by enhancing the cell body response to axotomy. J Neurosci Res. 2002;68:1–6. doi: 10.1002/jnr.10176. [DOI] [PubMed] [Google Scholar]
- 61.Novikova L, Novikov L, Kellerth JO. Survival effects of BDNF and NT-3 on axotomized rubrospinal neurons depend on the temporal pattern of neurotrophin administration. Eur J Neurosci. 2000;12:776–80. doi: 10.1046/j.1460-9568.2000.00978.x. [DOI] [PubMed] [Google Scholar]
- 62.Ming GL, Henley J, Tessier-Lavigne M, et al. Electrical activity modulates growth cone guidance by diffusible factors. Neuron. 2001;29:441–52. doi: 10.1016/s0896-6273(01)00217-3. [DOI] [PubMed] [Google Scholar]
- 63.Fournier A, Strittmatter S. Repulsive factors and axon regeneration in the CNS. Curr Opin Neurobiol. 2001;11:89–94. doi: 10.1016/s0959-4388(00)00178-1. [DOI] [PubMed] [Google Scholar]
- 64.Dickson B. Molecular mechanisms of axon guidance. Science. 2002;298:1959–64. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- 64a.Cheng H, Cao Y, Olson L. Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science. 1996;273:510–14. doi: 10.1126/science.273.5274.510. [DOI] [PubMed] [Google Scholar]
- 65.Horner P, Power A, Kempermann G, et al. Proliferation and differentiation of progenitor cells throughout the intact adult spinal cord. J Neurosci. 2000;20:2218–28. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nunes M, Roy N, Keyoung H, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–47. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- 67.Wichterle H, Lieberam I, Jessell T. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–97. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 68.McDonald J, Liu XZ, Qu Y, et al. Transplanted embryonic stem cells survive, differentiate, and promote recovery in injured rat spinal cord. Nat Med. 1999;5:1410–12. doi: 10.1038/70986. [DOI] [PubMed] [Google Scholar]
- 69.Cao Q, Howard R, Dennison J, et al. Differentiation of engrafted neuronal-restricted precursor cells is inhibited in the traumatically injured spinal cord. Exp Neurol. 2002;177:349–59. doi: 10.1006/exnr.2002.7981. [DOI] [PubMed] [Google Scholar]
- 69a.Lu P, Jones LL, Snyder EY, Tuszynski M. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003;181:115–29. doi: 10.1016/s0014-4886(03)00037-2. [DOI] [PubMed] [Google Scholar]
- 70.Wu P, Tarsenko Y, Gu Y, et al. Region-specific generation of cholinergic neurons from fetal human neural stem cells grafted in adult rat. Nat Neurosci. 2002;5:1271–78. doi: 10.1038/nn974. [DOI] [PubMed] [Google Scholar]
- 71.Takami T, Oudega M, Bates M, et al. Schwann cell but not olfactory ensheathing glia transplants improve hindlimb locomotor performance in the moderately contused adult rat thoracic spinal cord. J Neurosci. 2002;22:6670–81. doi: 10.1523/JNEUROSCI.22-15-06670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Field P, Raisman G. Regeneration of adult rat corticospinal axons induced by transplanted olfactory ensheathing cells. Neuroscience. 1998;18:10514–24. doi: 10.1523/JNEUROSCI.18-24-10514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramon-Cueto A, Cordero M, Santos-Benito F, et al. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron. 2000;25:425–36. doi: 10.1016/s0896-6273(00)80905-8. [DOI] [PubMed] [Google Scholar]
- 74.Li Y, Decherchi P, Raisman G. Transplantation of olfactory ensheathing cells into spinal cord lesions restores breathing and climbing. J Neurosci. 2003;23:727–31. doi: 10.1523/JNEUROSCI.23-03-00727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y, Kim D, Himes T. Transplants of fibroblasts genetically modified to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci. 1999;19:4370–87. doi: 10.1523/JNEUROSCI.19-11-04370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McTigue D, Horner P, Stokes B, et al. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci. 1998;18:5354–65. doi: 10.1523/JNEUROSCI.18-14-05354.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akiyama Y, Radtke C, Kocsis J. Remyelination of the rat spinal cord by transplantation of identified bone marrow stromal cells. J Neurosci. 2002;22:6623–30. doi: 10.1523/JNEUROSCI.22-15-06623.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akiyama Y, Honmou O, Kato T, et al. Transplantation of clonal neural precursor cells derived from adult human brain establishes functional peripheral myelin in the rat spinal cord. Exp Neurol. 2001;167:27–39. doi: 10.1006/exnr.2000.7539. [DOI] [PubMed] [Google Scholar]
- 79.Pluchino S, Quattrini A, Brambilla E, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:686–94. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- 80.Keirstead H, Hur T, Rogister B. Polysialylated neural cell adhesion molecule-positive CNS precursors generate both oligodendrocytes and Schwann cells to remyelinate the CNS after transplantation. J Neurosci. 1999;19:7529–36. doi: 10.1523/JNEUROSCI.19-17-07529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80a.Nieto J, Hoang TX, Tillakaratne NJK, Havton LA. Lumbosacral spinal cord neurons reinnervate implanted avulsed ventral roots in a cauda equina injury injury and repair model. Soc Neurosci Abstr. 2002;31:852. [Google Scholar]
- 81.Carlstedt T, Anand P, Hallin R, et al. Spinal nerve root repair and reimplantation of avulsed ventral roots into the spinal cord after brachial plexus injury. J Neurosurg. 2000;93:237–47. doi: 10.3171/spi.2000.93.2.0237. [DOI] [PubMed] [Google Scholar]
- 82.Cullheim S, Wallquist W, Mammarberg H, et al. Properties of motoneurons underlying their regenerative capacity after axon lesions in the ventral funiculus or at the surface of the spinal cord. Brain Res Rev. 2002;40:309–16. doi: 10.1016/s0165-0173(02)00213-8. [DOI] [PubMed] [Google Scholar]
- 83.Navarro X, Valero A, Gudino G. Ensheathing glia transplants promote dorsal root regeneration and spinal reflex restitution after multiple lumbar rhizotomy. Ann Neurol. 1999;45:207–15. doi: 10.1002/1531-8249(199902)45:2<207::aid-ana11>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 84.Pascual J, Gudino-Cabrera G, Insausti R, et al. Spinal transplants of olfactory ensheathing cells promote axon regeneration and bladder activity after bilateral lumbosacral dorsal rhizotomy in the adult rat. J Urol. 2002;167:1522–26. [PubMed] [Google Scholar]
- 85.Ji R, Woolf C. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2000:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 86.Ikeda H, Heinke B, Ruscheweyh R, et al. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–40. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- 87.Novikov L, Novikova L, Mosahebi A, et al. A novel biodegradable implant for neuronal rescue and regeneration after spinal cord injury. Biomaterials. 2002;23:3369–76. doi: 10.1016/s0142-9612(02)00037-6. [DOI] [PubMed] [Google Scholar]
- 88.Woerly S. Restorative surgery of the central nervous system by means of tissue engineering using NeuroGel implants. Neurosurg Rev. 2000;23:59–77. doi: 10.1007/pl00021694. [DOI] [PubMed] [Google Scholar]
- 89.Tillakaratne N, Hoang T, de Leon R, et al. Use-dependent modulation of inhibitory capacity in the feline lumbar spinal cord. J Neurosci. 2002;22:3130–43. doi: 10.1523/JNEUROSCI.22-08-03130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Capaday C. The special nature of human walking and its neural control. Trends Neurosci. 2002;25:370–76. doi: 10.1016/s0166-2236(02)02173-2. [DOI] [PubMed] [Google Scholar]
- 91.Chen X, Carp J, Chen L, Wolpaw J. Corticospinal tract transection prevents operantly conditioned H-reflex increase in rats. Exp Brain Res. 2002;144:88–94. doi: 10.1007/s00221-002-1026-8. [DOI] [PubMed] [Google Scholar]
- 92.Blight A. Animal models of spinal cord injury. Top Spinal Cord Inj Rehabil. 2000;6:1–13. [Google Scholar]
- 93.Benowitz L, Goldberg D, Madsen J, et al. Inosine stimulates extensive axon collateral growth in the rat corticospinal tract after injury. Proc Natl Acad Sci USA. 1999;96:13486–90. doi: 10.1073/pnas.96.23.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramer M, Harper G, Bradbury E. Progress in spinal cord research. Spinal Cord. 2000;38:449–72. doi: 10.1038/sj.sc.3101055. [DOI] [PubMed] [Google Scholar]
- 95.Bracken M, Shepard M, Holford T, et al. Results of the third acute spinal cord injury randomized controlled trial. J Neurosurg. 1998;89:699–706. doi: 10.3171/jns.1998.89.5.0699. [DOI] [PubMed] [Google Scholar]
- 96.Geisler F, Coleman W, Grieco G, et al. The Sygen multicenter acute spinal cord injury study. Spine. 2001;26:S87–S98. doi: 10.1097/00007632-200112151-00015. [DOI] [PubMed] [Google Scholar]