

Abstract
Purpose
Rapid premature release of lipophilic drugs from liposomal lipid bilayer to plasma proteins and biological membranes is a challenge for targeted drug delivery. The purpose of this study is to reduce premature release of lipophilic short-chain ceramides by encapsulating ceramides into liposomal aqueous interior with the aid of poly( lactic-coglycolicacid) (PLGA).
Methods
BODIPY FL labeled ceramide (FL-ceramide) and BODIPY-TR labeled ceramide (TR-ceramide) were encapsulated into carboxy-terminated PLGA nanoparticles. The negatively charged PLGA nanoparticles were then encapsulated into cationic liposomes to obtain PLGA/liposome hybrids. As a control, FL-ceramide and/or TR ceramide co-loaded liposomes without PLGA were prepared. The release of ceramides from PLGA/liposome hybrids and liposomes in rat plasma, cultured MDA-MB-231 cells, and rat blood circulation was compared using fluorescence resonance energy transfer (FRET) between FL-ceramide (donor) and TR-ceramide (acceptor).
Results
FRET analysis showed that FL-ceramide and TR-ceramide in liposomal lipid bilayer were rapidly released during incubation with rat plasma. In contrast, the FL-ceramide and TR-ceramide in PLGA/liposome hybrids showed extended release. FRET images of cells revealed that ceramides in liposomal bilayer were rapidly transferred to cell membranes. In contrast, ceramides in PLGA/liposome hybrids were internalized into cells with nanoparticles simultaneously. Upon intravenous administration to rats, ceramides encapsulated in liposomal bilayer were completely released in 2 minutes. In contrast, ceramides encapsulated in the PLGA core were retained in PLGA/liposome hybrids for 4 hours.
Conclusions
The PLGA/liposome hybrid nanoparticles reduced in vitro and in vivo premature release of ceramides and offer a viable platform for targeted delivery of lipophilic drugs.
Keywords: PLGA, liposome, ceramide, FRET, premature release
1. Introduction
Intravenously administered liposomal formulations have been successfully developed for targeted drug delivery. Weakly basic amphiphilic drugs such as doxorubicin which are remotely loaded into the aqueous interior of liposomes show sustained in vivo release(1). However, rapid premature release of lipophilic drugs from liposomal lipid bilayer to plasma proteins and biological membranes is still a challenge for targeted drug delivery (2). Many lipophilic drugs encapsulated in liposomes such as verteporfin(3), verapamil(4), SN-38(5), vinblastine and vinorelbine(6) have shown rapid premature release upon intravenous administration. Liposome is used as a solubilization carrier of a large class of lipophilic drugs for parenteral administration but is unable to efficiently deliver drug molecules to target tissues(1). The drug location in liposomes is critical for drug release rate and pharmacokinetics. For example, doxorubicin in liposomal bilayer showed a half-life < 0.2 hour in small rodents while doxorubicin in liposomal aqueous interior has a much longer in vivo half-life (20 hours) (7). Enlighten by the successful example of Doxil in which doxorubicin is loaded into the aqueous interior, we hypothesize that encapsulation of lipophilic drugs into liposomal interior could reduce premature drug release.
Ceramides are endogenous sphingolipids that have been found to play a crucial role as mediators of apoptotic signaling(8). Short-chain ceramides and long-chain ceramides have been demonstrated to have different physiological effects on cells (9). For example, short-chain C6-ceramide (IC50 3–14 μM) was reported to be more cytotoxic than long-chain C16-ceramide (IC50 > 100 μM) in MDA435/LCC6 human breast cancer cells and J774 mouse macrophage cells (10). Synthesized exogenous short-chain ceramide is cytotoxic to various cancer cells and inhibits tumor growth in mice (10, 11). However, the therapeutic effectiveness of short-chain ceramide is limited by the incapability of targeted delivery to tumor tissues. Short-chain ceramide was loaded into liposomal bilayer and its anti-tumor activity was evaluated in mouse xenograft tumor model (11–13). Unfortunately, upon intravenous administration, rapid release of ceramide from liposomal bilayer was observed, which compromised systemic delivery efficiency and specificity (4, 14). The objective of this study is to encapsulate lipophilic short-chain ceramides into the aqueous interior of liposomes with the aid of poly( lactic-co-glycolic acid) (PLGA) and to reduce premature release of ceramides from PLGA/liposome hybrids. Fluorescence resonance energy transfer (FRET) between BODIPY FL labeled ceramide (FL-ceramide, FRET donor) and BODIPY TR labeled ceramide (TR-ceramide, FRET acceptor) was employed to monitor the release of ceramides from PLGA/liposome hybrid nanoparticles.
2. Materials and methods
2.1 Materials
The lipids 1,2-distearyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-DSPE), and 3β-[N-(N′, N′-Dimethylaminoethane)-carbamoyl]Cholesterol Hydrochloride (DC-cholesterol) were purchased from Avanti Polar Lipids (Alabaster, AL). BODIPY FL labeled C5-ceramide (FL-ceramide) and BODIPY-TR labeled C5-ceramide (TR-ceramide) were obtained from Invitrogen (Carlsbad, CA). Poly(D, L-lactide–co–glycolide) (50/50) with terminal carboxyl acid (PLGA, 0.65 dL/g, MW 30 kDa) was obtained from Birmingham Polymers (Pelham, AL). Poly(vinyl alcohol) (PVA) (MW 30–70 kDa) and other chemicals were purchased from Sigma (St. Louis, MO). HiTrap SP FF cation exchange column was supplied by GE Life Sciences (Piscataway, NJ).
2.2 Preparation of ceramide-loaded PLGA nanoparticles
As shown in Figure 1A, ceramide-loaded PLGA nanoparticles were prepared using a nanoprecipitation method (15). Briefly, 60 mg of PLGA was completely dissolved in 3 ml tetrahydrafuran (THF). FL-ceramide (0.96 mg) and TR-ceramide (1.12 mg) were added to the PLGA solution and stirred for 5 min. This solution was drop wise added to 6 ml of aqueous solution containing 1% (w/v) of PVA over a period of 10 min on a magnetic stir plate operated at 800 rpm. This suspension was sonicated using a Misonix probe sonicator (Qsonica, Newtown, CT) with output amplitude of 20% for 5 min. Then, the suspension was stirred at room temperature for 24 h to remove THF completely. Large PLGA aggregates and ceramide aggregates were removed by centrifugation at 2655 g for 10 min. The supernatant was subjected to ultracentrifugation (Beckman L-80 ultracentrifuge equipped with a Ti-70 rotor) at 61, 700 g for 3 h at 20°C. The obtained PLGA nanoparticles were resuspended in 1.5 mL of PBS (pH 7.4) for PLGA/liposome hybrid preparation. Similarly, PLGA nanoparticles containing only FL-ceramide and only TR-ceramide were prepared using the same method.
Figure 1.

Preparation of FL-ceramide and TR-ceramide co-loaded (A) PLGA/liposome hybrids and (B) liposomes
2.3 Encapsulation of PLGA nanoparticles into liposomes
PLGA nanoparticles were passively loaded into liposomes using thin lipid film hydration method (Figure 1A). Briefly, DSPC (19.8), PEG-DSPE (14 mg) and DC-cholestrol (10.7 mg) were dissolved in chloroform at the required molar ratio (50: 10: 40), and dried to a thin film under a stream of nitrogen gas. Subsequently, the residual organic solvent was removed in a high vacuum for 6 h. The lipid films were hydrated in 1.5 mL of above freshly prepared PLGA nanoparticle suspension containing FL-ceramide and TR-ceramide by stirring at 65 °C for 2 h. The hydrated multilamellar vesicle suspension was extruded 8 times through two stacked 200 nm polycarbonate membranes at 65 °C using a low volume microfluidizer (Mcrofluidics, Newton, MA). Similarly, FL-ceramide and TR-ceramide individually loaded PLGA/liposome hybrids were prepared using the same method. As a control, FL-ceramide and TR-ceramide co-loaded liposomes without PLGA were also prepared (Figure 1B). FL-ceramide (0.12 mg) and TR-ceramide (0.14 mg) were dissolved in lipid chloroform solution containing DSPC (19.8 mg), PEG-DSPE (14 mg) and DC-cholestrol (10.7 mg). The lipid film was hydrated in 1.5 mL of PBS without PLGA. The rest procedures were the same as that for PLGA/liposome hybrid preparation.
2.4 Separation of unencapsulated PLGA nanoparticles
Unencapsulated PLGA nanoparticles and PLGA/liposome hybrids were separated using a HiTrap cation exchange column. After sample loading, the column was washed with 10 mL of phosphate buffer (pH 8, containing 44 mM Na2HPO4 and 2 mM NaH2PO4) to remove unencapsulated PLGA nanoparticles and then washed with 5 mL of elution buffer (44 mM Na2HPO4, 2 mM NaH2PO4 and 500 mM NaCl). The eluted fraction was subjected to buffer exchange by dialysis against PBS.
2.5 Characterization of nanoparticles
After dilution with distilled water, the hydrodynamic sizes of nanoparticles were determined by using a ZetaSizer (Malvern Instruments, Malvern, UK) based on a dynamic light scattering (DLS) technique and reported as intensity-weighted average ± standard deviation for three replicate samples. Zeta potentials of nanoparticles were also measured using the ZetaSizer. The samples were appropriately diluted with 1 mM HEPES buffer (adjusted to pH 7.4 with 1 M HCl) in order to maintain a constant ionic strength. The zeta-potentials were reported as the mean value ± standard deviation for three replicate samples. Fluorescence spectra of nanoparticles were measured on a Safire II microplate reader (Tecan Group Ltd, Morrisville, NC) with an excitation at 480 nm and an emission scan 500–700 nm.
PLGA nanoparticles and PLGA/liposome hybrids were characterized for size and morphology using a Hitachi 7650 transmission electron microscope (TEM) (Tokyo, Japan) operating at 80 kV. Samples for TEM analysis were dispersed in pure water and dropped onto a carbon-coated copper grid negatively stained with 2% phosphotungstic acid. The samples were dried in air at room temperature before loaded in the microscope.
To determine the contents of FL-ceramide and TR-ceramide in PLGA nanoparticles, three batches of FL-ceramide and TR-ceramide co-loaded PLGA nanoparticle suspensions were prepared following the same method described above. After ultracentrifugation at 61,700 g for 3 h, the PLGA nanoparticles were dried under vacuum and 10 mg of PLGA nanoparticles were dissolved in 20 mL of acetonitrile to release ceramides. The concentrations of FL-ceramide and TR-ceramide were determined using fluorescence intensity standard curves of FL-ceramide and TR-ceramide established in acetonitrile.
To estimate encapsulation efficiency of PLGA nanoparticles, the amount of PLGA nanoparticles was determined before and after cation exchange column purification. Briefly, PLGA nanoparticle suspensions were prepared following the method described above. After ultracentrifugation at 61,700 g for 3 h, the PLGA nanoparticles were dried under vacuum and weighed to obtain the mass of total PLGA nanoparticles. The PLGA nanoparticles were suspended in 1.5 mL of PBS and encapsulated into liposomes using the method described above. After removing unencapsulated PLGA nanoparticles using a cation exchange column, the PLGA/liposome hybrids were lysed with 1% (v/v) Triton X-100 and subsequently subjected to ultracentrifugation at 61,700 g for 3 h. The precipitates were washed with water and dried under vaccum to obtain the encapsulated PLGA nanoparticles. The mass ratio of encapsulated PLGA nanoparticles and total PLGA nanoparticles was the estimated encapsulation efficiency. This experiment was repeated in triplicate.
2.6 In vitro ceramide release
Time-resolved fluorescence study was performed to monitor ceramide release in PBS, DSPC vesicle solution and rat plasma. DSPC vesicles (2 mg/mL) were freshly prepared in PBS using thin film hydration method. 10 μL of liposomes containing both FL-ceramide and TR-ceramide were added to 100 μL of PBS, DSPC vesicle solution and rat plasma and incubated at 37 °C. Fluorescence spectra (excitation at 480 nm and an emission scan 500–700 nm) were recorded every minute over 10 minutes. Similarly, 10 μL of FL-ceramide and TR-ceramide co-loaded PLGA/liposome hybrids were incubated in 100 μL of PBS, DSPC vesicle solution and rat plasma at 37 °C and fluorescence spectra were recorded every 10 minutes over 100 minutes. FRET ratio at each time point was calculated as R = I632/(I518 + I632), where I632 and I518 are the fluorescence intensities at 632 nm and 518 nm, respectively(16).
2.7 Cell culture and FRET imaging
A breast cancer cell line MDA-MB-231 obtained from American Type Culture Collection (ATCC, Rockville, MD) was cultured in RPMI medium 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen Life Technologies, Carlsbad, CA). The cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C. Cells were cultured on 8-well Lab-Tek glass chamber slides (Thermo Fisher Scientific, Rochester, NY) for confocal microscope imaging. 1×104 cells per well were incubated for 2 days to allow cell adherence. 20 μL of liposomes or PLGA/liposome hybrids were added to cells in 0.4 ml of serum-free RPMI medium and incubated at 37°C for 4 hours before imaging.
The cells were washed with PBS 3 times and then FRET images were acquired using a Zeiss LSM 510 META confocal microscope (Oberkochen, Germany) with a 40X oil-immersion objective. Fluorescence of FL-ceramide was acquired under 488 nm excitation and 555–530 nm emission. Fluorescence of TR-ceramide was obtained under 543 nm excitation and 585–615 emission. FRET fluorescence was recorded under 488 nm excitation and 585 – 615 nm emission. The images were processed using software Olympus FV1000 Viewer (Ver.2.0c).
2.8 In vivo release study
The animal care and procedures were performed according to a protocol approved by the by the University Committee for the Use and Care of Animals (UCUCA) at the University of Michigan. Six double jugular catheterized female Sprague-Dawley (SD) rats (60 days old, 270–310 g) were purchased from Charles River Laboratories, Inc. (Raleigh, NC). One blood sample (100 μL) was collected from each rat before dose administration to measure the background fluorescence at 518 nm and 631 nm (excitation 480 nm). Dosing solutions of liposomes and PLGA/liposome hybrids were administered (equivalent to 20 μg/rat TR-ceramide) via the left jugular vein catheter (n = 3). Blood samples (100 μL) were collected via the right jugular catheters at 2, 10, 30, 60, 120, and 240 min postdose and placed in a lithium heparinized 96-well plate. The fluorescence intensities at 518 nm and 631 nm (excitation 480 nm) of each blood sample were measured immediately after collection using a Safire2 microplate reader. The FRET ratio of each blood sample was calculated as R = I632′/(I518′ + I632′), where I632′ and I518′ are the background subtracted fluorescence intensities at 632 nm and 518 nm, respectively.
3. Results
3.1 Preparation and characterization of PLGA/liposome hybrids
To load ceramides into the aqueous interior of liposomes, PLGA/liposome hybrid nanoparticles were prepared in two steps: preparation of PLGA nanoparticles and encapsulation of PLGA into liposomes (Figure 1A). The DLS particle sizes and zeta potentials of PLGA nanoparticles, liposomes, and PLGA/liposome hybrids were shown in Table 1. By using 1% (w/v) of PVA as a stabilizer, the average DLS particle size of PLGA particles was controlled to be 115 nm, consistent with the particle size observed in TEM image (Figure 1B). Due to the PLGA terminal carboxyl acid, PLGA nanoparticles showed an average negative surface charge of −28.4 mV in pH 7.4 HEPES buffer. Liposomes loaded with FL-ceramide and TR-ceramide showed an average DLS size of 130 nm and a positive zeta potential of 36.2 mV, which is attributed to the presence of 40 mol% positively charged DC-cholesterol. The encapsulation of PLGA nanoparticles into liposomes increased the average DLS sizes to 181–196 nm and decrease zeta potentials to 26.1–31.5 mV. The TEM image of FL-ceramide and TR-ceramide co-loaded PLGA/liposome hybrids showed the entrapment of PLGA particles in the aqueous interior of liposomes (Figure 2D).
Table 1.
Particle sizes and zeta potentials of nanoparticles
| Samples | Particle size ± SE (nm) | Zeta potential ± SE (mV) |
|---|---|---|
| PLGA nanoparticles | 114.5 ± 23.7 | −28.4 ± 3.5 |
| FL-ceramide/TR-ceramide co-loaded liposomes | 130.1 ± 8.9 | +36.2 ± 2.9 |
| FL-ceramide loaded PLGA/liposome hybrid | 195.6 ± 14.8 | +31.5 ± 4.4 |
| TR-ceramide loaded PLGA/liposome hybrid | 180.7 ± 13.0 | +26.1 ± 3.8 |
| FL-ceramide/TR-ceramide co-loaded | 187.9 ± 16.7 | +28.6 ± 5.3 |
| PLGA/liposome hybrid | ||
Figure 2.

Particle size and morphology of nanoparticles: (A) Dynamic light scattering (DLS) size measurement of FL-ceramide and TR-ceramide co-loaded liposomes; (B) TEM image of PLGA nanoparticles; (C) DLS size measurement and (D) TEM image of FL-ceramide and TR-ceramide co-loaded PLGA/liposome hybrids.
The average contents of FL-ceramide and TR-ceramide in PLGA nanoparticles were determined as 0.8 ± 0.1% and 0.9 ± 0.1% (w/w, n = 3), respectively. Unencapsulated PLGA nanoparticles and PLGA/liposome hybrids were separated using cation exchange chromatography. After removal of unencapsulated PLGA nanoparticles, 19.5 ± 4.7% (n = 3) of PLGA nanoparticles remained in the PLGA/liposome hybrid fraction, which was the average encapsulation efficiency of PLGA nanoparticles.
As shown in the supplementary Figure S1, due to the overlap of FL-ceramide (donor) emission spectrum and TR-ceramide (acceptor) absorption spectrum, FRET effect is expected if FL-ceramide and TR-ceramide are co-loaded into liposomes or PLGA/liposome hybrids (Distance between two ceramides is less than 10 nm)(17). Figure 3A and 3B showed the fluorescence spectra of ceramide(s)-loaded liposomes and PLGA/liposome hybrids suspended in PBS, respectively. When FL-ceramide and TR-ceramide co-loaded liposomes were excited at 480 nm, maximum FRET fluorescence and FL-ceramide fluorescence were detected at 632 nm and 518 nm, respectively (Figure 3A, solid line). The FRET ratio (I632/(I518 + I632) = 0.63) depends on the distance between FL-ceramide and TR-ceramide. The release of ceramides from liposomes will lead to a decrease of FRET ratio(16). In contrast, no FRET signal was detected from FL-ceramide loaded liposomes or TR-ceramide loaded liposomes. Similarly, FRET signal was detected from FL-ceramide and TR-ceramide co-loaded PLGA/liposome hybrids (FRET ratio = 0.45) but not FL-ceramide or TR-ceramide loaded PLGA/liposome hybrids (Figure 3B).
Figure 3.
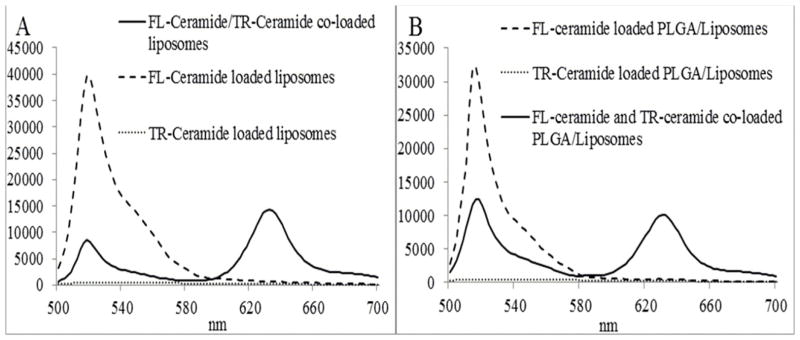
Fluorescence spectra of ceramide(s) loaded (A) liposomes and (B) PLGA/liposome hybrids in PBS. (Excitation 480 nm)
3.2 In vitro release in DSPC vesicle solution and rat plasma
The biological membranes in blood cells and tissues and lipoproteins in plasma have been reported to be a driving force to promote drug release from liposomes (4, 14). The release of ceramides from liposomes and PLGA/liposome hybrids was evaluated in DSPC vesicle solution (2 mg/mL) and rat plasma (Figure 4). Consistent with a previous report (4), rapid transfer of ceramides from liposomes to DSPC vesicles was observed. The FRET ratio decreased to a steady state of 0.1 in 5 minutes (Figure 4A), indicating a complete release of ceramides. The FRET ratio 0.1 was attributed to the recovery of FRET between FL-ceramide and TR-ceramide in the lipid bilayer of DSPC vesicles. Similarly, the release of ceramides in rat plasma led to a steady-state FRET ratio of 0.05 at 4 min. The FRET ratio 0.05 might be caused by the FRET between FL-ceramide and TR-ceramide bound to the same plasma protein. In contrast, the FRET ratio of PLGA/liposome hybrids gradually decreased in DSPC vesicle solution and rat plasma and did not reach a steady state even after incubation for 100 minutes (Figure 4B). After 100 minute incubation, the FRET ratio of PLGA/liposome hybrids in DSPC vesicle solution and rat plasma was 0.38 and 0.26, respectively, indicating an incomplete release of ceramides. The release of ceramides from PLGA/liposome hybrids did not follow zero-order kinetics. Due to the interaction between PLGA core and liposomal bilayer, a fraction of ceramides might have transferred from PLGA particle to lipid bilayer prior to incubation. The initial fast decrease of FRET ratio was likely due to the rapid release of ceramides from liposomal bilayer. After initial fast release, the release of ceramides was limited by the diffusion rate of ceramide in PLGA matrix and the degradation rate of PLGA.
Figure 4.
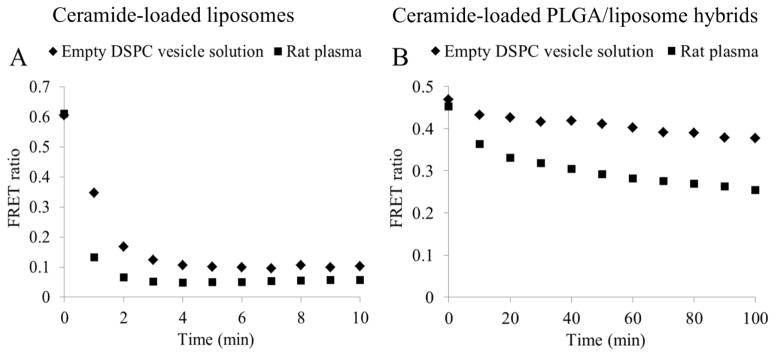
Release of ceramides from (A) liposomes and (B) PLGA/liposome hybrids incubated in DSPC vesicle solution and rat plasma
3.3 Incorporation of PLGA changed ceramide cellular uptake pathway
FRET microscope imaging revealed different ceramide cellular uptake pathways of liposomes and PLGA/liposome hybrids (Figure 5). Columns 1–4 showed bright-field images, TR-ceramide fluorescence, FL-ceramide fluorescence, and FRET fluorescence, respectively. For the cells treated with FL-ceramide and TR ceramide co-loaded PLGA/liposome hybrids, fluorescence of TR-ceramide (A2) and FL-ceramide (A3) was mainly detected from intracellular organelles. FRET signals were observed from intracellular organelles, suggesting that a majority of ceramides remained in PLGA particles and were internalized in to cells with PLGA/liposome hybrids simultaneously. In contrast, for cells treated with FL-ceramide and TR ceramide co-loaded liposomes, no remarkable FRET signals were detected (B4). Intensive fluorescence of TR-ceramide (B2) and FL-ceramide (B3) was detected on the cell membranes, suggesting that ceramides were transferred from liposomes to cell membrane and then internalized into cells. Cells treated with mixed FL-ceramide loaded PLGA/liposome hybrids and TR ceramide loaded PLGA/liposome hybrids did not show FRET signals (C4). Fluorescence of TR-ceramide (C2) and FL-ceramide (C3) was mainly observed in intracellular organelles, suggesting that a fraction of FL-ceramide and TR-ceramide remained in separate PLGA/liposome hybrids and were internalized into cells separately. In addition, rapid inter-liposome exchange of FL-ceramide and TR-ceramide was observed in PBS and serum-free RPMI medium (data not shown). Hence, cell incubation with mixed FL-ceramide liposomes and TR ceramide liposomes was not conducted.
Figure 5.

Images of MDA-MB-231 cells treated with FL-ceramide and TR ceramide co-loaded PLGA/liposome hybrids (A1–A4), FL-ceramide and TR ceramide co-loaded liposomes (B1–B4), and mixed FL-ceramide loaded PLGA/liposome hybrids and TR ceramide loaded PLGA/liposome hybrids (C1–C4, molar ratio of FL-ceramide and TR-ceramide = 1:1).
3.4 Incorporation of PLGA decreased in vivo release of ceramides
The release of ceramides in rat blood circulation was monitored by measuring FRET ratios of collected blood samples. Following i.v. administration of FL-ceramide and TR-ceramide co-loaded liposomes, the blood FRET ratio rapidly decreased from 0.63 to 0.04 and 0.02 in 2 minutes and 10 minutes respectively (Figure 6, diamonds). Steady-state FRET ratios (0.01–0.02) were observed between 10 – 240 minutes postdose which were attributed to the FRET between FL-ceramide and TR-ceramide bound to serum proteins or ceramides distributed into blood cell membranes. For the rats treated with FL-ceramide and TR-ceramide co-loaded PLGA/liposome hybrids, the blood FRET ratio showed a decrease from 0.45 to 0.26 in 2 minutes and then gradually decreased to 0.02 at 4 hours (Figure 6, squares), indicating a controlled release of ceramides within 4 hours. Consistent with in vitro release in DSPC vesicle solution and rat plasma, a rapid initial decrease of FRET ratio from 0.45 to 0.26 was observed in vivo.
Figure 6.

Rat blood FRET ratio-time profile after intravenous administration of FL-ceramide and TR-ceramide co-loaded liposomes and PLGA/liposome hybrids (n = 3).
4. Discussion
Ceramides have emerged as important intracellular signalling molecules which can mediate diverse cellular effects, such as cell apoptosis. Targeted delivery of exogenous ceramide to cancer cells is one of the therapeutic strategies to overcome chemo-resistance of cancer cells and induce cancer cell apoptosis(18). To be delivered to target tissues, short-chain ceramides have been encapsulated into liposomal bilayer for systemic administration (11, 12, 19, 20). However, upon intravenous administration, rapid release of ceramide was observed due to the sink effects of plasma proteins and a large pool of biological membranes (4, 14). To reduce the premature release, in this study, ceramides were encapsulated into PLGA nanoparticles which were entrapped in liposomal aqueous interior. Similar PLGA/liposome core-shell hybrid nanoparticles have been developed previously (21–26). It was reported that the lipid bilayer shell formed a barrier which decreased the diffusion and drug release from polymeric cores (22, 24). Compared with ceramides in conventional liposomal bilayers, ceramides in PLGA/liposome hybrids has two additional release barriers: 1) the diffusion processes in PLGA matrix and liposomal internal aqueous phase and 2) the degradation of PLGA, which might explain the gradual release of ceramides from PLGA/liposome hybrids.
The location of encapsulated ceramides in PLGA/liposome hybrids is critical for their extended release. One challenge for preparation of ceramide loaded PLGA/liposome hybrids is the selective encapsulation of ceramide into liposomal interior but not lipid bilayer. Single-step and one-pot synthesis approaches of PLGA/lipid hybrids have been previously reported (25, 27). In this study, however, ceramide-loaded PLGA/liposome hybrids prepared using a single-step approach did not show a release advantage compared with liposomal formulation (data not shown), suggesting that ceramides were mainly encapsulated into liposomal bilayer instead of PLGA particles. Hence, a two-step approach was used to prepare ceramide loaded PLGA particles and subsequently to encapsulate PLGA particles into liposomes.
Another challenge for PLGA/liposome hybrid preparation is the encapsulation efficiency (EE) of PLGA nanoparticles. Hydrophilic drugs can be encapsulated into liposomes using a passive loading approach, where EE is usually very low. The EE of passive loading depends on lipid concentration, media conditions, liposome size and lamellarity (28). Among them, lipid concentration and liposome size are the two most critical variables in determining drug EE. Hence, a small volume of aqueous suspension (1.5 mL) with a very high concentration of PLGA particles was used to hydrate lipid film. The small volume of the suspension resulted in a high lipid concentration and thus an increased EE, while the high concentration of PLGA particles increased the amount of encapsulated PLGA particles. Using a mathematical model developed by Xu and Burgess (http://www.liposomemodel.com/) and the following physicochemical parameters of PLGA/liposome hybrids (particle size 187.9 ± 16.7 nm, DSPC bilayer thickness 5.1 nm, average molecular surface area of DSPC/DC-cholesterol in PBS buffer 45.18 À2, a total concentration of DSPC and DC-cholesterol 30 mM, and sample volume 1.5 mL)(28), the EE of PLGA particles is predicted as 11.67%, which is lower than the experimental value (19.5 ± 4.7%). This difference could be explained by the electrostatic interaction between negatively charged PLGA particles and positively charged lipid film (DC-cholestrol), which facilitated the encapsulation of PLGA particles. Previous studies also revealed that electrostatic interactions between drugs and liposomes affected drug EE (29, 30). In future studies, efficient approaches for improving EE of PLGA particles include increasing the size of liposomes and enhancing electrostatic interaction between PLGA particles and lipids.
The FRET ratio is an indicator of the distance between FL-ceramide and TR-ceramide. In this study, the decrease of FRET ratio was utilized to monitor the release of ceramides. Both the current FRET method and previously reported dual radio-labeling method consistently revealed rapid in vitro and in vivo release of ceramides from conventional liposome formulation (4, 14). However, the FRET method provided a more accurate release rate of ceramides than the conventional dual radio-labeling method in which ceramide and liposome were labeled with two different radioactive elements separately. The steady-state FRET ratios (0.01–0.02) detected between 10 – 240 minutes postdose indicated a complete release of ceramides from liposomes within 10 minutes (Table 2). In contrast, rat pharmacokinetic studies of radio-labeled C6-ceramide showed 15.14% and 2.66–3.54% of injected ceramide in rat plasma at 2 min and 60 min postdose, indicating a period of release longer than 10 minutes. The period of ceramide release was overestimated in the radio-labeling pharmacokinetic studies because the radio- labeling method was unable to distinguish encapsulated ceramides and plasma protein-bound ceramides. The radioactivity detected in rat plasma at 60 and 120 minutes was likely attributed to plasma protein-bound ceramide instead of encapsulated ceramide. The long retention of ceramide in plasma demonstrated in radio-labeling pharmacokinetic studies was likely due to the redistribution of ceramide from tissues and blood cells to plasma.
Table 2.
In vivo release profiles of liposome ceramides determined by FRET study and conventional radio-labeling pharmacokinetic studies.
Besides dual radio-labeling method, LC-MS/MS is a very useful tool for quantitation of total ceramide in cells and plasma samples (31–33). However, similar to dual radio-labeling method, the published LC-MS/MS methods are unable to distinguish released free ceramide and encapsulated ceramide, making it difficult to monitor the release of ceramide in cells and blood stream. In contrast, the FRET method is able to detect the release of FL-ceramide and TR- ceramide in real-time. Hence, separation of release ceramide and encapsulated ceramide is unnecessary.
In this study, different in vitro and in vivo release rates of ceramides from PLGA/liposome hybrids were observed. Ceramide-loaded PLGA/liposome hybrids showed a faster decrease of FRET ratios in rats compared with in vitro plasma incubation. The different in vitro and in vivo release rates are mainly caused by the presence of blood cells and endothelial cells in rats. The large volume of cell membranes in rats accelerates the release of ceramides from PLGA/liposome hybrids. In addition, in vitro incubation is a closed and static system while a rat is an open and dynamic system. The dynamic blood flow and rapid distribution of ceramides to tissues increase the release of ceramides.
5. Conclusions
The developed PLGA/liposome hybrid nanoparticles showed gradual in vitro and in vivo release of ceramides, demonstrating advantages over conventional liposomal formulation. According to various requirements and purposes of drug delivery, the rate of drug release from PLGA/liposome hybrid nanoparticles can be further engineered by changing PLGA particle size, PLGA degradation, and the ratio of lactide to glycolide in PLGA polymer. The PLGA/liposome hybrid nanoparticles offer a viable platform for controlled release and targeted delivery of lipophilic drugs.
Supplementary Material
Acknowledgments
This work was partially supported by the National Institutes of Health (RO1 CA120023 and R21 CA143474); University of Michigan Cancer Center Research Grant (Munn); and University of Michigan Cancer Center Core Grant to DS.
Contributor Information
Peng Zou, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI 48108, United States.
Stephan T. Stern, Nanotechnology Characterization Laboratory, SAIC-Frederick, Inc., NCI-Frederick, Frederick, Maryland 21702, United States
Duxin Sun, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI 48108, United States.
References
- 1.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. Journal of pharmaceutical sciences. 2008 Nov;97(11):4696–740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 2.Fahr A, van Hoogevest P, May S, Bergstrand N, MLSL Transfer of lipophilic drugs between liposomal membranes and biological interfaces: consequences for drug delivery. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2005 Nov;26(3–4):251–65. doi: 10.1016/j.ejps.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhary RK, Shariff I, Dolphin D. Drug release characteristics of lipid based benzoporphyrin derivative. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques. 2003 Jan-Apr;6(1):13–9. [PubMed] [Google Scholar]
- 4.Shabbits JA, Chiu GN, Mayer LD. Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. Journal of controlled release : official journal of the Controlled Release Society. 2002 Dec 5;84(3):161–70. doi: 10.1016/s0168-3659(02)00294-8. [DOI] [PubMed] [Google Scholar]
- 5.Pal A, Khan S, Wang YF, Kamath N, Sarkar AK, Ahmad A, et al. Preclinical safety, pharmacokinetics and antitumor efficacy profile of liposome-entrapped SN-38 formulation. Anticancer research. 2005 Jan-Feb;25(1A):331–41. [PubMed] [Google Scholar]
- 6.Zhigaltsev IV, Maurer N, Akhong QF, Leone R, Leng E, Wang J, et al. Liposome-encapsulated vincristine, vinblastine and vinorelbine: a comparative study of drug loading and retention. Journal of controlled release : official journal of the Controlled Release Society. 2005 May 5;104(1):103–11. doi: 10.1016/j.jconrel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Amselem S, Cohen R, Barenholz Y. In vitro tests to predict in vivo performance of liposomal dosage forms. Chemistry and physics of lipids. 1993 Sep;64(1–3):219–37. doi: 10.1016/0009-3084(93)90067-d. [DOI] [PubMed] [Google Scholar]
- 8.Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annual review of physiology. 1998;60:643–65. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 9.Sot J, Aranda FJ, Collado MI, Goni FM, Alonso A. Different effects of long- and short-chain ceramides on the gel-fluid and lamellar-hexagonal transitions of phospholipids: a calorimetric, NMR, and x-ray diffraction study. Biophysical journal. 2005 May;88(5):3368–80. doi: 10.1529/biophysj.104.057851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shabbits JA, Mayer LD. Intracellular delivery of ceramide lipids via liposomes enhances apoptosis in vitro. Biochimica et biophysica acta. 2003 May 2;1612(1):98–106. doi: 10.1016/s0005-2736(03)00108-1. [DOI] [PubMed] [Google Scholar]
- 11.Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005 May 1;11(9):3465–74. doi: 10.1158/1078-0432.CCR-04-1770. [DOI] [PubMed] [Google Scholar]
- 12.Stover T, Kester M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. The Journal of pharmacology and experimental therapeutics. 2003 Nov;307(2):468–75. doi: 10.1124/jpet.103.054056. [DOI] [PubMed] [Google Scholar]
- 13.Shabbits JA, Mayer LD. High ceramide content liposomes with in vivo antitumor activity. Anticancer research. 2003 Sep-Oct;23(5A):3663–9. [PubMed] [Google Scholar]
- 14.Zolnik BS, Stern ST, Kaiser JM, Heakal Y, Clogston JD, Kester M, et al. Rapid distribution of liposomal short-chain ceramide in vitro and in vivo. Drug metabolism and disposition: the biological fate of chemicals. 2008 Aug;36(8):1709–15. doi: 10.1124/dmd.107.019679. [DOI] [PubMed] [Google Scholar]
- 15.Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. Journal of colloid and interface science. 2010 Nov 1;351(1):19–29. doi: 10.1016/j.jcis.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Forster resonance energy transfer imaging. Proceedings of the National Academy of Sciences of the United States of America. 2008 May 6;105(18):6596–601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophysical journal. 1998 May;74(5):2702–13. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barth BM, Cabot MC, Kester M. Ceramide-based therapeutics for the treatment of cancer. Anti-cancer agents in medicinal chemistry. 2011 Nov;11(9):911–9. doi: 10.2174/187152011797655177. [DOI] [PubMed] [Google Scholar]
- 19.Koshkaryev A, Piroyan A, Torchilin VP. Increased apoptosis in cancer cells in vitro and in vivo by ceramides in transferrin-modified liposomes. Cancer biology & therapy. 2012 Jan 1;13(1):50–60. doi: 10.4161/cbt.13.1.18871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khazanov E, Priev A, Shillemans JP, Barenholz Y. Physicochemical and biological characterization of ceramide-containing liposomes: paving the way to ceramide therapeutic application. Langmuir : the ACS journal of surfaces and colloids. 2008 Jun 1;24(13):6965–80. doi: 10.1021/la800207z. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Zhao P, Su W, Wang S, Liao Z, Niu R, et al. PLGA/polymeric liposome for targeted drug and gene co-delivery. Biomaterials. 2010 Nov;31(33):8741–8. doi: 10.1016/j.biomaterials.2010.07.082. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Han X, Li X, Luo Y, Zhao H, Yang M, et al. Core-shell hybrid liposomal vesicles loaded with panax notoginsenoside: preparation, characterization and protective effects on global cerebral ischemia/reperfusion injury and acute myocardial ischemia in rats. International journal of nanomedicine. 2012;7:4299–310. doi: 10.2147/IJN.S32385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences of the United States of America. 2011 Jul 5;108(27):10980–5. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aryal S, Hu CM, Fang RH, Dehaini D, Carpenter C, Zhang DE, et al. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine. 2013 Feb 14; doi: 10.2217/nnm.12.153. [DOI] [PubMed] [Google Scholar]
- 25.Zheng Y, Yu B, Weecharangsan W, Piao L, Darby M, Mao Y, et al. Transferrin-conjugated lipid-coated PLGA nanoparticles for targeted delivery of aromatase inhibitor 7alpha-APTADD to breast cancer cells. International journal of pharmaceutics. 2010 May 10;390(2):234–41. doi: 10.1016/j.ijpharm.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sunoqrot S, Bae JW, Jin SE, RMP, Liu Y, Hong S. Kinetically controlled cellular interactions of polymer-polymer and polymer-liposome nanohybrid systems. Bioconjugate chemistry. 2011 Mar 16;22(3):466–74. doi: 10.1021/bc100484t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang RH, Aryal S, Hu CM, Zhang L. Quick synthesis of lipid-polymer hybrid nanoparticles with low polydispersity using a single-step sonication method. Langmuir : the ACS journal of surfaces and colloids. 2010 Nov 16;26(22):16958–62. doi: 10.1021/la103576a. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Khan MA, Burgess DJ. Predicting hydrophilic drug encapsulation inside unilamellar liposomes. International journal of pharmaceutics. 2012 Feb 28;423(2):410–8. doi: 10.1016/j.ijpharm.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Mohammed AR, Weston N, Coombes AG, Fitzgerald M, Perrie Y. Liposome formulation of poorly water soluble drugs: optimisation of drug loading and ESEM analysis of stability. International journal of pharmaceutics. 2004 Nov 5;285(1–2):23–34. doi: 10.1016/j.ijpharm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Jiang X, Ashley C, Brinker CJ. Electrostatically mediated liposome fusion and lipid exchange with a nanoparticle-supported bilayer for control of surface charge, drug containment, and delivery. Journal of the American Chemical Society. 2009 Jun 10;131(22):7567–9. doi: 10.1021/ja902039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherer M, Leuthauser-Jaschinski K, Ecker J, Schmitz G, Liebisch G. A rapid and quantitative LC-MS/MS method to profile sphingolipids. Journal of lipid research. 2010 Jul;51(7):2001–11. doi: 10.1194/jlr.D005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mano N, Oda Y, Yamada K, Asakawa N, Katayama K. Simultaneous quantitative determination method for sphingolipid metabolites by liquid chromatography/ionspray ionization tandem mass spectrometry. Analytical biochemistry. 1997 Jan 15;244(2):291–300. doi: 10.1006/abio.1996.9891. [DOI] [PubMed] [Google Scholar]
- 33.Yoo HH, Son J, Kim DH. Liquid chromatography-tandem mass spectrometric determination of ceramides and related lipid species in cellular extracts. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2006 Nov 7;843(2):327–33. doi: 10.1016/j.jchromb.2006.06.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.