

Abstract
DNA polymerase mu (Pol μ) is the only template-dependent human DNA polymerase capable of repairing double strand DNA breaks (DSBs) with unpaired 3′-ends in non-homologous end joining (NHEJ). To probe this function, we structurally characterized Pol μ’s catalytic cycle for single nucleotide incorporation. These structures indicate that, unlike other template-dependent DNA polymerases, there are no large-scale conformational changes in protein subdomains, amino acid side chains, or DNA upon dNTP binding or catalysis. Instead, the only major conformational change is seen earlier in the catalytic cycle, when the flexible Loop1 region repositions upon DNA binding. Pol μ variants with changes in Loop1 have altered catalytic properties and are partially defective in NHEJ. The results indicate that specific Loop1 residues contribute to Pol μ’s unique ability to catalyze template-dependent NHEJ of DSBs with unpaired 3′-ends.
Keywords: Family X DNA polymerase, nonhomologous end joining, DNA double strand break repair, Loop1
Mammalian X Family polymerases β, λ, μ, and terminal deoxynucleotidyltransferase (TdT) perform short gap filling synthesis during repair of single- or double-stranded DNA breaks1. To fulfill this function, they are equipped with an 8kDa domain N-terminal to the polymerase domain that aids in filling short gaps in DNA by binding to the 5′ end of the break, allowing the enzyme to bridge both ends of the gap. In Pols β and λ, which are implicated in base excision repair (BER), the 8 kDa domain harbors a deoxyribose phosphate (dRP) lyase activity that is lacking in the 8 kDa domain of Pol μ and TdT1. Pol μ, TdT and Pol λ participate in nonhomologous end-joining (NHEJ) of broken DNA ends2, utilizing an N-terminal BRCT domain3,4 (similar to the BRCA-1 C-terminal protein-protein interaction domain5) that is absent in Pol β.
Pol μ can incorporate either ribo- or deoxyribonucleotides in a template-dependent manner during gap-filling synthesis6,7. Though Pol μ can efficiently fill small gaps in DNA caused by single-strand breaks6,8, it primarily functions in double-strand break (DSB) repair by NHEJ, specifically, preserving immunoglobulin gene sequence during V(D)J recombination2,9–11. Transgenic animals deficient in Pol μ expression exhibit impaired immunoglobulin κ light chain rearrangement, leading to reduced levels of mature B-cells9,12. In a similar fashion, Pol μ aids in repair of DJH rearrangements during embryonic development11 and is involved in repair of DNA breaks due to ionizing radiation12,13.
The DSB repair capability of Pol μ is directly related to its unique substrate specificity. In contrast to other polymerases, Pol μ can extend a 3′ primer overhang in a template-dependent manner, using as a template the non-complementary 3′ overhang of another DNA molecule, an activity used for accurate repair of DSBs lacking microhomology14. Pol μ also has limited ability to conduct template-independent synthesis when extending a DNA molecule whose 3′ end is not base paired15,16. Two structural determinants of synthesis from an unpaired 3′ primer terminus have been identified. The first is Loop1, which exists in all Family X polymerases, but varies in length and sequence. This loop is required for repair of DSBs lacking microhomology by Pol μ and for template-independent synthesis by Pol μ and TdT14,17,18. Loop1 may act as a surrogate for an absent or incomplete template strand. The second structural determinant consists of residues that stabilize the primer terminus, to assure its proper alignment for catalysis. Arg416 and His329 have been proposed to fill this role in Pol μ, with Arg416 anchoring the primer terminus to the body of the protein, and His329 bridging the primer terminal phosphate and the γ-phosphate of the incoming nucleotide19,20.
Multiple structures of Pols β and λ have provided insights into gap-filling synthesis21,22, and the checkpoint mechanisms preventing strand misalignment or incorporation of nucleotides containing an incorrect sugar or base23–28. In contrast, only one structure is available for murine Pol μ20. In order to better understand the mechanism by which Pol μ performs its unique function, we structurally characterized the catalytic cycle for canonical template-dependent gap-filling synthesis by human Pol μ. This was facilitated using a variant with decreased surface entropy and drastically improved crystallizability. We obtained four X-ray crystal structures of this variant that address the catalytic cycle from DNA binding through nucleotide incorporation. These structures indicate that repositioning of Loop1 within the DNA binding cleft is required for binding of duplex DNA substrates, after which no further major conformational changes of protein subdomains, DNA, or active site side chains are observed throughout the catalytic cycle. This is in striking contrast to other DNA polymerases that assemble a catalytically competent active site through choreographed movements of protein domains, DNA rearrangements, and nucleotide binding29–32. This analysis of catalysis by human Pol μ provides new insight into aspects of substrate selection that contribute to its unique biological function.
RESULTS
Truncation of human Pol μ Loop2 improved crystallization
Despite extensive efforts with both murine and human Pol μ, a pre-catalytic ternary complex of the catalytic domain (Pro132-Ala496) of murine Pol μ is the sole example of Pol μ structural information to date20. Here we overcome this limitation using a variant of the catalytic domain of hPol μ (Pro132-Ala494), that has increased crystallizablity and comparable gap-filling activity to the full-length enzyme (Supplementary Fig. 1). In this variant, the large disordered loop (Loop2) connecting β-strands 4 and 5 was truncated by removing residues Pro398-Pro410 and inserting a glycine (labeled Gly410) (Supplementary Fig. 2). The resulting variant (hPol μ Δ2) crystallized readily, with and without substrates bound.
Polymerization by hPol μ Δ2 was indistinguishable from wild type hPol μ for canonical template-dependent synthesis on a single-nucleotide gapped DNA substrate (Supplementary Fig. 3a, and Supplementary Table 1) and for template-independent synthesis on a single-stranded primer (Supplementary Fig. 3b). Loop2 truncation was also not detrimental to Pol μ’s ability to act in concert with its binding partners in a reconstituted NHEJ reaction (Supplementary Fig. 3c). Given that at least the length of Loop2 is conserved, it remains plausible that it could serve as an interaction surface for some cellular factor not present in the reconstituted NHEJ assay. These data indicated that hPol μ Δ2 serves as an appropriate model for X-ray crystallography studies.
Structural characterization of the hPol Δ2 catalytic cycle
hPol μ Δ2 was co-crystallized in the presence of a single-nucleotide gapped DNA substrate with a 5′-phosphate on the downstream end of the gap (Fig. 1a, binary complex, PDB ID code 4LZG), and in a pre-catalytic ternary complex with a nonhydrolyzable incoming nucleotide (PDB ID code 4M04, Fig. 1 and Table 1). Crystals of the ternary complex were then soaked with a hydrolyzable dTTP for in crystallo incorporation (PDB ID code 4M0A, Fig. 2) to obtain a post-catalytic nicked complex.
Figure 1. Structural features of the hPol μ Δ2 binary and pre-catalytic ternary complexes.

(a) Single-nucleotide gapped DNA substrate used for co-crystallization with hPol μ Δ2. (b) Ribbon diagram of the hPol μ Δ2 binary complex with substrate from (a), displaying the individual domains structures (8 kDa, fingers, palm, and thumb domains are shown in orange, green, red, and purple, respectively). Loop 1 is drawn in blue, and the truncated Loop 2 is indicated by a red asterisk. α-helices are labeled alphabetically, and β-strands numerically. The template (T), upstream primer (P), and downstream primer (D) strands are also labeled. (c) Hydrogen bonding network stabilizing the position of the primer terminus (light blue, with 3′-OH marked) in the hPol μ Δ2 binary complex (blue). (d) Binding of the 5′-phosphate on the downstream end of the gapped DNA substrate. Putative hydrogen bonding interactions between the hPol μ Δ2 8kDa subdomain (blue sticks) and the 5′-phosphorylated downstream primer (D). (e) Structural superposition of the hPol μ Δ2 binary (blue, DNA in light blue) and pre-catalytic ternary complexes (orange, DNA in yellow). (f) Superposition of the catalytic centers of the binary and ternary complexes (colored as in e). Movement of the primer terminal 3′-OH upon binding of the incoming nucleotide is shown as a solid green line. The distance between the primer terminal 3′-OH and the α-phosphate is shown as a dashed line. Mg2+ ions are shown as purple spheres. All structural figures were generated using PyMOL (Schrödinger, http://www.pymol.org).
Table 1.
Data collection and refinement statistics
| Apoprotein (4LZD)* | Binary (4LZG)* | Ternary (4M04)* | Nicked (4M0A)* | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P41212 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 124.19, 124.19, 50.41 | 60.00, 68.51, 110.31 | 59.89, 68.47, 109.77 | 60.02, 68.67, 110.44 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å)╪ | 50–1.85 (1.92–1.85) | 50–1.60 (1.63–1.60) | 50–1.90 (1.97–1.90) | 50–1.85 (1.88–1.85) |
| Rsym | 5.6 (41.4) | 6.2 (27.2) | 6.4 (31.6) | 7.1 (44.2) |
| I/σI | 34.4 (3.5) | 32.4 (3.2) | 24.1 (3.6) | 27.5 (2.7) |
| Completeness (%) | 99.3 (96.4) | 98.6 (83.1) | 99.7 (98.1) | 98.2 (92.1) |
| Redundancy | 9.0 (4.8) | 6.2 (2.4) | 6.2 (3.5) | 6.9 (4.1) |
| Refinement | ||||
| Resolution (Å) | 1.85 | 1.60 | 1.90 | 1.85 |
| No. reflections | 34070 | 59954 | 36188 | 38957 |
| Rwork/Rfree | 18.09/22.83 | 17.40/20.45 | 16.39/19.59 | 16.26/20.03 |
| No. atoms | ||||
| Protein | 2616 | 2551 | 2564 | 2540 |
| DNA | --- | 344 | 344 | 364 |
| Nucleotide | --- | --- | 28 | 9§ |
| Ions | 2 | 4 | 3 | 4 |
| Water | 328 | 512 | 380 | 398 |
| B-factors (Å2) | ||||
| Protein | 18.33 | 12.36 | 13.89 | 12.19 |
| DNA | --- | 11.11 | 12.28 | 10.98 |
| Nucleotide | --- | --- | 5.68 | 10.54§ |
| Ions | 37.00 | 17.66 | 7.32 | 13.63 |
| Water | 26.61 | 25.38 | 23.03 | 21.96 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.011 | 0.011 | 0.010 | 0.013 |
| Bond angles (°) | 1.280 | 1.449 | 1.325 | 1.522 |
A single crystal was used to collect each data set.
Values in parentheses are for highest resolution shell.
Inorganic pyrophosphate.
Figure 2. Structural characterization of the nucleotide incorporation by hPol μ Δ2.
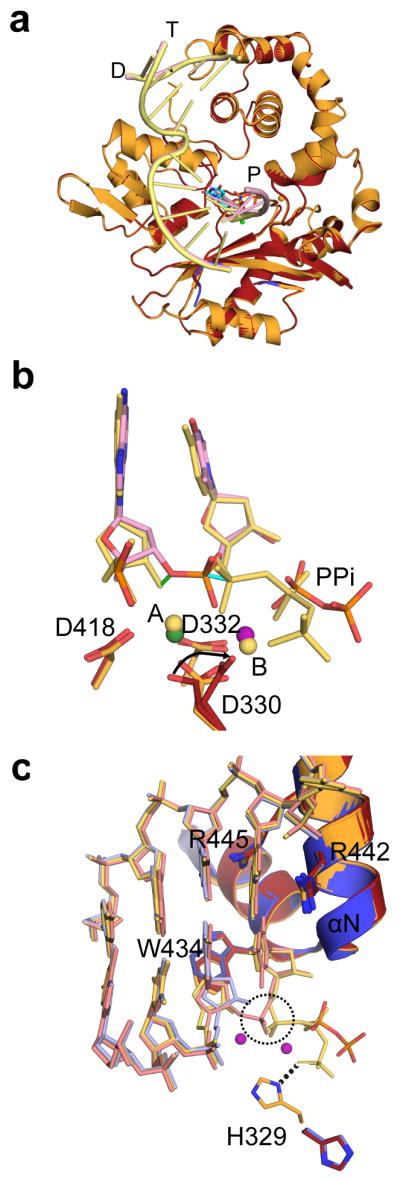
(a) Structural superposition of the hPol μ Δ2 pre-catalytic ternary (orange, DNA in yellow) and nicked (red, DNA in pink). (b) Superposition of the primer terminal and incoming nucleotides from the pre-catalytic hPol μ Δ2 ternary complex (yellow) with the newly incorporated base from the post-catalytic complex (pink). Movement of the primer terminal 3′-OH and the α-phosphates are indicated by green and cyan solid lines, respectively. Divalent metals from the ternary complex are shown as yellow spheres, while those from the post-catalytic complex are shown in green (Mn2+) and purple (Mg2+). (c) Superposition of the active centers of the binary (blue, DNA in light blue), pre-catalytic ternary (orange, DNA in yellow) and post-catalytic nicked (red, DNA in pink) complexes of hPol μ Δ2. Incorporation of the incoming nucleotide leads to inversion of the α-phosphate (dashed circle) and release of inorganic pyrophosphate.
hPol μ Δ2 displays the canonical left-handed subdomain structure common to the Family X polymerases, with fingers, palm, and thumb subdomains preceded by an 8 kDa domain1 (Fig. 1b). The upstream and downstream duplex regions are located distal from one another, an arrangement enforced by a 90° bend in the DNA backbone immediately 5′ of the unpaired templating nucleotide. This bent conformation is consistent with similar complexes of other Family X polymerases20,21,33. Protein/DNA interactions in the binary complex are primarily mediated through hydrogen bonding interactions between the phosphate backbone of the template DNA strand and positively charged amino acid side chains lining the substrate binding cleft, namely Arg442, Arg449, and Lys450. These electrostatic interactions are typical of sequence-nonspecific DNA-binding enzymes, and involve the phosphate backbone rather than the individual nucleotide bases.
Protein-DNA interactions with the upstream primer strand are mediated through hydrogen bonding interactions (Gly247 N and Thr250 OG1) with the phosphate backbone, and via a sodium ion coordinated by the helix-hairpin-helix (HhH) motif formed by α-helices F and G (HhH2, Glu232-Glu257) in the fingers (Fig. 3a)34. The side chain of Arg416 tethers the primer terminal phosphate to the palm, linking it to the backbone carbonyl of Cys390 through a hydrogen bonding network (Fig. 1c). This interaction is not required for single-nucleotide gap filling, but is critical for correct positioning of the primer terminus during NHEJ and template-independent synthesis19.
Figure 3. Electron density for hPol μ Δ2 structures.

2Fo-Fc electron density maps, contoured at 1σ, for (a) sodium ion coordinated by HhH2 in the hPol μ Δ2 binary complex, active site regions of the (b) pre-catalytic ternary complex, and (c) the post-catalytic complex. The Mg2+ ions are drawn as green spheres, the incoming nucleotide in the ternary complex is shown in cyan, and the pyrophosphate released from phosphodiester bond formation in orange. The anomalous difference map (violet) for the Mn2+ (dark purple) in the active site of the post-catalytic complex is contoured to 5σ. (d) Position and interactions involving Loop1 in the hPol μ Δ2 apoprotein.
On the downstream end of the DNA duplex, the 5′-phosphate is bound by multiple putative hydrogen bonding interactions with the N-terminal end of α-helix D in the HhH1 (Leu196-His219) motif in the 8 kDa domain (Fig. 1d). hPol μ Δ2 possesses a binding pocket for the 5′ end of the downstream primer, created by backbone hydrogen bonding interactions from His204, Gly206, and Ser209. The backbone and side chain of His208 position the 5′-terminal nucleotide in the pocket, where it also interacts with the side chain of Arg175. Arg175 has a pivotal role in end bridging of single- and double-strand breaks via interaction with the 5′-phosphate19,35.
In a departure from the strictly template-dependent Family X polymerases β and λ, which use structural rearrangements as proposed kinetic checkpoints for correct incorporation 36–38, the hPol μ Δ2/DNA complex undergoes minimal conformational change upon binding of the incoming nucleotide. A comparison of the binary and ternary complexes shows that the two structures are nearly indistinguishable (RMSD of 0.164 Å over 294 Cα, Table 1, Supplementary Table 2, Figs. 1e and 3b). All atoms in the active site of the ternary complex are present in catalytically relevant conformations, including two fully-occupied Mg2+ ions not observed in the binary complex. Compared with the binary complex, the primer terminal sugar is slightly shifted, moving the 3′-OH 1.9 Å to a position near the incoming nucleotide (Fig. 1f). In each complex, the primer-terminal nucleotide is observed with a C3′-endo sugar pucker. This differs from binary complex crystal structures of Pols β and λ, where the deoxyribose sugar pucker shifts from C2′-endo to C3′-endo only upon occupation of the metal A site by a divalent Mg2+ ion21,22. In the ternary complex, the sugar pucker of the incoming dUMPNPP is also C3′-endo, and a Mg2+ ion fully occupies the metal A site, positioning the 3′-OH inline with the α-phosphate and the bridging atom of the pyrophosphate leaving group, at a distance of 3.6 Å (Fig. 1f). The octahedral coordination spheres for the two Mg2+ ions are filled by interactions with Asp330, Asp332, Asp418, the triphosphate oxygens, and two water molecules. Subtle adjustments in the position of the side chains of Val420 and Trp434, as well as His329 and Asp330 correlate with incoming nucleotide binding and metal site occupation. The movement of the two latter side chains is consistent with previous molecular dynamics simulations of the mPol μ ternary complex structure39.
His329 interacts with both the primer terminus and the incoming triphosphate of the mouse pre-catalytic ternary complex20 (R.M.S.D of 0.580 Å over 284 Cα atoms). In hPol μ Δ2, His329 maintains a similar interaction with the γ-phosphate of the incoming nucleotide, but lies farther from the primer terminal phosphate (2.7 Å in mPol μ versus 3.5 Å in hPol μ Δ2). Alanine substitution of His329 suggests that the interactions of this side chain and either the primer terminal phosphate or the incoming triphosphate are not required for single-nucleotide gap-filling, but become critical during NHEJ with decreased template strand structural integrity20.
Crystals of the hPol μΔ2 ternary complex were soaked with hydrolyzable dTTP to catalyze phosphodiester bond formation in crystallo. Comparison of the pre- and post-catalytic complexes shows minimal conformational change associated with nucleotide incorporation (R.M.S.D. of 0.118 Å over 287 Cα atoms, Fig. 2, Table 1, and Supplementary Table 2). There is clear electron density for the newly formed bond between the 3′-O of the primer and the α-phosphate, along with stereochemical inversion of the α-phosphate (Fig. 3c). Subtle differences are observed in the active site as a result of catalysis—the primer terminal sugar is tilted slightly toward the incoming nucleotide, shifting the 3′-oxygen 1.2 Å from its original position in the pre-catalytic state to its final position in the post-catalytic state, and allowing the nascent bond to form (Figs. 2b and 2c). Asp330, which interacts with both of the divalent metal ions, displays multiple rotamer conformations, alternating between continuing to coordinate the ion still occupying the metal A site, and rotating away from it. In each alternate conformation, however, Asp330 maintains an interaction with the ion in the metal B site. Additionally, the side chain of His329 reverts to the conformation observed in the binary complex structure (Fig. 2c). There is no evidence of attempted translocation or dissociation.
Structure of hPol μ in the absence of bound DNA substrate
hPol μ Δ2 also crystallized in the absence of DNA (PDB ID code 4LZD, Table 1), in a ‘closed’ conformation similar to the DNA-bound forms (RMSD of 0.791–0.814 Å, Fig. 4a and Table 1). The only major difference between the apo and DNA-bound structures is in the position of Loop1 (Fig. 4b). In the DNA-bound structures, Loop1 is largely disordered, but the ordered ends of β-strands 3 and 4 show that the trajectory of Loop1 moves away from the DNA binding cleft and does not preclude binding of the template strand (Fig. 4b)20. In the absence of DNA, residues His365-Cys369 and His381-Asp383 of Loop1 are ordered, and β-strand 3 extends along the base of the palm. The N-terminal end of the loop does not follow a similar trajectory as when DNA is present. In addition, the C-terminal end of the loop infiltrates deeply into the active site before eventually rejoining β-strand 4 (Figs. 4b and 4c). This represents the first structural evidence that Loop1 exhibits variable conformations based on the presence or absence of DNA substrate.
Figure 4. Structural characteristics of the hPol μ apoprotein.

(a) Structural superposition of the hPol μ Δ2 apoprotein (green) with the pre-catalytic ternary (orange, DNA in yellow) complex. (b) Comparison of Loop1 conformations in hPol μ Δ2 apoprotein (green) and pre-catalytic ternary (orange, DNA in yellow), compared to the structurally homologous region in murine TdT (PDB ID code 1JMS41, gray) structures. Ordered residues at either end of Loop 1 are labeled. Hypothetical positions of disordered sections of the loop are shown as dashed lines. (c) Position of Loop1 in the hPol μ apoprotein (green), in relation to the bound single-nucleotide gapped DNA substrate (yellow) and incoming nucleotide (cyan) from the hPol μ Δ2 ternary complex (orange). (d) Location and composition of the binding pocket containing Phe385 in the hPol μ Δ2 apoprotein structure.
The conformation of Loop1 in the apoprotein structure places the side chain of Met382 in close proximity to Trp434, altering its position (Figs. 3d and 4c). This is reminiscent of the energetically stabilizing methionine-tryptophan aromatic interactions described by Valley, et a 40, and may represent a means of anchoring Loop1 in the DNA binding cleft in the absence of bound substrate. The residues occupying the active site (Met382-Phe385) are conserved in mammalian Pol μ orthologs, but not in other Family X polymerases (Supplementary Figs. 2c and 2d). The position of Loop1 in the apoprotein would block binding of either single- or double-stranded DNA. Loop1 must reposition to correctly bind DNA in a catalytically competent conformation, regardless of the structure of that substrate. In mTdT, this loop is often ordered and occupies a substantial portion of the DNA binding cleft (Fig. 4b)41,42. Such positioning would prevent binding of a template strand, consistent with an inability to perform template-dependent synthesis43,44. The binding site of Pol μ, however, accommodates structurally disparate substrates, in varying states of template presence or continuity. Thus, the flexibility of Loop1 could allow Pol μ to stabilize substrates of varying structures using different Loop1 conformations.
Implications of Loop1 flexibility for DSB repair
Using the structure of the pre-catalytic ternary complex, a model for a noncomplementary DSB substrate was generated by removing the templating base opposite the primer terminus (Supplementary Fig. 4a). If the overall conformation of the Pol μ polymerase domain behaves as a rigid scaffold for DSB substrate accommodation, the presence of a discontinuous template strand would likely have negligible effect on the protein structure. Interestingly, the protein component closest to the site of the DSB was not Loop1, which is critical for utilization of these substrates14, but β-strand 6 in the thumb. Given its trajectory in the pre-catalytic single-nucleotide gapped complex, Loop1 would not occupy a position capable of stabilizing the discontinuous template strand or the upstream primer terminus (Supplementary Fig. 4b, modeled in green), consistent with the fact that Loop1 is not required for synthesis on a single-strand break substrate17. Therefore, Loop1 would likely assume a different conformation in order to better stabilize a DSB substrate with a discontinuous, noncomplementary structure (Supplementary Fig. 4b, modeled in red). Hypothetically, Loop1 could assume a more extended conformation along the contour of the upstream template strand, potentially stabilizing template bases at the −2 and −3 positions. Such interactions could indirectly ensure that the primer terminus maintains a catalytically relevant position.
Role of Loop1 in substrate utilization by hPol μ
To investigate the role of Loop1 residues in catalysis and substrate selection, three conserved amino acids (His363, Met382, and Phe385) were mutated. His363 is ordered in all structures of Pol μ, and maintains its general position regardless of overall Loop1 conformation (Fig. 4b). Because the trajectory of Loop1 deviates immediately C-terminal to His363 depending on the presence or absence of bound DNA substrate, His363 may be a ‘hinge’ point on the N-terminal end of the loop. Consistent with the importance of His363, substitution with alanine or proline impaired single-nucleotide gap-filling activity (Fig. 5a and Supplementary Table 3), and H363P had decreased activity on a single-stranded substrate (Fig. 5b, Supplementary Table 4, and Supplementary Fig. 5). These mutants had nearly normal activity during NHEJ of a substrate with complementary ends (H363A, 93 ± 4 % and H363P, 84 ± 5 %; s.e, n = 3), but were less active with a substrate lacking complementarity (H363A, 57 ± 4 % and H363P, 25 ± 3%) (Figs. 5c, 5d, Supplementary Table 4 and Supplementary Fig. 5). Alanine substitution for Met382 slightly impaired single-nucleotide gap-filling activity but severely impaired template-independent synthesis (Fig. 5, Supplementary Table 3, and Supplementary Fig. 5). M382A displayed only 40% of wild type activity on a DSB substrate with complementary ends, and was even less active on DSB substrates lacking complementarity (16.1% of wild type activity). Thus, M382A impacts synthesis, regardless of DNA substrate structure. Phe385, on the C-terminal end of Loop1, occupies a small pocket between the palm and thumb formed by the backbone of residues Val421 and Ala422 and the side chains of Val420, Pro423, Gln426, and His459 (Figs. 4c and 4d). Upon binding of a DNA substrate, this portion of Loop1 shifts (5.9 Å for backbone CA atom, 10.2 Å for side chain CD1 atom), removing Phe385 from this pocket and exposing it to solvent (Fig. 4c). We hypothesize that Phe385 may anchor the position of Loop1 proximal to the active site in the unliganded protein, limiting large-scale mobility while allowing localized conformational sampling that could stabilize a variety of non-duplex DNA substrates. To test this hypothesis, Phe385 was replaced with alanine, which had little effect on gap filling, but reduced template-independent synthesis (Fig. 5a and 5b, Supplementary Table 3, and Supplementary Fig. 5). Additionally, F385A showed little difference on DSB substrates with complementary ends, but no activity was observed on noncomplementary ends (Fig. 5c, 5d, Supplementary Table 4 and Supplementary Fig. 5). These results are consistent with our hypothesis that Phe385 may serve as an anchor for the Loop1 position proximal to the active site. Overall, these results support the theory that specific Loop1 residues have important roles in catalysis and substrate selection. To further test the possibility that alterations to the ends of β-strands 3 and 4 might destabilize the globular fold of the palm subdomain and disrupt the DNA binding cleft, alanine substitutions of Gln364, His365, Gln366, and Glu386 were also generated. These mutations had no discernible effect on substrate utilization (unpublished data; A.F.M, K.B., and J.M.P.).
Figure 5. Biochemical characterization of wildtype and Loop1 mutants of hPol μ.

(a) Comparison of the catalytic efficiencies of hPol μ Loop1 mutants (3 nM), as determined by steady-state kinetics for template-dependent single-nucleotide incorporation. (b) Template-independent synthesis of wildtype and hPol μ Loop1 mutants (100 nM) on a single-stranded oligo dT15 DNA substrate. (c) Activity of hPol μ Loop1 mutants during in vitro NHEJ of DNA ends with partly complementary overhangs (top panel), or noncomplementary overhangs (bottom panel). Ku, XRCC4-ligase IV complex, and polymerase mutants (0.5nM) were added to linear substrates with end structures varied as noted in cartoons beside each panel. (d) Comparison of NHEJ end joining activity for hPol μ mutants, for either complementary (white bars) or noncomplementary (black bars) substrates, relative to joining observed in the presence of wildtype hPol μ. Error bars represent the standard error of the mean, for reactions performed in triplicate. Images of uncropped gels used in this study are found in Supplementary Fig. 5.
DISCUSSION
Pols β, λ, and μ differ substantially in the extent of conformational rearrangements required for catalysis, with regards to protein subdomains, DNA position, and active site side chains participating in assembly of the nascent base pair binding site (Table 2). While binding of the incoming dNTP induces ‘closing’ of the thumb in Pol β (Figs. 6a and 6b), Pol λ and Pol μ remain closed throughout the catalytic cycle (Figs. 6c, 6d, and Fig. 2c, respectively). Correctly positioning the templating nucleotide requires DNA movement in Pol β (5.2 Å) and Pol λ (5.5 Å) (Figs. 6a and 6c), but not for Pol μ (Figs. 2a and 2c). Loop1 in Pol β is too short to impede correct positioning of the DNA substrate, but in Pol λ, rearrangement of the template strand requires a 9.9 Å shift of Loop1 (Fig. 6c)33. Deletion of Loop1in Pol λ does not significantly affect catalytic efficiency, but reduces fidelity and discrimination against ribonucleotide incorporation 27,36, suggesting that it functions as a kinetic checkpoint to prevent misincorporation. It should be noted that although Loop1 in Pol λ may undergo a rearrangement similar to that seen in Pol μ, there are significant differences in both extent and timing of the movement. Loop1 in Pol λ is twelve residues shorter than Loop1 in Pol μ, so its range of motion may be more limited. In addition, Pol λ’s Loop1 shifts upon binding of the incoming nucleotide, aiding in active site assembly33. Therefore, the position of Loop1 in Pol λ may prevent the template strand from reaching the catalytically relevant conformation in the absence of incoming nucleotide. In contrast, Pol μ Loop1 movement occurs at an earlier stage of the catalytic cycle, repositioning to allow entry of the DNA substrate. This region in Pol μ is mostly disordered in both the binary and ternary complexes, but its overall trajectory does not appear to change upon dNTP binding.
Table 2.
Correlation between template-dependence and protein/DNA movements during catalysis by Family X polymerases
| Pol β | Pol λ | Pol μ | |
|---|---|---|---|
| Motion | |||
| Protein subdomains | + | − | − |
| Side chains | + | + | +/− |
| DNA | + | + | − |
| Loop1 | N/A | + | + |
| Template-dependence | |||
| Undamaged template | + | optional | optional |
| Paired primer terminus | + | + | optional |
| Single-stranded | − | − | + |
| NHEJ (complementary) | − | + | + |
| NHEJ (noncomplementary) | − | − | + |
Figure 6. Comparison of structural rigidity across the Family X polymerases.

(a) Superposition of binary (PDB ID code 3IBS57, gray; DNA in light gray) and pre-catalytic ternary (PDB ID code 2FMS21, purple; DNA in lavender) complexes of human Pol β, focusing on movement of the thumb subdomain and DNA strands upon incoming nucleotide (cyan) binding. (b) Side chain rearrangements occurring in the Pol β active site as a consequence of nucleotide binding. Dashed lines illustrate ‘open’ to ‘closed’ transitions. (c) Superposition of binary (PDB ID code 1XSL33, gray; DNA in light gray) and pre-catalytic ternary (PDB ID code 2PFO22, magenta; DNA in pink) complexes of human Pol λ. Movements of the DNA template strand (T) and Loop1 are emphasized. (d) Active site sidechain rearrangements on Pol λ α-helices M and N in preparation for catalysis. Perspective matches that for Pol β in panel (d). (e) Comparison of structural motion in Family X polymerases during active site assembly. inary and pre-catalytic ternary structures of Pol β (binary PDB ID code 3ISB57, ternary PDB ID code 2FMS21), Pol λ (binary PDB ID code 1XSL33, ternary PDB ID code 2PFO22), Pol μ, and TdT (PDB ID codes 4I2A and 4I2742) were superimposed, using the structurally conserved Cα atoms of the palm subdomain (see Online Methods for details of structural analysis). The extent of protein subdomain motion (purple bar), minor groove residues (red bar), the N-helix (orange bar), and DNA template strand (blue bar) movements were calculated in Å, using the measurement wizard in PyMOL (Schrödinger, http://www.pymol.org).
Finally, several side chain rearrangements that are induced by nucleotide binding to Pol β and Pol λ are not observed in Pol μ. For example, while the side chains of the YF motif in Pols β and λ (Tyr271-Phe272 in Pol β, and Tyr505-Phe506 in Pol λ) rotate into the minor groove (Figs. 6b and 6d), only one of the structurally homologous residues, Trp434, changes position, and then only slightly (1.3Å). For Pol β, thumb subdomain ‘closing’ and DNA rearrangements induce movements of α-helix N residues Lys280 and Arg283, forming the nascent base pair binding site and stabilizing the position of the templating base (Fig. 6b). Though Pol λ displays no motion of the thumb subdomain, side chains of the structurally homologous residues (Arg514 and Arg517) on α-helix N undergo rearrangements that place these residues in similar positions to those observed for Pol β (Fig. 6d). Arg517 stacks with the templating base in the binary structure, but then extends into the minor groove in the ternary complex. Upon DNA relocation to the active conformation, Arg514 stabilizes the templating base. There are no such side chain motions observed in the nascent base pair binding pocket of Pol μ. Arg442 (structurally homologous to Lys280 in Pol β, and Arg514 in Pol λ) is involved in a stacking interaction with the templating nucleotide, and Arg445 (Arg283 in Pol β and Arg517 in Pol λ) sustains a similar extended conformation along the minor groove in each of the DNA bound structures (Fig. 2c), even in the absence of DNA. Thus, there are no major conformational changes of protein subdomains, DNA, or active site side chains observed upon dNTP binding or incorporation by Pol μ (Fig. 2c and 5e). Given that template-dependent polymerases have historically been thought to achieve correct incorporation through kinetic checkpoints of structural rearrangement, Pol μ appears to be a substantial departure from the norm. Pol μ’s rigidity during catalysis is more similar to that of template-independent TdT, a striking observation, since Pol μ is primarily a template-dependent enzyme. This is also reminiscent of human Pol η, a translesion synthesis polymerase that has a largely pre-formed active site undergoing minimal subdomain movement upon dNTP binding and catalysis45,46.
In summary, numerous studies of DNA polymerases over the last 25 years have emphasized the critical importance of dNTP-induced conformational changes to template-dependent DNA synthesis29–32. In remarkable contrast to those polymerases, the present results indicate that dNTP binding has little structural consequence for catalysis by Pol μ. Instead, repositioning of Loop 1 to allow DNA binding appears to be largely sufficient for template-dependent synthesis. We suggest that structural rigidity after DNA binding is key to Pol μ’s ability to do what no other polymerase has been shown to do, catalyze template-dependent DNA synthesis using unstable 3′-ends that are not base paired. This suggests there may be an inverse relationship between the stability of the primer terminus and rigidity of the polymerase in the catalytic cycle. Pol μ is at the rigid end of a spectrum of conformational flexibility among polymerases whose catalytic cycle has been characterized structurally1.
METHODS
Expression and purification of human Pol μ constructs
Sequences encoding full-length (Met1-Ala494) or the truncated catalytic domain (Pro132-Ala494) of human Pol μ were cloned into the pGEXM vector, using the NotI and SalI restriction sites. This vector is a modified version of the pGEX4T3 vector backbone, where a Tobacco Etch Virus protease cleavage site has been inserted downstream of the thrombin cleavage site, and the sequence of the multicloning site has been replaced by that from the pMALX vector (Supplementary Fig. 6)47. Three TGA stop codons have been inserted downstream of the multicloning site, each utilizing a different reading frame. Loop2 residues Pro398-Pro410 were deleted using site-directed mutagenesis (hPol μ Δ2). A glycine residue (labeled Gly410 in these structures) was added in order to fuse the ends of β-strands 4 and 5. Loop1 residues Cys369-Phe385 were similarly deleted in the hPol μ Δ1 constructs to be used as controls for NHEJ assays. Wildtype, Δ1, or Δ2 human Pol μ constructs were expressed in Rosetta2 (DE3) cells, overnight at 18°C. The cells were lysed by sonication, and the lysate was cleared by centrifugation. Soluble protein was bound in-batch to glutathione 4B sepharose resin, and the Pol μ proteins were obtained by on-resin TEV cleavage overnight at 4°C. Pol μ was then purified by size exclusion chromatography on a Superdex 200 16/60 column, and ion exchange chromatography using a MonoQ HR 5/5 column. The final, purified full-length protein was concentrated in 25 mM Tris pH 8, 100 mM NaCl, 5% glycerol, 1 mM DTT, 10 mM MgCl2 buffer, and the catalytic domain was concentrated in 25 mM Tris pH 8, 80 mM NaCl, 5% glycerol, 1mM DTT. All proteins were flash frozen in liquid nitrogen and stored at −80°C. Individual amino acid substitutions were also generated using site-directed mutagenesis, and were expressed and purified in similar fashion to the wildtype proteins, lacking only the ion exchange step.
Crystallization of the hPol μ Δ2 apoprotein
Crystals of the hPol μ Δ2 catalytic domain were grown by mixing 1 μL of concentrated protein (10.7mg/mL) with 1 μL of mother liquor (50 mM imidazole pH 8, 0.8 M sodium citrate), at room temperature, using the hanging drop vapor diffusion technique48. The crystals reached usable size within 24 hours, and were then directly transferred to a cryoprotectant solution containing 50 mM imidazole pH 8, 75 mM NaCl, 1 M sodium citrate, and 10% glycerol. Following a room temperature overnight soak in cryoprotectant containing 8.84 mM rUTP, the crystals were flash frozen in liquid nitrogen and placed into a stream of nitrogen gas cooled to −180 ° C for data collection. The apoprotein crystallized in a tetragonal crystal form (P41212), rather than the orthorhombic form favored by the DNA-bound complexes, and contained a single molecule of hPol μ Δ2 per asymmetric unit (Table 1). The final model contained residues Trp137-Ala494, and residues 370–380 of Loop1 were disordered. Though the crystallization conditions contained the nucleotide UTP, in the attempt to obtain a structure similar to that of mTdT with bound ddATP (PDB ID code 1KEJ41), no electron density was visible for the UTP.
Crystallization of the hPol μ binary complex with a single-nucleotide gapped DNA substrate
The following DNA oligonucleotides were used to create the binary complex: template (5′-CGGCATACG-3′), upstream primer (5′-CGTA-3′), and a 5′-phosphorylated downstream primer (5′-pGCCG-3′). Oligonucleotides were mixed in equimolar ratios in 100 mM Tris pH 7.5 and 40 mM MgCl2. The DNA was annealed in a thermal cycler by denaturation at 94°C, followed by a slow temperature gradient from 90°C to 4°C. The annealed DNA was then mixed in a 4:1 molar ratio to concentrated hPol μ catalytic domain Δ2 and incubated on ice at 4°C for one hour. Crystals of the binary complex were grown at 4 °C by mixing 1 μL of the protein/DNA complex with 1 μL of mother liquor (85 mM MES pH 6.5, 85 mM CaCl2, 42.5 mM NaCl, 8.5 % PEG3350, 11 % glycerol), using the sitting drop vapor diffusion technique48. Crystals were transferred to a cryoprotectant solution containing 0.1 M MES pH 6.5, 0.1 M CaCl2, 50 mM NaCl, 10 mM MgCl2, 20 % PEG3350, and 15 % glycerol in three steps, then flash frozen in liquid nitrogen and placed into a stream of nitrogen gas cooled to −180 ° C for data collection.
Crystallization of the hPol μ ternary complex of a single-nucleotide gapped DNA substrate and an incoming deoxyribonucleotide
A DNA/protein mixture identical to that used for the binary complex was incubated on ice at 4 °C for two hours, when dUMPNPP was added to a final concentration of 1 mM. Crystals of the ternary complex were grown at room temperature, by mixing 1 μL ternary complex with 1 μL mother liquor (85–90 mM HEPES pH 7.5, 17–18 % PEG4000), using the sitting drop vapor diffusion technique. Crystals were transferred to a cryoprotectant solution containing 0.1 M HEPES pH 7.5, 10 mM MgCl2, 50 mM NaCl, 20 % PEG4000, 5 % glycerol, 15 % ethylene glycol, and 1 mM dUMPMPP in two steps. The crystals were then flash frozen in liquid nitrogen and placed into a stream of nitrogen gas cooled to −180 ° C for data collection. The post-catalytic nicked complex was obtained by soaking crystals of the ternary complex in cryoprotectant containing 10 mM dTTP for 16 hours, then backsoaking in cryoprotectant containing 2 mM pyrophosphate.
Data collection and structure solution
The data for the apoprotein and ternary complex structures were collected at a wavelength of 1.54 Å on an in-house rotating anode source, using a Rigaku 007HF X-ray generator and a Saturn92 CCD detector. Binary and nicked complex data were collected at a wavelength of 1.0 Å on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. A lower resolution data set of the apoprotein was first collected, and the structure was solved by molecular replacement, using Phaser49 in the PHENIX suite50. Murine Pol μ catalytic domain (PDB ID code 2IHM20, molecule B) was used as the starting model. The quality of the molecular replacement solution was improved by refinement in AutoBuild51, and this model was used for refinement of the high resolution apoprotein data set. All three DNA-bound crystal structures were observed in space group P212121, with a single molecule of protein/DNA per asymmetric unit (Table 1). Crystal packing is likely influenced by head-to-tail base stacking interactions at each end of the duplex DNA. The mPol μ structure was also used as a starting model for molecular replacement for the ternary complex and the refined ternary complex structure (with incoming nucleotide removed) was used as the starting model for refinement of the binary and nicked complexes. The same Rfree test reflections were used for the binary, ternary, and nicked data sets to avoid potential model bias. All structures were refined using iterative cycles of manual model building and refinement in COOT52,53 and Phenix50. Data refinement statistics are listed in Table 1. Ramachandran statistics were generated using MolProbity54.
Template-dependent primer extension reactions with a single-nucleotide gapped DNA substrate
Reactions (20 μl) contained: 50 mM Tris pH 7.5, 1 mM DTT, 4% glycerol, 0.1 mg/ml BSA, 5 mM MgCl2, 50 μM dNTPs and 200 nM DNA substrate with a 32P-5′ end-labeled upstream primer and 5′-phosphorylated downstream primer. Reactions were initiated by adding enzyme (wildtype or hPol μ Δ2 truncated catalytic domain) at 10 or 50 nM and incubated in 37°C. 10 μl aliquots were removed after 5 and 15 minutes incubation, and mixed with equal volume of quench solution (99% formamide, 5 mM EDTA, 0.1 % xylene cyanol, 0.1 % bromophenol blue). The products were resolved by 12% (v/v) PAGE and visualized by phosphorimagery.
Template-independent primer extension reactions on a single-stranded DNA substrate
Reactions (20 μl) contained: 50 mM Tris pH 7.5, 1 mM DTT, 4% glycerol, mg/ml BSA, 100 μM MnCl2, 200 nM T15-mer oligonucleotide substrate 32P -5′ end-labeled, and either 10 μM dNTPs or 50 μM dTTP. Reactions were initiated by adding enzyme (truncated catalytic domain constructs of wildtype Pol μ, Pol μ mutants, or Pol μ Δ2) at 100 nM and incubated in 37°C. The truncated catalytic domain construct was used for these reactions. 10 μl aliquots were removed after 5 and 20 minutes incubation, mixed with equal volume of quench solution (99% formamide, 5 mM EDTA, 0.1 % xylene cyanol, 0.1 % bromophenol blue and the products were resolved by 12% (v/v) PAGE and visualized by phosphorimagery. Original images of gels, autoradiographs and blots used in this study can be found in Supplementary Figure 5a.
Steady-state kinetics for single-nucleotide incorporation
The steady state measurements of single nucleotide incorporation were performed as described elsewhere27,36. DNA substrates were prepared by hybridizing a 32P-5′-end-labeled 14-nucleotide primer (P14A, 5′ GTCAGACTGACGTA) and a 14-nucleotide downstream primer (DP14b, 5′ GCCGGACGACGGAG) with a phosphate on the 5′ end to a 29-mer template (T-A, 5′ CTCCGTCGTCCGGCATACGTCAGTCTGAC) to create a one-nucleotide gap substrate. Reaction mixtures (10 μl) contained 50 mM Tris, pH 7.5, 1 mM dithiothreitol, 4% (v/v) glycerol, 0.1 mg/ml bovine serum albumin, 5 mM MgCl2, 200 nM DNA, and 3 nM enzyme (truncated catalytic domain constructs of wildtype Pol μ, Pol μ mutants, or Pol μ Δ2). The truncated catalytic domain construct was used for these reactions. Reactions were initiated by adding dTTP at one of the following concentrations 0.2, 0.5, 1.5, 5, 15, 45, 70, 100 μM. The reaction mixtures were incubated at 37 °C for 4 min. After adding an equal volume of loading dye (99% (v/v) formamide, 5 mM EDTA, 0.1% (w/v) xylene cyanol, and 0.1% (w/v) bromophenol blue), products were resolved on a 12% denaturing polyacrylamide gel and quantified by phosphor screen autoradiography. The data were fit to the Michaelis-Menten equation using nonlinear regression from KaleidaGraph software version 3.6 (Synergy Software, http://www.synergy.com.
Nonhomologous end-joining assays
The expression and purification of human Ku, XRCC4-Ligase IV complex, and polymerase β proteins were carried out as described previously55,56. Both 300 bp substrates were generated by PCR amplification of a fragment of the mouse Jk1 locus in the presence of Cy5 labeled dCTP, followed by restriction digestion of sites appended to this common core. The 3′ GCG overhang containing substrate was generated by amplification with the primers 5′ TTTTTGCCACGCTGGCTTAGCTGTATAGTCAGGGA and 5′ CACCTGCCTCGCTGGCACACCCATCTCAGACTGGC, and digested with BglI. The 3′-G overhang substrate was generated by amplification with primers 5′ CAAGTGGACCACATGTCTTAGCTGTATAGTCAGGGAAATC and 5′ CCGCCGACGCCATGTCACACCCATCTCAGACTGGCTACCC, and digested with AhdI. Complete digestion was validated by electrophoresis, and substrates further purified with a QIAquick cartridge. End joining reactions were carried out in a buffer containing: 25 mM Tris (pH 7.5), 1 mM DTT, 150 mM KCl, 4% glycerol, 40 μg/ml bovine serum albumin, 0.1 mM EDTA and 9% polyethyelene glycol (MW 8,000 kDa). Ligations were carried out by adding (final concentrations) 10 nM Ku, 20 nM XRCC4-LigaseIV complex, and 0.5 nM full-length wildtype or mutant polymerase, to a mixture containing 2 nM DNA substrate, 10 μM of each dNTP, 100 μM of each rNTP, 5 mM Mg2+, and 100 ng supercoiled DNA. All reactions were carried out at 37 °C for 10 minutes, stopped by addition of EDTA and SDS, and DNA extracted once with a 1:1 mixture of phenol and chloroform. Ligation products were resolved using a 5% native PAGE gel for qualitative comparisons (gel images), before quantification using qPCR and primers specific for substrate (5′ GGCACTCTCCAAGGCAAAGA and 5′ ACATGTCTAGCCTATTCCCGGCTT) and product (5′ CTTACGTTTGATTTCCCTGACTATACAG and 5′ GCAGGGTAGCCAGTCTGAGATG). Original images of gels, autoradiographs and blots used in this study can be found in Supplementary Figure 5b and 5c.
Comparison of structural motion in Family X polymerases during active site assembly
The binary and pre-catalytic ternary structures of Pol β (binary PDB ID code 3ISB57, ternary PDB ID code 2FMS21), Pol λ (binary PDB ID code 1XSL33, ternary PDB ID code 2PFO22), Pol μ, and TdT (PDB ID codes 4I2A and 4I2742) were superimposed, using the structurally conserved Cα atoms of the palm subdomain (Fig. 6). The extent of protein subdomain motion was quantified by measuring movement of the Cα atom of the N-helix arginine (Arg283 in Pol β, Arg517 in Pol λ, Arg445 in Pol μ, and Arg 458 in TdT, purple bar). Movement of the minor groove residues in the active site was measured by the equivalent CZ atoms of the YF motif (Phe271 and Phe505, respectively) for Pol β and λ, and by the equivalent CH2 atoms of the GW motif (Trp434 and Trp450, respectively) for Pol μ and TdT (red bar). Movement of the N-helix arginine (Arg283 in Pol β, Arg517 in Pol λ, Arg445 in Pol μ, and Arg 458 in TdT, orange bar) was measured using the coordinates of the CZ atom. Comparison of DNA template strand movement was quantified using the C1′ atom of the templating nucleotide in the nascent base pair site (blue bar). All distances were calculated in Å, using the measurement wizard in PyMOL (Schrödinger, http://www.pymol.org).
Supplementary Material
Acknowledgments
We thank G. Mueller and B. Beard for critical reading of the manuscript. This research was supported by the Division of Intramural Research of the National Institute of Environmental Health Sciences, National Institutes of Health (NIH) Grant 1ZIA ES102645-03 (L.C.P.), Grant Z01 ES065070 (T.A.K.), and NIH Grant CA097096 (D.A.R.). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences Contract W-31-109-Eng-38.
Footnotes
ACCESSION CODES:
Coordinates for human Pol μ Δ2 apoprotein (PDB ID code 4LZD), and in complex with bound DNA substrates (PDB ID codes 4LZG, 4M04, and 4M0A) have been deposited in the Protein Data Bank, www.pdb.org.
AUTHOR CONTRIBUTIONS:
A.F.M., L.C.P., K.B., and D.A.R., designed research; A.F.M., K.B., and J.M.P. performed research; and all authors contributed to data analysis and manuscript preparation. The authors declare no conflict of interest.
References
- 1.Moon AF, et al. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair (Amst) 2007;6:1709–25. doi: 10.1016/j.dnarep.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nick McElhinny SA, Ramsden DA. Sibling rivalry: competition between Pol X family members in V(D)J recombination and general double strand break repair. Immunol Rev. 2004;200:156–64. doi: 10.1111/j.0105-2896.2004.00160.x. [DOI] [PubMed] [Google Scholar]
- 3.DeRose EF, et al. Solution structure of polymerase mu’s BRCT Domain reveals an element essential for its role in nonhomologous end joining. Biochemistry. 2007;46:12100–10. doi: 10.1021/bi7007728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mueller GA, et al. A comparison of BRCT domains involved in nonhomologous end-joining: introducing the solution structure of the BRCT domain of polymerase lambda. DNA Repair (Amst) 2008;7:1340–51. doi: 10.1016/j.dnarep.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bork P, et al. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 6.Nick McElhinny SA, Ramsden DA. Polymerase mu is a DNA-directed DNA/RNA polymerase. Mol Cell Biol. 2003;23:2309–15. doi: 10.1128/MCB.23.7.2309-2315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz JF, et al. Lack of sugar discrimination by human Pol mu requires a single glycine residue. Nucleic Acids Res. 2003;31:4441–9. doi: 10.1093/nar/gkg637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roettger MP, Fiala KA, Sompalli S, Dong Y, Suo Z. Pre-steady-state kinetic studies of the fidelity of human DNA polymerase mu. Biochemistry. 2004;43:13827–38. doi: 10.1021/bi048782m. [DOI] [PubMed] [Google Scholar]
- 9.Bertocci B, De Smet A, Berek C, Weill JC, Reynaud CA. Immunoglobulin kappa light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu. Immunity. 2003;19:203–11. doi: 10.1016/s1074-7613(03)00203-6. [DOI] [PubMed] [Google Scholar]
- 10.Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006;25:31–41. doi: 10.1016/j.immuni.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Gozalbo-Lopez B, et al. A role for DNA polymerase mu in the emerging DJH rearrangements of the postgastrulation mouse embryo. Mol Cell Biol. 2009;29:1266–75. doi: 10.1128/MCB.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lucas D, et al. Altered hematopoiesis in mice lacking DNA polymerase mu is due to inefficient double-strand break repair. PLoS Genet. 2009;5:e1000389. doi: 10.1371/journal.pgen.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chayot R, Danckaert A, Montagne B, Ricchetti M. Lack of DNA polymerase mu affects the kinetics of DNA double-strand break repair and impacts on cellular senescence. DNA Repair (Amst) 2010;9:1187–99. doi: 10.1016/j.dnarep.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Nick McElhinny SA, et al. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol Cell. 2005;19:357–66. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Andrade P, Martin MJ, Juarez R, Lopez de Saro F, Blanco L. Limited terminal transferase in human DNA polymerase mu defines the required balance between accuracy and efficiency in NHEJ. Proc Natl Acad Sci U S A. 2009;106:16203–8. doi: 10.1073/pnas.0908492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez O, et al. DNA polymerase mu (Pol mu), homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J. 2000;19:1731–42. doi: 10.1093/emboj/19.7.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juarez R, Ruiz JF, Nick McElhinny SA, Ramsden D, Blanco L. A specific loop in human DNA polymerase mu allows switching between creative and DNA-instructed synthesis. Nucleic Acids Res. 2006;34:4572–82. doi: 10.1093/nar/gkl457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romain F, Barbosa I, Gouge J, Rougeon F, Delarue M. Conferring a template-dependent polymerase activity to terminal deoxynucleotidyltransferase by mutations in the Loop1 region. Nucleic Acids Res. 2009;37:4642–56. doi: 10.1093/nar/gkp460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin MJ, Juarez R, Blanco L. DNA-binding determinants promoting NHEJ by human Polmu. Nucleic Acids Res. 2012;40:11389–403. doi: 10.1093/nar/gks896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moon AF, et al. Structural insight into the substrate specificity of DNA Polymerase mu. Nat Struct Mol Biol. 2007;14:45–53. doi: 10.1038/nsmb1180. [DOI] [PubMed] [Google Scholar]
- 21.Batra VK, et al. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–66. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair (Amst) 2007;6:1333–40. doi: 10.1016/j.dnarep.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol Cell. 2008;30:315–24. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bebenek K, Pedersen LC, Kunkel TA. Replication infidelity via a mismatch with Watson-Crick geometry. Proc Natl Acad Sci U S A. 2011;108:1862–7. doi: 10.1073/pnas.1012825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavanaugh NA, et al. Molecular insights into DNA polymerase deterrents for ribonucleotide insertion. J Biol Chem. 2011;286:31650–60. doi: 10.1074/jbc.M111.253401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–42. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 27.Gosavi RA, Moon AF, Kunkel TA, Pedersen LC, Bebenek K. The catalytic cycle for ribonucleotide incorporation by human DNA Pol lambda. Nucleic Acids Res. 2012;40:7518–27. doi: 10.1093/nar/gks413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Picher AJ, et al. Promiscuous mismatch extension by human DNA polymerase lambda. Nucleic Acids Res. 2006;34:3259–66. doi: 10.1093/nar/gkl377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–67. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez AC, Park HW, Mao C, Beese LS. Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J Mol Biol. 2000;299:447–62. doi: 10.1006/jmbi.2000.3728. [DOI] [PubMed] [Google Scholar]
- 31.Wing RA, Bailey S, Steitz TA. Insights into the replisome from the structure of a ternary complex of the DNA polymerase III alpha-subunit. J Mol Biol. 2008;382:859–69. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu EY, Beese LS. The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J Biol Chem. 2011;286:19758–67. doi: 10.1074/jbc.M110.191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol lambda catalytic cycle. Nat Struct Mol Biol. 2005;12:97–8. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 34.Doherty AJ, Serpell LC, Ponting CP. The helix-hairpin-helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996;24:2488–97. doi: 10.1093/nar/24.13.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis BJ, Havener JM, Ramsden DA. End-bridging is required for pol mu to efficiently promote repair of noncomplementary ends by nonhomologous end joining. Nucleic Acids Res. 2008;36:3085–94. doi: 10.1093/nar/gkn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bebenek K, Garcia-Diaz M, Zhou RZ, Povirk LF, Kunkel TA. Loop 1 modulates the fidelity of DNA polymerase lambda. Nucleic Acids Res. 2010;38:5419–31. doi: 10.1093/nar/gkq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radhakrishnan R, et al. Regulation of DNA repair fidelity by molecular checkpoints: “gates” in DNA polymerase beta’s substrate selection. Biochemistry. 2006;45:15142–56. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Beard WA, Wilson SH, Broyde S, Schlick T. Polymerase beta simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions per se. J Mol Biol. 2002;317:651–71. doi: 10.1006/jmbi.2002.5450. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Schlick T. Modeling DNA polymerase mu motions: subtle transitions before chemistry. Biophys J. 2010;99:3463–72. doi: 10.1016/j.bpj.2010.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valley CC, et al. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287:34979–91. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delarue M, et al. Crystal structures of a template-independent DNA polymerase: murine terminal deoxynucleotidyltransferase. EMBO J. 2002;21:427–39. doi: 10.1093/emboj/21.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouge J, Rosario S, Romain F, Beguin P, Delarue M. Structures of Intermediates along the Catalytic Cycle of Terminal Deoxynucleotidyltransferase: Dynamical Aspects of the Two-Metal Ion Mechanism. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Bollum FJ. Thermal conversion of nonpriming deoxyribonucleic acid to primer. J Biol Chem. 1959;234:2733–4. [PubMed] [Google Scholar]
- 44.Kato KI, Goncalves JM, Houts GE, Bollum FJ. Deoxynucleotide-polymerizing enzymes of calf thymus gland. II. Properties of the terminal deoxynucleotidyltransferase. J Biol Chem. 1967;242:2780–9. [PubMed] [Google Scholar]
- 45.Biertumpfel C, et al. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–8. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ummat A, et al. Human DNA polymerase eta is pre-aligned for dNTP binding and catalysis. J Mol Biol. 2012;415:627–34. doi: 10.1016/j.jmb.2011.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon AF, Mueller GA, Zhong X, Pedersen LC. A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein Sci. 2010;19:901–13. doi: 10.1002/pro.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chayen NE. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallogr D Biol Crystallogr. 1998;54:8–15. doi: 10.1107/s0907444997005374. [DOI] [PubMed] [Google Scholar]
- 49.Zwart PH, et al. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–35. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–9. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovell SC, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 55.Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol. 2000;20:2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prasad R, Kumar A, Widen SG, Casas-Finet JR, Wilson SH. Identification of residues in the single-stranded DNA-binding site of the 8-kDa domain of rat DNA polymerase beta by UV cross-linking. J Biol Chem. 1993;268:22746–55. [PubMed] [Google Scholar]
- 57.Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH. DNA polymerase beta substrate specificity: side chain modulation of the “A-rule”. J Biol Chem. 2009;284:31680–9. doi: 10.1074/jbc.M109.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.