

Abstract
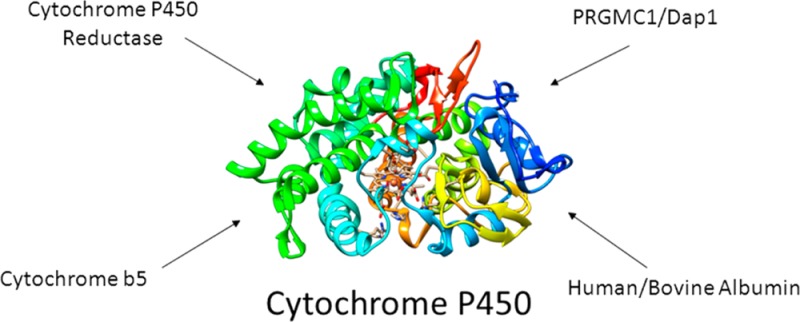
Through their unique oxidative chemistry, cytochrome P450 monooxygenases (CYPs) catalyze the elimination of most drugs and toxins from the human body. Protein–protein interactions play a critical role in this process. Historically, the study of CYP–protein interactions has focused on their electron transfer partners and allosteric mediators, cytochrome P450 reductase and cytochrome b5. However, CYPs can bind other proteins that also affect CYP function. Some examples include the progesterone receptor membrane component 1, damage resistance protein 1, human and bovine serum albumin, and intestinal fatty acid binding protein, in addition to other CYP isoforms. Furthermore, disruption of these interactions can lead to altered paths of metabolism and the production of toxic metabolites. In this review, we summarize the available evidence for CYP protein–protein interactions from the literature and offer a discussion of the potential impact of future studies aimed at characterizing noncanonical protein–protein interactions with CYP enzymes.
1. Introduction
Protein–protein interactions are essential to the function of the ubiquitous xenobiotic-metabolizing cytochrome P450 monooxygenases, or CYPs.1 Classic studies have demonstrated the importance of CYP interactions with the accessory proteins cytochrome P450 reductase (CPR) and cytochrome b5 (b5) for both electron transfer and allosteric modulation.2−4 However, it has recently become clear that CYP enzymes may participate in additional interactions with other protein partners, such as progesterone receptor membrane component 1 (PGRMC1), human serum albumin (HSA), and even other CYP isoforms. These interactions have been shown to alter CYP function and, in some cases, lead to the production of toxic metabolites. In this review, we attempt to summarize the available evidence from the primary literature and offer some future perspectives on the study of noncanonical protein–protein interactions with cytochrome P450 enzymes.
2. Cytochrome P450 Interactions with Cytochrome P450 Reductase and Cytochrome b5
2.1. Cytochrome P450 Reductase
Cytochrome P450 reductase (CPR) was identified as the primary CYP electron transfer partner by the late 1960s.2,5 The term cytochrome P450 reductase is a bit of a misnomer, as this enzyme participates as a reducing partner in a number of different reactions, including those catalyzed by heme oxygenases I and II, squalene monooxygenase, and certain fatty acid desaturases.6−9 Initially characterized as a cytochrome c reductase, CPR was eventually found to be localized in the endoplasmic reticulum, where it functions as a redox partner with CYPs that are also located there. CPR is a diflavin (FMN, FAD) containing reductase that receives its electron-reducing equivalents from NADPH.10−13 The CPR electron transfer protein shuttles two electrons to the CYP during the reaction cycle, with the second electron transfer thought to be the rate-limiting step.1,14,15 CPR is known to be post-translationally modified and has an apparent molecular weight between 76 and 80 kDa, depending on post-translational modifications, the species-specific isoform, and detergent solubilization procedures.2,13 In addition to the FMN, FAD, and NADPH binding domains, CPR also contains a 60 amino acid hydrophobic N-terminal membrane anchoring region, which is essential for full activity with native redox partners, such as cytochrome P450 enzymes.3
A number of important crystal structures of CPR and several of its mutants have been produced, including those demonstrating important conformational changes that take place upon reduction and CYP binding.10,16−20 The first crystal structure of CPR was obtained using an N-terminally truncated sequence of the rat isoform.10,16 This structure demonstrated that the NADPH–FAD binding domain also contained an important linker sequence that could help position the FMN and FAD domains in close proximity for efficient electron transfer. The linker sequence distinguishes CPR from the closely related, single-domain ferrodoxin reductases. It is worthwhile to note that this particular N-terminally truncated construct was not able to support CYP reduction, even though it successfully reduced cytochrome c. More recent X-ray crystal structures have provided some insight into CYP–CPR interactions.6,18 The crystal structure of a CPR mutant with a four amino acid deletion in the hinge region between the FMN binding domain and the rest of the protein demonstrated remarkable flexibility in this region by crystallizing in three different conformations.18 Although this particular mutant was not able to catalyze intramolecular electron transfer between the FAD and FMN prosthetic groups, it was able to successfully reduce CYPs, given enough reducing equivalents. The three different conformations observed suggest that CPR inherently possesses a high degree of conformational plasticity, especially in the C terminus of the hinge region, allowing it to efficiently interact with a variety of CYP isoforms and other enzymes. Other structures obtained from protein X-ray crystallography and NMR have confirmed this conformational flexibility and the ability of CPR to transition between closed and open forms,6,21,22 which had also been predicted by computational molecular dynamics studies.23,24 Another intriguing possibility is that subtle structural differences between CYP isoforms may regulate interaction with CPR and cytochrome b5.25,26
Seminal work examining structural and functional variation in the CPR gene has been conducted by both the Masters10,16,17,27,28 and Kranendonk29−31 groups, underscoring the importance of CPR in drug disposition and disease. Several mutations in the gene encoding CPR are known to be associated with specific disease states, such as a rare form of congenital adrenal hyperplasia known as Antley–Bixler syndrome and reduced steroidogenesis.32,33 Amino acid changes that are implicated in these conditions include Y181D, A457H, Y459H, V492E, and R616X.
Surprisingly, despite the large number of CYPs present in the human genome (57, excluding possible pseudogenes; http://drnelson.uthsc.edu/hum.html), there is only one gene encoding CPR. Additionally, the total CYP content of the human liver is estimated to be between 5- and 25-fold higher than that of CPR, suggesting differential expression.34−36 The fact that CYPs and CPR interact in a 1:1 stoichiometry raises an unusual conundrum: exactly how do CYPs and CPR interact to promote efficient oxidation of substrates? From a structural perspective, it stands to reason that all of the different CYP isoforms must share a common functional CPR binding surface. Indeed, several studies have proposed that the binding interaction between CYP and CPR takes place between the positively charged proximal surface near the CYP heme and the negatively charged FMN and FAD/NADPH domains of CPR, suggesting that their interaction is primarily driven by electrostatics.37−39 However, there is some controversy in this regard, as other studies have suggested that hydrophobic interactions are more predominant.40,41 These discrepancies may be the result of the different experimental approaches employed (e.g., X-ray crystallography vs site-directed mutagenesis and stopped-flow fluorescence spectroscopy).
Surprisingly, the Kd for the CYP–CPR complex is remarkably low, between 5 and 110 nM, depending on the particular isoform used and whether substrate is present, implying a very high-affinity interaction.42,43 Upon initial examination, a tight-binding interaction may seem counterintuitive; however, a rapid kon or koff rate would allow the system to work more efficiently due to swift equilibration.35 Clearly, the interaction can be modulated by the presence of substrates and/or inhibitors, as well as other CYP isoforms, as noted below. Another compounding factor is the variety of methods, often indirect, that have been used to determine CYP–CPR affinities, making it difficult to compare various experimental conditions.
Separate from its function as an electron transfer partner, CPR may also act as an allosteric effector to induce conformational changes in CYPs that modulate the rate of electron transfer.44−46 A number of useful structural studies conducted with the bacterial model CYP isoform, CYP101 (P450cam), have been useful to inform our understanding of how CPR might interact with mammalian CYPs in order to modulate electron transfer. Pochapsky and colleagues identified an X-Pro cis–trans isomerization redox switch in CYP101 (P450cam) that is triggered by binding to the reduced form of CYP101’s cognate electron transfer partner, putidaredoxin (Pdx).47 These conformational changes account for most of the significant structural perturbations observed by NMR upon complex formation.47 Conformational plasticity may be one mechanism by which the binding affinity of a CYP and its redox partner are modulated: if the subset of conformational states occupied by the reduced electron transfer protein is the same as that selected upon binding to a cognate CYP, then the entropic cost (i.e., free energy) of binding would be lowered, resulting in a higher affinity.48
In a unique approach to examine CYP–electron transfer partner interactions, Hiruma and colleagues utilized paramagnetic NMR in conjunction with a site-specific lanthanide label to solve the solution structure of the CYP101–Pdx complex.45 This CYP101–Pdx complex structure was, in turn, validated by simultaneous determination of the X-ray crystal structure of the same complex. In contrast to reports of the interaction between mammalian CYPs and CPR, here hydrophobic networks dominated the binding interface between the two proteins. The data suggested a minor conformational substate (or substates) that could represent an encounter complex between the two proteins, as postulated by an earlier study.46 Additionally, the retrieved structural parameters were consistent with an electron transfer rate theoretically faster than that observed experimentally, suggesting the involvement of a gating process in electron transfer.24,45 Similar allosterically induced conformational changes have also been observed with mammalian CYPs and cytochrome b5.44 Taken together, these studies underscore the dynamic nature between CYPs and their electron transfer partners, both from a functional standpoint as well as a structural one.
Seemingly small structural changes, such as a single amino acid substitution, could have dramatic functional consequences on the ability of CPR to efficiently deliver its electrons to a particular CYP isoform. Recent studies have examined how genetic variation in the human CPR gene49,50 can manifest itself in the form of alterations in the isoform dependent CYP-mediated metabolism of certain substrates (Table 1). Interestingly, the effects of specific CPR mutations seem to be CYP isoform dependent, suggesting a unique interaction surface between individual CYP isoforms and CPR. In a study examining the effect of the 35 most common CPR mutations on the activity of CYP1A2 and CYP2C19, Miller and colleagues discovered that while most mutations either decreased the activity of both CYP1A2 and CYP2C19, or eliminated activity all together (most notably the A287P and R457H mutations), certain mutations enhanced CYP activity.51 In most cases, the enhancements were specific to the CYP isoform examined and the particular CPR mutation. For CYP1A2, activity enhancement was observed with CPR mutants G213E, R406H, and A462T. In the case of CYP2C19, increased activity was reported for CPR mutants M263V and G504R. Most surprisingly, the Q153R CPR mutation, a rare disease-causing mutation that inhibits steroidogenic activity, increased the activity of both CYP1A2 and CYP2C19 to 144 and 284%, respectively. The authors rationalized these results by suggesting that many of the CPR mutants with reduced activity had decreased FAD binding or lay in a region known to be important for CPR–CYP contacts. The CPR gain-of-function mutants were more difficult to explain, but Miller and colleagues suggest that, at least in the case of the Q153R mutant, electron transfer rates to the CYP may be affected by the close proximity of glutamine residue 153 to the FMN moiety.51 The change in electrostatic charge in the Q153R mutant may accelerate electron transfer from CPR to the CYP. Whatever the mechanism involved, it is clear that there are important functional consequences for subtle alterations in the interactions between CPR and CYP isoforms. A follow-up study examined the effects of CPR mutations on the ability of CYP2D6 to metabolize different substrates.52 Interestingly, for any particular CPR mutation, different effects were seen depending on the substrate used. The Q153R CPR mutation mentioned above had a slight stimulatory effect on the CYP2D6-mediated metabolism of 2H-1-benzopyran-3-carbonitrile,7-(ethoxy-methoxy)-2-oxo-(9Cl) (EOMCC), yet it almost doubled the rate of metabolism of dextromethorphan. Similar effects were seen with other CPR mutant/substrate combinations. This observation may speak to the promiscuous nature of drug-metabolizing CYPs and the multiple substrate binding sites within the active site.53,54 Additional work with the CYP3A4 isoform also indicated that it was susceptible to functional variation due to CPR mutations.32,55,56 Consistent with the previous findings for CYP2D6 and CYP1A2, CYP3A4 activity is decreased in combination with most of the CPR mutants examined, but it is increased with the Q153R mutant.55 This effect was again substrate-specific for the substrates testosterone and quinidine, and nominal activity was observed for midazolam and erythromycin oxidation.
Table 1. Effect of Human P450 Oxidoreductase (CPR) Genetic Variants on Cytochrome P450 Enzyme Activities with Different P450 Substratesa.
| CYP1A251 | CYP2C1951 | CYP2D652 |
CYP3A455 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| single POR mutations | EOMCCb | EOMCC | EOMCC | dextramethorphan | bufuralol | testosterone (6β-hydroxylation) | midazolam (1-hydroxylation) | quinidine | erythromycin |
| A115V | NDc | ND | |||||||
| Q153R | 144 | 284 | 128 | 198 | 153 | 129 | 94 | 150 | 76 |
| Y181D | ND | ND | |||||||
| A287P | ND | ND | ND | 27 | 24 | 17 | 17 | 3 | ND |
| R457H | ND | ND | ND | ND | ND | ND | ND | 1 | ND |
| V492E | ND | ND | |||||||
| A503V | 85 | 113 | 85 | 62 | 53 | 77 | 61 | 89 | 97 |
| C569Y | 6 | ND | |||||||
| V608F | 5 | ND | |||||||
Values represent percent of wild-type.
EOMCC: 2H-1-benzopyran-3-carbonitrile,7-(ethoxy-methoxy)-2-oxo-(9Cl).
ND, evaluated but undetectable.
A study utilizing the full-length CYP and CPR mutant isoforms simultaneously expressed in insect Sf9 cells via a baculovirus vector recapitulated the results obtained with the bacterially expressed isoforms (referenced above).56 A result unique to this study was the observation that some of the reduced-function CPR mutants regained activity when coexpressed with CYP isoforms in the Sf9 insect cells. This suggests that mutant CPR association with a CYP may help to stabilize the CPR protein into an active conformation, thereby rescuing activity. This is an intriguing possibility, given the postulate by Pochapsky regarding the ability of a CYP to select the active conformation of a particular electron transfer partner.46
Additional studies have pointed to the importance of the N-terminal hydrophobic anchoring sequence in supporting CYP-mediated oxidation. Hayashi and colleagues found that an N-terminally truncated form of rat CPR failed to support CYP1A1-mediated 7-ethoxycoumarin O-deethylation, although it efficiently reduced cytochrome c and rat heme oxygenase I.57 Furthermore, structural studies have demonstrated that crystallized, yet catalytically active, yeast CPR has a substantially different structure58 than the conventional N-terminally truncated catalytically inactive rat enzyme,10,59 reinforcing the importance of this sequence in supporting efficient electron transfer from CPR to CYP.
CPR genetic variants may also have effects on different CYP isoform alleles as well.60 In a study examining the CYP2C9 alleles CYP2C9.1, CYP2C9.2, and CYP2C9.3, Tracy and colleagues found increased metabolic oxidation of the substrates diclofenac, flurbiprofen, and tolbutamide with four different mutant CPR enzymes tested (Q153R, A287P, R457H, and A503V).60 This was in contrast to the predominate effect seen previously with CYP1A2, CYP2D6, CYP2B6, and CYP3A4, where a decrease in activity was reported with these CPR mutants. The authors noted that since the effects they observed led to an increase in the Vmax, but not a change in the Km, it was most likely that the increase in activity observed was due to enhanced electron transfer and not an increase in CYP substrate affinity.60
Variation in the CPR gene can lead directly to toxicity for certain drugs that are metabolized through reduction.61−63 The herbicide paraquat produces toxicity through the generation of superoxide anion during redox cycling.64 This redox cycling process is predominately mediated through CPR.61 Han et al. examined the effect of six common CPR mutations in patients with impaired steroidogenesis on the redox cycling of paraquat in a CHO cell model system.61 In five out of the six CPR mutants examined, paraquat toxicity was diminished. Although not explicitly confirmed in this study, the authors inferred that the mutants either affected the catalytic activity or expression levels of CPR, thereby reducing its ability to activate paraquat. In the C569Y mutant, the observed paraquat toxicity was equivalent to that with the wild-type enzyme, implying a rescue of function with this particular mutation. The authors suggest that variation in the CPR gene may be a primary susceptibility determinant in paraquat toxicity. Similarly, in a study investigating the toxicity of mitomycin C, Wang et al. found that CPR mutations may be useful as biomarkers to predict the therapeutic response to this anticancer drug.62 Mitomycin C is a common chemotherapeutic agent that is metabolically activated by either a one- or two-electron reduction step.65 CPR is the primary enzyme responsible for the one electron reduction, which produces the semiquinone anion radical that is thought cause damage to cellular DNA. Of six CPR mutants that were examined (Y181D, A287P, R457H, V492E, C569Y, and V608F), cellular toxicity of mitomycin C was decreased in all with the exception of the C569Y mutant, demonstrating that a high degree of CPR functional activity is needed to activate mitomycin C.62 In particular, the V608F and Y181D mutants lacked any ability to activate mitomycin C, presumably due to altered association with the flavin moiety.
In addition to mitomycin C, the impact of CPR variants has also been examined with tacrolimus,66 warfarin,67 and other drugs, illustrating the importance of this particular enzyme in drug metabolism and elimination.
2.2. Cytochrome b5
Cytochrome b5 (b5) is a small (∼17 kDa) heme-containing integral membrane protein located on the cytosolic side of the endoplasmic reticulum4,68,69 and was originally identified in 1950 in silkworm larva.70 Although the majority of b5 is membrane-bound, a certain fraction is present as a soluble, cytosolic form in erythrocytes, where it catalyzes the reduction of methemoglobin.71 Although mutations in the b5 gene itself are quite rare, a few have been traced to the congenital abnormality of 17,20-lyase deficiency.72,73 Despite the fact that it can act as an electron donor to CYP enzymes, b5 is primarily involved in lipid biosynthesis, delivering electrons to microsomal desaturases that synthesize steroids, fatty acids, and plasmalogens.69 In the early 1970s, Estabrook and Mannering first demonstrated that b5 stimulated the metabolism of several CYP substrates.74,75 However, at this time, the functional role b5 played in CYP-catalyzed oxidation was still somewhat unclear. Early reports suggested that it could be involved in delivery of the second electron in the CYP catalytic cycle, thereby accelerating the rate-limiting step in CYP-mediated catalysis.4,69,74 Remarkably, a more contemporary report from the Wolf laboratory in a hepatic CPR deletion mouse model established that b5 and b5 reductase can act as efficient CYP electron donors, without the need of CPR.76 Despite its role in electron transfer, the effects seen from b5 are highly variable and dependent on the particular reaction conditions used, as addition of b5 can either inhibit or have no effect on some reactions rather than stimulate them.42,68,77−82 Furthermore, several studies have demonstrated that addition of apo (heme free) b5 can stimulate the activities of many CYPs, including CYP3A4, CYP2C9, and CYP17.7,83−85 It has been postulated that, in certain cases at least, b5 exerts the majority of its effects in the absence of electron transfer.77,86−89 It is still not completely clear if b5 functions solely as an electron donor, allosteric modulator, electron sink, or an uncoupling inhibitor.75,84,90−93 It is entirely plausible that its function may be contextual, depending on the particular isoform/substrate combination with which it is interacting.
A number of elegant studies over the years have established that electrostatic interactions are the basis for formation of the CYP–b5 complex, much like that for CYP–CPR.37,94−97 Waskell and colleagues mapped out the binding interface between CYP2B4 and b5 through extensive site-directed mutagenesis of both proteins and identified R122, R126, R133, K139, and K433 of CYP2B4 as residues critical to their interaction.37
Recently, a number of structural and dynamic 2D NMR studies have provided additional insight into the CYP–b5 interaction. In a pioneering NMR structural examination of b5 embedded in lipid bicells, Ramamoorthy, Waskell, and colleagues were able to take advantage of rotating frame separated local field (SLF) NMR.100 The authors employed two-dimensional proton evolved local-field (2D PELF) pulse sequences to accurately measure a broad range of heteronuclear dipolar couplings, allowing for a complete mapping of b5 protein dynamics in a native lipid bilayer environment.98 Notably, the authors discovered that the hydrophobic N-terminal α helix of b5 precessed in a cone with an approximate 8° fluctuation in the tilt of the helix, which is estimated to be 13°, suggesting that the lipid-embedded hydrophobic N-terminus is more mobile than originally thought.98 This added flexibility may allow this region of the protein to interact with a diverse variety of CYP isoforms. There is a growing appreciation of how inherent protein flexibility can allow proteins to adopt non-native states, which may allow them to gain new functions or interact with different protein partners.99 Similar to the approach employed by Soong et al., Ramamoorthy and colleagues utilized a modified INEPT technique, dipolar enhanced polarization transfer (DREPT), to detect an increased range of residual dipolar couplings from isotopically labeled b5 that allowed them to characterize the amino acid side chain motions of b5 while embedded in lipid bicells.100
Two-dimensional NMR methodologies have also been useful in providing explanations for the specific rate enhancements seen with certain CYP–substrate pairs. The bifunctional steroidogenic enzyme CYP17A1 performs both steroid hydroxylation, which is unaffected by b5, and an androgen-forming lyase reaction that is enhanced 10-fold in its presence.101,102 Work by the Scott group examining the interactions between an isotopically labeled b5 construct and CYP17A1 led to the observation that the CYP17A1–b5 interaction is stronger when the hydroxylase substrate pregnenolone is present in the CYP17A1 active site than when the lyase substrate 17α-hydroxypregnenolone is bound.44 This suggests that, at least in this case, the b5 reaction rate enhancement may be primarily driven by substrate affinity.44 In a follow-up study, in which CYP17A1 was isotopically labeled and the b5 construct was unlabeled, titration of b5 into CYP17A1–pregnenolone complex induced a set of conformational substates closely resembling those of the CYP17A1–17α-hydroxypregnenolone complex without b5.103 This result implied that b5 may also be able to allosterically induce catalytically productive conformations in CYP17A1, even in the absence of substrate.
These studies as well as others98,104−106 have culminated in a complete structural and catalytic model for the CYP–b5 interaction.94 Ahuja et al. determined the structure of full-length, membrane bound b5 in the presence of CYP2B4.94 This structure, determined for the first time in the presence of a phospholipid bilayer, confirmed the electrostatic nature of the CYP–b5 interaction, revealing a large number of charge pairings between surface residues on CYP2B4 and b5 that facilitate the interaction between the two proteins. However, it also hinted at the importance of hydrophobic interactions for the complex that are mediated by the phospholipid bilayer. Interestingly, the authors described an increase in the affinity between CYP2B4 and b5 in the presence of a small molecule substrate or inhibitor, similar to that observed by Estrada et al. for the CYP17A1–b5 interaction.44 Additionally, their results suggest a pathway for electron transfer between CYP2B4 and b5 mediated through a salt bridge with Arg125 of CYP2B4 and the heme propionates of b5.94 This study illustrates the power of 2D NMR techniques to obtain detailed structural data for the interactions between two protein partners.
Finn and colleagues created a liver-specific b5 conditional knockout mouse in order to better define the functional importance of the CYP–b5 interaction.78In vitro metabolism studies demonstrated the complete lack of NADH-mediated metabolism and a 50 to 90% decrease in NADPH-mediated metabolism for most of the drug substrates examined. Additionally, a dramatic increase in the half-life for these particular drug substrates was noted, thus establishing that b5 is a major contributor to the in vivo metabolism of many drugs and xenobiotics, albeit in a substrate- and isoform-selective manner.78 A follow-up study demonstrated altered clearance of several anticancer agents in the same knockout mouse model.107 Therefore, mutant b5 alleles that are deficient in their ability to interact with, and transfer electrons to, hepatic CYPs could lead to altered clearance rates for certain drugs, which may contribute to drug-induced toxicities.
Furthermore, varying levels of b5 function important for drug oxidation can also produce altered metabolite ratios, potentially increasing the production of toxic products in certain instances. In a study conducted by the Hollenberg group, b5 was determined to be essential for the formation of protein adducts to CYP3A5 from metabolic activation of the contraceptive agent 17α-ethynylestradiol.79 In the absence of b5, the reactive 17α-oxirene-related species was not formed, leading to CYP enzyme protection. An additional concern from a toxicological point of view is the potential for the generation of reactive oxygen species (ROS) due to increased uncoupling.108 Indeed, using a reconstituted system containing both lipid and CPR, b5 has been shown to generate superoxide and hydroxyl radicals in the presence of Cr(VI), leading to double-stranded DNA breaks in vitro.(109,110) The fact that CPR was needed to observe the effect suggests the importance of the interaction between b5 and CPR to generate these ROS. Additionally, ROS generated by b5 have been demonstrated to increase lipid peroxidation in reconstituted biological membranes, which may in turn lead to cellular damage and death.111 Therefore, there are multiple mechanisms through which altered b5 levels and/or function can exert a toxicological effect.
3. Cytochrome P450 Interactions with Other CYP Isoforms
Surprisingly, CYPs also exhibit protein–protein interactions with other CYPs. In the early 1990s, research by Coon, Davydov, and others demonstrated that CYPs can form homomeric complexes.112−116 Work in the ensuing 2 decades has established that multiple CYP isoforms can interact with one another in both homomeric and heteromeric complexes and that these complexes can often have significant effects on CYP-mediated oxidation of substrates87,117−122 (for recent reviews, please see references (117) and (118)). For simplicity, we have grouped the CYP–CYP interactions into those described by homomeric (two or more molecules of the same isoform) and heteromeric (two or more molecules of different isoforms).
3.1. Homomeric CYP–CYP Interactions
Pioneering studies of homomeric CYP–CYP interactions were conducted by Davydov in the early to mid-1990s, examining the response of CYP2B4 to pressure induced conformational changes.112−114 Davydov and his colleagues discovered that approximately 35% of the ferrous CYP2B4 CO-bound complex was refractory to conversion into the inactive P420 form upon application of pressures in excess of 2 kbar.113 However, this refractory population was reduced to less than 5% upon addition of 0.2% Triton N-101, a detergent that created conditions known to produce monomers. These results implied that a significant portion of active CYP (i.e., CYP able to be reduced and bind CO) was present in the oligomeric state. Later work by this same group would demonstrate that sodium dithionite or CPR induced reduction kinetics in CYP3A4 are multiphasic under conditions favoring CYP3A4 oligomers but monophasic under conditions favoring monomers.123 These studies are suggestive of active CYP arranged in homomeric oligomers in the phospholipid bilayer.
Confirmation of a direct physical interaction between individual CYPs in a live cell membrane was achieved in seminal studies by Kemper and colleagues using fluorescently labeled CYP2E1 and CYP2C2 in cells transfected with murine CYP cDNA.124 They found that while fluorescence resonance energy transfer (FRET) occurred between individual CYP2C2 molecules in a membrane, it could not be detected between CYP2E1 monomers, representing a homomeric self-association with CYP2C2 but, interestingly, not CYP2E1.124 The CYP2C2 dimers were later confirmed to exist in murine hepatocyte endoplasmic reticulum membranes, indicating the in vivo relevance of such associations.125
Taken together, these studies laid the groundwork for analysis of the functional consequences of CYP–CYP homomeric interactions. In an elegant kinetic study conducted by Jamakhandi et al., the authors found that metabolism data for CYP2E1 fit best to either a quaternary (CYP–CPR–CYP–CPR), trimeric (CYP–CYP–CPR), or dimeric (CYP–CYP) complex model where only the binary (CYP–CPR) complex was active.126 The authors found that simply varying the individual concentrations of CYP or CPR in a reconstitution system produced contradictory results, which did not correspond to the simplest functional (1:1) complex. Simultaneously, Job titration analysis revealed an asymmetric plot, indicative of higher-order molecular complexes. In an attempt to identify the catalytically relevant species, both CYP and CPR concentrations were titrated simultaneously, resulting in a two-dimensional data surface. When the data were globally fit to 32 different candidate models (including binary, ternary, and quaternary CYP–CPR complexes), it revealed that the most likely model was one composed of the binary complex (CYP–CPR), the quaternary complex (CYP–CPR–CYP–CPR), and the homodimer (CYP–CYP). A second, although less likely, possibility was a model that involved a weakly bound ternary complex (CYP-CYP–CPR). Interestingly, the results implied that the binary complex was the only catalytically active state. Therefore, in this case, even though CYP2E1 was shown to be capable of forming CYP–CYP complexes that could have an effect on catalytic activity, the binary CYP–CPR complex was still shown to be the only active protein–protein complex.
More recently, a comprehensive study by the Backes group examining the effect of homomeric CYP complex formation on the activity of CYP1A2, CYP2B4, and CYP2E1 found that, at least in the case of CYP1A2, CYP–CYP homomeric complexes contribute to altered catalytic activity seen with this enzyme under conditions that would promote dimer formation.127 To understand how CYP homomeric complexes might affect function, the catalytic activities of several individual CYPs, including CYP2B4, CYP2E1, and CYP1A2, were assessed in reconstituted systems as a function of CYP concentration. Interestingly, although oxidation mediated by CYP2B4 as a function of CYP–CPR best fit to a simple hyperbolic Michaelis–Menten equation, CYP2E1 and CYP1A2 demonstrated atypical (non-Michaelis–Menten) kinetics that fit best to a sigmoidal curve, indicating a high degree of cooperativity with these particular isoforms. This non-Michaelis–Menten kinetic behavior could be converted back to a hyperbola simply by increasing the ionic strength of the buffer, suggesting that the CYP–CYP interaction may have a strong electrostatic character. The authors further went on to verify the CYP–CYP complex in the membrane using a variety of biophysical techniques, including chemical cross-linking and bioluminescence resonance energy transfer (BRET).127
In yet another study, CYP–CYP interaction was found to affect enzymatic function by a direct modulation of allosteric (non-Michaelis–Menten) enzyme kinetics. Both homotropic and heterotropic cooperativity have long been known to occur with hepatic drug-metabolizing CYP isoforms, particularly CYP3A4.53,54,128 Recently, Davydov and colleagues reported that stimulation of CYP3A4-mediated oxidation of 7-benzyloxy-4-(trifluoromethyl)coumarin (7-BFC) by α-naphthoflavone in human liver microsomes (HLM) was highly dependent on the degree of CYP3A4 oligomerization.129 The amount of enzyme activation seen was directly correlated with the surface density of the enzyme. While no activation was detectable at high lipid/CYP ratios (≥750), activation reached greater than 225% at a lipid/CYP ratio of 140:1. This suggests that allosteric activation requires at least some degree of homomeric CYP–CYP interaction.
Although a preponderance of physical and functional evidence supports homomeric CYP–CYP complexes, at least in the dimeric form, it is still not entirely clear what role these complexes may play in vivo in the detoxification and elimination of xenobiotic agents, especially under conditions of CYP induction or endoplasmic reticulum (ER) stress. One suggested physiological function basis for the formation of CYP–CYP complexes is their ability to prompt a hypothetical “functional allosteric mechanism” that could lead to a rapid response for detoxification of toxic xenobiotics.118,130 Future studies with relevant in vivo model systems may shed some light in this area.
3.2. Heteromeric CYP–CYP Interactions
The identification of the existence of homomeric CYP complexes has led to interest in the possibility of heteromeric CYP complexes as well. Using the same methodology that revealed homomeric interactions between individual CYP2C2 molecules, Kemper and colleagues identified additional heteromeric interactions between CYP2C2 and CYP2E1.131 This implied that differences in homo- and heteromeric oligomerization state between isoforms may help explain some of the relative differences seen in activity. Indeed, this was found to be the case in complexes formed between CYP1A2 and CYP2B4 in reconstituted systems.120,121 In a reconstituted system containing CPR, lipid, and both CYP1A2 and CYP2B4 enzymes, a small increase in catalytic activity for the CYP2B4 substrate benzphetamine was detected. However, activity against another CYP2B4 substrate, 7-pentoxyresorufin-O-dealkylation (PROD), was dramatically inhibited.121 Inhibition of PROD was also dependent on CPR levels, with the inhibitory effect being more pronounced at subsaturating CPR concentrations, suggesting competition for reductase binding between the two isoforms. In a follow-up study, Backes and colleagues determined that it was likely that a ternary complex containing CPR, CYP1A2, and CYP2B4 was formed, for CYP2B4 activity was dramatically reduced at subsaturating reductase concentrations.120 This result suggested an alternative model where, under substoichiometric conditions, CPR is specifically bound to CYP1A2 and at higher CPR concentrations, reductase binding to CYP1A2 becomes saturated, resulting in the formation of a quaternary complex in which CPR binds to both CYP1A2 and CYP2B4 enzymes.120
In a more recent study examining the interaction between CYP2C9 and CYP2C19 in reconstituted systems, Kupfer and colleagues found evidence of a metabolic interaction between even these highly related CYP isoforms.132 Previously, using in vitro systems reconstituted with purified enzyme, CYP2C19 was the most active isoform for methoxychlor-O-demethylation.133,134 However, in HLM, CYP2C9 performed the bulk of methoxychlor-O-demethylation. Interestingly, while an equimolar ratio of CPR to CYP2C9 supported maximal rates of methoxychlor-O-demethylation, the rate of methoxychlor-O-demethylation by CYP2C19 was not fully saturated in the same system; even when reconstituted with a 9-fold molar excess of CPR, CYP2C19 methoxychlor-O-demethylation and S-mephenytoin hydroxylation kinetics were not fully saturated (as compared with that in HLM). However, when a binary reconstitution system was prepared by mixing CYP2C9 and CYP2C19 enzymes, methoxychlor-O-demethylation and S-mephenytoin hydroxylation by CYP2C19 were inhibited dramatically, with the amount of inhibition dependent on the amount of CPR and substrate used, yet increasing concentrations of CPR were generally able to overcome the inhibitory effects. By contrast, in the incubation containing only CYP2C9, diclofenac hydroxylation was activated by the presence of CYP2C19, thus demonstrating that CYP–CYP interactions can modulate the catalytic rates of a variety of oxidation reactions but that this modulation is highly dependent on the substrate undergoing metabolism, with some substrates activating metabolism and others inhibiting it.132,135
Surprisingly, Subramanian and colleagues found that while addition of CYP2D6 to a reconstituted incubation of CYP2C9 and (S)-flurbiprofen increased the Ks by 20-fold, it had no effect on the Km, but it decreased the Vmax by 50%.136 One possible explanation for this discrepancy is that affinity constants determined by heme spin state (i.e., optical spectrum) perturbation may mask total ligand binding because they measure only substrate interactions that perturb the heme iron.137 It is possible that, in the presence of an additional CYP isoform, the spin state shift may be less complete, thereby not reflecting the effects of additional ligand binding. No effects were observed on the metabolism of CYP2D6 substrates. The authors additionally noted that the effects on metabolism were dependent on the order of addition of the enzymatic constituents, with the greatest rate enhancements observed when CPR was added before the second CYP, again suggesting a competition for reductase binding.136 In yet another study, Yamazaki et al. discovered similar types of enzymatic stimulation between CYP1A2 and other isoforms, including CYP3A4, CYP2C9, and CYP2D6, which indicate a more generalizable phenomenon among the hepatic CYPs.87
As more substantial biophysical and functional evidence has accrued demonstrating heteromeric interaction between individual CYP isoforms, attention has shifted to structurally defining the mechanism of this interaction. Two different studies conducted in 2010 by the Backes and Tracy laboratories were instructive in this regard. The Backes group set out to answer the question of whether two different CYP isoforms need to be located in the same membrane to functionally interact, thereby potentially excluding long-range allosteric effects.138 Their experimental system consisted of incorporating CYP1A2 and CYP2B4 into vesicles with CPR individually and together as a CYP–CYP complex. If the two CYPs functionally interacted in the same membrane, then metabolic stimulation (or inhibition) would be observed only when both CYPs were present in the same vesicle, and a mixture of both the individual CYP1A2–CPR and CYP2B4–CPR vesicles would simply produce an additive effect. Indeed, this is exactly what the authors observed prior to confirming this functional interaction between these two CYPs with cross-linking and coimmunoprecipitation experiments.
Work from the Tracy lab examined the effect of the hydrophobic N-terminal anchor sequence on mediating the interaction between CYP2C9 and CYP3A4.139 Using a reconstituted enzyme system containing both CYP2C9 and CYP3A4, the authors demonstrated that CYP2C9 mediated (S)-naproxen metabolism was inhibited as much as 80% by the presence of CYP3A4, while the Km remained unchanged. The degree of inhibition was directly proportional to the CYP3A4 concentration and indirectly proportional to the CPR concentration, suggesting a competition for reductase. Oddly, CYP2C9 did not seem to alter CYP3A4-mediated testosterone metabolism. In order to study the role of the hydrophobic N-terminal anchor sequence, the experiments were performed with both N-terminally truncated and full-length CYP isoforms. There was no evidence for metabolic inhibition when the full-length CYP3A4 and CYP2C9 isoforms were incubated in the presence of the truncated form of the other CYP isoform, but evidence for inhibition was found only when both full length forms were present. This indicated that interaction between the hydrophobic N-termini of the two CYP isoforms was required for functional inhibition. It is possible that, in this context, the hydrophobic N-termini may orient the CYPs in the membrane. Finally, the authors conducted coimmunoprecipitation studies to demonstrate that the two CYPs were directly interacting in the membrane.
It has now been firmly established that CYP’s form both homomeric and heteromeric complexes with functional consequences. The difficult work of defining how these complexes interact structurally and what their in vivo roles may be now lies ahead.
4. Cytochrome P450 Interactions with Other Proteins
Of all the protein–protein interactions in which CYPs have been documented to participate, perhaps the most enigmatic is that with nonenzymatic proteins, including the progesterone receptor membrane component 1 (PGRMC1)140 and bovine and human serum albumin (BSA/HSA).141 Although these proteins do not provide reducing equivalents to the CYP catalytic cycle, they are still capable of modulating CYP-mediated metabolism. Presently, these mechanisms remain to be fully elucidated; intriguingly, they may involve direct delivery of a drug substrate to the particular CYP. Below, we provide a brief summary of what is currently known regarding these CYP protein–protein interactions.
4.1. PGRMC1–CYP Interactions
Despite its rather precise identification, the progesterone receptor membrane component 1 is a somewhat misleading name, as it neither binds progesterone directly nor does it share any homology with progesterone receptors.142,143 In contrast, PGRMC1 shares a high degree of structural homology with cytochrome b5.143 PGRMC1 has been demonstrated to either increase or decrease the rate of CYP-mediated metabolism, depending on the CYP–substrate pair being examined, although perhaps not directly through electron transfer.144 PGRMC1 also seems to play a role in the development of certain cancers, as recent reports suggest that it may be the inscrutable “sigma-2” receptor, a known biomarker of tumor cell proliferation.145,146 In mammals, there exist two additional PGRMC1 family isoforms, PRGMC2/Dg6147 and neudesin,148 although it is not known if they also interact with CYP enzymes. PRGMC1 is highly expressed in the human liver and typically colocalized in the smooth endoplasmic reticulum (SER) with CYP proteins. It has been shown to interact directly with CYP enzymes, including CYP3A4, CYP2C9, CYP7A1, CYP21A2, and CYP51.140 PGRMC1 activates progesterone oxidation mediated by CYP21A2142,149 and is essential for CYP51 activity in the cholesterol synthesis pathway.140 Min and colleagues reported that the coexpression of PGRMC1 with CYP21A1 enhanced progesterone 21-hydroxylation in COS-7 cells, while a heme-deficient PGRMC1 mutant had no effect.142 In an experiment with the yeast homologue, Dap1, Hugues et al. showed that both CYP51A1 and CYP61A1 can be positively regulated by the presence of Dap1.140
Remarkably, PRGMC1 has been revealed to exhibit isoform-selective effects in its interaction with CYPs.150,151 Oda found that PGRMC1 generally reduced CYP activities, either through decreased Vmax and Km values (in the case of CYP3A4) or simply through a decreased Vmax (in the case of CYP2C9); however, in the case of CYP2E1, it had no effect.151 Coimmunoprecipitation studies suggested that the mechanism behind the differential effects may be differences in binding affinity between PGRMC1 and individual CYP isoforms. Szczesna-Skorupa and Kemper found similar results in their studies of the interactions of PGRMC1 with CYP2C2, CYP2C8, and CYP3A4 in transfected HepG2 cells.150 In each case, PGRMC1 downregulated CYP activities, which could be rescued by increased expression of CPR. PGRMC1 was found to have a high affinity for CPR, but only in the absence of CYP2C2, indicating competition between PGRMC1 and CYP2C2 for binding to CPR. In contrast to what was observed with the drug-metabolizing CYP isoforms, CYP51 activity was reduced with decreasing levels of PGRMC1, indicating that a separate mechanism may be in place for the interaction of PGRMC1 with CYPs involved in steroid synthesis.
The mechanism of CYP activation or inhibition by PGRMC1 is highly debatable; however, it is conceivable that it may involve drug delivery or sequestration, as a hydrophobic binding pocket has been identified on PGRMC1.152 In any case, much work remains to elucidate the structural and functional details of the CYP–PGRMC1 interaction.
4.2. Albumin–CYP Interactions
Human serum albumin (HSA) is a 65 kDa monomeric, slightly basic globular protein that circulates as a major component of blood plasma and is the most abundant protein in the human body.141,153−155 HSA functions primarily as a carrier for steroids, fatty acids, and thyroid hormone. Additionally, it plays a role in stabilizing osmotic pressure by regulating extracellular fluid volume. HSA binds a large variety of drugs and xenobiotic agents and has a major impact on their pharmacokinetics and disposition.141 Under normal physiological circumstances, HSA does not cross from the portal circulation into hepatocytes. However, it is synthesized in the liver and is present in the SER of hepatocytes,155−157 so it is conceivable that HSA could come into direct contact with CYP isoforms.
Moreover, the extent of drug or toxin protein binding has long been known to effect in vitro-to-in vivo extrapolations of disposition.141,158 Therefore, in an effort to improve in vitro-to-in vivo predictions, researchers began to add either HSA or bovine serum albumin (BSA, a cheaper and more well-characterized substitute) to both microsomal incubations and recombinant baculovirus-expressed enzymes.159 This accomplished the desired effect of improving pharmacokinetic predictions by increasing the in vitro intrinsic clearance rate, CLint (Vmax/Km) to more closely match what is seen in vivo. This effect is often mediated through a decrease in the substrate Km, and it has been observed for a number of different CYP isoform/drug combinations, including CYP2C8,160 CYP2C9,161,162 and CYP1A2.163 However, for some CYP isoforms, including CYP2C19 and CYP2D6, BSA was found to be inhibitory.164,165 Xu et al. established that the degree of activation or inhibition observed was dependent on the concentration of BSA used, with lower concentrations activating and higher concentrations being inhibitory.164 In addition, BSA’s CYP activation/inhibition profile was demonstrated to deviate from that observed with HSA, indicating the importance of the albumin source for comparison studies and in vitro-to-in vivo extrapolations.164 A number of theories have been proposed to account for the effect seen with albumin, including direct drug delivery, allosterically induced conformational change, and inhibitory long-chain unsaturated fatty acid sequestration. Although no single theory fits the data precisely, recent evidence seems to lend credence to long-chain unsaturated fatty acid sequestration.160,162,166 Of course, because none of these theories are mutually exclusive, it may be that multiple mechanisms are involved rather than a single predominant mechanism with any particular drug/CYP isoform. Lastly, other proteins related to albumin, such as human intestinal fatty acid binding protein (IFABP), have also been demonstrated to improve in vitro-to-in vivo predictions based on microsomal incubations,167 suggesting a more generalizable phenomena with this class of proteins.
In addition to clearly defining the particular mechanisms of albumin–CYP interaction, future work in this area will also need to delineate exactly which members of this protein family interact with individual CYP isoforms. A comprehensive understanding of this protein–protein interaction will no doubt further enhance our ability to correlate in vitro metabolism to that observed in vivo.
5. Conclusions and Future Perspectives
The CYP pathway of oxidative transformation of drugs and other xenobiotics is critical for the elimination of toxicity in the host organism. Proper functioning of this pathway relies on effective formation of protein–protein complexes among the CYPs, their electron transfer partners, and other proteins that can influence their function. Mutations in either the CYP or an interacting protein that effect the structure or stability of the CYP or its protein partner can lead to deficiencies or alternative pathways of biotransformation, which in turn may produce toxic metabolites. Therefore, a comprehensive understanding of the many different types and functions of protein–protein interactions that CYPs undergo is necessary to predict and treat such toxicities. New techniques to understand protein–protein interactions that are beginning to be developed will be a boon for research in this area. Additionally, from a pharmacological point of view, modulating protein–protein interactions may provide an avenue for therapeutic intervention.168 It is likely that additional CYP protein–protein interactions remain to be discovered. Conceivably, CYPs might also interact with phase II, or drug conjugating, enzymes such as glucuronidases, sulfotransferases, or gluthione-S-transferases to modulate their activity as well. These sorts of interactions would be logical from a synergistic point of view.
As our understanding of CYP protein–protein interactions increases, we will not only improve our ability to predict and prevent toxic CYP–drug interactions but also gain a new appreciation for the many unique attributes of this versatile enzyme family.
Acknowledgments
The authors would like to gratefully acknowledge Dr. Kip Conner for his thorough review and insightful critique of the manuscript.
Glossary
Abbreviations
- CYP
cytochrome P450
- CPR
cytochrome P450 reductase
- b5
cytochrome b5
- HLM
human liver microsomes
- 2D NMR
two-dimensional nuclear magnetic resonance
- Pdx
putidaredoxin
- EOMCC
2H-1-benzopyran-3-carbonitrile,7-(ethoxy-methoxy)-2-oxo-(9Cl)
- SLF
separated local field
- 2D PELF
two-dimensional proton evolved local field
- INEPT
insensitive nuclei enhanced by polarization transfer
- DREPT
dipolar enhanced polarization transfer
- BRET
bioluminescence resonance energy transfer
- ER
endoplasmic reticulum
- SER
smooth endoplasmic reticulum
- 7-BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- PROD
7-pentoxyresorufin-O-dealkylation
- PGRMC1
progesterone receptor membrane component 1
- HSA
human serum albumin
- BSA
bovine serum albumin
- IFABP
intestinal fatty acid binding protein
This work was supported in part by grants from the National Center for Research Resources [grant P20-RR021940-07], the National Institute of General Medical Sciences [grant P20-GM103549-07], and the Kansas IDeA Network of Biomedical Research Excellence [grant QH846868-K-INBRE] (J.N.L.) from the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- (2005) Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., Ed.) 3rd ed., Kluwer Academic/Plenum Publishers, New York. [Google Scholar]
- Lu A. Y.; Junk K. W.; Coon M. J. (1969) Resolution of the cytochrome P-450-containing omega-hydroxylation system of liver microsomes into three components. J. Biol. Chem. 244, 3714–3721. [PubMed] [Google Scholar]
- Black S. D.; French J. S.; Williams C. H. Jr.; Coon M. J. (1979) Role of a hydrophobic polypeptide in the N-terminal region of NADPH-cytochrome P-450 reductase in complex formation with P-450LM. Biochem. Biophys. Res. Commun. 91, 1528–1535. [DOI] [PubMed] [Google Scholar]
- Schenkman J. B.; Jansson I. (2003) The many roles of cytochrome b5. Pharmacol. Ther. 97, 139–152. [DOI] [PubMed] [Google Scholar]
- Lu A. Y.; Coon M. J. (1968) Role of hemoprotein P-450 in fatty acid omega-hydroxylation in a soluble enzyme system from liver microsomes. J. Biol. Chem. 243, 1331–1332. [PubMed] [Google Scholar]
- Sugishima M., Sato H., Higashimoto Y., Harada J., Wada K., Fukuyama K. Noguchi M. (2014) Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. U.S.A. 111, 2524–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H.; Shimada T.; Martin M. V.; Guengerich F. P. (2001) Stimulation of cytochrome P450 reactions by apo-cytochrome b5: evidence against transfer of heme from cytochrome P450 3A4 to apo-cytochrome b5 or heme oxygenase. J. Biol. Chem. 276, 30885–30891. [DOI] [PubMed] [Google Scholar]
- Laden B. P.; Tang Y.; Porter T. D. (2000) Cloning, heterologous expression, and enzymological characterization of human squalene monooxygenase. Arch. Biochem. Biophys. 374, 381–388. [DOI] [PubMed] [Google Scholar]
- Laguna J. C.; Nagi M. N.; Cook L.; Cinti D. L. (1989) Action of ebselen on rat hepatic microsomal enzyme-catalyzed fatty acid chain elongation, desaturation, and drug biotransformation. Arch. Biochem. Biophys. 269, 272–283. [DOI] [PubMed] [Google Scholar]
- Wang M.; Roberts D. L.; Paschke R.; Shea T. M.; Masters B. S.; Kim J. J. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova I. F.; Li H.; Zhang H.; Peterson J. A.; Poulos T. L. (1999) Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyanagi T.; Mason H. S. (1973) Some properties of hepatic reduced nicotinamide adenine dinucleotide phosphate-cytochrome c reductase. Biochemistry 12, 2297–2308. [DOI] [PubMed] [Google Scholar]
- Yasukochi Y.; Masters B. S. (1976) Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J. Biol. Chem. 251, 5337–5344. [PubMed] [Google Scholar]
- Strobel H. W.; Nadler S. G.; Nelson D. R. (1989) Cytochrome P-450: cytochrome P-450 reductase interactions. Drug Metab. Rev. 20, 519–533. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A. K.; Hurley J. K.; Tollin G.; Waskell L. (1994) Investigation of the rate limiting step for electron transfer from NADPH:cytochrome P450 reductase to cytochrome b5: a laser flash-photolysis study. Arch. Biochem. Biophys. 310, 318–324. [DOI] [PubMed] [Google Scholar]
- Djordjevic S.; Roberts D. L.; Wang M.; Shea T.; Camitta M. G.; Masters B. S.; Kim J. J. (1995) Crystallization and preliminary X-ray studies of NADPH-cytochrome P450 reductase. Proc. Natl. Acad. Sci. U.S.A 92, 3214–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman L. J.; McLain J.; Masters B. S. (2003) Chimeric enzymes of cytochrome P450 oxidoreductase and neuronal nitric-oxide synthase reductase domain reveal structural and functional differences. J. Biol. Chem. 278, 25700–25707. [DOI] [PubMed] [Google Scholar]
- Hamdane D.; Xia C.; Im S. C.; Zhang H.; Kim J. J.; Waskell L. (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 284, 11374–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J.; Gutierrez A.; Barsukov I. L.; Huang W. C.; Grossmann J. G.; Roberts G. C. (2009) Domain motion in cytochrome P450 reductase: conformational equilibria revealed by NMR and small-angle X-ray scattering. J. Biol. Chem. 284, 36628–36637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner M.; Ellis J.; Huang W. C.; Lloyd Raven E.; Roberts G. C.; Oldham N. J. (2011) Detection of a protein conformational equilibrium by electrospray ionisation-ion mobility-mass spectrometry. Angew. Chem., Int. Ed. 50, 8291–8294. [DOI] [PubMed] [Google Scholar]
- Vincent B., Morellet N., Fatemi F., Aigrain L., Truan G., Guittet E. Lescop E. (2012) The closed and compact domain organization of the 70-kDa human cytochrome P450 reductase in its oxidized state as revealed by NMR. J. Mol. Biol. 420, 296–309. [DOI] [PubMed] [Google Scholar]
- Laursen T., Singha A., Rantzau N., Tutkus M., Borch J., Hedegård P., Stamou D., Møller B. L. Hatzakis N. S. (2014) Single molecule activity measurements of cytochrome P450 oxidoreductase reveal the existence of two discrete functional states. ACS Chem. Biol. 9, 630–634. [DOI] [PubMed] [Google Scholar]
- Sundermann A., Oostenbrink C. (2013) Molecular dynamics simulations give insight into the conformational change, complex formation, and electron transfer pathway for cytochrome P450 reductase. Protein Sci. 22, 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C.; Hamdane D.; Shen A. L.; Choi V.; Kasper C. B.; Pearl N. M.; Zhang H.; Im S. C.; Waskell L.; Kim J. J. (2011) Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 286, 16246–16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q.; Doneanu C. E.; Shaffer S. A.; Adman E. T.; Goodlett D. R.; Nelson S. D. (2006) Identification of the interactions between cytochrome P450 2E1 and cytochrome b5 by mass spectrometry and site-directed mutagenesis. J. Biol. Chem. 281, 20404–20417. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Liu Y.; Yan J.; Cao S.; Bai F.; Yang Y.; Huang S.; Yao L.; Anzai Y.; Kato F.; Podust L. M.; Sherman D. H.; Li S. (2014) New reactions and products resulting from alternative interactions between the P450 enzyme and redox partners. J. Am. Chem. Soc. 136, 3640–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanasami R.; Horowitz P. M.; Masters B. S. (1995) Flavin-binding and protein structural integrity studies on NADPH-cytochrome P450 reductase are consistent with the presence of distinct domains. Arch. Biochem. Biophys. 316, 267–274. [DOI] [PubMed] [Google Scholar]
- Kim J. J.; Roberts D. L.; Djordjevic S.; Wang M.; Shea T. M.; Masters B. S. (1996) Crystallization studies of NADPH-cytochrome P450 reductase. Methods Enzymol. 272, 368–377. [DOI] [PubMed] [Google Scholar]
- Kranendonk M.; Marohnic C. C.; Panda S. P.; Duarte M. P.; Oliveira J. S.; Masters B. S.; Rueff J. (2008) Impairment of human CYP1A2-mediated xenobiotic metabolism by Antley–Bixler syndrome variants of cytochrome P450 oxidoreductase. Arch. Biochem. Biophys. 475, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutinho D.; Marohnic C. C.; Panda S. P.; Rueff J.; Masters B. S.; Kranendonk M. (2012) Altered human CYP3A4 activity caused by Antley–Bixler syndrome-related variants of NADPH-cytochrome P450 oxidoreductase measured in a robust in vitro system. Drug Metab. Dispos. 40, 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marohnic C. C.; Panda S. P.; McCammon K.; Rueff J.; Masters B. S.; Kranendonk M. (2010) Human cytochrome P450 oxidoreductase deficiency caused by the Y181D mutation: molecular consequences and rescue of defect. Drug Metab. Dispos. 38, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluck C. E.; Mullis P. E.; Pandey A. V. (2010) Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochem. Biophys. Res. Commun. 401, 149–153. [DOI] [PubMed] [Google Scholar]
- Pandey A. V.; Fluck C. E. (2013) NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol. Ther. 138, 229–254. [DOI] [PubMed] [Google Scholar]
- Reed J. R.; Cawley G. F.; Backes W. L. (2011) Inhibition of cytochrome P450 1A2-mediated metabolism and production of reactive oxygen species by heme oxygenase-1 in rat liver microsomes. Drug Metab. Lett. 5, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. A.; Ebel R. E.; O’Keeffe D. H.; Matsubara T.; Estabrook R. W. (1976) Temperature dependence of cytochrome P-450 reduction. A model for NADPH-cytochrome P-450 reductase:cytochrome P-450 interaction. J. Biol. Chem. 251, 4010–4016. [PubMed] [Google Scholar]
- Estabrook R. W.; Hildebrandt A. G.; Baron J.; Netter K. J.; Leibman K. (1971) A new spectral intermediate associated with cytochrome P-450 function in liver microsomes. Biochem. Biophys. Res. Commun. 42, 132–139. [DOI] [PubMed] [Google Scholar]
- Bridges A.; Gruenke L.; Chang Y. T.; Vakser I. A.; Loew G.; Waskell L. (1998) Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J. Biol. Chem. 273, 17036–17049. [DOI] [PubMed] [Google Scholar]
- Shen S.; Strobel H. W. (1993) Role of lysine and arginine residues of cytochrome P450 in the interaction between cytochrome P4502B1 and NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 304, 257–265. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Tateishi T.; Hatano M.; Fujii-Kuriyama Y. (1991) Probing the role of lysines and arginines in the catalytic function of cytochrome P450d by site-directed mutagenesis. Interaction with NADPH-cytochrome P450 reductase. J. Biol. Chem. 266, 3372–3375. [PubMed] [Google Scholar]
- Kenaan C.; Zhang H.; Shea E. V.; Hollenberg P. F. (2011) Uncovering the role of hydrophobic residues in cytochrome P450-cytochrome P450 reductase interactions. Biochemistry 50, 3957–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voznesensky A. I.; Schenkman J. B. (1992) The cytochrome P450 2B4-NADPH cytochrome P450 reductase electron transfer complex is not formed by charge-pairing. J. Biol. Chem. 267, 14669–14676. [PubMed] [Google Scholar]
- Shimada T.; Mernaugh R. L.; Guengerich F. P. (2005) Interactions of mammalian cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b(5) enzymes. Arch. Biochem. Biophys. 435, 207–216. [DOI] [PubMed] [Google Scholar]
- French J. S.; Guengerich F. P.; Coon M. J. (1980) Interactions of cytochrome P-450, NADPH-cytochrome P-450 reductase, phospholipid, and substrate in the reconstituted liver microsomal enzyme system. J. Biol. Chem. 255, 4112–4119. [PubMed] [Google Scholar]
- Estrada D. F.; Laurence J. S.; Scott E. E. (2013) Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 288, 17008–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma Y.; Hass M. A.; Kikui Y.; Liu W. M.; Olmez B.; Skinner S. P.; Blok A.; Kloosterman A.; Koteishi H.; Lohr F.; Schwalbe H.; Nojiri M.; Ubbink M. (2013) The structure of the cytochrome p450cam-putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography. J. Mol. Biol. 425, 4353–4365. [DOI] [PubMed] [Google Scholar]
- Pochapsky S. S.; Pochapsky T. C.; Wei J. W. (2003) A model for effector activity in a highly specific biological electron transfer complex: the cytochrome P450(cam)-putidaredoxin couple. Biochemistry 42, 5649–5656. [DOI] [PubMed] [Google Scholar]
- OuYang B.; Pochapsky S. S.; Dang M.; Pochapsky T. C. (2008) A functional proline switch in cytochrome P450cam. Structure 16, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochapsky S. S.; Dang M.; OuYang B.; Simorellis A. K.; Pochapsky T. C. (2009) Redox-dependent dynamics in cytochrome P450cam. Biochemistry 48, 4254–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Agrawal V.; Giacomini K. M.; Miller W. L. (2008) Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc. Natl. Acad. Sci. U.S.A. 105, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Pandey A. V.; Agrawal V.; Reardon W.; Lapunzina P. D.; Mowat D.; Jabs E. W.; Van Vliet G.; Sack J.; Fluck C. E.; Miller W. L. (2005) Diversity and function of mutations in p450 oxidoreductase in patients with Antley–Bixler syndrome and disordered steroidogenesis. Am. J. Hum. Genet. 76, 729–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal V.; Huang N.; Miller W. L. (2008) Pharmacogenetics of P450 oxidoreductase: effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet. Genomics 18, 569–576. [DOI] [PubMed] [Google Scholar]
- Sandee D.; Morrissey K.; Agrawal V.; Tam H. K.; Kramer M. A.; Tracy T. S.; Giacomini K. M.; Miller W. L. (2010) Effects of genetic variants of human P450 oxidoreductase on catalysis by CYP2D6 in vitro. Pharmacogenet. Genomics 20, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov D. R.; Halpert J. R. (2008) Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both?. Expert Opin. Drug Metab. Toxicol. 4, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins W. M. (2006) Current views on the fundamental mechanisms of cytochrome P450 allosterism. Expert Opin. Drug Metab. Toxicol. 2, 573–579. [DOI] [PubMed] [Google Scholar]
- Agrawal V.; Choi J. H.; Giacomini K. M.; Miller W. L. (2010) Substrate-specific modulation of CYP3A4 activity by genetic variants of cytochrome P450 oxidoreductase. Pharmacogenet. Genomics 20, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Pan L. Q.; Naranmandura H.; Zeng S.; Chen S. Q. (2012) Influence of various polymorphic variants of cytochrome P450 oxidoreductase (POR) on drug metabolic activity of CYP3A4 and CYP2B6. PLoS One 7, e38495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S.; Omata Y.; Sakamoto H.; Hara T.; Noguchi M. (2003) Purification and characterization of a soluble form of rat liver NADPH-cytochrome P-450 reductase highly expressed in Escherichia coli. Protein Expression Purif. 29, 1–7. [DOI] [PubMed] [Google Scholar]
- Lamb D. C.; Kim Y.; Yermalitskaya L. V.; Yermalitsky V. N.; Lepesheva G. I.; Kelly S. L.; Waterman M. R.; Podust L. M. (2006) A second FMN binding site in yeast NADPH-cytochrome P450 reductase suggests a mechanism of electron transfer by diflavin reductases. Structure 14, 51–61. [DOI] [PubMed] [Google Scholar]
- Hubbard P. A.; Shen A. L.; Paschke R.; Kasper C. B.; Kim J. J. (2001) NADPH-cytochrome P450 oxidoreductase. Structural basis for hydride and electron transfer. J. Biol. Chem. 276, 29163–29170. [DOI] [PubMed] [Google Scholar]
- Subramanian M.; Agrawal V.; Sandee D.; Tam H. K.; Miller W. L.; Tracy T. S. (2012) Effect of P450 oxidoreductase variants on the metabolism of model substrates mediated by CYP2C9.1, CYP2C9.2, and CYP2C9.3. Pharmacogenet. Genomics 22, 590–597. [DOI] [PubMed] [Google Scholar]
- Han J. F.; Wang S. L.; He X. Y.; Liu C. Y.; Hong J. Y. (2006) Effect of genetic variation on human cytochrome p450 reductase-mediated paraquat cytotoxicity. Toxicol. Sci. 91, 42–48. [DOI] [PubMed] [Google Scholar]
- Wang S. L.; Han J. F.; He X. Y.; Wang X. R.; Hong J. Y. (2007) Genetic variation of human cytochrome p450 reductase as a potential biomarker for mitomycin C-induced cytotoxicity. Drug Metab. Dispos. 35, 176–179. [DOI] [PubMed] [Google Scholar]
- Iyanagi T.; Xia C.; Kim J. J. (2012) NADPH-cytochrome P450 oxidoreductase: prototypic member of the diflavin reductase family. Arch. Biochem. Biophys. 528, 72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussell K. C.; Udasin R. G.; Gray J. P.; Mishin V.; Smith P. J.; Heck D. E.; Laskin J. D. (2011) Redox cycling and increased oxygen utilization contribute to diquat-induced oxidative stress and cytotoxicity in Chinese hamster ovary cells overexpressing NADPH-cytochrome P450 reductase. Free Radical Biol. Med. 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow H. A.; Penketh P. G.; Baumann R. P.; Sartorelli A. C. (2004) Bioactivation and resistance to mitomycin C. Methods Enzymol. 382, 221–233. [DOI] [PubMed] [Google Scholar]
- de Jonge H.; Metalidis C.; Naesens M.; Lambrechts D.; Kuypers D. R. (2011) The P450 oxidoreductase *28 SNP is associated with low initial tacrolimus exposure and increased dose requirements in CYP3A5-expressing renal recipients. Pharmacogenomics 12, 1281–1291. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li L.; Ding X.; Kaminsky L. S. (2011) Identification of cytochrome P450 oxidoreductase gene variants that are significantly associated with the interindividual variations in warfarin maintenance dose. Drug Metab. Dispos. 39, 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im S. C.; Waskell L. (2011) The interaction of microsomal cytochrome P450 2B4 with its redox partners, cytochrome P450 reductase and cytochrome b(5). Arch. Biochem. Biophys. 507, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergeres G.; Waskell L. (1995) Cytochrome b5, its functions, structure and membrane topology. Biochimie 77, 604–620. [DOI] [PubMed] [Google Scholar]
- Sanborn R. C.; Williams C. M. (1950) The cytochrome system in the cecropia silkworm, with special reference to the properties of a new component. J. Gen. Physiol. 33, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegesh E.; Hegesh J.; Kaftory A. (1986) Congenital methemoglobinemia with a deficiency of cytochrome b5. N. Engl. J. Med. 314, 757–761. [DOI] [PubMed] [Google Scholar]
- Kok R. C., Timmerman M. A., Wolffenbuttel K. P., Drop S. L. de Jong F. H. (2010) Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J. Clin. Endocrinol. Metab. 95, 994–999. [DOI] [PubMed] [Google Scholar]
- Idkowiak J.; Randell T.; Dhir V.; Patel P.; Shackleton C. H.; Taylor N. F.; Krone N.; Arlt W. (2012) A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J. Clin. Endocrinol. Metab. 97, E465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt A.; Estabrook R. W. (1971) Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch. Biochem. Biophys. 143, 66–79. [DOI] [PubMed] [Google Scholar]
- Correia M. A.; Mannering G. J. (1973) Reduced diphosphopyridine nucleotide synergism of the reduced triphosphopyridine nucleotide-dependent mixed-function oxidase system of hepatic microsomes. I. Effects of activation and inhibition of the fatty acyl coenzyme A desaturation system. Mol. Pharmacol. 9, 455–469. [PubMed] [Google Scholar]
- Henderson C. J.; McLaughlin L. A.; Wolf C. R. (2013) Evidence that cytochrome b5 and cytochrome b5 reductase can act as sole electron donors to the hepatic cytochrome P450 system. Mol. Pharmacol. 83, 1209–1217. [DOI] [PubMed] [Google Scholar]
- Peng H. M.; Auchus R. J. (2014) Two surfaces of cytochrome b5 with major and minor contributions to CYP3A4-catalyzed steroid and nifedipine oxygenation chemistries. Arch. Biochem. Biophys. 541, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; McLaughlin L. A.; Ronseaux S.; Rosewell I.; Houston J. B.; Henderson C. J.; Wolf C. R. (2008) Defining the in vivo role for cytochrome b5 in cytochrome P450 function through the conditional hepatic deletion of microsomal cytochrome b5. J. Biol. Chem. 283, 31385–31393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. L.; Hollenberg P. F. (2007) The inactivation of cytochrome P450 3A5 by 17α-ethynylestradiol is cytochrome b5-dependent: metabolic activation of the ethynyl moiety leads to the formation of glutathione conjugates, a heme adduct, and covalent binding to the apoprotein. J. Pharmacol. Exp. Ther. 321, 276–287. [DOI] [PubMed] [Google Scholar]
- Reed J. R.; Hollenberg P. F. (2003) Examining the mechanism of stimulation of cytochrome P450 by cytochrome b5: the effect of cytochrome b5 on the interaction between cytochrome P450 2B4 and P450 reductase. J. Inorg. Biochem. 97, 265–275. [DOI] [PubMed] [Google Scholar]
- Gruenke L. D.; Konopka K.; Cadieu M.; Waskell L. (1995) The stoichiometry of the cytochrome P-450-catalyzed metabolism of methoxyflurane and benzphetamine in the presence and absence of cytochrome b5. J. Biol. Chem. 270, 24707–24718. [DOI] [PubMed] [Google Scholar]
- Morgan E. T.; Coon M. J. (1984) Effects of cytochrome b5 on cytochrome P-450-catalyzed reactions. Studies with manganese-substituted cytochrome b5. Drug Metab. Dispos. 12, 358–364. [PubMed] [Google Scholar]
- Yamazaki H.; Nakamura M.; Komatsu T.; Ohyama K.; Hatanaka N.; Asahi S.; Shimada N.; Guengerich F. P.; Shimada T.; Nakajima M.; Yokoi T. (2002) Roles of NADPH-P450 reductase and apo- and holo-cytochrome b5 on xenobiotic oxidations catalyzed by 12 recombinant human cytochrome P450s expressed in membranes of Escherichia coli. Protein Expression Purif. 24, 329–337. [DOI] [PubMed] [Google Scholar]
- Yamazaki H.; Nakano M.; Imai Y.; Ueng Y. F.; Guengerich F. P.; Shimada T. (1996) Roles of cytochrome b5 in the oxidation of testosterone and nifedipine by recombinant cytochrome P450 3A4 and by human liver microsomes. Arch. Biochem. Biophys. 325, 174–182. [DOI] [PubMed] [Google Scholar]
- Soucy P.; Luu-The V. (2002) Assessment of the ability of type 2 cytochrome b5 to modulate 17,20-lyase activity of human P450c17. J. Steroid Biochem. Mol. Biol. 80, 71–75. [DOI] [PubMed] [Google Scholar]
- Yamazaki H.; Johnson W. W.; Ueng Y. F.; Shimada T.; Guengerich F. P. (1996) Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4. Characterization of a reconstituted cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies with apo-cytochrome b5. J. Biol. Chem. 271, 27438–27444. [DOI] [PubMed] [Google Scholar]
- Yamazaki H.; Gillam E. M.; Dong M. S.; Johnson W. W.; Guengerich F. P.; Shimada T. (1997) Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: effects of cytochrome P450-P450 and cytochrome P450-b5 interactions. Arch. Biochem. Biophys. 342, 329–337. [DOI] [PubMed] [Google Scholar]
- Auchus R. J.; Lee T. C.; Miller W. L. (1998) Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165. [DOI] [PubMed] [Google Scholar]
- Lee-Robichaud P.; Akhtar M. E.; Wright J. N.; Sheikh Q. I.; Akhtar M. (2004) The cationic charges on Arg347, Arg358 and Arg449 of human cytochrome P450c17 (CYP17) are essential for the enzyme’s cytochrome b5-dependent acyl-carbon cleavage activities. J. Steroid Biochem. Mol. Biol. 92, 119–130. [DOI] [PubMed] [Google Scholar]
- Bell L. C.; Guengerich F. P. (1997) Oxidation kinetics of ethanol by human cytochrome P450 2E1. Rate-limiting product release accounts for effects of isotopic hydrogen substitution and cytochrome b5 on steady-state kinetics. J. Biol. Chem. 272, 29643–29651. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Davydov D. R.; Halpert J. R. (2005) Role of cytochrome B5 in modulating peroxide-supported cyp3a4 activity: evidence for a conformational transition and cytochrome P450 heterogeneity. Drug Metab. Dispos. 33, 1131–1136. [DOI] [PubMed] [Google Scholar]
- Schenkman J. B.; Voznesensky A. I.; Jansson I. (1994) Influence of ionic strength on the P450 monooxygenase reaction and role of cytochrome b5 in the process. Arch. Biochem. Biophys. 314, 234–241. [DOI] [PubMed] [Google Scholar]
- Perret A.; Pompon D. (1998) Electron shuttle between membrane-bound cytochrome P450 3A4 and b5 rules uncoupling mechanisms. Biochemistry 37, 11412–11424. [DOI] [PubMed] [Google Scholar]
- Ahuja S.; Jahr N.; Im S. C.; Vivekanandan S.; Popovych N.; Le Clair S. V.; Huang R.; Soong R.; Xu J.; Yamamoto K.; Nanga R. P.; Bridges A.; Waskell L.; Ramamoorthy A. (2013) A model of the membrane-bound cytochrome b5-cytochrome P450 complex from NMR and mutagenesis data. J. Biol. Chem. 288, 22080–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Hamdane D.; Im S. C.; Waskell L. (2008) Cytochrome b5 inhibits electron transfer from NADPH-cytochrome P450 reductase to ferric cytochrome P450 2B4. J. Biol. Chem. 283, 5217–5225. [DOI] [PubMed] [Google Scholar]
- Omata Y.; Robinson R. C.; Gelboin H. V.; Pincus M. R.; Friedman F. K. (1994) Specificity of the cytochrome P-450 interaction with cytochrome b5. FEBS Lett. 346, 241–245. [DOI] [PubMed] [Google Scholar]
- Stayton P. S.; Sligar S. G. (1990) The cytochrome P-450cam binding surface as defined by site-directed mutagenesis and electrostatic modeling. Biochemistry 29, 7381–7386. [DOI] [PubMed] [Google Scholar]
- Soong R.; Smith P. E.; Xu J.; Yamamoto K.; Im S. C.; Waskell L.; Ramamoorthy A. (2010) Proton-evolved local-field solid-state NMR studies of cytochrome b5 embedded in bicelles, revealing both structural and dynamical information. J. Am. Chem. Soc. 132, 5779–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. W.; Wang J. (2013) Structure and function of heme proteins in non-native states: a mini-review. J. Inorg. Biochem. 129, 162–171. [DOI] [PubMed] [Google Scholar]
- Xu J.; Soong R.; Im S. C.; Waskell L.; Ramamoorthy A. (2010) INEPT-based separated-local-field NMR spectroscopy: a unique approach to elucidate side-chain dynamics of membrane-associated proteins. J. Am. Chem. Soc. 132, 9944–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri M.; Suhara K.; Shiroo M.; Fujimura Y. (1982) Role of cytochrome b5 in the cytochrome P-450-mediated C21-steroid 17,20-lyase reaction. Biochem. Biophys. Res. Commun. 108, 379–384. [DOI] [PubMed] [Google Scholar]
- Onoda M.; Hall P. F. (1982) Cytochrome b5 stimulates purified testicular microsomal cytochrome P-450 (C21 side-chain cleavage). Biochem. Biophys. Res. Commun. 108, 454–460. [DOI] [PubMed] [Google Scholar]
- Estrada D. F.; Skinner A. L.; Laurence J. S.; Scott E. E. (2014) Human cytochrome P450 17A1 conformational selection: modulation by ligand and cytochrome b5. J. Biol. Chem. 289, 14310–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Durr U. H.; Xu J.; Im S. C.; Waskell L.; Ramamoorthy A. (2013) Dynamic interaction between membrane-bound full-length cytochrome P450 and cytochrome b5 observed by solid-state NMR spectroscopy. Sci. Rep. 3, 2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan S.; Ahuja S.; Im S. C.; Waskell L.; Ramamoorthy A. (2013) 1H, 13C and 15N resonance assignments for the full-length mammalian cytochrome b in a membrane environment. Biomol. NMR Assignments 10.1007/s12104-013-9528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey M. K.; Vivekanandan S.; Ahuja S.; Pichumani K.; Im S. C.; Waskell L.; Ramamoorthy A. (2012) Determination of 15N chemical shift anisotropy from a membrane-bound protein by NMR spectroscopy. J. Phys. Chem. B 116, 7181–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C. J.; McLaughlin L. A.; Finn R. D.; Ronseaux S.; Kapelyukh Y.; Wolf C. R. (2014) A role for cytochrome b5 in the in vivo disposition of anticancer and cytochrome P450 probe drugs in mice. Drug Metab. Dispos. 42, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locuson C. W.; Wienkers L. C.; Jones J. P.; Tracy T. S. (2007) CYP2C9 protein interactions with cytochrome b(5): effects on the coupling of catalysis. Drug Metab. Dispos. 35, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthiry G. R.; Antholine W. E.; Kalyanaraman B.; Myers J. M.; Myers C. R. (2007) Reduction of hexavalent chromium by human cytochrome b5: generation of hydroxyl radical and superoxide. Free Radical Biol. Med. 42, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthiry G. R.; Antholine W. E.; Myers J. M.; Myers C. R. (2008) Addition of DNA to Cr(VI) and cytochrome b5 containing proteoliposomes leads to generation of DNA strand breaks and Cr(III) complexes. Chem. Biodiversity 5, 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev L. F. (2007) The involvement of lipid radical cycles and the adenine nucleotide translocator in neurodegenerative diseases. J. Alzheimer’s Dis. 11, 183–190. [DOI] [PubMed] [Google Scholar]
- Davydov D. R.; Deprez E.; Hoa G. H.; Knyushko T. V.; Kuznetsova G. P.; Koen Y. M.; Archakov A. I. (1995) High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution: evidence for conformational inhomogeneity in the oligomers. Arch. Biochem. Biophys. 320, 330–344. [DOI] [PubMed] [Google Scholar]
- Davydov D. R.; Knyushko T. V.; Hoa G. H. (1992) High pressure induced inactivation of ferrous cytochrome P-450 LM2 (IIB4) CO complex: evidence for the presence of two conformers in the oligomer. Biochem. Biophys. Res. Commun. 188, 216–221. [DOI] [PubMed] [Google Scholar]
- Davydov D. R.; Darovsky B. V.; Dedinsky I. R.; Kanaeva I. P.; Bachmanova G. I.; Blinov V. M.; Archakov A. I. (1992) Cytochrome C (Fe2+) as a competitive inhibitor of NADPH-dependent reduction of cytochrome P450 LM2: locating protein-protein interaction sites in microsomal electron carriers. Arch. Biochem. Biophys. 297, 304–313. [DOI] [PubMed] [Google Scholar]
- Schwarz D.; Pirrwitz J.; Meyer H. W.; Coon M. J.; Ruckpaul K. (1990) Membrane topology of microsomal cytochrome P-450: saturation transfer EPR and freeze-fracture electron microscopy studies. Biochem. Biophys. Res. Commun. 171, 175–181. [DOI] [PubMed] [Google Scholar]
- Myasoedova K. N.; Berndt P. (1990) Cytochrome P-450LM2 oligomers in proteoliposomes. FEBS Lett. 275, 235–238. [DOI] [PubMed] [Google Scholar]
- Reed J. R.; Connick J. P.; Cheng D.; Cawley G. F.; Backes W. L. (2012) Effect of homomeric P450–P450 complexes on P450 function. Biochem. J. 446, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov D. R. (2011) Microsomal monooxygenase as a multienzyme system: the role of P450–P450 interactions. Expert Opin. Drug Metab. Toxicol. 7, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley G. F.; Zhang S.; Kelley R. W.; Backes W. L. (2001) Evidence supporting the interaction of CYP2B4 and CYP1A2 in microsomal preparations. Drug Metab. Dispos. 29, 1529–1534. [PubMed] [Google Scholar]
- Backes W. L.; Batie C. J.; Cawley G. F. (1998) Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4–1A2 complex with a high affinity for NADPH-cytochrome P450 reductase. Biochemistry 37, 12852–12859. [DOI] [PubMed] [Google Scholar]
- Cawley G. F.; Batie C. J.; Backes W. L. (1995) Substrate-dependent competition of different P450 isozymes for limiting NADPH-cytochrome P450 reductase. Biochemistry 34, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Alston K.; Robinson R. C.; Park S. S.; Gelboin H. V.; Friedman F. K. (1991) Interactions among cytochromes P-450 in the endoplasmic reticulum. Detection of chemically cross-linked complexes with monoclonal antibodies. J. Biol. Chem. 266, 735–739. [PubMed] [Google Scholar]
- Davydov D. R.; Fernando H.; Baas B. J.; Sligar S. G.; Halpert J. R. (2005) Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: heterogeneity of the enzyme caused by its oligomerization. Biochemistry 44, 13902–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna-Skorupa E.; Mallah B.; Kemper B. (2003) Fluorescence resonance energy transfer analysis of cytochromes P450 2C2 and 2E1 molecular interactions in living cells. J. Biol. Chem. 278, 31269–31276. [DOI] [PubMed] [Google Scholar]
- Li B.; Yau P.; Kemper B. (2011) Identification of cytochrome P450 2C2 protein complexes in mouse liver. Proteomics 11, 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamakhandi A. P.; Kuzmic P.; Sanders D. E.; Miller G. P. (2007) Global analysis of protein–protein interactions reveals multiple CYP2E1-reductase complexes. Biochemistry 46, 10192–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J. R.; Backes W. L. (2012) Formation of P450. P450 complexes and their effect on P450 function. Pharmacol. Ther. 133, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert E.; Tracy T. S. (2014) Different enzyme kinetic models. Methods Mol. Biol. 1113, 23–35. [DOI] [PubMed] [Google Scholar]
- Davydov D. R.; Davydova N. Y.; Sineva E. V.; Kufareva I.; Halpert J. R. (2013) Pivotal role of P450–P450 interactions in CYP3A4 allostery: the case of alpha-naphthoflavone. Biochem. J. 453, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins W. M.; Lu W. D.; Cook D. L. (2002) Is there a toxicological advantage for non-hyperbolic kinetics in cytochrome P450 catalysis? Functional allostery from “distributive catalysis”. J. Biol. Chem. 277, 33258–33266. [DOI] [PubMed] [Google Scholar]
- Ozalp C.; Szczesna-Skorupa E.; Kemper B. (2005) Bimolecular fluorescence complementation analysis of cytochrome p450 2c2, 2e1, and NADPH-cytochrome p450 reductase molecular interactions in living cells. Drug Metab. Dispos. 33, 1382–1390. [DOI] [PubMed] [Google Scholar]
- Hazai E.; Kupfer D. (2005) Interactions between CYP2C9 and CYP2C19 in reconstituted binary systems influence their catalytic activity: possible rationale for the inability of CYP2C19 to catalyze methoxychlor demethylation in human liver microsomes. Drug Metab. Dispos. 33, 157–164. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Kupfer D. (2002) Metabolism of the endocrine disruptor pesticide-methoxychlor by human P450s: pathways involving a novel catechol metabolite. Drug Metab. Dispos. 30, 1035–1042. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Krausz K.; Gelboin H. V.; Kupfer D. (2004) CYP2C subfamily, primarily CYP2C9, catalyses the enantioselective demethylation of the endocrine disruptor pesticide methoxychlor in human liver microsomes: use of inhibitory monoclonal antibodies in P450 identification. Xenobiotica 34, 117–132. [DOI] [PubMed] [Google Scholar]
- Hazai E.; Bikadi Z.; Simonyi M.; Kupfer D. (2005) Association of cytochrome P450 enzymes is a determining factor in their catalytic activity. J. Comput.-Aided Mol. Des. 19, 271–285. [DOI] [PubMed] [Google Scholar]
- Subramanian M.; Low M.; Locuson C. W.; Tracy T. S. (2009) CYP2D6–CYP2C9 protein–protein interactions and isoform-selective effects on substrate binding and catalysis. Drug Metab. Dispos. 37, 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes W. L.; Sligar S. G.; Schenkman J. B. (1982) Kinetics of hepatic cytochrome P-450 reduction: correlation with spin state of the ferric heme. Biochemistry 21, 1324–1330. [DOI] [PubMed] [Google Scholar]
- Reed J. R.; Eyer M.; Backes W. L. (2010) Functional interactions between cytochromes P450 1A2 and 2B4 require both enzymes to reside in the same phospholipid vesicle: evidence for physical complex formation. J. Biol. Chem. 285, 8942–8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian M.; Tam H.; Zheng H.; Tracy T. S. (2010) CYP2C9–CYP3A4 protein–protein interactions: role of the hydrophobic N terminus. Drug Metab. Dispos. 38, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A. L.; Powell D. W.; Bard M.; Eckstein J.; Barbuch R.; Link A. J.; Espenshade P. J. (2007) Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab. 5, 143–149. [DOI] [PubMed] [Google Scholar]
- Yamasaki K.; Chuang V. T.; Maruyama T.; Otagiri M. (2013) Albumin–-drug interaction and its clinical implication. Biochim. Biophys. Acta 1830, 5435–5443. [DOI] [PubMed] [Google Scholar]
- Min L.; Strushkevich N. V.; Harnastai I. N.; Iwamoto H.; Gilep A. A.; Takemori H.; Usanov S. A.; Nonaka Y.; Hori H.; Vinson G. P.; Okamoto M. (2005) Molecular identification of adrenal inner zone antigen as a heme-binding protein. FEBS J. 272, 5832–5843. [DOI] [PubMed] [Google Scholar]
- Mifsud W.; Bateman A. (2002) Membrane-bound progesterone receptors contain a cytochrome b5-like ligand-binding domain. Genome Biol. 3, research0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso J. J.; Liu X.; Saunders M. M.; Claffey K. P.; Phoenix K. (2008) Regulation of ovarian cancer cell viability and sensitivity to cisplatin by progesterone receptor membrane component-1. J. Clin. Endocrinol. Metab. 93, 1592–1599. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zeng C.; Chu W.; Pan F.; Rothfuss J. M.; Zhang F.; Tu Z.; Zhou D.; Zeng D.; Vangveravong S.; Johnston F.; Spitzer D.; Chang K. C.; Hotchkiss R. S.; Hawkins W. G.; Wheeler K. T.; Mach R. H. (2011) Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat. Commun. 2, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed I. S.; Chamberlain C.; Craven R. J. (2012) S2R(Pgrmc1): the cytochrome-related sigma-2 receptor that regulates lipid and drug metabolism and hormone signaling. Expert Opin. Drug Metab. Toxicol. 8, 361–370. [DOI] [PubMed] [Google Scholar]
- Gerdes D.; Wehling M.; Leube B.; Falkenstein E. (1998) Cloning and tissue expression of two putative steroid membrane receptors. Biol. Chem. 379, 907–911. [DOI] [PubMed] [Google Scholar]
- Kimura I.; Nakayama Y.; Yamauchi H.; Konishi M.; Miyake A.; Mori M.; Ohta M.; Itoh N.; Fujimoto M. (2008) Neurotrophic activity of neudesin, a novel extracellular heme-binding protein, is dependent on the binding of heme to its cytochrome b5-like heme/steroid-binding domain. J. Biol. Chem. 283, 4323–4331. [DOI] [PubMed] [Google Scholar]
- Min L.; Takemori H.; Nonaka Y.; Katoh Y.; Doi J.; Horike N.; Osamu H.; Raza F. S.; Vinson G. P.; Okamoto M. (2004) Characterization of the adrenal-specific antigen IZA (inner zone antigen) and its role in the steroidogenesis. Mol. Cell. Endocrinol. 215, 143–148. [DOI] [PubMed] [Google Scholar]
- Szczesna-Skorupa E.; Kemper B. (2011) Progesterone receptor membrane component 1 inhibits the activity of drug-metabolizing cytochromes P450 and binds to cytochrome P450 reductase. Mol. Pharmacol. 79, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda S.; Nakajima M.; Toyoda Y.; Fukami T.; Yokoi T. (2011) Progesterone receptor membrane component 1 modulates human cytochrome p450 activities in an isoform-dependent manner. Drug Metab. Dispos. 39, 2057–2065. [DOI] [PubMed] [Google Scholar]
- Rohe H. J.; Ahmed I. S.; Twist K. E.; Craven R. J. (2009) PGRMC1 (progesterone receptor membrane component 1): a targetable protein with multiple functions in steroid signaling, P450 activation and drug binding. Pharmacol. Ther. 121, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz F. (2014) A clinical update of using albumin as a drug vehicle—a commentary. J. Controlled Release 190, 331–336. [DOI] [PubMed] [Google Scholar]
- Fanali G.; di Masi A.; Trezza V.; Marino M.; Fasano M.; Ascenzi P. (2012) Human serum albumin: from bench to bedside. Mol. Aspects Med. 33, 209–290. [DOI] [PubMed] [Google Scholar]
- Peters J. T. (1996) All about Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, CA. [Google Scholar]
- Witke W. F.; Gibbs P. E.; Zielinski R.; Yang F.; Bowman B. H.; Dugaiczyk A. (1993) Complete structure of the human Gc gene: differences and similarities between members of the albumin gene family. Genomics 16, 751–754. [DOI] [PubMed] [Google Scholar]
- Bowman B. H. (1993) Hepatic Plasma Proteins: Mechanisms of Function and Regulation, Academic Press, San Diego, CA. [Google Scholar]
- Poulin P. (2013) Prediction of total hepatic clearance by combining metabolism, transport, and permeability data in the in vitro–in vivo extrapolation methods: emphasis on an apparent fraction unbound in liver for drugs. J. Pharm. Sci. 102, 2085–2095. [DOI] [PubMed] [Google Scholar]
- (2004) Applications of Pharmacokinetic Principles in Drug Development (Krishna R., Ed.) Kluwer Academic/Plenum Press, New York. [Google Scholar]
- Wattanachai N.; Polasek T. M.; Heath T. M.; Uchaipichat V.; Tassaneeyakul W.; Miners J. O. (2011) In vitro–in vivo extrapolation of CYP2C8-catalyzed paclitaxel 6α-hydroxylation: effects of albumin on in vitro kinetic parameters and assessment of interindividual variability in predicted clearance. Eur. J. Clin. Pharmacol. 67, 815–824. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Matsumoto S.; Ding L. R.; Fischer N. E.; Inaba T. (2004) The comparative interaction of human and bovine serum albumins with CYP2C9 in human liver microsomes. Life Sci. 75, 2145–2155. [DOI] [PubMed] [Google Scholar]
- Rowland A.; Elliot D. J.; Knights K. M.; Mackenzie P. I.; Miners J. O. (2008) The “albumin effect” and in vitro–in vivo extrapolation: sequestration of long-chain unsaturated fatty acids enhances phenytoin hydroxylation by human liver microsomal and recombinant cytochrome P450 2C9. Drug Metab. Dispos. 36, 870–877. [DOI] [PubMed] [Google Scholar]
- Wattanachai N.; Tassaneeyakul W.; Rowland A.; Elliot D. J.; Bowalgaha K.; Knights K. M.; Miners J. O. (2012) Effect of albumin on human liver microsomal and recombinant CYP1A2 activities: impact on in vitro–in vivo extrapolation of drug clearance. Drug Metab. Dispos. 40, 982–989. [DOI] [PubMed] [Google Scholar]
- Xu B. Q.; Ishii M.; Ding L. R.; Fischer N. E.; Inaba T. (2003) Interaction of serum proteins with CYP isoforms in human liver microsomes: inhibitory effects of human and bovine albumin, alpha-globulins, alpha-1-acid glycoproteins and gamma-globulins on CYP2C19 and CYP2D6. Life Sci. 72, 1953–1962. [DOI] [PubMed] [Google Scholar]
- Ishii M.; Xu B. Q.; Ding L. R.; Fischer N. E.; Inaba T. (2001) Interaction of plasma proteins with cytochromes P450 mediated metabolic reactions: inhibition by human serum albumin and alpha-globulins of the debrisoquine 4-hydroxylation (CYP2D) in liver microsomes of human, hamster and rat. Toxicol. Lett. 119, 219–225. [DOI] [PubMed] [Google Scholar]
- Yamazaki H.; Shimada T. (1999) Effects of arachidonic acid, prostaglandins, retinol, retinoic acid and cholecalciferol on xenobiotic oxidations catalysed by human cytochrome P450 enzymes. Xenobiotica 29, 231–241. [DOI] [PubMed] [Google Scholar]
- Rowland A.; Knights K. M.; Mackenzie P. I.; Miners J. O. (2009) Characterization of the binding of drugs to human intestinal fatty acid binding protein (IFABP): potential role of IFABP as an alternative to albumin for in vitro–in vivo extrapolation of drug kinetic parameters. Drug Metab. Dispos. 37, 1395–1403. [DOI] [PubMed] [Google Scholar]
- Metz A.; Ciglia E.; Gohlke H. (2012) Modulating protein-protein interactions: from structural determinants of binding to druggability prediction to application. Curr. Pharm. Des. 18, 4630–4647. [DOI] [PubMed] [Google Scholar]